

Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics
 , , , , ,
, , , , , 
Abstract
1. Introduction
2. Clinical Evidence of Neurological Involvement in Long COVID
2.1. Epidemiology
2.2. Risk Factors
2.3. Outcomes
3. Mechanisms of Neurological Long COVID and Review of Therapeutics
3.1. Viral Neuroinvasion and Persistent Viral Shedding
3.1.1. Invasion of Olfactory Epithelium
3.1.2. Dysbiosis and Brain–Gut Axis
3.1.3. Reactivation of Herpesviruses
3.1.4. Related Long COVID Therapeutics: Antivirals
3.1.5. Related Long COVID Therapies: Anosmia
3.2. Abnormal Systemic and Neurologic Immunological Response
3.2.1. Systemic Inflammation
3.2.2. Monocyte Expansion and T Cell Dysfunction
3.2.3. Autoantibody Generation
3.2.4. Related Long COVID Therapies: Anti-Inflammatory Therapy
3.2.5. Neural Glial Cell Reactivity
3.3. Coaguloapathies and Endotheliopathy-Associated Neurovascular Injury
3.3.1. Microclot Formation
3.3.2. Antiphospholipid Antibodies
3.3.3. Endotheliopathy
3.3.4. Blood–Brain Barrier Disruption
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sudre, C.H.; Murray, B.; Varsavsky, T. Attributes and predictors of long COVID [published correction appears in Nat Med]. Nat. Med. 2021, 27, 626–631. [Google Scholar] [CrossRef]
- Groff, D.; Sun, A.; Ssentongo, A.E. Short-term and Long-term Rates of Postacute Sequelae of SARS-CoV-2 Infection: A Systematic Review. JAMA Netw. Open 2021, 4, e2128568. [Google Scholar] [CrossRef]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Alkodaymi, M.S.; Omrani, O.A.; Fawzy, N.A. Prevalence of post-acute COVID-19 syndrome symptoms at different follow-up periods: A systematic review and meta-analysis. Clin. Microbiol. Infect. 2022, 28, 657–666. [Google Scholar] [CrossRef]
- Premraj, L.; Kannapadi, N.V.; Briggs, J. Mid and long-term neurological and neuropsychiatric manifestations of post-COVID-19 syndrome: A meta-analysis. J. Neurol. Sci. 2022, 434, 120162. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Huang, L.; Wang, Y. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Stefanou, M.I.; Palaiodimou, L.; Bakola, E. Neurological manifestations of long-COVID syndrome: A narrative review. Ther. Adv. Chronic Dis. 2022, 13, 20406223221076890. [Google Scholar] [CrossRef]
- Nersesjan, V.; Amiri, M.; Lebech, A.M. Central and peripheral nervous system complications of COVID-19: A prospective tertiary center cohort with 3-month follow-up. J. Neurol. 2021, 268, 3086–3104. [Google Scholar] [CrossRef]
- Beghi, E.; Giussani, G.; Westenberg, E. Acute and post-acute neurological manifestations of COVID-19: Present findings, critical appraisal, and future directions. J. Neurol. 2022, 269, 2265–2274. [Google Scholar] [CrossRef]
- Estiri, H.; Strasser, Z.H.; Brat, G.A. Evolving Phenotypes of non-hospitalized Patients that Indicate Long COVID. medRxiv 2021. [Google Scholar] [CrossRef]
- Taquet, M.; Dercon, Q.; Luciano, S.; Geddes, J.R.; Husain, M.; Harrison, P.J. Incidence, co-occurrence, and evolution of long-COVID features: A 6-month retrospective cohort study of 273,618 survivors of COVID-19. PLoS Med. 2021, 18, e1003773. [Google Scholar] [CrossRef]
- Graham, E.L.; Clark, J.R.; Orban, Z.S.; Lim, P.H.; Szymanski, A.L.; Taylor, C.; DiBiase, R.M.; Jia, D.T.; Balabanov, R.; Ho, S.U.; et al. Persistent neurologic symptoms and cognitive dysfunction in non-hospitalized COVID-19 “long haulers”. Ann. Clin. Transl. Neurol. 2021, 8, 1073–1085. [Google Scholar] [CrossRef]
- Bai, F.; Tomasoni, D.; Falcinella, C. Female gender is associated with long COVID syndrome: A prospective cohort study. Clin. Microbiol. Infect. 2022, 28, 611.e9–611.e16. [Google Scholar] [CrossRef]
- Hill, E.; Mehta, H.; Sharma, S. Risk Factors Associated with Post-Acute Sequelae of SARS-CoV-2 in an EHR Cohort: A National COVID Cohort Collaborative (N3C) Analysis as Part of the NIH RECOVER Program. medRxiv 2022. medRxiv:2022.08.15.22278603. [Google Scholar] [CrossRef]
- Staffolani, S.; Iencinella, V.; Cimatti, M.; Tavio, M. Long COVID-19 syndrome as a fourth phase of SARS-CoV-2 infection. Infez. Med. 2022, 30, 22–29. [Google Scholar] [CrossRef]
- Yoo, S.M.; Liu, T.C.; Motwani, Y. Factors Associated with Post-Acute Sequelae of SARS-CoV-2 (PASC) After Diagnosis of Symptomatic COVID-19 in the Inpatient and Outpatient Setting in a Diverse Cohort. J. Gen. Intern. Med. 2022, 37, 1988–1995. [Google Scholar] [CrossRef]
- Parotto, M.; Myatra, S.N.; Munblit, D.; Elhazmi, A.; Ranzani, O.T.; Herridge, M.S. Recovery after prolonged ICU treatment in patients with COVID-19. Lancet Respir. Med. 2021, 9, 812–814. [Google Scholar] [CrossRef]
- Graham, E.L.; Koralnik, I.J.; Liotta, E.M. Therapeutic Approaches to the Neurologic Manifestations of COVID-19. Neurotherapeutics 2022, 19, 1435–1466. [Google Scholar] [CrossRef]
- Iosifescu, A.L.; Hoogenboom, W.S.; Buczek, A.J.; Fleysher, R.; Duong, T.Q. New-onset and persistent neurological and psychiatric sequelae of COVID-19 compared to influenza: A retrospective cohort study in a large New York City healthcare network. Int. J. Methods Psychiatr. Res. 2022, 31, e1914. [Google Scholar] [CrossRef]
- Su, Y.; Yuan, D.; Chen, D.G. Multiple early factors anticipate post-acute COVID-19 sequelae. Cell 2022, 185, 881–895. [Google Scholar] [CrossRef]
- Scherer, P.E.; Kirwan, J.P.; Rosen, C.J. Post-acute sequelae of COVID-19: A metabolic perspective. Elife 2022, 11, e78200. [Google Scholar] [CrossRef]
- Peluso, M.J.; Deeks, S.G. Early clues regarding the pathogenesis of long-COVID. Trends Immunol. 2022, 43, 268–270. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, H.; Fokaidis, V. Identifying Contextual and Spatial Risk Factors for Post-Acute Sequelae of SARS-CoV-2 Infection: An EHR-Based Cohort Study from the RECOVER Program. medRxiv 2022. medRxiv:2022.10.13.22281010. [Google Scholar] [CrossRef]
- Mainous, A.G., III; Rooks, B.J.; Wu, V.; Orlando, F.A. COVID-19 Post-acute Sequelae Among Adults: 12 Month Mortality Risk. Front. Med. 2021, 8, 2351. [Google Scholar] [CrossRef]
- Meza-Torres, B.; Delanerolle, G.; Okusi, C. Differences in Clinical Presentation with Long COVID after Community and Hospital Infection and Associations with All-Cause Mortality: English Sentinel Network Database Study. JMIR Public Health Surveill. 2022, 8, e37668. [Google Scholar] [CrossRef]
- Gao, P.; Liu, J.; Liu, M. Effect of COVID-19 Vaccines on Reducing the Risk of Long COVID in the Real World: A Systematic Review and Meta-Analysis. Int. J. Environ. Res. Public Health 2022, 19, 12422. [Google Scholar] [CrossRef]
- Strain, W.D.; Sherwood, O.; Banerjee, A.; Togt, V.; Hishmeh, L.; Rossman, J. The Impact of COVID Vaccination on Symptoms of Long COVID: An International Survey of People with Lived Experience of Long COVID. Vaccines 2022, 10, 652. [Google Scholar] [CrossRef]
- Bakhshandeh, B.; Sorboni, S.G.; Javanmard, A.R.; Mottaghi, S.S.; Mehrabi, M.R.; Sorouri, F.; Abbasi, A.; Jahanafrooz, Z. Variants in ACE2; potential influences on virus infection and COVID-19 severity. Infect. Genet. Evol. J. Mol. Epidemiol. Evol. Genet. Infect. Dis. 2021, 90, 104773. [Google Scholar] [CrossRef]
- Möhlendick, B.; Schönfelder, K.; Breuckmann, K.; Elsner, C.; Babel, N.; Balfanz, P.; Dahl, E.; Dreher, M.; Fistera, D.; Herbstreit, F.; et al. ACE2 polymorphism and susceptibility for SARS-CoV-2 infection and severity of COVID-19. Pharm. Genom. 2021, 31, 165–171. [Google Scholar] [CrossRef]
- Maglietta, G.; Diodati, F.; Puntoni, M.; Lazzarelli, S.; Marcomini, B.; Patrizi, L.; Caminiti, C. Prognostic Factors for Post-COVID-19 Syndrome: A Systematic Review and Meta-Analysis. J. Clin. Med. 2022, 11, 1541. [Google Scholar] [CrossRef]
- Fernández-de-Las-Peñas, C.; Arendt-Nielsen, L.; Díaz-Gil, G.; Gómez-Esquer, F.; Gil-Crujera, A.; Gómez-Sánchez, S.M.; Ambite-Quesada, S.; Palomar-Gallego, M.A.; Pellicer-Valero, O.J.; Giordano, R. Genetic Association between ACE2 (rs2285666 and rs2074192) and TMPRSS2 (rs12329760 and rs2070788) Polymorphisms with Post-COVID Symptoms in Previously Hospitalized COVID-19 Survivors. Genes 2022, 13, 1935. [Google Scholar] [CrossRef]
- Gallagher, T.M.; Buchmeier, M.J. Coronavirus spike proteins in viral entry and pathogenesis. Virology 2001, 279, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pöhlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003, 426, 450–454. [Google Scholar] [CrossRef]
- Song, E.; Zhang, C.; Israelow, B. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef]
- Meinhardt, J.; Radke, J.; Dittmayer, C. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Brann, D.H.; Tsukahara, T.; Weinreb, C. Non-neuronal expression of SARS-CoV-2 entry genes in the olfactory system suggests mechanisms underlying COVID-19-associated anosmia. Sci. Adv. 2020, 6, eabc5801. [Google Scholar] [CrossRef] [PubMed]
- Kandemirli, S.G.; Altundag, A.; Yildirim, D.; Tekcan Sanli, D.E.; Saatci, O. Olfactory Bulb MRI and Paranasal Sinus CT Findings in Persistent COVID-19 Anosmia. Acad. Radiol. 2021, 28, 28–35. [Google Scholar] [CrossRef]
- Butowt, R.; Meunier, N.; Bryche, B.; Bartheld, C.S. The olfactory nerve is not a likely route to brain infection in COVID-19: A critical review of data from humans and animal models. Acta Neuropathol. 2021, 141, 809–822. [Google Scholar] [CrossRef]
- Pellegrini, L.; Albecka, A.; Mallery, D.L. SARS-CoV-2 Infects the Brain Choroid Plexus and Disrupts the Blood-CSF Barrier in Human Brain Organoids. Cell Stem Cell 2020, 27, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Muhl, L.; He, L.; Sun, Y. The SARS-CoV-2 receptor ACE2 is expressed in mouse pericytes but not endothelial cells: Implications for COVID-19 vascular research. Stem Cell Rep. 2022, 17, 1089–1104. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.; Frontera, J.; Placantonakis, D.G.; Galetta, S.; Balcer, L.; Melmed, K.R. Cerebrospinal fluid from COVID-19 patients with olfactory/gustatory dysfunction: A review. Clin. Neurol. Neurosurg. 2021, 207, 106760. [Google Scholar] [CrossRef]
- Song, E.; Bartley, C.M.; Chow, R.D. Divergent and self-reactive immune responses in the CNS of COVID-19 patients with neurological symptoms. Cell Rep. Med. 2021, 2, 100288. [Google Scholar] [CrossRef]
- Matschke, J.; Lütgehetmann, M.; Hagel, C. Neuropathology of patients with COVID-19 in Germany: A post-mortem case series. Lancet Neurol. 2020, 19, 919–929. [Google Scholar] [CrossRef]
- Thakur, K.T.; Miller, E.H.; Glendinning, M.D. COVID-19 neuropathology at Columbia University Irving Medical Center/New York Presbyterian Hospital. Brain 2021, 144, 2696–2708. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.C.; Kern, F.; Losada, P.M. Dysregulation of brain and choroid plexus cell types in severe COVID-19 [published correction appears in Nature. Nature 2021, 595, 565–571. [Google Scholar] [CrossRef]
- Ye, Q.; Zhou, J.; He, Q. SARS-CoV-2 infection in the mouse olfactory system. Cell Discov. 2021, 7, 49. [Google Scholar] [CrossRef]
- Tsivgoulis, G.; Fragkou, P.C.; Lachanis, S. Olfactory bulb and mucosa abnormalities in persistent COVID-19-induced anosmia: A magnetic resonance imaging study. Eur. J. Neurol. 2021, 28, e6–e8. [Google Scholar] [CrossRef]
- Yamagishi, M.; Hasegawa, S.; Nakano, Y. Examination and classification of human olfactory mucosa in patients with clinical olfactory disturbances. Arch. Otorhinolaryngol. 1988, 245, 316–320. [Google Scholar] [CrossRef]
- Vaira, L.A.; Hopkins, C.; Sandison, A. Olfactory epithelium histopathological findings in long-term coronavirus disease 2019 related anosmia. J. Laryngol. Otol. 2020, 134, 1123–1127. [Google Scholar] [CrossRef]
- de Melo, G.D.; Lazarini, F.; Levallois, S.; Hautefort, C.; Michel, V.; Larrous, F.; Verillaud, B.; Aparicio, C.; Wagner, S.; Gheusi, G.; et al. COVID-19-related anosmia is associated with viral persistence and inflammation in human olfactory epithelium and brain infection in hamsters. Sci. Transl. Med. 2021, 13, eabf8396. [Google Scholar] [CrossRef]
- Wu, Y.; Guo, C.; Tang, L. Prolonged presence of SARS-CoV-2 viral RNA in faecal samples. Lancet Gastroenterol. Hepatol. 2020, 5, 434–435. [Google Scholar] [CrossRef]
- Sun, J.; Xiao, J.; Sun, R. Prolonged Persistence of SARS-CoV-2 RNA in Body Fluids. Emerg. Infect. Dis. 2020, 26, 1834–1838. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y. Alterations in Gut Microbiota of Patients with COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef]
- Nataf, S.; Pays, L. Molecular Insights into SARS-CoV2-Induced Alterations of the Gut/Brain Axis. Int. J. Mol. Sci. 2021, 22, 10440. [Google Scholar] [CrossRef]
- Daulagala, S.W.P.L.; Noordeen, F. Epidemiology and factors influencing varicella infections in tropical countries including Sri Lanka. Virusdisease 2018, 29, 277–284. [Google Scholar] [CrossRef]
- Smatti, M.K.; Al-Sadeq, D.W.; Ali, N.H.; Pintus, G.; Abou-Saleh, H.; Nasrallah, G.K. Epstein-Barr Virus Epidemiology, Serology, and Genetic Variability of LMP-1 Oncogene Among Healthy Population: An Update. Front. Oncol. 2018, 8, 211. [Google Scholar] [CrossRef]
- Meng, M.; Zhang, S.; Dong, X.; Sun, W.; Deng, Y.; Li, W.; Li, R.; Annane, D.; Wu, Z.; Chen, D. COVID-19 associated EBV reactivation and effects of ganciclovir treatment. Immun. Inflamm. Dis. 2022, 10, e597. [Google Scholar] [CrossRef] [PubMed]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of Long COVID Prevalence and Its Relationship to Epstein-Barr Virus Reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Pablos, M.; Paiva, B.; Montero-Mateo, R.; Garcia, N.; Zabaleta, A. Epstein-Barr Virus and the Origin of Myalgic Encephalomyelitis or Chronic Fatigue Syndrome. Front. Immunol. 2021, 12, 656797. [Google Scholar] [CrossRef] [PubMed]
- Monje, M.; Iwasaki, A. The neurobiology of long COVID. Neuron 2022, 110, 3484–3496. [Google Scholar] [CrossRef]
- Klein, J.; Wood, J.; Jaycox, J.; Lu, P.; Dhodapkar, R.M.; Gehlhausen, J.R.; Tabachnikova, A.; Tabacof, L.; Malik, A.A.; Kamath, K.; et al. Distinguishing features of Long COVID identified through immune profiling. medRxiv 2022. medRxiv:2022.08.09.22278592. [Google Scholar] [CrossRef]
- Martinez-Reviejo, R.; Tejada, S.; Adebanjo, G.A.R.; Chello, C.; Machado, M.C.; Parisella, F.R.; Campins, M.; Tammaro, A.; Rello, J. Varicella-Zoster virus reactivation following severe acute respiratory syndrome coronavirus 2 vaccination or infection: New insights. Eur. J. Intern. Med. 2022, 104, 73–79. [Google Scholar] [CrossRef]
- Wen, W.; Chen, C.; Tang, J. Efficacy and safety of three new oral antiviral treatment (molnupiravir, fluvoxamine and Paxlovid) for COVID-19: A meta-analysis. Ann. Med. 2022, 54, 516–523. [Google Scholar] [CrossRef]
- Hammond, J.; Leister-Tebbe, H.; Gardner, A. Oral Nirmatrelvir for High-Risk, Nonhospitalized Adults with COVID-19. N. Engl. J. Med. 2022, 386, 1397–1408. [Google Scholar] [CrossRef] [PubMed]
- Dawood, A.A. The efficacy of Paxlovid against COVID-19 is the result of the tight molecular docking between Mpro and antiviral drugs (nirmatrelvir and ritonavir) [published online ahead of print, 2 November 2022]. Adv. Med. Sci. 2022, 68, 1–9. [Google Scholar] [CrossRef]
- Xie, Y.; Choi, T.; Al-Aly, Z. Nirmatrelvir and the Risk of Post-Acute Sequelae of COVID-19. Infectious Diseases (except HIV/AIDS). medRixv 2022. [Google Scholar] [CrossRef]
- Singh, C.V.; Jain, S.; Parveen, S. The outcome of fluticasone nasal spray on anosmia and triamcinolone oral paste in dysgeusia in COVID-19 patients. Am. J. Otolaryngol. 2021, 42, 102892. [Google Scholar] [CrossRef]
- Abdelalim, A.A.; Mohamady, A.A.; Elsayed, R.A.; Elawady, M.A.; Ghallab, A.F. Corticosteroid nasal spray for recovery of smell sensation in COVID-19 patients: A randomized controlled trial. Am. J. Otolaryngol. 2021, 42, 102884. [Google Scholar] [CrossRef] [PubMed]
- O’Kelly, B.; Vidal, L.; McHugh, T.; Woo, J.; Avramovic, G.; Lambert, J.S. Safety and efficacy of low dose naltrexone in a long covid cohort; an interventional pre-post study. Brain Behav. Immun. Health 2022, 24, 100485. [Google Scholar] [CrossRef] [PubMed]
- Mohamad, S.A.; Badawi, A.M.; Mansour, H.F. Insulin fast-dissolving film for intranasal delivery via olfactory region, a promising approach for the treatment of anosmia in COVID-19 patients: Design, in-vitro characterization and clinical evaluation. Int. J. Pharm. 2021, 601, 120600. [Google Scholar] [CrossRef] [PubMed]
- Rezaeian, A. Effect of Intranasal Insulin on Olfactory Recovery in Patients with Hyposmia: A Randomized Clinical Trial. Otolaryngol. Head Neck Surg. 2018, 158, 1134–1139. [Google Scholar] [CrossRef]
- Glynne, P.; Tahmasebi, N.; Gant, V.; Gupta, R. Long COVID following mild SARS-CoV-2 infection: Characteristic T cell alterations and response to antihistamines. J. Investig. Med. Off. Publ. Am. Fed. Clin. Res. 2022, 70, 61–67. [Google Scholar] [CrossRef]
- Nguyen, L.C.; Yang, D.; Nicolaescu, V.; Best, T.J.; Ohtsuki, T.; Chen, S.N.; Friesen, J.B.; Drayman, N.; Mohamed, A.; Dann, C.; et al. Cannabidiol Inhibits SARS-CoV-2 Replication and Promotes the Host Innate Immune Response. bioRxiv 2021. bioRxiv:2021.03.10.432967. [Google Scholar] [CrossRef]
- Esposito, G.; Pesce, M.; Seguella, L.; Sanseverino, W.; Lu, J.; Corpetti, C.; Sarnelli, G. The potential of cannabidiol in the COVID-19 pandemic. Br. J. Pharmacol. 2020, 177, 4967–4970. [Google Scholar] [CrossRef] [PubMed]
- Campos, A.C.; Fogaça, M.V.; Sonego, A.B.; Guimarães, F.S. Cannabidiol, neuroprotection and neuropsychiatric disorders. Pharmacol. Res. 2016, 112, 119–127. [Google Scholar] [CrossRef]
- Castillo, A.; Tolón, M.R.; Fernández-Ruiz, J.; Romero, J.; Martinez-Orgado, J. The neuroprotective effect of cannabidiol in an in vitro model of newborn hypoxic-ischemic brain damage in mice is mediated by CB(2) and adenosine receptors. Neurobiol. Dis. 2010, 37, 434–440. [Google Scholar] [CrossRef]
- Degerman, E.; Rauch, U.; Lindberg, S.; Caye-Thomasen, P.; Hultgårdh, A.; Magnusson, M. Expression of insulin signalling components in the sensory epithelium of the human saccule. Cell Tissue Res. 2013, 352, 469–478. [Google Scholar] [CrossRef]
- Khani, E.; Khiali, S.; Beheshtirouy, S.; Entezari-Maleki, T. Potential pharmacologic treatments for COVID-19 smell and taste loss: A comprehensive review. Eur. J. Pharmacol. 2021, 912, 174582. [Google Scholar] [CrossRef] [PubMed]
- Ryan, F.J.; Hope, C.M.; Masavuli, M.G. Long-term perturbation of the peripheral immune system months after SARS-CoV-2 infection. BMC Med. 2022, 20, 26. [Google Scholar] [CrossRef]
- Phetsouphanh, C.; Darley, D.R.; Wilson, D.B. Immunological dysfunction persists for 8 months following initial mild-to-moderate SARS-CoV-2 infection. Nat. Immunol. 2022, 23, 210–216. [Google Scholar] [CrossRef]
- Queiroz, M.A.F.; Neves, P.F.M.d.; Lima, S.S.; Lopes, J.D.; Torres, M.K.; Vallinoto, I.M.; Bichara, C.D.; Santos, E.F.; de Brito, M.T.; Da Silva, A.L.; et al. Cytokine Profiles Associated with Acute COVID-19 and Long COVID-19 Syndrome. Front. Cell. Infect. Microbiol. 2022, 12, 922422. [Google Scholar] [CrossRef]
- Arun, S.; Storan, A.; Myers, B. Mast cell activation syndrome and the link with long COVID. Br. J. Hosp. Med. 2022, 83, 1–10. [Google Scholar] [CrossRef]
- Weinstock, L.B.; Brook, J.B.; Walters, A.S.; Goris, A.; Afrin, L.B.; Molderings, G.J. Mast cell activation symptoms are prevalent in Long-COVID. Int. J. Infect. Dis. 2021, 112, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Peluso, M.J.; Deitchman, A.N.; Torres, L. Long-term SARS-CoV-2-specific immune and inflammatory responses in individuals recovering from COVID-19 with and without post-acute symptoms. Cell Rep. 2021, 36, 109518. [Google Scholar] [CrossRef]
- Vibholm, L.K.; Nielsen, S.S.F.; Pahus, M.H. SARS-CoV-2 persistence is associated with antigen-specific CD8 T-cell responses. EBioMedicine 2021, 64, 103230. [Google Scholar] [CrossRef] [PubMed]
- Patterson, B.K.; Francisco, E.B.; Yogendra, R. Persistence of SARS CoV-2 S1 Protein in CD16+ Monocytes in Post-Acute Sequelae of COVID-19 (PASC) up to 15 Months Post-Infection. Front. Immunol. 2022, 12, 5526. [Google Scholar] [CrossRef] [PubMed]
- Etter, M.M.; Martins, T.A.; Kulsvehagen, L.; Pössnecker, E.; Duchemin, W.; Hogan, S.; Sanabria-Diaz, G.; Müller, J.; Chiappini, A.; Rychen, J.; et al. Severe Neuro-COVID is associated with peripheral immune signatures, autoimmunity and neurodegeneration: A prospective cross-sectional study. Nat. Commun. 2022, 13, 6777. [Google Scholar] [CrossRef]
- Heming, M.; Li, X.; Räuber, S. Neurological Manifestations of COVID-19 Feature T Cell Exhaustion and Dedifferentiated Monocytes in Cerebrospinal Fluid. Immunity 2021, 54, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Townsend, L.; Dyer, A.H.; Naughton, A. Longitudinal Analysis of COVID-19 Patients Shows Age-Associated T Cell Changes Independent of Ongoing Ill-Health. Front. Immunol. 2021, 12, 1593. [Google Scholar] [CrossRef]
- Yi, J.S.; Cox, M.A.; Zajac, A.J. T-cell exhaustion: Characteristics, causes and conversion. Immunology 2010, 129, 474–481. [Google Scholar] [CrossRef]
- Visvabharathy, L.; Hanson, B.; Orban, Z. Neuro-COVID long-haulers exhibit broad dysfunction in T cell memory generation and responses to vaccination. medRxiv 2021. medRxiv:2021(08.08.21261763). [Google Scholar] [CrossRef]
- Schwabenland, M.; Salié, H.; Tanevski, J. Deep spatial profiling of human COVID-19 brains reveals neuroinflammation with distinct microanatomical microglia-T-cell interactions. Immunity 2021, 54, 1594–1610. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Fu, L.; Gonzales, D.M.; Lavi, E. Coronavirus neurovirulence correlates with the ability of the virus to induce proinflammatory cytokine signals from astrocytes and microglia. J. Virol. 2004, 78, 3398–3406. [Google Scholar] [CrossRef]
- Wallukat, G.; Hohberger, B.; Wenzel, K. Functional autoantibodies against G-protein coupled receptors in patients with persistent Long-COVID-19 symptoms. J. Transl. Autoimmun. 2021, 4, 100100. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.E.; Feng, A.; Meng, W. New-onset IgG autoantibodies in hospitalized patients with COVID-19. Nat. Commun. 2021, 12, 5417. [Google Scholar] [CrossRef]
- Arthur, J.M.; Forrest, J.C.; Boehme, K.W. Development of ACE2 autoantibodies after SARS-CoV-2 infection. PLoS ONE 2021, 16, e0257016. [Google Scholar] [CrossRef]
- Sacchi, M.C.; Tamiazzo, S.; Stobbione, P. SARS-CoV-2 infection as a trigger of autoimmune response. Clin. Transl. Sci. 2021, 14, 898–907. [Google Scholar] [CrossRef]
- Allahyari, F.; Hosseinzadeh, R.; Nejad, J.H.; Heiat, M.; Ranjbar, R. A case report of simultaneous autoimmune and COVID-19 encephalitis. J. Neurovirol. 2021, 27, 504–506. [Google Scholar] [CrossRef]
- Burr, T.; Barton, C.; Doll, E.; Lakhotia, A.; Sweeney, M. N-Methyl-d-Aspartate Receptor Encephalitis Associated with COVID-19 Infection in a Toddler. Pediatr. Neurol. 2021, 114, 75–76. [Google Scholar] [CrossRef]
- Flannery, P.; Yang, I.; Keyvani, M.; Sakoulas, G. Acute Psychosis Due to Anti-N-Methyl D-Aspartate Receptor Encephalitis Following COVID-19 Vaccination: A Case Report. Front. Neurol. 2021, 12, 764197. [Google Scholar] [CrossRef] [PubMed]
- Durovic, E.; Bien, C.; Bien, C.G.; Isenmann, S. MOG antibody-associated encephalitis secondary to COVID-19: Case report. BMC Neurol. 2021, 21, 414. [Google Scholar] [CrossRef]
- da Costa, M.D.; Rato, M.L.; Cruz, D.; Valadas, A.; Antunes, A.P.; Albuquerque, L. Longitudinally extensive transverse myelitis with anti-myelin oligodendrocyte glycoprotein antibodies following SARS-CoV-2 infection. J. Neuroimmunol. 2021, 361, 577739. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.C.; Walker, T.A.; Truong, A.D. Evidence of Persisting Autoreactivity in Post-Acute Sequelae of SARS-CoV-2 Infection. medRxiv 2021. medRxiv:2021.09.21.21263845. [Google Scholar] [CrossRef]
- Paterson, R.W.; Brown, R.L.; Benjamin, L. The emerging spectrum of COVID-19 neurology: Clinical, radiological and laboratory findings. Brain 2020, 143, 3104–3120. [Google Scholar] [CrossRef]
- Peluso, M.J.; Anglin, K.; Durstenfeld, M.S. Effect of Oral Nirmatrelvir on Long COVID Symptoms: 4 Cases and Rationale for Systematic Studies. Pathog. Immun. 2022, 7, 95–103. [Google Scholar] [CrossRef]
- Ayoubkhani, D.; Bermingham, C.; Pouwels, K.B. Trajectory of long covid symptoms after covid-19 vaccination: Community based cohort study. BMJ 2022, 377, e069676. [Google Scholar] [CrossRef]
- Dale, A.M.; Strickland, J.; Gardner, B.; Symanzik, J.; Evanoff, B.A. Assessing agreement of self-reported and observed physical exposures of the upper extremity. Int. J. Occup. Environ. Health 2010, 16, 1–10. [Google Scholar] [CrossRef]
- Spuch, C.; López-García, M.; Rivera-Baltanás, T. Efficacy and Safety of Lithium Treatment in SARS-CoV-2 Infected Patients. Front. Pharmacol. 2022, 13, 1081. [Google Scholar] [CrossRef]
- Fisher, B.; Barone, F.; Jobling, K. Syndrome Oeor 132 Ofipwps. Results of a Phase II Randomised, Double-Blind, Placebo-Controlled, Proof of Concept Study. Ann. Rheum. Dis. 2019, 78, 177. [Google Scholar] [CrossRef]
- Wenzel, J.; Lampe, J.; Müller-Fielitz, H. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. [Google Scholar] [CrossRef] [PubMed]
- Buckingham, S.C.; Campbell, S.L.; Haas, B.R.; Montana, V.; Robel, S.; Ogunrinu, T.; Sontheimer, H. Glutamate release by primary brain tumors induces epileptic activity. Nat. Med. 2011, 17, 1269–1274. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V. Astrogliopathology in neurological, neurodevelopmental and psychiatric disorders. Neurobiol. Dis. 2016, 85, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Castañeda, A.; Lu, P.; Geraghty, A.C. Mild respiratory SARS-CoV-2 infection can cause multi-lineage cellular dysregulation and myelin loss in the brain. bioRxiv 2022. bioRxiv:2022(01.07.475453). [Google Scholar] [CrossRef]
- Oliveira, A.O.; Malwade, S.; Rufino de Sousa, N.; Goparaju, S.K.; Gracias, J.; Orhan, F.; Steponaviciute, L.; Schalling, M.; Sheridan, S.D.; Perlis, R.H.; et al. SARS-CoV-2 promotes microglial synapse elimination in human brain organoids. Mol. Psychiatry 2022, 27, 3939–3950. [Google Scholar] [CrossRef]
- Pretorius, E.; Vlok, M.; Venter, C. Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc. Diabetol. 2021, 20, 172. [Google Scholar] [CrossRef]
- Grobbelaar, L.M.; Venter, C.; Vlok, M. SARS-CoV-2 spike protein S1 induces fibrin(ogen) resistant to fibrinolysis: Implications for microclot formation in COVID-19. Biosci. Rep. 2021, 41, BSR20210611. [Google Scholar] [CrossRef]
- Ryu, J.K.; Sozmen, E.G.; Dixit, K. SARS-CoV-2 Spike Protein Induces Abnormal Inflammatory Blood Clots Neutralized by Fibrin Immunotherapy. bioRxiv 2021. bioRxiv:2021.10.12.464152. [Google Scholar] [CrossRef]
- Bouck, E.G.; Denorme, F.; Holle, L.A. COVID-19 and Sepsis Are Associated with Different Abnormalities in Plasma Procoagulant and Fibrinolytic Activity. Arter. Thromb. Vasc. Biol. 2021, 41, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Jana, A.K.; Greenwood, A.B.; Hansmann, U.H.E. Presence of a SARS-CoV-2 protein enhances Amyloid Formation of Serum Amyloid A. bioRxiv 2021. bioRxiv:2021(05.18.444723). [Google Scholar] [CrossRef] [PubMed]
- Zuo, Y.; Estes, S.K.; Ali, R.A. Prothrombotic autoantibodies in serum from patients hospitalized with COVID-19. Sci. Transl. Med. 2020, 12, eabd3876. [Google Scholar] [CrossRef]
- Tung, M.L.; Tan, B.; Cherian, R.; Chandra, B. Anti-phospholipid syndrome and COVID-19 thrombosis: Connecting the dots. Rheumatol. Adv. Pract. 2021, 5, rkaa081. [Google Scholar] [CrossRef]
- Passos, F.R.S.; Heimfarth, L.; Monteiro, B.S. Oxidative stress and inflammatory markers in patients with COVID-19: Potential role of RAGE, HMGB1, GFAP and COX-2 in disease severity. Int. Immunopharmacol. 2022, 104, 108502. [Google Scholar] [CrossRef] [PubMed]
- Fogarty, H.; Townsend, L.; Morrin, H. Persistent endotheliopathy in the pathogenesis of long COVID syndrome. J. Thromb. Haemost. 2021, 19, 2546–2553. [Google Scholar] [CrossRef]
- Poyatos, P.; Luque, N.; Eizaguirre, S. Post-COVID-19 patients show an increased endothelial progenitor cell production. Transl. Res. 2022, 243, 14–20. [Google Scholar] [CrossRef]
- Christensen, R.H.; Berg, R.M.G. Vascular Inflammation as a Therapeutic Target in COVID-19 “Long Haulers”: HIITing the Spot? Front. Cardiovasc. Med. 2021, 8, 187. [Google Scholar] [CrossRef]
- Lee, M.H.; Perl, D.P.; Steiner, J. Neurovascular injury with complement activation and inflammation in COVID-19. Brain 2022, 145, 2555–2568. [Google Scholar] [CrossRef]
- Lambadiari, V.; Mitrakou, A.; Kountouri, A. Association of COVID-19 with impaired endothelial glycocalyx, vascular function and myocardial deformation 4 months after infection. Eur. J. Heart Fail. 2021, 23, 1916–1926. [Google Scholar] [CrossRef]
- Buzhdygan, T.P.; DeOre, B.J.; Baldwin-Leclair, A. The SARS-CoV-2 spike protein alters barrier function in 2D static and 3D microfluidic in-vitro models of the human blood-brain barrier. Neurobiol. Dis. 2020, 146, 105131. [Google Scholar] [CrossRef] [PubMed]
- Osiaevi, I.; Schulze, A.; Evers, G. Persistent capillary rarefication in long COVID syndrome [published online ahead of print, 2022 Aug 11. Angiogenesis 2022, 26, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Guedj, E.; Campion, J.Y.; Dudouet, P. 18F-FDG brain PET hypometabolism in patients with long COVID. Eur. J. Nucl. Med. Mol. Imaging 2021, 48, 2823–2833. [Google Scholar] [CrossRef]
- Yong, S.J. Persistent Brainstem Dysfunction in Long-COVID: A Hypothesis. ACS Chem. Neurosci. 2021, 12, 573–580. [Google Scholar] [CrossRef]
- Barnden, L.R.; Shan, Z.Y.; Staines, D.R. Hyperintense sensorimotor T1 spin echo MRI is associated with brainstem abnormality in chronic fatigue syndrome. Neuroimage Clin. 2018, 20, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Barnden, L.R.; Crouch, B.; Kwiatek, R. A brain MRI study of chronic fatigue syndrome: Evidence of brainstem dysfunction and altered homeostasis. NMR Biomed. 2011, 24, 1302–1312. [Google Scholar] [CrossRef]
- Barnden, L.R.; Shan, Z.Y.; Staines, D.R. Intra brainstem connectivity is impaired in chronic fatigue syndrome. Neuroimage Clin. 2019, 24, 102045. [Google Scholar] [CrossRef]




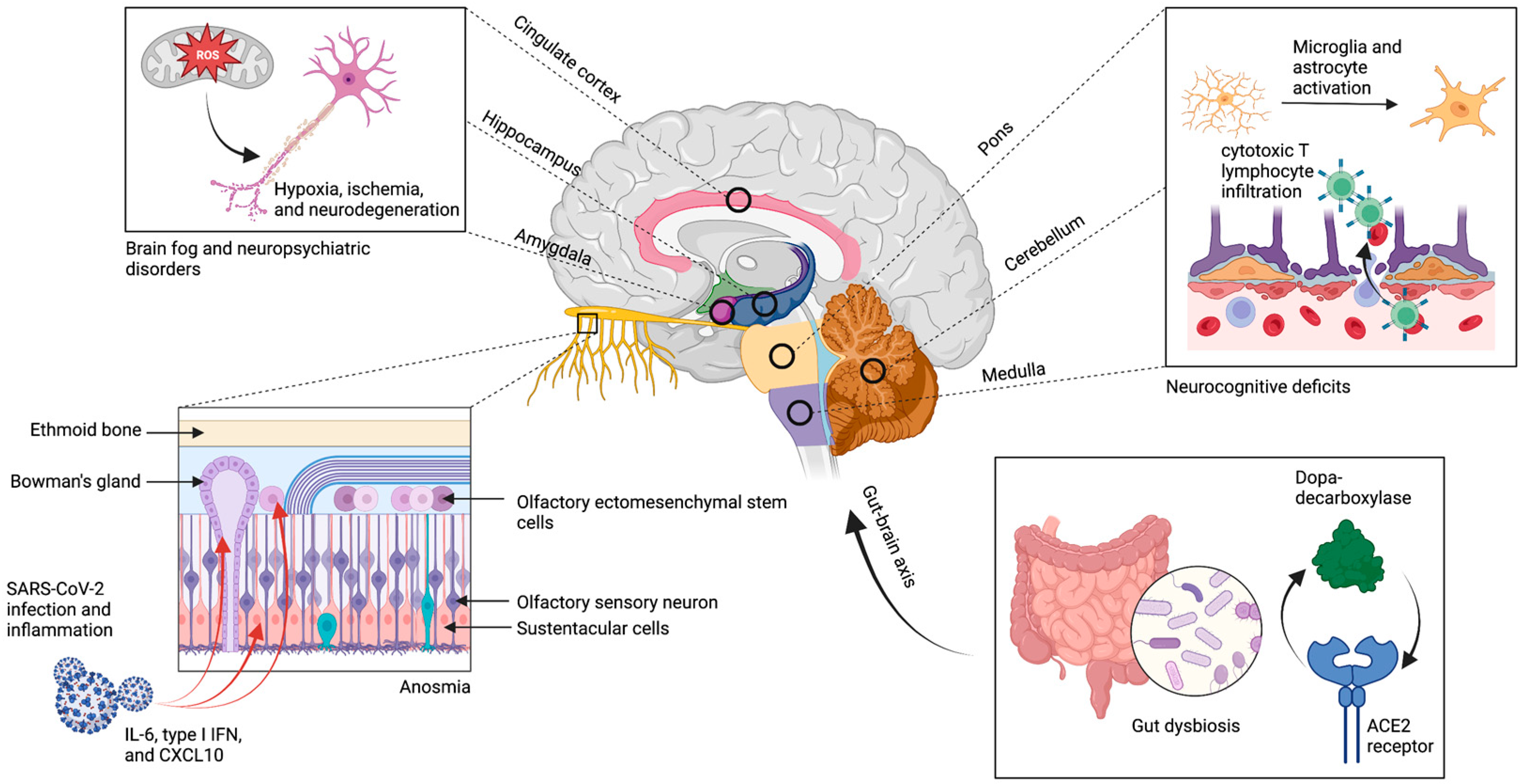{kind=link}
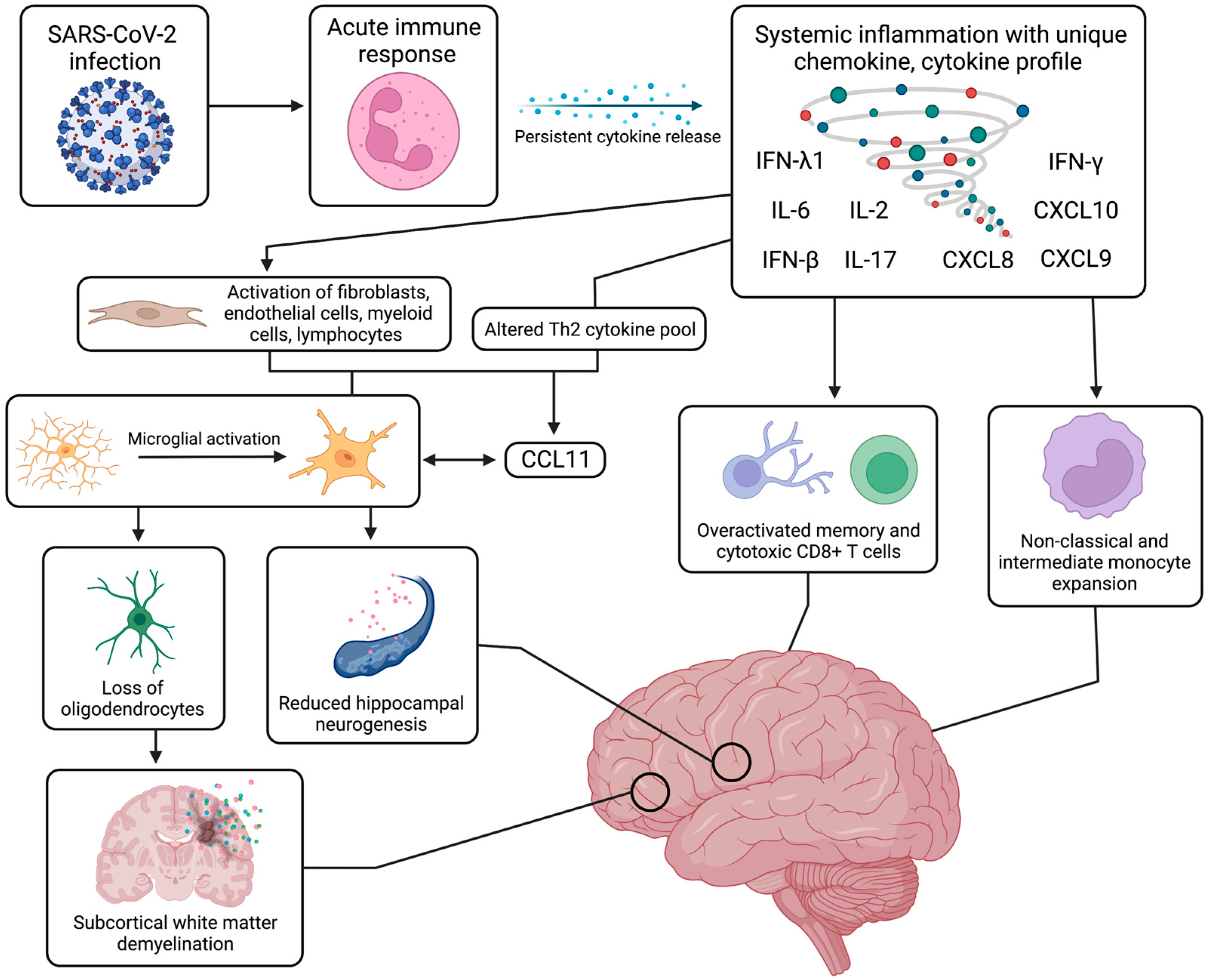{kind=link}
{kind=link}
| Population Characteristics | ||||||||
|---|---|---|---|---|---|---|---|---|
| Reference/Country | Study Design | Sample Size (n) | Inclusion Criteria/Long COVID Diagnosis | Acute COVID Diagnosis | Comorbidities | Intervention | Comparator | Outcomes |
| Xie et al. [69] United States | Quasi-experimental with no blinding | 56,340 | At least one post-acute sequela from a set of 12 prespecified post-acute sequelae of SARS-CoV-2 infection 30 days after positive test | Laboratory confirmed | 15.02% had cancer, 22.95% had chronic lung disease, 8.09% had dementia, 33.96% had type 2 diabetes mellitus, 30.92% had cardiovascular disease, 80.99% had hyperlipidemia | Nirmatrelvir | None | Reduced risk of fatigue and neurocognitive impairment |
| Singh et al. [70] India | Unclear | 120 | Mild to moderate COVID-19 | Positive RT-PCR test | Not mentioned | Nasal fluticasone spray | None | Significant improvement in recognizing odors |
| Abdelalim et al. [71] Egypt | Randomized double blind | 100 | Unclear | Positive rRT–PCR test by nasopharyngeal swab | 16% had diabetes mellitus, 14% had hypertension | Mometasone furoate nasal spray and olfactory training | Only olfactory training | Treatment showed no significant improvement in recovery rates and duration of anosmia |
| O’Kelly et al. [72] Ireland | Quasi-experimental with no blinding | 52 | Ongoing symptoms 12 or more weeks after initial infection | Laboratory Confirmed | 9.6% had hypertension, 9.6% had dyslipidemia, 3.8% had diabetes mellitus | Low dose naltrexone | None | Improvement in activities of daily living, energy levels, pain levels, levels of concentration and sleep disturbance |
| Mohamad et al. [73] Egypt | Randomized single blind parallel design | 40 | Post-COVID olfactory loss verified by endoscopic examination with anosmia lasting for at least two weeks | Unclear | Not mentioned | Insulin films | Plain films | Significant improvement in olfactory detection scores and olfactory values discrimination |
| Rezaeian [74] Iran | Randomized double blind | 38 | Hyposmic patients with a Connecticut Chemosensory Clinical Research Center (CCCRC) score between 2 and 5.75 | NA | 7.9% had hypertension, 2.6% had cardiovascular disease | Insulin therapy | Normal saline therapy | Administration of intranasal insulin significantly improved hyposmia with higher mean CCCRC scores |
| Glynne et al. [75] United Kingdom | Quasi-experimental with no blinding | 49 | Either PCR, serological evidence, or suffered acute illness | Positive RT–PCR test | Not mentioned | Loratadine 10 mg two times per day or fexofenadine 180 mg two times per day and loratadine 10 mg two times per day or fexofenadine 180 mg two times per day | Supportive care | Decrease in fatigue, neurologic, and neuropsychiatric symptoms among many others |
| Population Characteristics | ||||||||
|---|---|---|---|---|---|---|---|---|
| Trial Registration Number/Country | Study Design | Recruitment Target (n), Age Range (Years) | Definition of Long COVID | Method of Acute COVID-19 Diagnosis | Comorbidities | Intervention | Comparator | Neurological Outcomes |
| NCT0557666 United States | Randomized double blind | 200, >18 | Post-COVID-19 symptoms persisting greater than three months with at least two post-COVID symptoms of moderate or severe intensity | Self-reported history of confirmed COVID-19 infection preceding post-COVID symptoms | Excluded individuals with severe liver disease, HIV infection, suspected or confirmed pregnancy or breastfeeding, or other medical conditions that would compromise patient’s safety or compliance | Nirmatrelvir with Ritonavir (Paxlovid) | Placebo with Ritonavir | Severity of brain fog, PROMIS cognitive function abilities score |
| NCT05430152 Canada | Randomized double blind | 160, 19–69 | Symptoms lasting three to six months post-acute infection | Positive test result or clinical confirmation by a physician | Excluded participants with any use of opioid medications | Low dose naltrexone | Placebo | Changes in fatigue intensity score, pain severity score, and symptom severity score |
| NCT05597722 United States | Randomized open label | 120, 21–65 | COVID-symptoms that persist three months or longer and a moderate cognitive impairment (MOCA) score ≤18 for at least three months | Positive PCR or home antigen test | Excluded individuals with pre-existing cardiac or kidney condition, severe hypertension, glaucoma, hyperthyroidism, and advanced arteriosclerosis | Dextroamphetamine-Amphetamine | Stimulant medication | Changes in cognitive impairment |
| NCT0494412 United States | Randomized quadruple blind | 70, 18–75 | Confirmed SARS-CoV-2 infection by PCR at least 24 weeks prior to baseline with a raw score of at least a score of 21 on PROMIS Fatigue SF 7a at screening | Laboratory-confirmed SARS-CoV-2 PCR test | Excluded individuals with orthostatic hypertension, tachycardia, use of sedating medications, lab abnormalities that may cause fatigue, history of anaphylaxis, previous chronic fatigue syndrome, previous fibromyalgia, previous lupus, previous Sjogren’s syndrome, or diagnosis of sleep apnea | RSLV-132 | Placebo | PROMIS fatigue 7a T-score, FACIT fatigue questionnaire, severity of brain fog, performance on concentration task |
| NCT05350774 United States | Randomized double blind | 60, >18 | Persistent neurological symptoms exceeding 12 weeks from acute infection | Patient reported positive antigen test for SARS-CoV-2 followed by confirmatory nucleocapsid antibody testing or a positive SARS-CoV-2 PCR test result | Excluded individuals with ventricular arrhythmias, coronary artery disease, or a condition prior to COVID-19 infection that would confound interpretation | IV immunoglobulin, IV methylprednisone | Placebo | Proportion of participants with clinically meaningful improvement in the Health Utilities Index Mark 3 (HUI3) four weeks after start of study |
| NCT0561858 United States | Randomized quadruple blind | 50, 18–80 | Reported fatigue and/or brain fog for less than four weeks prior to enrollment on questionnaire after SARS-CoV-2 infection | Documented or self-reported positive test for COVID-19 less than four weeks prior to enrollment | Excluded individuals with current alcohol abuse, history of fibromyalgia, chronic fatigue syndrome, or progressive cognitive disorder, any active medical, psychiatric, or social problems that would interfere with study procedure, or use of any tobacco, marijuana, or illicit drug products | Lithium | Placebo | Severity of brain fog, frequency of anxiety, frequency of headaches, severity of insomnia, change in sense of smell and taste, and performance on cognitive tests |
| NCT05104424 Saudi Arabia | Randomized open label | 44, >18 | Unclear | Unclear | Excluded patients with any nasal deviation, congenital anomalies, or allergic sinusitis | Intranasal insulin, zinc, gabapentin, ice cube stimulation | Zinc only | Improvements in hyposmia (Sniffin Sticks test), dysgeusia, and self-assessment of hyposmia (Dynachron-olfaction questionnaire) |
| NCT04997395 United Kingdom | Open Label | 12, ≥18 | Confirmed by GP triage clinic and assessment by a long COVID clinic. Participant undertook the clinical assessment and investigations as recommended by the NICE guidance on long COVID | Unclear | Excluded individuals with ongoing mental and/or psychiatric illnesses/disorders that require active treatment during the trial period, use of anti-coagulant drugs, use of cannabinoids or cannabinoid-based medicine within three months prior to study, known dependence on cannabis, alcohol, or any other drug, history of chronic liver failure, history of allergy to tree nuts | MediCabilis Cannabis sativa 50 | Discontinuing cannabidiol for three-weeks following 21 week period of taking cannabidiol | Changes in cognition, fatigue, self-reported quality of life, pain score, and anxiety/depression |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leng, A.; Shah, M.; Ahmad, S.A.; Premraj, L.; Wildi, K.; Li Bassi, G.; Pardo, C.A.; Choi, A.; Cho, S.-M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells 2023, 12, 816. https://doi.org/10.3390/cells12050816
Leng A, Shah M, Ahmad SA, Premraj L, Wildi K, Li Bassi G, Pardo CA, Choi A, Cho S-M. Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells. 2023; 12(5):816. https://doi.org/10.3390/cells12050816
Chicago/Turabian StyleLeng, Albert, Manuj Shah, Syed Ameen Ahmad, Lavienraj Premraj, Karin Wildi, Gianluigi Li Bassi, Carlos A. Pardo, Alex Choi, and Sung-Min Cho. 2023. "Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics" Cells 12, no. 5: 816. https://doi.org/10.3390/cells12050816
APA StyleLeng, A., Shah, M., Ahmad, S. A., Premraj, L., Wildi, K., Li Bassi, G., Pardo, C. A., Choi, A., & Cho, S.-M. (2023). Pathogenesis Underlying Neurological Manifestations of Long COVID Syndrome and Potential Therapeutics. Cells, 12(5), 816. https://doi.org/10.3390/cells12050816