

Intermittent Lead Exposure Induces Behavioral and Cardiovascular Alterations Associated with Neuroinflammation
 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model Development
2.2. Behavioral Evaluation
2.2.1. Open-Field Exploration Test
2.2.2. Novel Object Recognition Test
2.3. Functional Evaluation
2.3.1. Metabolic Evaluation
2.3.2. Acute Physiological and Autonomic Evaluation
2.3.3. Data Acquisition and Analysis
2.3.4. Baro- and Chemoreceptor Reflex Evaluation
2.4. Immunohistochemistry (IHC)
2.5. Statistical Analysis
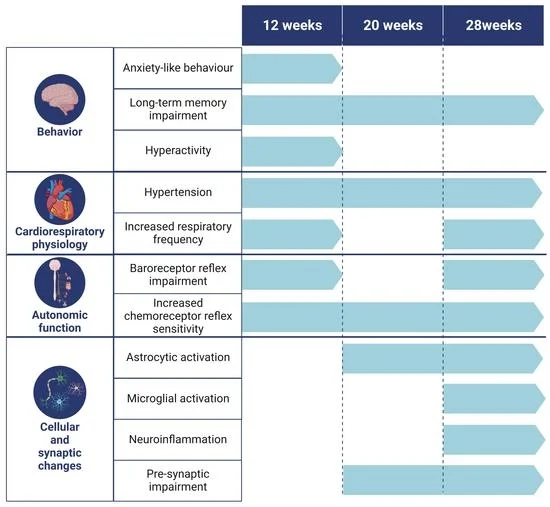
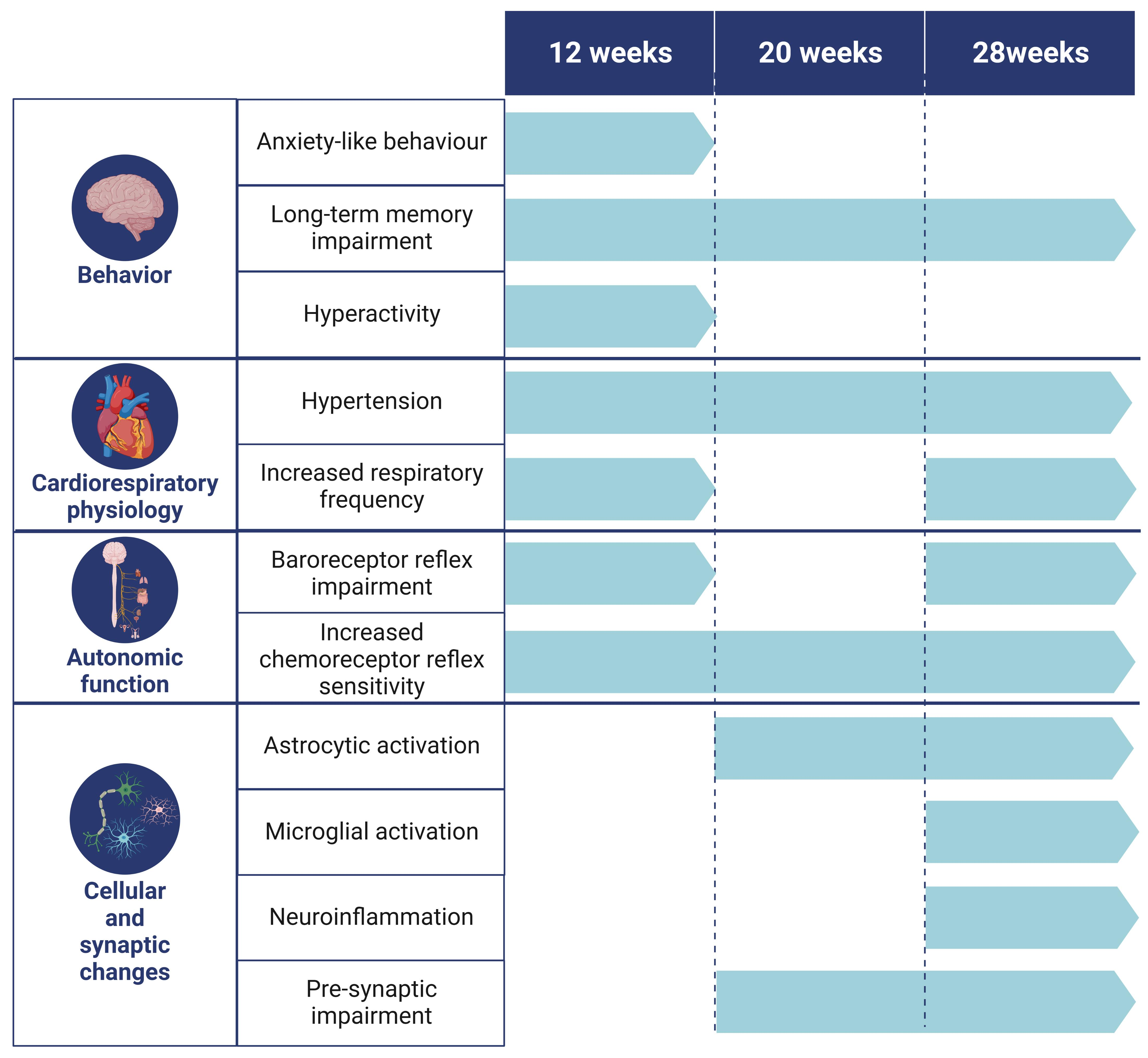
3. Results
3.1. Behavioral Changes
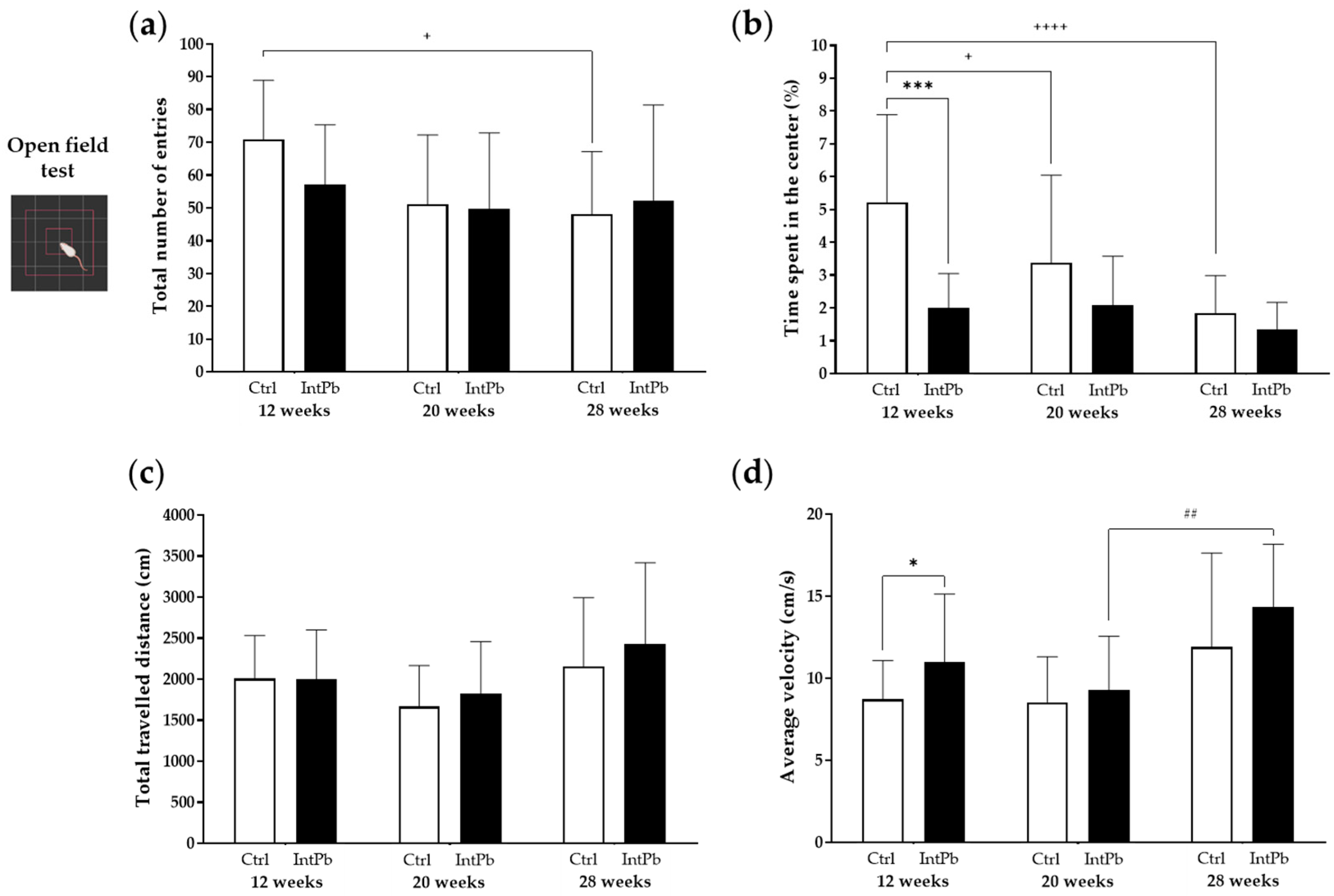
3.1.1. Intermittent Lead Exposure Does Not Cause Significant Changes in Locomotion and Anxiety-like Behavior
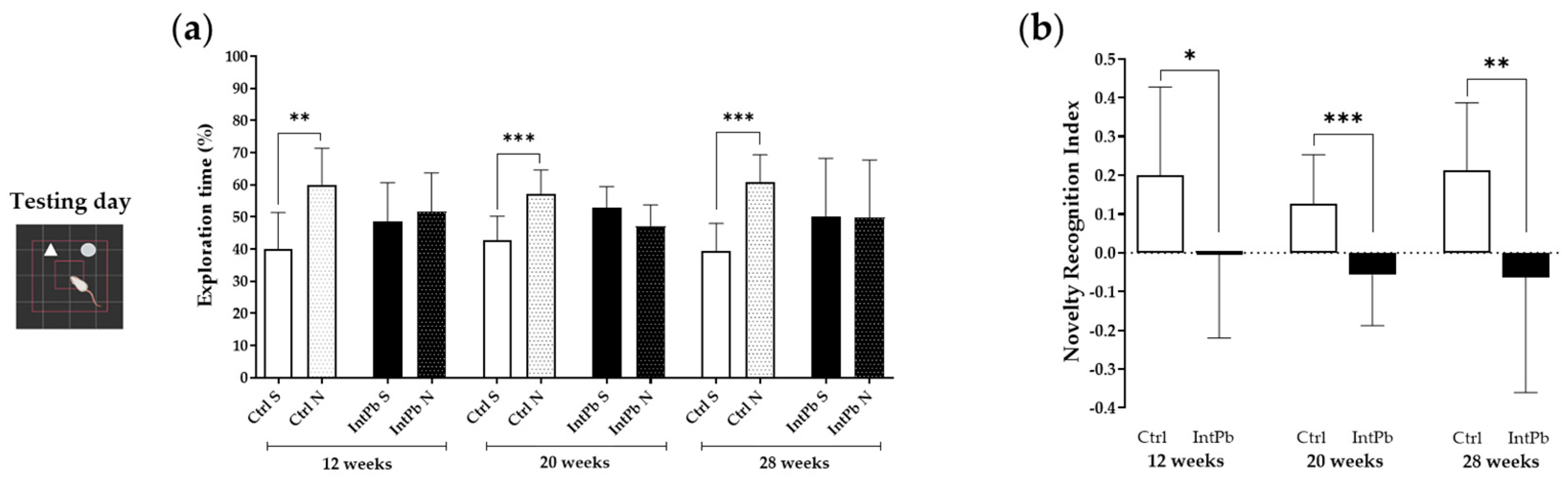
3.1.2. Animals Exposed to Lead Show Impaired Recognition of the Novel Object with a Strong Decrease in the Novel Object Recognition Index within All Evaluated Time Points
3.2. Physiological and Autonomic Changes
3.2.1. Animals Exposed to Lead Intermittently Show a Significant Increase in Blood Lead Levels, without Effects on Metabolic Parameters
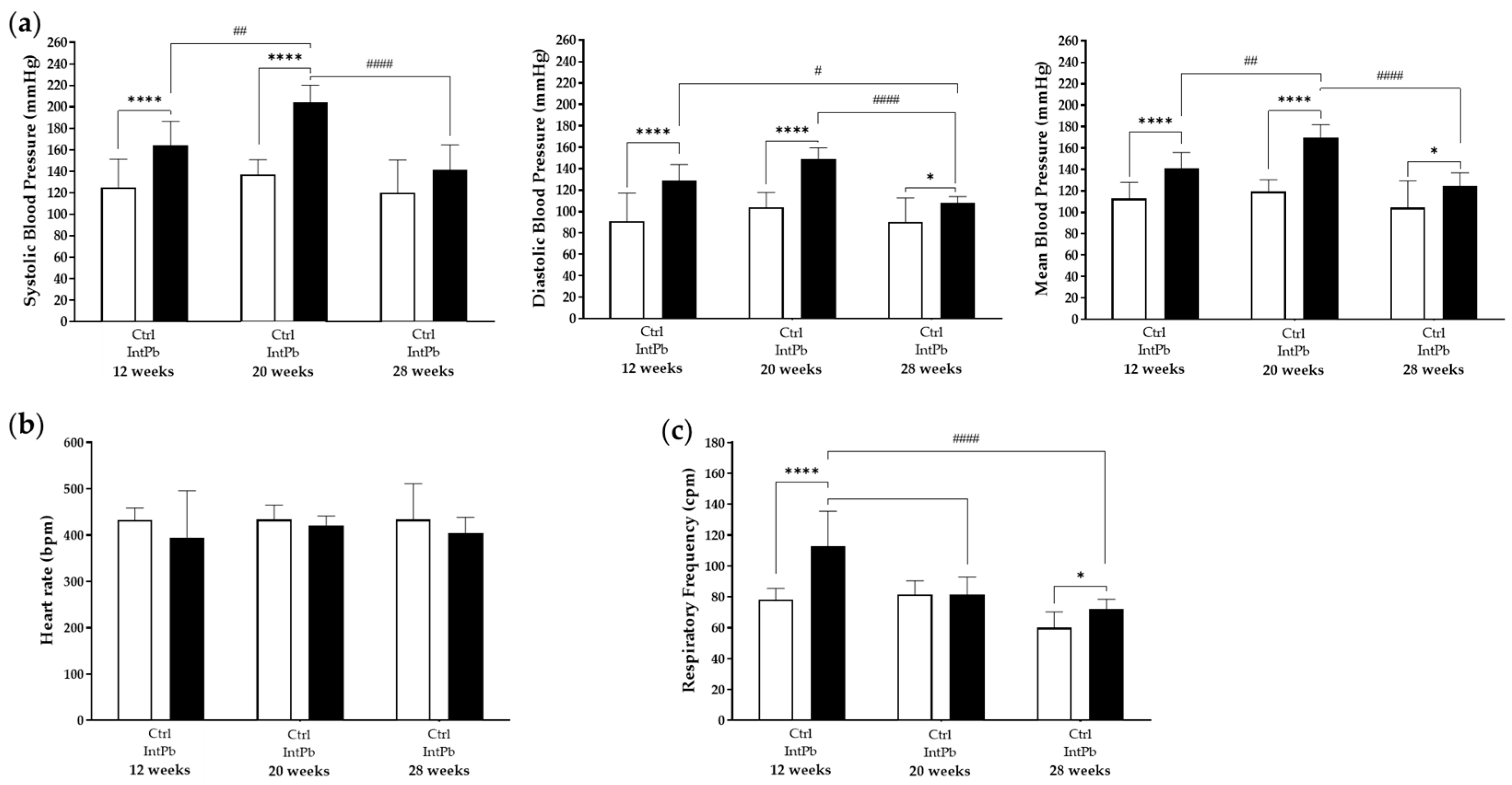
3.2.2. Lead-Exposed Animals Presented an Increase in Blood Pressure and Respiratory Frequency without Changes in Heart Rate
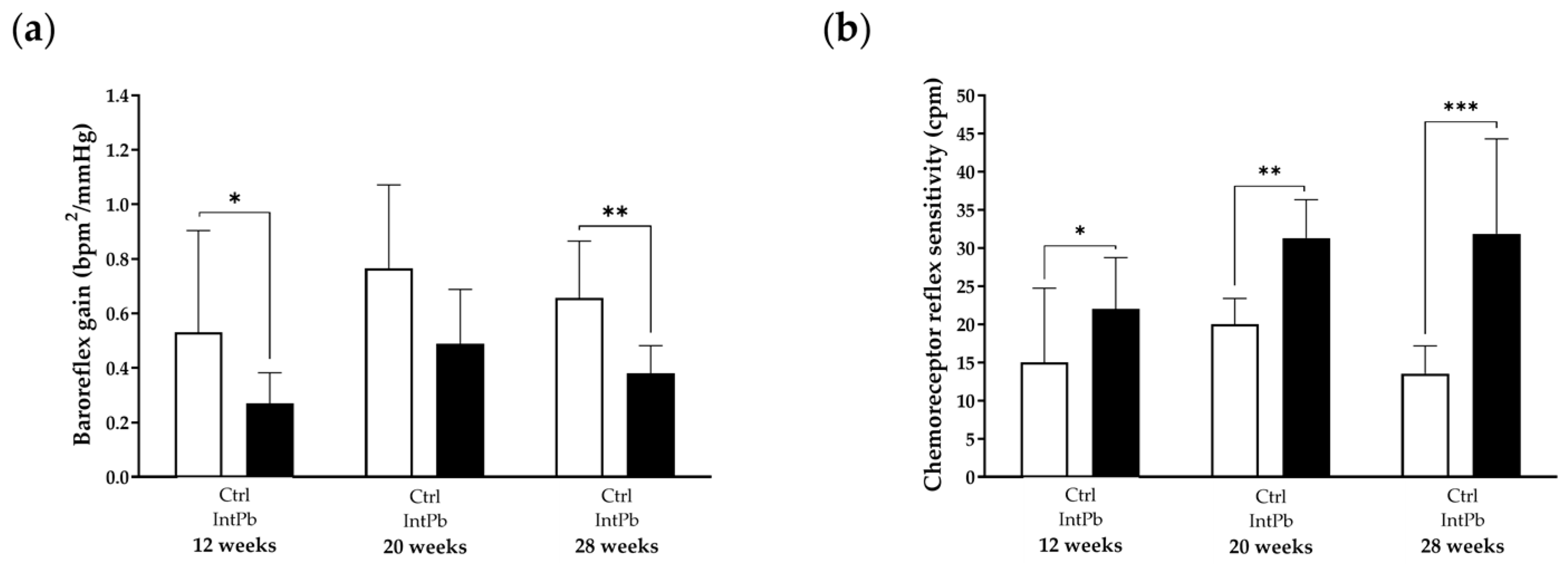
3.2.3. Presence of Lead Caused Strong Baroreflex Impairment and Increased Chemoreceptor Reflex Sensitivity
3.3. Molecular Changes
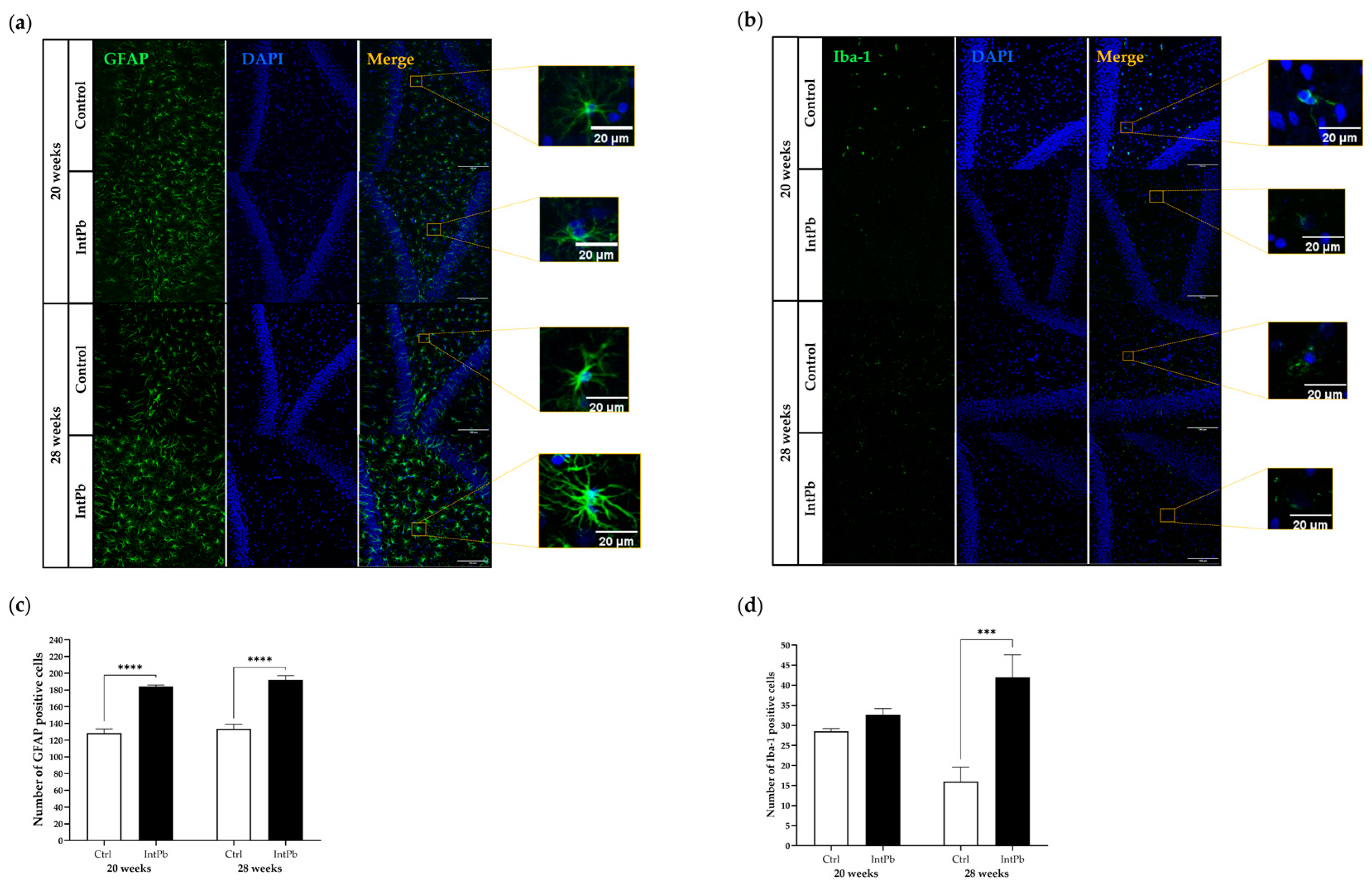
3.3.1. Intermittent Lead Exposure Caused an Increased Number of Astrocytes and Microglia with Underlying Morphological Changes
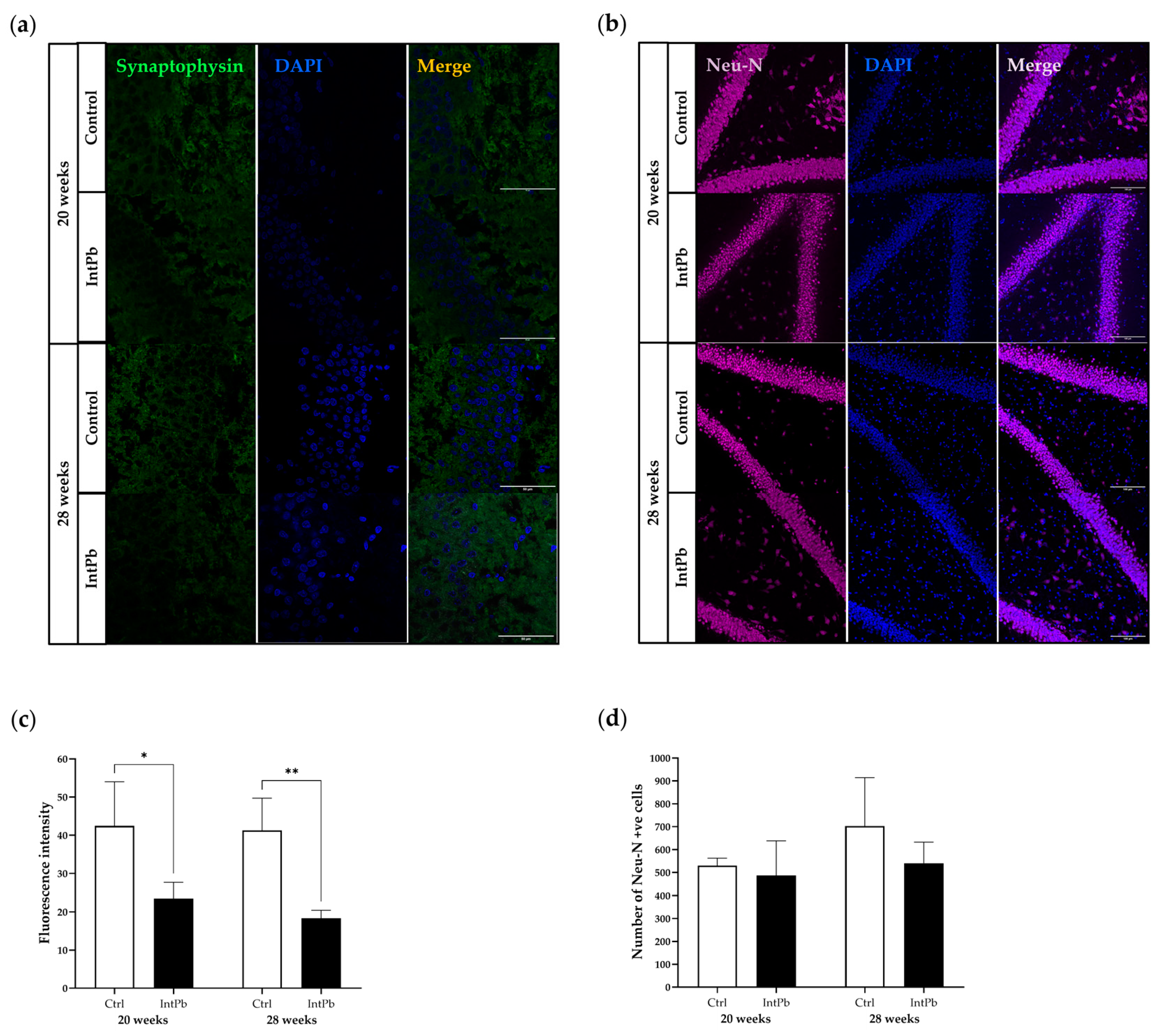
3.3.2. Intermittent Lead Exposure Causes a Decrease in the Synaptic Marker
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mason, L.H.; Harp, J.P.; Han, D.Y.; Mason, L.H.; Harp, J.P.; Han, D.Y. Pb Neurotoxicity: Neuropsychological Effects of Lead Toxicity. BioMed Res. Int. 2014, 2014, 840547. [Google Scholar] [CrossRef]
- Toscano, C.D.; Guilarte, T.R. Lead Neurotoxicity: From Exposure to Molecular Effects. Brain Res. Rev. 2005, 49, 529–554. [Google Scholar] [CrossRef] [PubMed]
- Lidsky, T.I.; Schneider, J.S. Lead Neurotoxicity in Children: Basic Mechanisms and Clinical Correlates. Brain 2003, 126, 5–19. [Google Scholar] [CrossRef] [PubMed]
- Ortega, D.R.; Esquivel, D.F.G.; Ayala, T.B.; Pineda, B.; Manzo, S.G.; Quino, J.M.; Mora, P.C.; Cruz, V.P. de la Cognitive Impairment Induced by Lead Exposure during Lifespan: Mechanisms of Lead Neurotoxicity. Toxics 2021, 9, 23. [Google Scholar] [CrossRef]
- Bellinger, D.C. Very Low Lead Exposures and Children’s Neurodevelopment. Curr. Opin. Pediatr. 2008, 20, 172–177. [Google Scholar] [CrossRef]
- Koller, K.; Brown, T.; Spurgeon, A.; Levy, L. Recent Developments in Low-Level Lead Exposure and Intellectual Impairment in Children. Environ. Health Perspect. 2004, 112, 987–994. [Google Scholar] [CrossRef]
- Valent, F.; Little, D.; Tamburlini, G.; Barbone, F. Burden of Disease Attributable to Selected Environmental Factors and Injuries among Europe’s Children and Adolescents. WHO Environ. Burd. Dis. Ser. 2004, 363, 2032–2039. [Google Scholar]
- Virgolini, M.B.; Aschner, M. Molecular Mechanisms of Lead Neurotoxicity. In Advances in Neurotoxicology; Elsevier Inc.: Amsterdam, The Netherlands, 2021; Volume 5, pp. 159–213. ISBN 9780128237755. [Google Scholar]
- Rocha, A.; Trujillo, K.A. Neurotoxicity of Low-Level Lead Exposure: History, Mechanisms of Action, and Behavioral Effects in Humans and Preclinical Models. Neurotoxicology 2019, 73, 58–80. [Google Scholar] [CrossRef]
- Chen, L.-W.; Dong, M.-H.; Kuang, F.; Liu, J.-T.; Zhang, J.-Q.; Bai, Y. Microglia and Astroglia: The Role of Neuroinflammation in Lead Toxicity and Neuronal Injury in the Brain. Neuroimmunol. Neuroinflamm. 2015, 2, 131. [Google Scholar] [CrossRef]
- Selvin-Tesla, A.; Lopez-Costa, J.J.; Nessi de Avinon, A.C.; Pecci Saavedra, J. Astroglial Alterations in Rat Hippocampus during Chronic Lead Exposure. Glia 1991, 4, 384–392. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W. Early Life Exposure to Lead (Pb) and Changes in DNA Methylation: Relevance to Alzheimer’s Disease. Rev. Environ. Health 2019, 34, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Zawia, N.H. Enhanced Taupathy and AD-like Pathology in Aged Primate Brains Decades after Infantile Exposure to Lead (Pb). Neurotoxicology 2013, 39, 95–101. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Eid, A.; Zawia, N.H. Lead Exposure and Tau Hyperphosphorylation: An In-Vitro Study. Neurotoxicology 2017, 62, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Bihaqi, S.W.; Bahmani, A.; Adem, A.; Zawia, N.H. NeuroToxicology Infantile Postnatal Exposure to Lead (Pb) Enhances Tau Expression in the Cerebral Cortex of Aged Mice: Relevance to AD. Neurotoxicology 2014, 44, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Eid, A.; Bihaqi, S.W.; Renehan, W.E.; Zawia, N.H. Developmental Lead Exposure and Lifespan Alterations in Epigenetic Regulators and Their Correspondence to Biomarkers of Alzheimer’s Disease. Alzheimer’s Dement. Diagn. Assess. Dis. Monit. 2016, 2, 123–131. [Google Scholar] [CrossRef]
- Bihaqi, S.W.; Alansi, B.; Masoud, A.M.; Mushtaq, F.; Subaiea, G.M.; Zawia, N.H. Influence of Early Life Lead (Pb) Exposure on α-Synuclein, GSK-3β and Caspase-3 Mediated Tauopathy: Implications on Alzheimer’s Disease. Curr. Alzheimer Res 2018, 15, 1114–1122. [Google Scholar] [CrossRef]
- Liu, J.T.; Chen, B.Y.; Zhang, J.Q.; Kuang, F.; Chen, L.W. Lead Exposure Induced Microgliosis and Astrogliosis in Hippocampus of Young Mice Potentially by Triggering TLR4-MyD88-NFκB Signaling Cascades. Toxicol. Lett. 2015, 239, 97–107. [Google Scholar] [CrossRef]
- Kraft, A.D.; Harry, G.J. Features of Microglia and Neuroinflammation Relevant to Environmental Exposure and Neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar] [CrossRef]
- Struzyńska, L.; Da̧browska-Bouta, B.; Koza, K.; Sulkowski, G. Inflammation-like Glial Response in Lead-Exposed Immature Rat Brain. Toxicol. Sci. 2007, 95, 156–162. [Google Scholar] [CrossRef]
- Liu, M.C.; Liu, X.Q.; Wang, W.; Shen, X.F.; Che, H.L.; Guo, Y.Y.; Zhao, M.G.; Chen, J.Y.; Luo, W.J. Involvement of Microglia Activation in the Lead Induced Long-Term Potentiation Impairment. PLoS ONE 2012, 7, e43924. [Google Scholar] [CrossRef]
- Kasten-Jolly, J.; Pabello, N.; Bolivar, V.J.; Lawrence, D.A. Developmental Lead Effects on Behavior and Brain Gene Expression in Male and Female BALB/CAnNTac Mice. Neurotoxicology 2012, 33, 1005–1020. [Google Scholar] [CrossRef] [PubMed]
- Kasten-Jolly, J.; Heo, Y.; Lawrence, D.A. Central Nervous System Cytokine Gene Expression: Modulation by Lead. J. Biochem. Mol. Toxicol. 2011, 25, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, K.L.; Kaushik, D.K.; Goswami, P.; Basu, A. Acute Exposure to Lead Acetate Activates Microglia and Induces Subsequent Bystander Neuronal Death via Caspase-3 Activation. Neurotoxicology 2014, 41, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Verstraeten, S.V.; Aimo, L.; Oteiza, P.I. Aluminium and Lead: Molecular Mechanisms of Brain Toxicity. Arch. Toxicol. 2008, 82, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Flora, G.; Gupta, D.; Tiwari, A. Toxicity of Lead: A Review with Recent Updates. Interdiscip. Toxicol. 2012, 5, 47–58. [Google Scholar] [CrossRef]
- Flora, S.J.S.; Flora, G.; Saxena, G.; Mishra, M. Arsenic and Lead Induced Free Radical Generation and Their Reversibility Following Chelation. Cell. Mol. Biol. 2007, 53, 26–47. [Google Scholar] [CrossRef]
- Saxena, G.; Pathak, U.; Flora, S.J.S. Beneficial Role of Monoesters of Meso-2,3-Dimercaptosuccinic Acid in the Mobilization of Lead and Recovery of Tissue Oxidative Injury in Rats. Toxicology 2005, 214, 39–56. [Google Scholar] [CrossRef]
- Mastitskaya, S.; Turovsky, E.; Marina, N.; Theparambil, S.M.; Hadjihambi, A.; Kasparov, S.; Teschemacher, A.G.; Ramage, A.G.; Gourine, A.V.; Hosford, P.S. Astrocytes Modulate Baroreflex Sensitivity at the Level of the Nucleus of the Solitary Tract. J. Neurosci. 2020, 40, 3052–3062. [Google Scholar] [CrossRef]
- Wu, K.L.H.; Chan, S.H.H.; Chan, J.Y.H. Neuroinflammation and Oxidative Stress in Rostral Ventrolateral Medulla Contribute to Neurogenic Hypertension Induced by Systemic Inflammation. J. Neuroinflamm. 2012, 9, 212. [Google Scholar] [CrossRef]
- Levick, S.P.; Murray, D.B.; Janicki, J.S.; Brower, G.L. Sympathetic Nervous System Modulation of Inflammation and Remodeling in the Hypertensive Heart. Hypertensionaha 2010, 55, 270–276. [Google Scholar] [CrossRef]
- Shi, P.; Diez-Freire, C.; Jun, J.Y.; Qi, Y.; Katovich, M.J.; Li, Q.; Sriramula, S.; Francis, J.; Sumners, C.; Raizada, M.K. Brain Microglial Cytokines in Neurogenic Hypertension. Hypertensionaha 2010, 56, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, B.; Liu, X.; Shen, X.Z.; Shi, P. Microglia, Autonomic Nervous System, Immunity and Hypertension: Is There a Link? Pharm. Res. 2020, 155, 104451. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Pan, W.; Xu, Y.; Zhang, J.; Wan, J.; Jiang, H. Microglia-Mediated Neuroinflammation: A Potential Target for the Treatment of Cardiovascular Diseases. J. Inflamm. Res. 2022, 15, 3083. [Google Scholar] [CrossRef]
- Shen, X.Z.; Li, Y.; Li, L.; Shah, K.H.; Bernstein, K.E.; Lyden, P.; Shi, P. Microglia Participate in Neurogenic Regulation of Hypertension. Hypertensionaha 2015, 66, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Waseem, S.; Bahmani, A.; Subaiea, G.M.; Zawia, N.H. Infantile Exposure to Lead and Late-Age Cognitive Decline: Relevance to AD. Alzheimer’s Dement. 2014, 10, 187–195. [Google Scholar] [CrossRef]
- Nigg, J.T.; Knottnerus, G.M.; Martel, M.M.; Nikolas, M.; Cavanagh, K.; Karmaus, W.; Rappley, M.D. Low Blood Lead Levels Associated with Clinically Diagnosed Attention-Deficit/Hyperactivity Disorder and Mediated by Weak Cognitive Control. Biol. Psychiatry 2008, 63, 325–331. [Google Scholar] [CrossRef]
- Sanders, T.; Liu, Y.; Buchner, V.; Tchounwou, P.B. Neurotoxic Effects and Biomarkers of Lead Exposure: A Review. Rev. Environ. Health 2009, 24, 15–45. [Google Scholar] [CrossRef] [PubMed]
- White, L.D.; Cory-Slechta, D.A.; Gilbert, M.E.; Tiffany-Castiglioni, E.; Zawia, N.H.; Virgolini, M.; Rossi-George, A.; Lasley, S.M.; Qian, Y.C.; Basha, M.R. New and Evolving Concepts in the Neurotoxicology of Lead. Toxicol. Appl. Pharm. 2007, 225, 1–27. [Google Scholar] [CrossRef]
- Goyer, R.A. Lead Toxicity: From Overt to Subclinical to Subtle Health Effects. Environ. Health Perspect. 1990, 86, 177–181. [Google Scholar] [CrossRef]
- Altmann, L.; Weinsberg, F.; Sveinsson, K.; Lilienthal, H.; Wiegand, H.; Winneke, G. Impairment of Long-Term Potentiation and Learning Following Chronic Lead Exposure. Toxicol. Lett. 1993, 66, 105–112. [Google Scholar] [CrossRef]
- Ordemann, J.M.; Austin, R.N. Lead Neurotoxicity: Exploring the Potential Impact of Lead Substitution in Zinc-Finger Proteins on Mental Health. Metallomics 2016, 8, 579–588. [Google Scholar] [CrossRef]
- Kuhlmann, A.C.; McGlothan, J.L.; Guilarte, T.R. Developmental Lead Exposure Causes Spatial Learning Deficits in Adult Rats. Neurosci. Lett. 1997, 233, 101–104. [Google Scholar] [CrossRef]
- Shvachiy, L. Comparative Study on the Physiological Dynamics Evoked by Different Profiles of Lead Exposure. Ph.D. Thesis, Universidade de Lisboa, Lisboa, Portugal, 2018. [Google Scholar]
- Shvachiy, L.; Amaro-Leal, Â.; Outeiro, T.F.; Rocha, I.; Geraldes, V. From Molecular to Functional Effects of Different Environmental Lead Exposure Paradigms. Biology 2022, 11, 1164. [Google Scholar] [CrossRef]
- Shvachiy, L.; Geraldes, V.; Amaro-Leal, Â.; Rocha, I. Intermittent Low-Level Lead Exposure Provokes Anxiety, Hypertension, Autonomic Dysfunction and Neuroinflammation. Neurotoxicology 2018, 69, 307–319. [Google Scholar] [CrossRef]
- Geraldes, V.; Carvalho, M.; Goncalves-Rosa, N.; Tavares, C.; Laranjo, S.; Rocha, I. Lead Toxicity Promotes Autonomic Dysfunction with Increased Chemoreceptor Sensitivity. Neurotoxicology 2016, 54, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, D.; Carvalho, M.L.; Geraldes, V.; Rocha, I.; Alves, L.C.; Santos, J.P. Lead in Liver and Kidney of Exposed Rats: Aging Accumulation Study. J. Trace Elem. Med. Biol. 2012, 26, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Shvachiy, L.; Geraldes, V.; Amaro-Leal, Â.; Rocha, I. Persistent Effects on Cardiorespiratory and Nervous Systems Induced by Long-Term Lead Exposure: Results from a Longitudinal Study. Neurotox. Res 2020, 37, 857–870. [Google Scholar] [CrossRef] [PubMed]
- dos Santos Alves, R.; de Souza, A.S. The Open Field Test. Igarss 2014, 2014, 1–5. [Google Scholar] [CrossRef]
- Antunes, M.; Biala, G. The Novel Object Recognition Memory: Neurobiology, Test Procedure, and Its Modifications. Cogn. Process 2012, 13, 93–110. [Google Scholar] [CrossRef]
- Costa, R.; Tamascia, M.L.; Nogueira, M.D.; Casarini, D.E.; Marcondes, F.K. Handling of Adolescent Rats Improves Learning and Memory and Decreases Anxiety. J. Am. Assoc. Lab. Anim. Sci. 2012, 51, 548–553. [Google Scholar]
- Buccafusco, J.J. Methods of Behavior Analysis in Neurosciences; CRC Tress LLC: Boca Raton, FL, USA, 2001; Volume 3, ISBN 084930704X. [Google Scholar]
- Ramos, A. Animal Models of Anxiety: Do I Need Multiple Tests? Trends Pharm. Sci 2008, 29, 493–498. [Google Scholar] [CrossRef]
- Moreira, E.G.; Vassilieff, I.; Vassilieff, V.S. Developmental Lead Exposure: Behavioral Alterations in the Short and Long Term. Neurotoxicol. Teratol. 2001, 23, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Mouro, F.M.; Batalha, V.L.; Ferreira, D.G.; Coelho, J.E.; Baqi, Y.; Müller, C.E.; Lopes, L.V.; Ribeiro, J.A.; Sebastião, A.M. Chronic and Acute Adenosine A2A Receptor Blockade Prevents Long-Term Episodic Memory Disruption Caused by Acute Cannabinoid CB1 Receptor Activation. Neuropharmacology 2017, 117, 316–327. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Sun, Z.; Lee, T.; Fusco, F.R.; Kimble, T.D.; Meade, C.A.; Cuthbertson, S.; Reiner, A. A Simple and Sensitive Antigen Retrieval Method for Free-Floating and Slide-Mounted Tissue Sections. J. Neurosci. Methods 1999, 93, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Meth. 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and Pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Hol, E.M.; Pekny, M. Glial Fibrillary Acidic Protein (GFAP) and the Astrocyte Intermediate Filament System in Diseases of the Central Nervous System. Curr. Opin. Cell. Biol. 2015, 32, 121–130. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Allan, S.M.; Rothwell, N.J. Inflammation in Central Nervous System Injury. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 1669–1677. [Google Scholar] [CrossRef]
- Liu, L.; Chan, C. The Role of Inflammasome in Alzheimer’s Disease. Ageing Res. Rev. 2014, 15, 6–15. [Google Scholar] [CrossRef]
- Chen, C.; Jiang, Z.Y.; Fu, X.; Yu, D.; Huang, H.; Tasker, J.G. Astrocytes Amplify Neuronal Dendritic Volume Transmission Stimulated by Norepinephrine. Cell. Rep. 2019, 29, 4349–4361.e4. [Google Scholar] [CrossRef]
- Martin-Fernandez, M.; Jamison, S.; Robin, L.M.; Zhao, Z.; Martin, E.D.; Aguilar, J.; Benneyworth, M.A.; Marsicano, G.; Araque, A. Synapse-Specific Astrocyte Gating of Amygdala-Related Behavior. Nat. Neurosci. 2017, 20, 1540–1548. [Google Scholar] [CrossRef]
- Bernal, G.M.; Peterson, D.A. Phenotypic and Gene Expression Modification with Normal Brain Aging in GFAP-Positive Astrocytes and Neural Stem Cells. Aging Cell. 2011, 10, 466–482. [Google Scholar] [CrossRef] [PubMed]
- Karimi-Abdolrezaee, S.; Billakanti, R. Reactive Astrogliosis after Spinal Cord Injury-Beneficial and Detrimental Effects. Mol. Neurobiol. 2012, 46, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Chekhonin, V.P.; Lebedev, S.V.; Dmitrieva, T.B.; Blinov, D.V.; Gurina, O.I.; Semenova, A.V.; Volodin, N.N. Enzyme Immunoassay of NSE and GFAP as the Criterion of Dynamic Evaluation of the Rat Blood-Brain Barrier in Perinatal Hypoxic Ischemic Injury of the CNS. Bull. Exp. Biol. Med. 2003, 136, 261–265. [Google Scholar] [CrossRef]
- Repko, J. Behavioral Toxicology of Inorganic Lead Exposure. In Health Effects of Occupational Lead and Arsenic Exposure, A Symposium, Division of Surveillance, Hazard Evaluation and Field Studies; Contract No. 210-75-0026; NIOSH: Cincinnati, OH, USA, 1976; pp. 59–72. [Google Scholar]
- Driscoll, J.W.; Stegner, S.E. Behavioral Effects of Chronic Lead Ingestion on Laboratory Rats. Pharm. Biochem. Behav. 1976, 4, 411–417. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Ma, Y.; Ni, L.; Zhao, S.; Li, L.; Zhang, J.; Fan, M.; Liang, C.; Cao, J.; Xu, L. Lead Exposure through Gestation-Only Caused Long-Term Learning/Memory Deficits in Young Adult Offspring. Exp. Neurol. 2003, 184, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, G.A.; Musabegovic, A.; Liu, X.; Kline, J.; Factor-Litvak, P.; Graziano, J.H. Lead Exposure and Motor Functioning in 4½-Year-Old Children: The Yugoslavia Prospective Study. J. Pediatr. 2000, 137, 555–561. [Google Scholar] [CrossRef]
- Lasley, S.M.; Gilbert, M.E. Presynaptic Glutamatergic Function in Dentate Gyrus in vivo Is Diminished by Chronic Exposure to Inorganic Lead. Brain Res. 1996, 736, 125–134. [Google Scholar] [CrossRef]
- Baghurst, P.A.; McMichael, A.J.; Wigg, N.R.; Vimpani, G.V.; Robertson, E.F.; Roberts, R.J.; Tong, S.-L. Environmental Exposure to Lead and Children’s Intelligence at the Age of Seven Years. N. Engl. J. Med. 2010, 327, 1279–1284. [Google Scholar] [CrossRef]
- Hu, F.; Xu, L.; Liu, Z.-H.; Ge, M.-M.; Ruan, D.-Y.; Wang, H.-L. Developmental Lead Exposure Alters Synaptogenesis through Inhibiting Canonical Wnt Pathway in vivo and in vitro. PLoS ONE 2014, 9, e101894. [Google Scholar] [CrossRef] [PubMed]
- Lasley, S.M.; Gilbert, M.E. Impairment of Synaptic Function by Exposure to Lead. Mol. Neurobiol. 2004, 42, 217–241. [Google Scholar]
- Chibowska, K.; Baranowska-Bosiacka, I.; Falkowska, A.; Gutowska, I.; Goschorska, M.; Chlubek, D. Molecular Sciences Effect of Lead (Pb) on Inflammatory Processes in the Brain. Int. J. Mol. Sci. 2016, 17, 2140. [Google Scholar] [CrossRef]
- Şahin, T.D.; Karson, A.; Balci, F.; Yazir, Y.; Bayramgürler, D.; Utkan, T. TNF-Alpha Inhibition Prevents Cognitive Decline and Maintains Hippocampal BDNF Levels in the Unpredictable Chronic Mild Stress Rat Model of Depression. Behav. Brain Res. 2015, 292, 233–240. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Benarroch, E.E. Central Control of Autonomic Function and Involvement in Neurodegenerative Disorders. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2013; Volume 117, pp. 45–57. [Google Scholar]
- Geraldes, V.; Goncalves-Rosa, N.; Liu, B.; Paton, J.F.R.; Rocha, I. Essential Role of RVL Medullary Neuronal Activity in the Long Term Maintenance of Hypertension in Conscious SHR. Auton. Neurosci. 2014, 186, 22–31. [Google Scholar] [CrossRef]
- Dampney, R.A.L. Central Neural Control of the Cardiovascular System: Current Perspectives. Adv. Physiol. Educ. 2016, 40, 283–296. [Google Scholar] [CrossRef]
- LeBouef, T.; Yaker, Z.; Whited, L. Physiology, Autonomic Nervous System. StatPearls. 2022. Available online: https://www.ncbi.nlm.nih.gov/books/NBK538516/ (accessed on 14 January 2023).
- Bankenahally, R.; Da, M.; Fcai, F.; Krovvidi, H.; Frca, M.M. Autonomic Nervous System: Anatomy, Physiology, and Relevance in Anaesthesia and Critical Care Medicine. BJA Educ. 2016, 16, 381–387. [Google Scholar] [CrossRef]
- Mccorry, L.K. Physiology of the Autonomic Nervous System. Am. J. Pharm. Educ. 2007, 71, 78. [Google Scholar] [CrossRef]
- Perez, C.M.; Hazari, M.S.; Farraj, A.K. Role of Autonomic Reflex Arcs in Cardiovascular Responses to Air Pollution Exposure. Cardiovasc. Toxicol. 2015, 15, 69–78. [Google Scholar] [CrossRef]
- Davidson, N.S.; Goldner, S.; McCloskey, D.I. Respiratory Modulation of Barareceptor and Chemoreceptor Reflexes Affecting Heart Rate and Cardiac Vagal Efferent Nerve Activity. J. Physiol. 1976, 259, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Segiet, A.; Smykiewicz, P.; Kwiatkowski, P.; Żera, T. Tumour Necrosis Factor and Interleukin 10 in Blood Pressure Regulation in Spontaneously Hypertensive and Normotensive Rats. Cytokine 2019, 113, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Santisteban, M.M.; Ahmari, N.; Carvajal, J.M.; Zingler, M.B.; Qi, Y.; Kim, S.; Joseph, J.; Garcia-Pereira, F.; Johnson, R.D.; Shenoy, V.; et al. Involvement of Bone Marrow Cells and Neuroinflammation in Hypertension. Circ. Res. 2015, 117, 178–191. [Google Scholar] [CrossRef]
- Boscolo, P.; Carmignani, M. Neurohumoral Blood Pressure Regulation in Lead Exposure. Environ. Health Perspect. 1988, 78, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, V.; Goncalves-Rosa, N.; Tavares, C.; Liu, B.; Paton, J.; Rocha, I. 6B.02: Hypothalamic and Medullar Mechanisms for Long-Term Autonomic Regulation of Arterial Blood Pressure. J. Hypertens. 2015, 33, e76. [Google Scholar] [CrossRef]
- Misselwitz, B.; Mühler, A.; Weinmann, H.J. A Toxicologic Risk for Using Manganese Complexes? A Literature Survey of Existing Data through Several Medical Specialties. Investig. Radiol. 1995, 30, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Geraldes, V.; Gonçalves-Rosa, N.; Liu, B.; Paton, J.F.R.; Rocha, I. Chronic Depression of Hypothalamic Paraventricular Neuronal Activity Produces Sustained Hypotension in Hypertensive Rats. Exp. Physiol. 2014, 99, 89–100. [Google Scholar] [CrossRef]
- Rocha, I.; Rosário, L.B.; de Oliveira, E.I.; Barros, M.A.; Silva-Carvallho, L. Enhancement of Carotid Chemoreceptor Reflex and Cardiac Chemosensitive Reflex in the Acute Phase of Myocardial Infarction of the Anesthetized Rabbit. Basic Res. Cardiol. 2003, 98, 175–180. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Age | Group | Blood Lead Levels (μg/dL) | Weight (g) | Food Intake (g) | Liquid Intake (mL) | Produced Feces (g) | Produced Urine (mL) |
|---|---|---|---|---|---|---|---|
| 12 weeks | Ctrl | <0.1 | 358 ± 95 | 25 ± 2 | 33 ± 7 | 12 ± 4 | 19 ± 4 |
| IntPb | 24.0 ± 3.1 | 345 ± 92 ns | 22 ± 3 ns | 22 ± 2 * | 9 ± 3 ns | 15 ± 4 ns | |
| 20 weeks | Ctrl | <0.1 | 386 ± 116 | 23 ± 9 | 39 ± 8 | 12 ± 3 | 17 ± 6 |
| IntPb | 5.8 ± 0.7 | 390 ± 99 ns | 25 ± 3 ns | 35 ± 0 ns | 8 ± 2 ns | 14 ± 4 ns | |
| 28 weeks | Ctrl | <0.1 | 333 ± 101 | 23 ± 5 | 24 ± 1 | 8 ± 1 | 11 ± 3 |
| IntPb | 20.5 ± 2.7 | 428 ± 123 ns | 24± 4 ns | 25 ± 5 ns | 12 ± 5 ns | 11 ± 1 ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shvachiy, L.; Amaro-Leal, Â.; Outeiro, T.F.; Rocha, I.; Geraldes, V. Intermittent Lead Exposure Induces Behavioral and Cardiovascular Alterations Associated with Neuroinflammation. Cells 2023, 12, 818. https://doi.org/10.3390/cells12050818
Shvachiy L, Amaro-Leal Â, Outeiro TF, Rocha I, Geraldes V. Intermittent Lead Exposure Induces Behavioral and Cardiovascular Alterations Associated with Neuroinflammation. Cells. 2023; 12(5):818. https://doi.org/10.3390/cells12050818
Chicago/Turabian StyleShvachiy, Liana, Ângela Amaro-Leal, Tiago F. Outeiro, Isabel Rocha, and Vera Geraldes. 2023. "Intermittent Lead Exposure Induces Behavioral and Cardiovascular Alterations Associated with Neuroinflammation" Cells 12, no. 5: 818. https://doi.org/10.3390/cells12050818
APA StyleShvachiy, L., Amaro-Leal, Â., Outeiro, T. F., Rocha, I., & Geraldes, V. (2023). Intermittent Lead Exposure Induces Behavioral and Cardiovascular Alterations Associated with Neuroinflammation. Cells, 12(5), 818. https://doi.org/10.3390/cells12050818