

The Tobacco Smoke Component, Acrolein, as a Major Culprit in Lung Diseases and Respiratory Cancers: Molecular Mechanisms of Acrolein Cytotoxic Activity



Abstract
1. Introduction
2. Methodology
3. Cigarette Smoke—Health Effects and Risks
4. Acrolein and Cigarette Smoke and Smoking
5. Acrolein and E-Cigarettes—A Healthier Alternative?
6. Acrolein Is One of the Main Risk Factors for Chronic Obstructive Pulmonary Disease
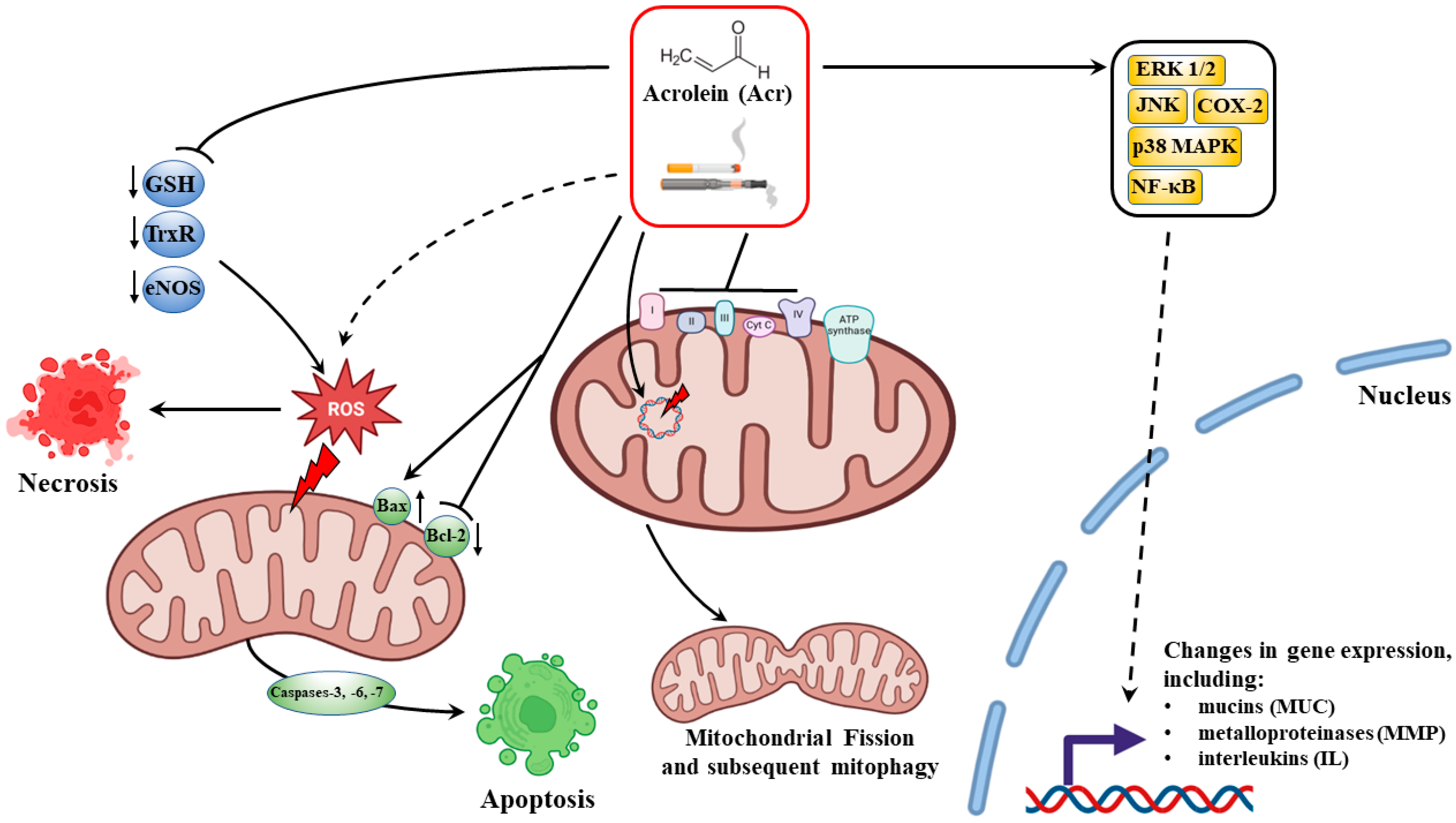
6.1. Acrolein Cytoxicity
6.2. Acrolein and Dysregulation of Mitochondrial Homeostasis
6.3. Acrolein-Dependent Oxidative Stress in Smokers
6.4. Mucus Hypersecretion
6.5. Acrolein-Dependent Tissue Modification and Degradation in Emphysema
6.6. Immunomodulatory Properties of Acrolein

{kind=link}
{kind=link}
| Target | Experimental Approach | Effect | References |
|---|---|---|---|
| Cytotoxic Properties of Acrolein | |||
| In Vitro Studies | |||
| Human umbilical vein endothelial cells (HUVEC) | Acrolein exposure | ↑ ROS, ↑ 8-OHdG—DNA damage, ↑ Bax, ↓ Bcl-2, ↑ caspase-3—apoptosis | [69] |
| Primary nasal epithelial cell cultures (PNECs) from healthy non-smokers | Cigarette smoke extract | ↑ caspase-3—apoptosis, | [72] |
| Bone marrow-derived GM-CSF-dependent immature macrophages (GM-IMs) | Acrolein exposure | ↑ ROS, ↑ caspase-3—apoptosis | [73] |
| Lewis lung carcinoma (LLC) cells | [73] | ||
| Human bronchial epithelial cell line (HBE1) | Acrolein exposure | ↓ GSH, ↑ ROS, phosphatidylserine (PS) externalization and DNA fragmentation—apoptosis | [74] |
| Human bronchial epithelial cell line (BEAS-2B) | Acrolein exposure | ↑ ROS, ↑ 8-OHdG, ↑ γ-H2AX—DNA damage, ↑ Bax, ↓ Bcl-2, ↑ caspase-3—apoptosis, G2/M cell cycle blockade | [75] |
| Murine FL5.12 pro B lymphoid progenitor cells | Acrolein exposure | ↓ Caspase-9, -8 and -3—apoptosis inhibition, oncotic/necrotic response | [81] |
| Human lung adenocarcinoma cells (A549) | Acrolein exposure | ↑ ROS, cytochrome c release, ↑ Bax, ↑ Bad, ↓ Bcl-2, ↑ apoptosis inducing factor AIF, ↑ caspase-9, -7, -6, -3—apoptosis | [82] |
| Human lung adenocarcinoma cells (A549), Human umbilical vein endothelial cells (HUVEC) | Cigarette smoke condensate | ↓ Caspase-9, and -3 —apoptosis inhibition, necrotic response | [83] |
| Human tracheobronchial epithelial cells (hTBE) | Cigarette smoke extract | ↓ Caspase-7, and -3 —apoptosis inhibition, necrotic response | [84] |
| Human alveolar epithelial type I-like (ATI-like) cells | Cigarette smoke extract | ↑ Caspase-7, and -3- apoptosis, necrotic response | [103] |
| In vivo studies | |||
| Lungs isolated from male albino rats | Acrolein inhalation | ↑ Proinflammatory TNF-α, ↑ Il-6 ↑ Bax, ↓ Bcl-2—apoptosis | [70] |
| Dysregulation of mitochondrial homeostasis | |||
| In vitro studies | |||
| Normal human lung fibroblasts (MRC-5), lung adenocarcinoma cells (A549) | Acrolein exposure | ↓ PARP-1, ↑ ROS, ↑ Caspase-9, and -3 —apoptosis, ↓ mitochondrial membrane potential, ↓ ATP, ↑ mtDNA damage, ↑ mitochondrial fission and mitophagy | [85] |
| Lung alveolar cell line RLE-6TN, Clara cell-like human bronchialepithelial cell line (H441), pAT2 cells isolated from male A/J mice (Jackson Laboratories) | Acrolein exposure | ↑ Mitochondrial metabolism of palmitate, ↑ glucose-6-phosphate dehydrogenase activity, ↑ phospholipase A2, ↓ phosphatidylcholine, inhibition of glycolysis | [87] |
| Human lung fibroblast cells (IMR-90) | Acrolein exposure | ↑ β-galactosidase, ↑ p53, ↑ p21, ↓ DNA synthesis, ↓ Sirt1, ↑ ROS, ↓ mitochondrial membrane potential, ↓ mitochondrial biogenesis regulator PGC-1, of subunits of the electron transport chain complexes | [88] |
| In vivo studies | |||
| Lung tissue isolated from male Wistar rats | Acrolein inhalation | ↓ Transcript abundance of subunits of the electron transport chain complexes, ↓ PPARGC1A regulators associated with mitochondrial biogenesis | [77] |
| SM/J (sensitive) and 129 × 1/SvJ (resistant) inbred mouse strains | Acrolein inhalation | ↓ Transcript abundance of subunits of the electron transport chain complexes, ↑ activity of glycolytic enzymes | [86] |
| Redox homeostasis imbalance, carbonylation of antioxidant enzymes | |||
| In vitro studies | |||
| Human gingival fibroblasts cells (HGFs) | Cigarette smoke | ↑ ROS, ↑ protein carbonylation, ↑ GSH-acrolein adducts, ↓ -SH groups | [91] |
| Lung adenocarcinoma cells (A549) | Cigarette smoke extract | ↑ protein carbonylation, ↑ GSH-acrolein adducts, ↓ -SH groups | [92,93] |
| Human ALI airway tissue models | Acrolein exposure | ↑ protein carbonylation, ↑ GSH-acrolein adducts, ↑ HMOX-1 | [14] |
| Normal human primary bronchial epithelial cells (NHBE), human mucoepidermoid pulmonary carcinoma cells (NCI-H292) | Acrolein exposure | ↑ protein carbonylation, ↑ GSH-acrolein adducts | [94] |
| Human bronchial epithelial cell line (BEAS-2B) | Acrolein exposure | ↑ Trx1 and Trx2 oxidation, ↑ peroxidases Prx1 and Prx3 oxidation, ↓ TrxR, | [95] |
| Human bronchial epithelial cell line (HBE1) | Acrolein exposure | ↑ protein carbonylation, ↑ GSH-acrolein adducts, ↑ Trx1 oxidation, ↑ Prx1 oxidation, ↑ GSH-acrolein adducts | [97] |
| Human umbilical vein endothelial EA.hy926 cells | Cigarette smoke extract | ↓ eNOS, ↓ NO | [98] |
| Murine bone marrow-derived macrophage cells, Lewis lung carcinoma cells (LLC) | Acrolein exposure | ↑ ROS, ↑ GADD34 | [99] |
| Bovine aortic endothelial cells (BAECs) | Acrolein exposure | ↓ GSH, ↑ ROS, ↑ Heme oxygenase-1 (HO-1), ↑ Nrf2 signaling pathway | [100] |
| Lung adenocarcinoma cells (A549) | Acrolein exposure | ↓ GSH, ↑ Nrf2 signaling pathway | [101] |
| Human alveolar epithelial type I-like (ATI-like) cells | Cigarette smoke extract | ↑ HO-1, ↑ Nrf2 signaling pathway, ↑ lipid peroxidation | [103] |
| In vivo studies | |||
| Female wild-type C57BL/6 mice | Acrolein inhalation | ↑ ROS, ↑ GADD34 | [99] |
| Mucus hypersecretion and tissue degradation in emphysema | |||
| In vitro studies | |||
| Human pulmonary mucoepidermoid carcinoma cell line NCI-H292 | Cigarette smoke extract | ↑ MUC7, ↑ MUC5AC | [112,113] |
| Human pulmonary mucoepidermoid cells NCI-H292 | Acrolein exposure | ↑ MUC5AC, ↑ MMP-9, ↑ MMP-14 | [123,124] |
| Human ALI airway tissue models | Acrolein exposure | ↑ MUC5AC, ↑ MMP-7, -10, -12, and -13 | [14] |
| Human aortic vascular smooth muscle cells | Cigarette smoke extract | ↑ MMP-1 | [126] |
| Primary human lung micro-vascular endothelial cells, fusion of human umbilical vascular endothelial cells with the lung carcinoma cell line A549 | Acrolein exposure | ↑ CLDN5 | [130] |
| The murine macrophage cell line, J774A.1 | Acrolein exposure | ↑ MMP-9, ↑ 5-lipoxygenase | [135] |
| In vivo studies | |||
| MUC7 transgenic mouse tissues | Cigarette smoke extract | ↑ MUC7 | [112] |
| Female Han Wistar rats | Cigarette smoke extract | ↑ MUC5AC | [113] |
| Gene-targeted Mmp9(-/-) mice, Mmp9(+/+) mice, | Acrolein exposure | ↑ MUC5AC, ↑ MMP-9 | [117] |
| Specific pathogen-free grade male C57BL/6 Mice | Airway remodeling model—acrolein inhalation | ↑ MUC5AC | [118] |
| Human bronchial epithelial cells and induced sputum from e-cigarette users | E-cigarette exposure | ↑ MUC4, ↑ MUC5AC, ↑ MMP-8, ↑ MMP-9 | [119,120] |
| A/J mice (Jackson Laboratories) | E-cigarette exposure | ↑ MUC5AC | [122] |
| Pathogen-free male Kunming mice | Acrolein intraperitoneal injection | ↑ MUC5AC, ↑ MMP-9 | [125] |
| Inbred mouse strains | Acrolein exposure | ↑ CLDN5, acute lung injury (ALI) | [130] |
| C57BL/6J mice | Cigarette smoke extract | ↓ Elastin, ↑ collagen | [132] |
| Airway smooth muscle from smokers | Cigarette smoke extract | ↑ MMP-1, -3 and -10, ↑ collagen VIII alpha 1 (COL8A1) | [136] |
| Sputum from smokers | Cigarette smoke extract | ↑ MMP-12 | [137] |
| Bronchoscopies on cigarette smokers, and e-cigarette users | Cigarette smoke extract, e-cigarette exposure | ↑ MMP-2, ↑ MMP-9 | [139] |
| Immunomodulatory properties | |||
| In vitro studies | |||
| Human ALI airway tissue models | Acrolein exposure | ↑ IL-1b, ↑ IL-6, ↑ IL-8, ↑ TNF-α, ↑ IFN-γ | [14] |
| Primary nasal epithelial cell cultures (PNECs) from healthy non-smokers | Cigarette smoke extract (CSE) containing acrolein | ↑ IL-8 | [72] |
| Bone marrow-derived GM-CSF-dependent immature macrophages (GM-IMs), Lewis lung carcinoma (LLC) cells | Acrolein exposure | ↑ NF-κB, ↑ TNF-α, ↑ IL-6, ↑ IL-12, ↑ CD11c+ F4/80 high macrophages | [73] |
| Human small airway epithelial cells (SAECs), normal human bronchial epithelial cells (NHBEs), normal human lung fibroblasts (NHLF) | Acrolein exposure | Influence on the p38MAPK/MK2 and ERK 1/2 signaling pathway, ↑ IL-8 | [76,150] |
| Rat lung epithelial cells (LE) | Acrolein exposure | Influence on the Raf-1/ERK signaling pathway, ↑ NF-κB, ↑ COX-2 | [151] |
| In vivo studies | |||
| Lungs isolated from male albino rats | Acrolein inhalation | ↑ TNF-α, ↑ Il-6 | [70] |
| Male C57BL/6J mice, peripheral blood was drawn from healthy, non-smoking adult volunteers | Acrolein inhalation and exposure | ↓ NF-κB, ↓ IL-2, ↓ IL-10, ↓ TNF-α, ↓ INF-γ | [145,148,149] |
| Female C57BL/6 mice | Cigarette smoke extract | ↓ SP-A, ↓ SP-D | [152,153] |
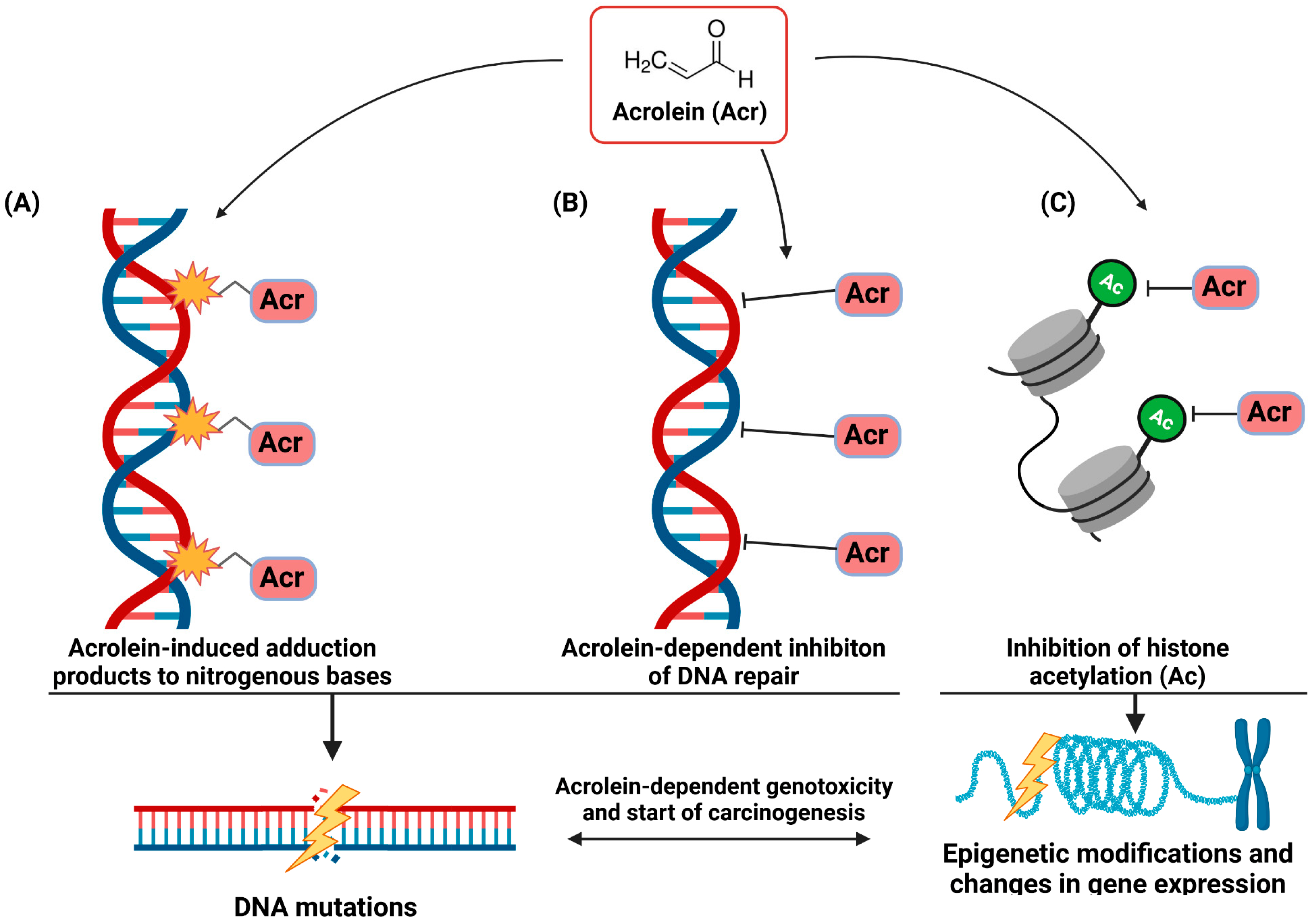
7. Acrolein in the Development of Lung and Oral Cancer
8. Goals for the Future
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/tobacco (accessed on 15 May 2021).
- Li, Y.; Hecht, S.S. Carcinogenic components of tobacco and tobacco smoke: A 2022 update. Food Chem. Toxicol. 2022, 165, 113179. [Google Scholar] [CrossRef] [PubMed]
- Omolaoye, T.S.; El Shahawy, O.; Skosana, B.T.; Boillat, T.; Loney, T.; Du Plessis, S.S. The mutagenic effect of tobacco smoke on male fertility. Environ. Sci. Pollut. Res. Int. 2022, 29, 62055–62066. [Google Scholar] [CrossRef] [PubMed]
- Rodgman, A.; Perfetti, T.A. The Chemical Components of Tobacco and Tobacco Smoke, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 9781466515482. [Google Scholar]
- Roberts, D.L. Nature tobacco flavor. Recent Adv. Tobacco Sci. 1988, 14, 49–81. [Google Scholar]
- Hoffmann, D.; Hoffmann, I. The Changing Cigarette: Chemical. In Risks Associated with Smoking Cigarettes with Low Machine-Measured Yields of Tar and Nicotine; National Institutes of Health (Autor), National Cancer Institute (Autor), U S Department of Health Human: Bethesda, Maryland, USA, 2001. [Google Scholar]
- Jassem, J. Tobacco smoking after diagnosis of cancer: Clinical aspects. Transl. Lung Cancer Res. 2019, 8, S50–S58. [Google Scholar] [CrossRef]
- Talhout, R.; Schulz, T.; Florek, E.; van Benthem, J.; Wester, P.; Opperhuizen, A. Hazardous compounds in tobacco smoke. Int. J. Environ. Res. Public Health 2011, 8, 613–628. [Google Scholar] [CrossRef]
- Pennings, J.L.A.; Cremers, J.W.J.M.; Becker, M.J.A.; Klerx, W.N.M.; Talhout, R. Aldehyde and Volatile Organic Compound Yields in Commercial Cigarette Mainstream Smoke Are Mutually Related and Depend on the Sugar and Humectant Content in Tobacco. Nicotine Tob. Res. 2020, 22, 1748–1756. [Google Scholar] [CrossRef]
- Talhout, R.; Opperhuizen, A.; van Amsterdam, J.G.C. Sugars as tobacco ingredient: Effects on mainstream smoke composition. Food Chem. Toxicol. 2006, 44, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Stevens, J.F.; Maier, C.S. Acrolein: Sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol. Nutr. Food Res. 2008, 52, 7–25. [Google Scholar] [CrossRef] [PubMed]
- Aizenbud, D.; Aizenbud, I.; Reznick, A.Z.; Avezov, K. Acrolein-an α,β-Unsaturated Aldehyde: A Review of Oral Cavity Exposure and Oral Pathology Effects. Rambam Maimonides Med. J. 2016, 7, e0024. [Google Scholar] [CrossRef]
- Moretto, N.; Volpi, G.; Pastore, F.; Facchinetti, F. Acrolein effects in pulmonary cells: Relevance to chronic obstructive pulmonary disease. Ann. N. Y. Acad. Sci. 2012, 1259, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Xiong, R.; Wu, Q.; Muskhelishvili, L.; Davis, K.; Shemansky, J.M.; Bryant, M.; Rosenfeldt, H.; Healy, S.M.; Cao, X. Evaluating Mode of Action of Acrolein Toxicity in an In Vitro Human Airway Tissue Model. Toxicol. Sci. 2018, 166, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Yeager, R.P.; Kushman, M.; Chemerynski, S.; Weil, R.; Fu, X.; White, M.; Callahan-Lyon, P.; Rosenfeldt, H. Proposed Mode of Action for Acrolein Respiratory Toxicity Associated with Inhaled Tobacco Smoke. Toxicol. Sci. 2016, 151, 347–364. [Google Scholar] [CrossRef]
- Liu, Q.; Lou, H.; Zhang, X.; Yang, Q. Association between acrolein exposure and respiratory hazards: A systematic review and meta-analysis. Atmos. Pollut. Res. 2023, 14, 101633. [Google Scholar] [CrossRef]
- Tang, M.; Lee, H.-W.; Weng, M.; Wang, H.-T.; Hu, Y.; Chen, L.-C.; Park, S.-H.; Chan, H.-W.; Xu, J.; Wu, X.-R.; et al. DNA damage, DNA repair and carcinogenicity: Tobacco smoke versus electronic cigarette aerosol. Mutat. Res. Rev. Mutat. Res. 2022, 789, 108409. [Google Scholar] [CrossRef]
- Nardone, N.; Jain, S.; Addo, N.; St Helen, G.; Jacob, P.; Benowitz, N.L. Sources and Biomarkers of Secondhand Tobacco Smoke Exposure in Urban Adolescents. Acad. Pediatr. 2020, 20, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Mallock, N.; Pieper, E.; Hutzler, C.; Henkler-Stephani, F.; Luch, A. Heated Tobacco Products: A Review of Current Knowledge and Initial Assessments. Front. Public Health 2019, 7, 287. [Google Scholar] [CrossRef] [PubMed]
- Znyk, M.; Jurewicz, J.; Kaleta, D. Exposure to Heated Tobacco Products and Adverse Health Effects, a Systematic Review. Int. J. Environ. Res. Public Health 2021, 18, 6651. [Google Scholar] [CrossRef] [PubMed]
- Hackshaw, A.; Morris, J.K.; Boniface, S.; Tang, J.-L.; Milenković, D. Low cigarette consumption and risk of coronary heart disease and stroke: Meta-analysis of 141 cohort studies in 55 study reports. BMJ 2018, 360, j5855. [Google Scholar] [CrossRef]
- Aredo, J.V.; Luo, S.J.; Gardner, R.M.; Sanyal, N.; Choi, E.; Hickey, T.P.; Riley, T.L.; Huang, W.-Y.; Kurian, A.W.; Leung, A.N.; et al. Tobacco Smoking and Risk of Second Primary Lung Cancer. J. Thorac. Oncol. 2021, 16, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Santucci, C.; Bosetti, C.; Peveri, G.; Liu, X.; Bagnardi, V.; Specchia, C.; Gallus, S.; Lugo, A. Dose-risk relationships between cigarette smoking and ovarian cancer histotypes: A comprehensive meta-analysis. Cancer Causes Control 2019, 30, 1023–1032. [Google Scholar] [CrossRef] [PubMed]
- Cumberbatch, M.G.; Rota, M.; Catto, J.W.F.; La Vecchia, C. The Role of Tobacco Smoke in Bladder and Kidney Carcinogenesis: A Comparison of Exposures and Meta-analysis of Incidence and Mortality Risks. Eur. Urol. 2016, 70, 458–466. [Google Scholar] [CrossRef]
- Islami, F.; Moreira, D.M.; Boffetta, P.; Freedland, S.J. A systematic review and meta-analysis of tobacco use and prostate cancer mortality and incidence in prospective cohort studies. Eur. Urol. 2014, 66, 1054–1064. [Google Scholar] [CrossRef] [PubMed]
- Lugo, A.; Peveri, G.; Bosetti, C.; Bagnardi, V.; Crippa, A.; Orsini, N.; Rota, M.; Gallus, S. Strong excess risk of pancreatic cancer for low frequency and duration of cigarette smoking: A comprehensive review and meta-analysis. Eur. J. Cancer 2018, 104, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Pizzol, D.; Foresta, C.; Garolla, A.; Demurtas, J.; Trott, M.; Bertoldo, A.; Smith, L. Pollutants and sperm quality: A systematic review and meta-analysis. Environ. Sci. Pollut. Res. Int. 2021, 28, 4095–4103. [Google Scholar] [CrossRef]
- Kassem, N.O.F.; Kassem, N.O.; Liles, S.; Zarth, A.T.; Jackson, S.R.; Daffa, R.M.; Chatfield, D.A.; Carmella, S.G.; Hecht, S.S.; Hovell, M.F. Acrolein Exposure in Hookah Smokers and Non-Smokers Exposed to Hookah Tobacco Secondhand Smoke: Implications for Regulating Hookah Tobacco Products. Nicotine Tob. Res. 2018, 20, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Carmella, S.G.; Chen, M.; Zhang, Y.; Zhang, S.; Hatsukami, D.K.; Hecht, S.S. Quantitation of acrolein-derived (3-hydroxypropyl)mercapturic acid in human urine by liquid chromatography-atmospheric pressure chemical ionization tandem mass spectrometry: Effects of cigarette smoking. Chem. Res. Toxicol. 2007, 20, 986–990. [Google Scholar] [CrossRef] [PubMed]
- Carmines, E.L.; Gaworski, C.L. Toxicological evaluation of glycerin as a cigarette ingredient. Food Chem. Toxicol. 2005, 43, 1521–1539. [Google Scholar] [CrossRef] [PubMed]
- Roemer, E.; Schorp, M.K.; Piadé, J.-J.; Seeman, J.I.; Leyden, D.E.; Haussmann, H.-J. Scientific assessment of the use of sugars as cigarette tobacco ingredients: A review of published and other publicly available studies. Crit. Rev. Toxicol. 2012, 42, 244–278. [Google Scholar] [CrossRef]
- Ding, Y.S.; Richter, P.; Hearn, B.; Zhang, L.; Bravo, R.; Yan, X.; Perez, J.J.; Chan, M.; Hughes, J.; Chen, P.; et al. Chemical Characterization of Mainstream Smoke from SPECTRUM Variable Nicotine Research Cigarettes. Tob. Regul. Sci. 2017, 3, 81–94. [Google Scholar] [CrossRef]
- Jain, V.; Alcheva, A.; Huang, D.; Caruso, R.; Jain, A.; Lay, M.; O’Connor, R.; Stepanov, I. Comprehensive Chemical Characterization of Natural American Spirit Cigarettes. Tob. Regul. Sci. 2019, 5, 381–399. [Google Scholar] [CrossRef]
- International Agency for Research on Cancer. Acrolein, Crotonaldehyde, and Arecoline: IARC Monographs on the Identification of Carcinogenic Hazards to Humans; International Agency for Research on Cancer: Lyon, France, 2021; Volume 128, ISBN 978-92-832-0168-7. [Google Scholar]
- Bein, K.; Leikauf, G.D. Acrolein—A pulmonary hazard. Mol. Nutr. Food Res. 2011, 55, 1342–1360. [Google Scholar] [CrossRef]
- Faroon, O.; Roney, N.; Taylor, J.; Ashizawa, A.; Lumpkin, M.H.; Plewak, D.J. Acrolein environmental levels and potential for human exposure. Toxicol. Ind. Health 2008, 24, 543–564. [Google Scholar] [CrossRef] [PubMed]
- Faroon, O.; Roney, N.; Taylor, J.; Ashizawa, A.; Lumpkin, M.H.; Plewak, D.J. Acrolein health effects. Toxicol. Ind. Health 2008, 24, 447–490. [Google Scholar] [CrossRef] [PubMed]
- Jerzyński, T.; Stimson, G.V.; Shapiro, H.; Król, G. Estimation of the global number of e-cigarette users in 2020. Harm Reduct. J. 2021, 18, 109. [Google Scholar] [CrossRef]
- Bandi, P.; Cahn, Z.; Goding Sauer, A.; Douglas, C.E.; Drope, J.; Jemal, A.; Fedewa, S.A. Trends in E-Cigarette Use by Age Group and Combustible Cigarette Smoking Histories, U.S. Adults, 2014–2018. Am. J. Prev. Med. 2021, 60, 151–158. [Google Scholar] [CrossRef] [PubMed]
- Park-Lee, E.; Ren, C.; Sawdey, M.D.; Gentzke, A.S.; Cornelius, M.; Jamal, A.; Cullen, K.A. Notes from the Field: E-Cigarette Use Among Middle and High School Students—National Youth Tobacco Survey, United States, 2021. MMWR Morb. Mortal. Wkly. Rep. 2021, 70, 1387–1389. [Google Scholar] [CrossRef]
- Lorkiewicz, P.; Keith, R.; Lynch, J.; Jin, L.; Theis, W.; Krivokhizhina, T.; Riggs, D.; Bhatnagar, A.; Srivastava, S.; Conklin, D.J. Electronic Cigarette Solvents, JUUL E-Liquids, and Biomarkers of Exposure: In Vivo Evidence for Acrolein and Glycidol in E-Cig-Derived Aerosols. Chem. Res. Toxicol. 2022, 35, 283–292. [Google Scholar] [CrossRef] [PubMed]
- de Medeiros, K.S.; Pacheco, B.F.P.; de Oliveira, P.E.; de Góis Nogueira, I.L.; Beserra Diógenes, V.R.; Fernandes, F.G.; Fernandes, G.C.; de Moura Santos, E.; do Rêgo, A.C.M.; Araújo-Filho, I. Impact of e-cigarettes as cancer risk: A protocol for systematic review and meta-analysis. Medicine 2023, 102, e32233. [Google Scholar] [CrossRef]
- Chen, M.; Carmella, S.G.; Lindgren, B.R.; Luo, X.; Ikuemonisan, J.; Niesen, B.; Thomson, N.M.; Murphy, S.E.; Hatsukami, D.K.; Hecht, S.S. Increased Levels of the Acrolein Metabolite 3-Hydroxypropyl Mercapturic Acid in the Urine of e-Cigarette Users. Chem. Res. Toxicol. 2022. [Google Scholar] [CrossRef]
- Cheng, G.; Guo, J.; Carmella, S.G.; Lindgren, B.; Ikuemonisan, J.; Niesen, B.; Jensen, J.; Hatsukami, D.K.; Balbo, S.; Hecht, S.S. Increased acrolein-DNA adducts in buccal brushings of e-cigarette users. Carcinogenesis 2022, 43, 437–444. [Google Scholar] [CrossRef]
- Guo, J.; Hecht, S.S. DNA damage in human oral cells induced by use of e-cigarettes. Drug Test. Anal. 2022. [Google Scholar] [CrossRef] [PubMed]
- Jaccard, G.; Djoko, D.T.; Korneliou, A.; Stabbert, R.; Belushkin, M.; Esposito, M. Mainstream smoke constituents and in vitro toxicity comparative analysis of 3R4F and 1R6F reference cigarettes. Toxicol. Rep. 2019, 6, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Papoušek, R.; Pataj, Z.; Nováková, P.; Lemr, K.; Barták, P. Determination of Acrylamide and Acrolein in Smoke from Tobacco and E-Cigarettes. Chromatographia 2014, 77, 1145–1151. [Google Scholar] [CrossRef]
- Conklin, D.J.; Ogunwale, M.A.; Chen, Y.; Theis, W.S.; Nantz, M.H.; Fu, X.-A.; Chen, L.-C.; Riggs, D.W.; Lorkiewicz, P.; Bhatnagar, A.; et al. Electronic cigarette-generated aldehydes: The contribution of e-liquid components to their formation and the use of urinary aldehyde metabolites as biomarkers of exposure. Aerosol Sci. Technol. 2018, 52, 1219–1232. [Google Scholar] [CrossRef]
- Fagan, P.; Pokhrel, P.; Herzog, T.A.; Moolchan, E.T.; Cassel, K.D.; Franke, A.A.; Li, X.; Pagano, I.; Trinidad, D.R.; Sakuma, K.-L.K.; et al. Sugar and Aldehyde Content in Flavored Electronic Cigarette Liquids. Nicotine Tob. Res. 2018, 20, 985–992. [Google Scholar] [CrossRef]
- Ogunwale, M.A.; Li, M.; Ramakrishnam Raju, M.V.; Chen, Y.; Nantz, M.H.; Conklin, D.J.; Fu, X.-A. Aldehyde Detection in Electronic Cigarette Aerosols. ACS Omega 2017, 2, 1207–1214. [Google Scholar] [CrossRef]
- Li, Y.; Burns, A.E.; Tran, L.N.; Abellar, K.A.; Poindexter, M.; Li, X.; Madl, A.K.; Pinkerton, K.E.; Nguyen, T.B. Impact of e-Liquid Composition, Coil Temperature, and Puff Topography on the Aerosol Chemistry of Electronic Cigarettes. Chem. Res. Toxicol. 2021, 34, 1640–1654. [Google Scholar] [CrossRef] [PubMed]
- Vreeke, S.; Korzun, T.; Luo, W.; Jensen, R.P.; Peyton, D.H.; Strongin, R.M. Dihydroxyacetone levels in electronic cigarettes: Wick temperature and toxin formation. Aerosol Sci. Technol. 2018, 52, 370–376. [Google Scholar] [CrossRef]
- Samburova, V.; Bhattarai, C.; Strickland, M.; Darrow, L.; Angermann, J.; Son, Y.; Khlystov, A. Aldehydes in Exhaled Breath during E-Cigarette Vaping: Pilot Study Results. Toxics 2018, 6, 46. [Google Scholar] [CrossRef]
- McRobbie, H.; Phillips, A.; Goniewicz, M.L.; Smith, K.M.; Knight-West, O.; Przulj, D.; Hajek, P. Effects of Switching to Electronic Cigarettes with and without Concurrent Smoking on Exposure to Nicotine, Carbon Monoxide, and Acrolein. Cancer Prev. Res. 2015, 8, 873–878. [Google Scholar] [CrossRef]
- Davis, L.C.; Sapey, E.; Thickett, D.R.; Scott, A. Predicting the pulmonary effects of long-term e-cigarette use: Are the clouds clearing? Eur. Respir. Rev. 2022, 31, 210121. [Google Scholar] [CrossRef] [PubMed]
- Quaderi, S.A.; Hurst, J.R. The unmet global burden of COPD. Glob. Health Epidemiol. Genom. 2018, 3, e4. [Google Scholar] [CrossRef]
- Kaur, M.; Chandel, J.; Malik, J.; Naura, A.S. Particulate matter in COPD pathogenesis: An overview. Inflamm. Res. 2022, 71, 797–815. [Google Scholar] [CrossRef] [PubMed]
- Adeloye, D.; Song, P.; Zhu, Y.; Campbell, H.; Sheikh, A.; Rudan, I. Global, regional, and national prevalence of, and risk factors for, chronic obstructive pulmonary disease (COPD) in 2019: A systematic review and modelling analysis. Lancet Respir. Med. 2022, 10, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Celli, B.R.; Wedzicha, J.A. Update on Clinical Aspects of Chronic Obstructive Pulmonary Disease. N. Engl. J. Med. 2019, 381, 1257–1266. [Google Scholar] [CrossRef]
- Stolz, D.; Mkorombindo, T.; Schumann, D.M.; Agusti, A.; Ash, S.Y.; Bafadhel, M.; Bai, C.; Chalmers, J.D.; Criner, G.J.; Dharmage, S.C.; et al. Towards the elimination of chronic obstructive pulmonary disease: A Lancet Commission. Lancet 2022, 400, 921–972. [Google Scholar] [CrossRef]
- Burcham, P.C. Acrolein and Human Disease: Untangling the Knotty Exposure Scenarios Accompanying Several Diverse Disorders. Chem. Res. Toxicol. 2017, 30, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Bhatnagar, A.; Pierce, W.M. Protein modification by acrolein: Formation and stability of cysteine adducts. Chem. Res. Toxicol. 2009, 22, 708–716. [Google Scholar] [CrossRef] [PubMed]
- Kaminskas, L.M.; Pyke, S.M.; Burcham, P.C. Michael addition of acrolein to lysinyl and N-terminal residues of a model peptide: Targets for cytoprotective hydrazino drugs. Rapid Commun. Mass Spectrom. 2007, 21, 1155–1164. [Google Scholar] [CrossRef]
- Ou, J.; Zheng, J.; Huang, J.; Ho, C.-T.; Ou, S. Interaction of Acrylamide, Acrolein, and 5-Hydroxymethylfurfural with Amino Acids and DNA. J. Agric. Food Chem. 2020, 68, 5039–5048. [Google Scholar] [CrossRef]
- Masaki, H.; Sinomiya, D.; Okano, Y.; Yoshida, M.; Iwabuchi, T. Impact of protein carbonylation on the chemical characteristics of the hair surface. Int. J. Cosmet. Sci. 2021, 43, 764–771. [Google Scholar] [CrossRef] [PubMed]
- Avezov, K.; Reznick, A.Z.; Aizenbud, D. Time and dose effects of cigarette smoke and acrolein on protein carbonyl formation in HaCaT keratinocytes. Adv. Exp. Med. Biol. 2015, 849, 57–64. [Google Scholar] [CrossRef]
- Chen, H.-J.C.; Cheng, S.-W.; Chen, N.-Y.; Wu, D.-C. Characterization and Quantification of Acrolein-Induced Modifications in Hemoglobin by Mass Spectrometry—Effect of Cigarette Smoking. Chem. Res. Toxicol. 2022, 35, 2260–2270. [Google Scholar] [CrossRef]
- Moghe, A.; Ghare, S.; Lamoreau, B.; Mohammad, M.; Barve, S.; McClain, C.; Joshi-Barve, S. Molecular mechanisms of acrolein toxicity: Relevance to human disease. Toxicol. Sci. 2015, 143, 242–255. [Google Scholar] [CrossRef]
- Liu, D.; Cheng, Y.; Mei, X.; Xie, Y.; Tang, Z.; Liu, J.; Cao, X. Mechanisms of acrolein induces toxicity in human umbilical vein endothelial cells: Oxidative stress, DNA damage response, and apoptosis. Environ. Toxicol. 2022, 37, 708–719. [Google Scholar] [CrossRef]
- Rashad, W.A.; Sakr, S.; Domouky, A.M. Comparative study of oral versus parenteral crocin in mitigating acrolein-induced lung injury in albino rats. Sci. Rep. 2022, 12, 10233. [Google Scholar] [CrossRef]
- Abraham, K.; Andres, S.; Palavinskas, R.; Berg, K.; Appel, K.E.; Lampen, A. Toxicology and risk assessment of acrolein in food. Mol. Nutr. Food Res. 2011, 55, 1277–1290. [Google Scholar] [CrossRef] [PubMed]
- Comer, D.M.; Elborn, J.S.; Ennis, M. Inflammatory and cytotoxic effects of acrolein, nicotine, acetylaldehyde and cigarette smoke extract on human nasal epithelial cells. BMC Pulm. Med. 2014, 14, 32. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ito, S.; Nishio, N.; Tanaka, Y.; Chen, N.; Isobe, K.-I. Acrolein induced both pulmonary inflammation and the death of lung epithelial cells. Toxicol. Lett. 2014, 229, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Nardini, M.; Finkelstein, E.I.; Reddy, S.; Valacchi, G.; Traber, M.; Cross, C.E.; van der Vliet, A. Acrolein-induced cytotoxicity in cultured human bronchial epithelial cells. Modulation by alpha-tocopherol and ascorbic acid. Toxicology 2002, 170, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Cheng, Y.; Tang, Z.; Mei, X.; Cao, X.; Liu, J. Toxicity mechanism of acrolein on DNA damage and apoptosis in BEAS-2B cells: Insights from cell biology and molecular docking analyses. Toxicology 2022, 466, 153083. [Google Scholar] [CrossRef] [PubMed]
- Moretto, N.; Bertolini, S.; Iadicicco, C.; Marchini, G.; Kaur, M.; Volpi, G.; Patacchini, R.; Singh, D.; Facchinetti, F. Cigarette smoke and its component acrolein augment IL-8/CXCL8 mRNA stability via p38 MAPK/MK2 signaling in human pulmonary cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L929–L938. [Google Scholar] [CrossRef] [PubMed]
- Tulen, C.B.M.; Snow, S.J.; Leermakers, P.A.; Kodavanti, U.P.; van Schooten, F.J.; Opperhuizen, A.; Remels, A.H.V. Acrolein inhalation acutely affects the regulation of mitochondrial metabolism in rat lung. Toxicology 2022, 469, 153129. [Google Scholar] [CrossRef]
- Sun, L.; Luo, C.; Long, J.; Wei, D.; Liu, J. Acrolein is a mitochondrial toxin: Effects on respiratory function and enzyme activities in isolated rat liver mitochondria. Mitochondrion 2006, 6, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Tulen, C.B.M.; Opperhuizen, A.; van Schooten, F.-J.; Remels, A.H.V. Disruption of the Molecular Regulation of Mitochondrial Metabolism in Airway and Lung Epithelial Cells by Cigarette Smoke: Are Aldehydes the Culprit? Cells 2023, 12, 299. [Google Scholar] [CrossRef] [PubMed]
- Conklin, D.J.; Haberzettl, P.; Jagatheesan, G.; Kong, M.; Hoyle, G.W. Role of TRPA1 in acute cardiopulmonary toxicity of inhaled acrolein. Toxicol. Appl. Pharmacol. 2017, 324, 61–72. [Google Scholar] [CrossRef]
- Kern, J.C.; Kehrer, J.P. Acrolein-induced cell death: A caspase-influenced decision between apoptosis and oncosis/necrosis. Chem. Biol. Interact. 2002, 139, 79–95. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Pallepati, P.; Bettaieb, A.; Tanel, A.; Averill-Bates, D.A. Acrolein induces a cellular stress response and triggers mitochondrial apoptosis in A549 cells. Chem. Biol. Interact. 2009, 181, 154–167. [Google Scholar] [CrossRef]
- Wickenden, J.A.; Clarke, M.C.H.; Rossi, A.G.; Rahman, I.; Faux, S.P.; Donaldson, K.; MacNee, W. Cigarette smoke prevents apoptosis through inhibition of caspase activation and induces necrosis. Am. J. Respir. Cell Mol. Biol. 2003, 29, 562–570. [Google Scholar] [CrossRef]
- Groskreutz, D.J.; Monick, M.M.; Babor, E.C.; Nyunoya, T.; Varga, S.M.; Look, D.C.; Hunninghake, G.W. Cigarette smoke alters respiratory syncytial virus-induced apoptosis and replication. Am. J. Respir. Cell Mol. Biol. 2009, 41, 189–198. [Google Scholar] [CrossRef]
- Wang, H.-T.; Lin, J.-H.; Yang, C.-H.; Haung, C.-H.; Weng, C.-W.; Maan-Yuh Lin, A.; Lo, Y.-L.; Chen, W.-S.; Tang, M. Acrolein induces mtDNA damages, mitochondrial fission and mitophagy in human lung cells. Oncotarget 2017, 8, 70406–70421. [Google Scholar] [CrossRef] [PubMed]
- Fabisiak, J.P.; Medvedovic, M.; Alexander, D.C.; McDunn, J.E.; Concel, V.J.; Bein, K.; Jang, A.S.; Berndt, A.; Vuga, L.J.; Brant, K.A.; et al. Integrative metabolome and transcriptome profiling reveals discordant energetic stress between mouse strains with differential sensitivity to acrolein-induced acute lung injury. Mol. Nutr. Food Res. 2011, 55, 1423–1434. [Google Scholar] [CrossRef]
- Agarwal, A.R.; Yin, F.; Cadenas, E. Metabolic shift in lung alveolar cell mitochondria following acrolein exposure. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2013, 305, L764–L773. [Google Scholar] [CrossRef] [PubMed]
- Luo, C.; Li, Y.; Yang, L.; Feng, Z.; Li, Y.; Long, J.; Liu, J. A cigarette component acrolein induces accelerated senescence in human diploid fibroblast IMR-90 cells. Biogerontology 2013, 14, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.; Song, Y.; Xie, J.; Xie, J.; Chen, Y.; Li, P.; Liu, D.; Hu, X.; Yu, Q. Acrolein Promotes Aging and Oxidative Stress via the Stress Response Factor DAF-16/FOXO in Caenorhabditis elegans. Foods 2022, 11, 1590. [Google Scholar] [CrossRef]
- Yasuo, M.; Droma, Y.; Kitaguchi, Y.; Ito, M.; Imamura, H.; Kawakubo, M.; Hanaoka, M. The relationship between acrolein and oxidative stress in COPD: In systemic plasma and in local lung tissue. Int. J. Chron. Obstruct. Pulmon. Dis. 2019, 14, 1527–1537. [Google Scholar] [CrossRef]
- Colombo, G.; Dalle-Donne, I.; Orioli, M.; Giustarini, D.; Rossi, R.; Clerici, M.; Regazzoni, L.; Aldini, G.; Milzani, A.; Butterfield, D.A.; et al. Oxidative damage in human gingival fibroblasts exposed to cigarette smoke. Free Radic. Biol. Med. 2012, 52, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Nishimoto-Kusunose, S.; Sawa, M.; Inaba, Y.; Ushiyama, A.; Ishii, K.; Hattori, K.; Ogasawara, Y. Exposure to aerosol extract from heated tobacco products causes a drastic decrease of glutathione and protein carbonylation in human lung epithelial cells. Biochem. Biophys. Res. Commun. 2022, 589, 92–99. [Google Scholar] [CrossRef]
- van der Toorn, M.; Smit-de Vries, M.P.; Slebos, D.-J.; de Bruin, H.G.; Abello, N.; van Oosterhout, A.J.M.; Bischoff, R.; Kauffman, H.F. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 293, L1156–L1162. [Google Scholar] [CrossRef]
- Xiong, R.; Wu, Q.; Bryant, M.; Rosenfeldt, H.; Healy, S.; Cao, X. In vitro dosimetry analyses for acrolein exposure in normal human lung epithelial cells and human lung cancer cells. Environ. Toxicol. Pharmacol. 2021, 83, 103576. [Google Scholar] [CrossRef]
- Myers, C.R.; Myers, J.M. The effects of acrolein on peroxiredoxins, thioredoxins, and thioredoxin reductase in human bronchial epithelial cells. Toxicology 2009, 257, 95–104. [Google Scholar] [CrossRef]
- Myers, C.R.; Myers, J.M.; Kufahl, T.D.; Forbes, R.; Szadkowski, A. The effects of acrolein on the thioredoxin system: Implications for redox-sensitive signaling. Mol. Nutr. Food Res. 2011, 55, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Spiess, P.C.; Deng, B.; Hondal, R.J.; Matthews, D.E.; van der Vliet, A. Proteomic profiling of acrolein adducts in human lung epithelial cells. J. Proteom. 2011, 74, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Horinouchi, T.; Mazaki, Y.; Terada, K.; Miwa, S. Cigarette Smoke Extract and Its Cytotoxic Factor Acrolein Inhibit Nitric Oxide Production in Human Vascular Endothelial Cells. Biol. Pharm. Bull. 2020, 43, 1804–1809. [Google Scholar] [CrossRef]
- Sun, Y.; Ito, S.; Nishio, N.; Tanaka, Y.; Chen, N.; Liu, L.; Isobe, K.-I. Enhancement of the acrolein-induced production of reactive oxygen species and lung injury by GADD34. Oxid. Med. Cell. Longev. 2015, 2015, 170309. [Google Scholar] [CrossRef]
- Wu, C.C.; Hsieh, C.W.; Lai, P.H.; Lin, J.B.; Liu, Y.C.; Wung, B.S. Upregulation of endothelial heme oxygenase-1 expression through the activation of the JNK pathway by sublethal concentrations of acrolein. Toxicol. Appl. Pharmacol. 2006, 214, 244–252. [Google Scholar] [CrossRef]
- Tirumalai, R.; Rajesh Kumar, T.; Mai, K.H.; Biswal, S. Acrolein causes transcriptional induction of phase II genes by activation of Nrf2 in human lung type II epithelial (A549) cells. Toxicol. Lett. 2002, 132, 27–36. [Google Scholar] [CrossRef]
- Gao, J.; Zou, X.; Yang, L.; Feng, Z.; Liu, J. Hydroxytyrosol protects against acrolein induced preosteoblast cell toxicity: Involvement of Nrf2/Keap1 pathway. J. Funct. Foods 2015, 19, 28–38. [Google Scholar] [CrossRef]
- Kosmider, B.; Messier, E.M.; Chu, H.W.; Mason, R.J. Human alveolar epithelial cell injury induced by cigarette smoke. PLoS ONE 2011, 6, e26059. [Google Scholar] [CrossRef] [PubMed]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, K.S.; Afzal, O.; Almalki, W.H.; Kazmi, I.; Javed Shaikh, M.A.; Thangavelu, L.; Gulati, M.; Singh, S.K.; Jha, N.K.; Gupta, P.K.; et al. Nuclear factor-kappa B (NF-κB) inhibition as a therapeutic target for plant nutraceuticals in mitigating inflammatory lung diseases. Chem. Biol. Interact. 2022, 354, 109842. [Google Scholar] [CrossRef] [PubMed]
- Valacchi, G.; Pagnin, E.; Phung, A.; Nardini, M.; Schock, B.C.; Cross, C.E.; van der Vliet, A. Inhibition of NFkappaB activation and IL-8 expression in human bronchial epithelial cells by acrolein. Antioxid. Redox Signal. 2005, 7, 25–31. [Google Scholar] [CrossRef]
- Wang, B.; Yu, L.; Liu, W.; Yang, M.; Fan, L.; Zhou, M.; Ma, J.; Wang, X.; Nie, X.; Cheng, M.; et al. Cross-sectional and longitudinal associations of acrolein exposure with pulmonary function alteration: Assessing the potential roles of oxidative DNA damage, inflammation, and pulmonary epithelium injury in a general adult population. Environ. Int. 2022, 167, 107401. [Google Scholar] [CrossRef] [PubMed]
- Park, S.L.; Le Marchand, L.; Cheng, G.; Balbo, S.; Chen, M.; Carmella, S.G.; Thomson, N.M.; Lee, Y.; Patel, Y.M.; Stram, D.O.; et al. Quantitation of DNA Adducts Resulting from Acrolein Exposure and Lipid Peroxidation in Oral Cells of Cigarette Smokers from Three Racial/Ethnic Groups with Differing Risks for Lung Cancer. Chem. Res. Toxicol. 2022, 35, 1914–1922. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Shirai, T.; Hirai, K.; Nakayasu, H.; Takahashi, S.; Kishimoto, Y.; Akamatsu, T.; Asada, K.; Kato, S. Mucus Plugs and Small Airway Dysfunction in Asthma, COPD, and Asthma-COPD Overlap. Allergy Asthma Immunol. Res. 2022, 14, 196–209. [Google Scholar] [CrossRef] [PubMed]
- Symmes, B.A.; Stefanski, A.L.; Magin, C.M.; Evans, C.M. Role of mucins in lung homeostasis: Regulated expression and biosynthesis in health and disease. Biochem. Soc. Trans. 2018, 46, 707–719. [Google Scholar] [CrossRef]
- Thai, P.; Loukoianov, A.; Wachi, S.; Wu, R. Regulation of airway mucin gene expression. Annu. Rev. Physiol. 2008, 70, 405–429. [Google Scholar] [CrossRef]
- Fan, H.; Bobek, L.A. Regulation of Human MUC7 Mucin Gene Expression by Cigarette Smoke Extract or Cigarette Smoke and Pseudomonas aeruginosa Lipopolysaccharide in Human Airway Epithelial Cells and in MUC7 Transgenic Mice. Open Respir. Med. J. 2010, 4, 63–70. [Google Scholar] [CrossRef]
- Baginski, T.K.; Dabbagh, K.; Satjawatcharaphong, C.; Swinney, D.C. Cigarette smoke synergistically enhances respiratory mucin induction by proinflammatory stimuli. Am. J. Respir. Cell Mol. Biol. 2006, 35, 165–174. [Google Scholar] [CrossRef]
- Lyon, J.P.; Jenkins, L.J.; Jones, R.A.; Coon, R.A.; Siegel, J. Repeated and continuous exposure of laboratory animals to acrolein. Toxicol. Appl. Pharmacol. 1970, 17, 726–732. [Google Scholar] [CrossRef]
- Leikauf, G.D.; Borchers, M.T.; Prows, D.R.; Simpson, L.G. Mucin apoprotein expression in COPD. Chest 2002, 121, 166S–182S. [Google Scholar] [CrossRef] [PubMed]
- Costa, D.L.; Kutzman, R.S.; Lehmann, J.R.; Drew, R.T. Altered lung function and structure in the rat after subchronic exposure to acrolein. Am. Rev. Respir. Dis. 1986, 133, 286–291. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Shaver, C.; Case, L.M.; Dietsch, M.; Wesselkamper, S.C.; Hardie, W.D.; Korfhagen, T.R.; Corradi, M.; Nadel, J.A.; Borchers, M.T.; et al. Acrolein-activated matrix metalloproteinase 9 contributes to persistent mucin production. Am. J. Respir. Cell Mol. Biol. 2008, 38, 446–454. [Google Scholar] [CrossRef]
- Chen, P.; Wang, X.; Li, Y.; Liu, H. An Inhibitor of Nuclear Factor-Kappa B Pathway Attenuates the Release of TGF-β1 and Inhibits the Fibrogenic Progress in a Model of Airway Remodeling Induced by Acrolein. Comput. Math. Methods Med. 2022, 2022, 4984634. [Google Scholar] [CrossRef]
- Ghosh, A.; Coakley, R.C.; Mascenik, T.; Rowell, T.R.; Davis, E.S.; Rogers, K.; Webster, M.J.; Dang, H.; Herring, L.E.; Sassano, M.F.; et al. Chronic E-Cigarette Exposure Alters the Human Bronchial Epithelial Proteome. Am. J. Respir. Crit. Care Med. 2018, 198, 67–76. [Google Scholar] [CrossRef]
- Reidel, B.; Radicioni, G.; Clapp, P.W.; Ford, A.A.; Abdelwahab, S.; Rebuli, M.E.; Haridass, P.; Alexis, N.E.; Jaspers, I.; Kesimer, M. E-Cigarette Use Causes a Unique Innate Immune Response in the Lung, Involving Increased Neutrophilic Activation and Altered Mucin Secretion. Am. J. Respir. Crit. Care Med. 2018, 197, 492–501. [Google Scholar] [CrossRef]
- Chung, S.; Baumlin, N.; Dennis, J.S.; Moore, R.; Salathe, S.F.; Whitney, P.L.; Sabater, J.; Abraham, W.M.; Kim, M.D.; Salathe, M. Electronic Cigarette Vapor with Nicotine Causes Airway Mucociliary Dysfunction Preferentially via TRPA1 Receptors. Am. J. Respir. Crit. Care Med. 2019, 200, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Arcos, I.; Geraghty, P.; Baumlin, N.; Campos, M.; Dabo, A.J.; Jundi, B.; Cummins, N.; Eden, E.; Grosche, A.; Salathe, M.; et al. Chronic electronic cigarette exposure in mice induces features of COPD in a nicotine-dependent manner. Thorax 2016, 71, 1119–1129. [Google Scholar] [CrossRef]
- Deshmukh, H.S.; Case, L.M.; Wesselkamper, S.C.; Borchers, M.T.; Martin, L.D.; Shertzer, H.G.; Nadel, J.A.; Leikauf, G.D. Metalloproteinases mediate mucin 5AC expression by epidermal growth factor receptor activation. Am. J. Respir. Crit. Care Med. 2005, 171, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, H.S.; McLachlan, A.; Atkinson, J.J.; Hardie, W.D.; Korfhagen, T.R.; Dietsch, M.; Liu, Y.; Di, P.Y.P.; Wesselkamper, S.C.; Borchers, M.T.; et al. Matrix metalloproteinase-14 mediates a phenotypic shift in the airways to increase mucin production. Am. J. Respir. Crit. Care Med. 2009, 180, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-S.; Wang, T.; Han, S.-X.; Dong, J.-J.; Liao, Z.-L.; He, G.-M.; Chen, L.; Chen, Y.-J.; Xu, D.; Hou, Y.; et al. p38 MAPK and MMP-9 cooperatively regulate mucus overproduction in mice exposed to acrolein fog. Int. Immunopharmacol. 2009, 9, 1228–1235. [Google Scholar] [CrossRef]
- Lemaître, V.; Dabo, A.J.; D’Armiento, J. Cigarette smoke components induce matrix metalloproteinase-1 in aortic endothelial cells through inhibition of mTOR signaling. Toxicol. Sci. 2011, 123, 542–549. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, T.E.; Zheng, Y.-T.; Hellmann, J.; Conklin, D.J.; Barski, O.; Bhatnagar, A. Acrolein activates matrix metalloproteinases by increasing reactive oxygen species in macrophages. Toxicol. Appl. Pharmacol. 2009, 236, 194–201. [Google Scholar] [CrossRef]
- Chen, Y.-J.; Chen, P.; Wang, H.-X.; Wang, T.; Chen, L.; Wang, X.; Sun, B.-B.; Liu, D.-S.; Xu, D.; An, J.; et al. Simvastatin attenuates acrolein-induced mucin production in rats: Involvement of the Ras/extracellular signal-regulated kinase pathway. Int. Immunopharmacol. 2010, 10, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.S.; Kim, W.J. The Role of Matrix Metalloproteinase in Inflammation with a Focus on Infectious Diseases. Int. J. Mol. Sci. 2022, 23, 546. [Google Scholar] [CrossRef]
- Jang, A.S.; Concel, V.J.; Bein, K.; Brant, K.A.; Liu, S.; Pope-Varsalona, H.; Dopico, R.A.; Di, Y.P.P.; Knoell, D.L.; Barchowsky, A.; et al. Endothelial dysfunction and claudin 5 regulation during acrolein-induced lung injury. Am. J. Respir. Cell Mol. Biol. 2011, 44, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-Y.; Wang, M.; Zhang, J.; Barve, S.S.; McClain, C.J.; Joshi-Barve, S. Acrolein Disrupts Tight Junction Proteins and Causes Endoplasmic Reticulum Stress-Mediated Epithelial Cell Death Leading to Intestinal Barrier Dysfunction and Permeability. Am. J. Pathol. 2017, 187, 2686–2697. [Google Scholar] [CrossRef] [PubMed]
- Shifren, A.; Durmowicz, A.G.; Knutsen, R.H.; Hirano, E.; Mecham, R.P. Elastin protein levels are a vital modifier affecting normal lung development and susceptibility to emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2007, 292, L778–L787. [Google Scholar] [CrossRef]
- Puchelle, E.; Zahm, J.-M.; Tournier, J.-M.; Coraux, C. Airway epithelial repair, regeneration, and remodeling after injury in chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2006, 3, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Oikonomidi, S.; Kostikas, K.; Tsilioni, I.; Tanou, K.; Gourgoulianis, K.I.; Kiropoulos, T.S. Matrix metalloproteinases in respiratory diseases: From pathogenesis to potential clinical implications. Curr. Med. Chem. 2009, 16, 1214–1228. [Google Scholar] [CrossRef]
- Kim, C.E.; Lee, S.J.; Seo, K.W.; Park, H.M.; Yun, J.W.; Bae, J.U.; Bae, S.S.; Kim, C.D. Acrolein increases 5-lipoxygenase expression in murine macrophages through activation of ERK pathway. Toxicol. Appl. Pharmacol. 2010, 245, 76–82. [Google Scholar] [CrossRef]
- Chen, L.; Ge, Q.; Tjin, G.; Alkhouri, H.; Deng, L.; Brandsma, C.-A.; Adcock, I.; Timens, W.; Postma, D.; Burgess, J.K.; et al. Effects of cigarette smoke extract on human airway smooth muscle cells in COPD. Eur. Respir. J. 2014, 44, 634–646. [Google Scholar] [CrossRef]
- Chaudhuri, R.; McSharry, C.; Brady, J.; Donnelly, I.; Grierson, C.; McGuinness, S.; Jolly, L.; Weir, C.J.; Messow, C.M.; Spears, M.; et al. Sputum matrix metalloproteinase-12 in patients with chronic obstructive pulmonary disease and asthma: Relationship to disease severity. J. Allergy Clin. Immunol. 2012, 129, 655–663.e8. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melén, E.; Söderhäll, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Coakley, R.D.; Ghio, A.J.; Muhlebach, M.S.; Esther, C.R.; Alexis, N.E.; Tarran, R. Chronic E-Cigarette Use Increases Neutrophil Elastase and Matrix Metalloprotease Levels in the Lung. Am. J. Respir. Crit. Care Med. 2019, 200, 1392–1401. [Google Scholar] [CrossRef]
- Cho, W.K.; Lee, C.G.; Kim, L.K. COPD as a Disease of Immunosenescence. Yonsei Med. J. 2019, 60, 407–413. [Google Scholar] [CrossRef]
- Endo, R.; Uchiyama, K.; Lim, S.-Y.; Itakura, M.; Adachi, T.; Uchida, K. Recognition of acrolein-specific epitopes by B cell receptors triggers an innate immune response. J. Biol. Chem. 2021, 296, 100648. [Google Scholar] [CrossRef]
- Wells, J.M.; O’Reilly, P.J.; Szul, T.; Sullivan, D.I.; Handley, G.; Garrett, C.; McNicholas, C.M.; Roda, M.A.; Miller, B.E.; Tal-Singer, R.; et al. An aberrant leukotriene A4 hydrolase-proline-glycine-proline pathway in the pathogenesis of chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2014, 190, 51–61. [Google Scholar] [CrossRef]
- Noerager, B.D.; Xu, X.; Davis, V.A.; Jones, C.W.; Okafor, S.; Whitehead, A.; Blalock, J.E.; Jackson, P.L. A Potential Role for Acrolein in Neutrophil-Mediated Chronic Inflammation. Inflammation 2015, 38, 2279–2287. [Google Scholar] [CrossRef]
- Lee, J.-S.; Lee, J.Y.; Lee, M.Y.; Hwang, D.H.; Youn, H.S. Acrolein with an alpha, beta-unsaturated carbonyl group inhibits LPS-induced homodimerization of toll-like receptor 4. Mol. Cells 2008, 25, 253–257. [Google Scholar] [PubMed]
- Hristova, M.; Spiess, P.C.; Kasahara, D.I.; Randall, M.J.; Deng, B.; van der Vliet, A. The tobacco smoke component, acrolein, suppresses innate macrophage responses by direct alkylation of c-Jun N-terminal kinase. Am. J. Respir. Cell Mol. Biol. 2012, 46, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Burcham, P.; Thompson, C.; Henry, P. Acrolein and the Lung: Chemical, Molecular, and Pathological Aspects in Advances in Molecular Toxicology; Elsevier: Amsterdam, The Netherlands, 2010; Volume 4, ISBN 9780444535849. [Google Scholar]
- Lambert, C.; McCue, J.; Portas, M.; Ouyang, Y.; Li, J.; Rosano, T.G.; Lazis, A.; Freed, B.M. Acrolein in cigarette smoke inhibits T-cell responses. J. Allergy Clin. Immunol. 2005, 116, 916–922. [Google Scholar] [CrossRef] [PubMed]
- Lambert, C.; Li, J.; Jonscher, K.; Yang, T.-C.; Reigan, P.; Quintana, M.; Harvey, J.; Freed, B.M. Acrolein inhibits cytokine gene expression by alkylating cysteine and arginine residues in the NF-kappaB1 DNA binding domain. J. Biol. Chem. 2007, 282, 19666–19675. [Google Scholar] [CrossRef]
- Kasahara, D.I.; Poynter, M.E.; Othman, Z.; Hemenway, D.; van der Vliet, A. Acrolein inhalation suppresses lipopolysaccharide-induced inflammatory cytokine production but does not affect acute airways neutrophilia. J. Immunol. 2008, 181, 736–745. [Google Scholar] [CrossRef]
- Moretto, N.; Facchinetti, F.; Southworth, T.; Civelli, M.; Singh, D.; Patacchini, R. α, β-Unsaturated aldehydes contained in cigarette smoke elicit IL-8 release in pulmonary cells through mitogen-activated protein kinases. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 296, L839–L848. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, P.; Hayes, B.E. Induction of COX-2 by acrolein in rat lung epithelial cells. Mol. Cell. Biochem. 2007, 301, 191–199. [Google Scholar] [CrossRef]
- Takamiya, R.; Takahashi, M.; Maeno, T.; Saito, A.; Kato, M.; Shibata, T.; Uchida, K.; Ariki, S.; Nakano, M. Acrolein in cigarette smoke attenuates the innate immune responses mediated by surfactant protein D. Biochim. Biophys. Acta Gen. Subj. 2020, 1864, 129699. [Google Scholar] [CrossRef]
- Takamiya, R.; Uchida, K.; Shibata, T.; Maeno, T.; Kato, M.; Yamaguchi, Y.; Ariki, S.; Hasegawa, Y.; Saito, A.; Miwa, S.; et al. Disruption of the structural and functional features of surfactant protein A by acrolein in cigarette smoke. Sci. Rep. 2017, 7, 8304. [Google Scholar] [CrossRef]
- Pastva, A.M.; Wright, J.R.; Williams, K.L. Immunomodulatory roles of surfactant proteins A and D: Implications in lung disease. Proc. Am. Thorac. Soc. 2007, 4, 252–257. [Google Scholar] [CrossRef]
- Tsou, H.-H.; Hu, C.-H.; Liu, J.-H.; Liu, C.-J.; Lee, C.-H.; Liu, T.-Y.; Wang, H.-T. Acrolein Is Involved in the Synergistic Potential of Cigarette Smoking- and Betel Quid Chewing-Related Human Oral Cancer. Cancer Epidemiol. Biomark. Prev. 2019, 28, 954–962. [Google Scholar] [CrossRef]
- Lee, H.-W.; Wang, H.-T.; Weng, M.; Chin, C.; Huang, W.; Lepor, H.; Wu, X.-R.; Rom, W.N.; Chen, L.-C.; Tang, M. Cigarette side-stream smoke lung and bladder carcinogenesis: Inducing mutagenic acrolein-DNA adducts, inhibiting DNA repair and enhancing anchorage-independent-growth cell transformation. Oncotarget 2015, 6, 33226–33236. [Google Scholar] [CrossRef]
- Hong, J.-H.; Tong, Z.-J.; Wei, T.-E.; Lu, Y.-C.; Huang, C.-Y.; Huang, C.-Y.; Chiang, C.-H.; Jaw, F.-S.; Cheng, H.-W.; Wang, H.-T. Cigarette Smoke Containing Acrolein Contributes to Cisplatin Resistance in Human Bladder Cancers through the Regulation of HER2 Pathway or FGFR3 Pathway. Mol. Cancer Ther. 2022, 21, 1010–1019. [Google Scholar] [CrossRef]
- Matsumoto, M.; Yamano, S.; Senoh, H.; Umeda, Y.; Hirai, S.; Saito, A.; Kasai, T.; Aiso, S. Carcinogenicity and chronic toxicity of acrolein in rats and mice by two-year inhalation study. Regul. Toxicol. Pharmacol. 2021, 121, 104863. [Google Scholar] [CrossRef]
- Peterson, L.A.; Seabloom, D.; Smith, W.E.; Vevang, K.R.; Seelig, D.M.; Zhang, L.; Wiedmann, T.S. Acrolein Increases the Pulmonary Tumorigenic Activity of the Tobacco-Specific Nitrosamine 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK). Chem. Res. Toxicol. 2022, 35, 1831–1839. [Google Scholar] [CrossRef] [PubMed]
- Paiano, V.; Maertens, L.; Guidolin, V.; Yang, J.; Balbo, S.; Hecht, S.S. Quantitative Liquid Chromatography-Nanoelectrospray Ionization-High-Resolution Tandem Mass Spectrometry Analysis of Acrolein-DNA Adducts and Etheno-DNA Adducts in Oral Cells from Cigarette Smokers and Nonsmokers. Chem. Res. Toxicol. 2020, 33, 2197–2207. [Google Scholar] [CrossRef]
- Chen, H.-J.C. Mass Spectrometry Analysis of DNA and Protein Adducts as Biomarkers in Human Exposure to Cigarette Smoking: Acrolein as an Example. Chem. Res. Toxicol. 2023, 36, 132–140. [Google Scholar] [CrossRef]
- Feng, Z.; Hu, W.; Hu, Y.; Tang, M. Acrolein is a major cigarette-related lung cancer agent: Preferential binding at p53 mutational hotspots and inhibition of DNA repair. Proc. Natl. Acad. Sci. USA 2006, 103, 15404–15409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Balbo, S.; Wang, M.; Hecht, S.S. Analysis of acrolein-derived 1,N2-propanodeoxyguanosine adducts in human leukocyte DNA from smokers and nonsmokers. Chem. Res. Toxicol. 2011, 24, 119–124. [Google Scholar] [CrossRef]
- Wang, H.-T.; Hu, Y.; Tong, D.; Huang, J.; Gu, L.; Wu, X.-R.; Chung, F.-L.; Li, G.-M.; Tang, M. Effect of carcinogenic acrolein on DNA repair and mutagenic susceptibility. J. Biol. Chem. 2012, 287, 12379–12386. [Google Scholar] [CrossRef]
- Choudhury, S.; Dyba, M.; Pan, J.; Roy, R.; Chung, F.-L. Repair kinetics of acrolein- and (E)-4-hydroxy-2-nonenal-derived DNA adducts in human colon cell extracts. Mutat. Res. 2013, 751–752, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Fang, L.; Li, H.; Tang, M.; Jin, C. Cigarette smoke component acrolein modulates chromatin assembly by inhibiting histone acetylation. J. Biol. Chem. 2013, 288, 21678–21687. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Chen, D.; Yu, C.; Li, H.; Brocato, J.; Huang, L.; Jin, C. Mechanisms Underlying Acrolein-Mediated Inhibition of Chromatin Assembly. Mol. Cell. Biol. 2016, 36, 2995–3008. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhou, J.; Wang, J.; Li, S.; Fukunaga, A.; Yodoi, J.; Tian, H. Progress in the mechanism and targeted drug therapy for COPD. Signal Transduct. Target. Ther. 2020, 5, 248. [Google Scholar] [CrossRef]
- Davies, S.S.; Zhang, L.S. Reactive Carbonyl Species Scavengers-Novel Therapeutic Approaches for Chronic Diseases. Curr. Pharmacol. Rep. 2017, 3, 51–67. [Google Scholar] [CrossRef]
- Rahman, I. Pharmacological antioxidant strategies as therapeutic interventions for COPD. Biochim. Biophys. Acta 2012, 1822, 714–728. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Matsuoka, Y.; Sakai, K.; Fujiya, N.; Fujii, H.; Mano, J. Catechins in green tea powder (matcha) are heat-stable scavengers of acrolein, a lipid peroxide-derived reactive carbonyl species. Food Chem. 2021, 355, 129403. [Google Scholar] [CrossRef] [PubMed]
- Aldini, G.; Dalle-Donne, I.; Facino, R.M.; Milzani, A.; Carini, M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med. Res. Rev. 2007, 27, 817–868. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Chen, P.; Zhang, C.; Chen, J.-B.; Wu, J. Oral N-acetylcysteine attenuates pulmonary emphysema and alveolar septal cell apoptosis in smoking-induced COPD in rats. Respirology 2009, 14, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Stav, D.; Raz, M. Effect of N-acetylcysteine on air trapping in COPD: A randomized placebo-controlled study. Chest 2009, 136, 381–386. [Google Scholar] [CrossRef]
- Moitra, S. N-acetylcysteine (NAC) in COPD: Benefits often lost in trials. QJM 2019, 112, 387–388. [Google Scholar] [CrossRef]
- Decramer, M.; Rutten-van Mölken, M.; Dekhuijzen, P.N.R.; Troosters, T.; van Herwaarden, C.; Pellegrino, R.; van Schayck, C.P.O.; Olivieri, D.; Del Donno, M.; de Backer, W.; et al. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): A randomised placebo-controlled trial. Lancet 2005, 365, 1552–1560. [Google Scholar] [CrossRef]
- Kode, A.; Rajendrasozhan, S.; Caito, S.; Yang, S.-R.; Megson, I.L.; Rahman, I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L478–L488. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Li, T.; Li, J.-H.; Miao, S.-Y.; Xiao, X.-Z. The Effects of Resveratrol on Inflammation and Oxidative Stress in a Rat Model of Chronic Obstructive Pulmonary Disease. Molecules 2017, 22, 1529. [Google Scholar] [CrossRef] [PubMed]
- Balstad, T.R.; Carlsen, H.; Myhrstad, M.C.W.; Kolberg, M.; Reiersen, H.; Gilen, L.; Ebihara, K.; Paur, I.; Blomhoff, R. Coffee, broccoli and spices are strong inducers of electrophile response element-dependent transcription in vitro and in vivo—Studies in electrophile response element transgenic mice. Mol. Nutr. Food Res. 2011, 55, 185–197. [Google Scholar] [CrossRef]
- Di Lorenzo, C.; Colombo, F.; Biella, S.; Stockley, C.; Restani, P. Polyphenols and Human Health: The Role of Bioavailability. Nutrients 2021, 13, 273. [Google Scholar] [CrossRef] [PubMed]
- Burcham, P.C. Potentialities and pitfalls accompanying chemico-pharmacological strategies against endogenous electrophiles and carbonyl stress. Chem. Res. Toxicol. 2008, 21, 779–786. [Google Scholar] [CrossRef]
- Jiang, K.; Huang, C.; Liu, F.; Zheng, J.; Ou, J.; Zhao, D.; Ou, S. Origin and Fate of Acrolein in Foods. Foods 2022, 11, 1976. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hikisz, P.; Jacenik, D. The Tobacco Smoke Component, Acrolein, as a Major Culprit in Lung Diseases and Respiratory Cancers: Molecular Mechanisms of Acrolein Cytotoxic Activity. Cells 2023, 12, 879. https://doi.org/10.3390/cells12060879
Hikisz P, Jacenik D. The Tobacco Smoke Component, Acrolein, as a Major Culprit in Lung Diseases and Respiratory Cancers: Molecular Mechanisms of Acrolein Cytotoxic Activity. Cells. 2023; 12(6):879. https://doi.org/10.3390/cells12060879
Chicago/Turabian StyleHikisz, Pawel, and Damian Jacenik. 2023. "The Tobacco Smoke Component, Acrolein, as a Major Culprit in Lung Diseases and Respiratory Cancers: Molecular Mechanisms of Acrolein Cytotoxic Activity" Cells 12, no. 6: 879. https://doi.org/10.3390/cells12060879
APA StyleHikisz, P., & Jacenik, D. (2023). The Tobacco Smoke Component, Acrolein, as a Major Culprit in Lung Diseases and Respiratory Cancers: Molecular Mechanisms of Acrolein Cytotoxic Activity. Cells, 12(6), 879. https://doi.org/10.3390/cells12060879