
Host Cell Restriction Factors Blocking Efficient Vector Transduction: Challenges in Lentiviral and Adeno-Associated Vector Based Gene Therapies


Abstract
1. Introduction
2. Lentiviral Vectors
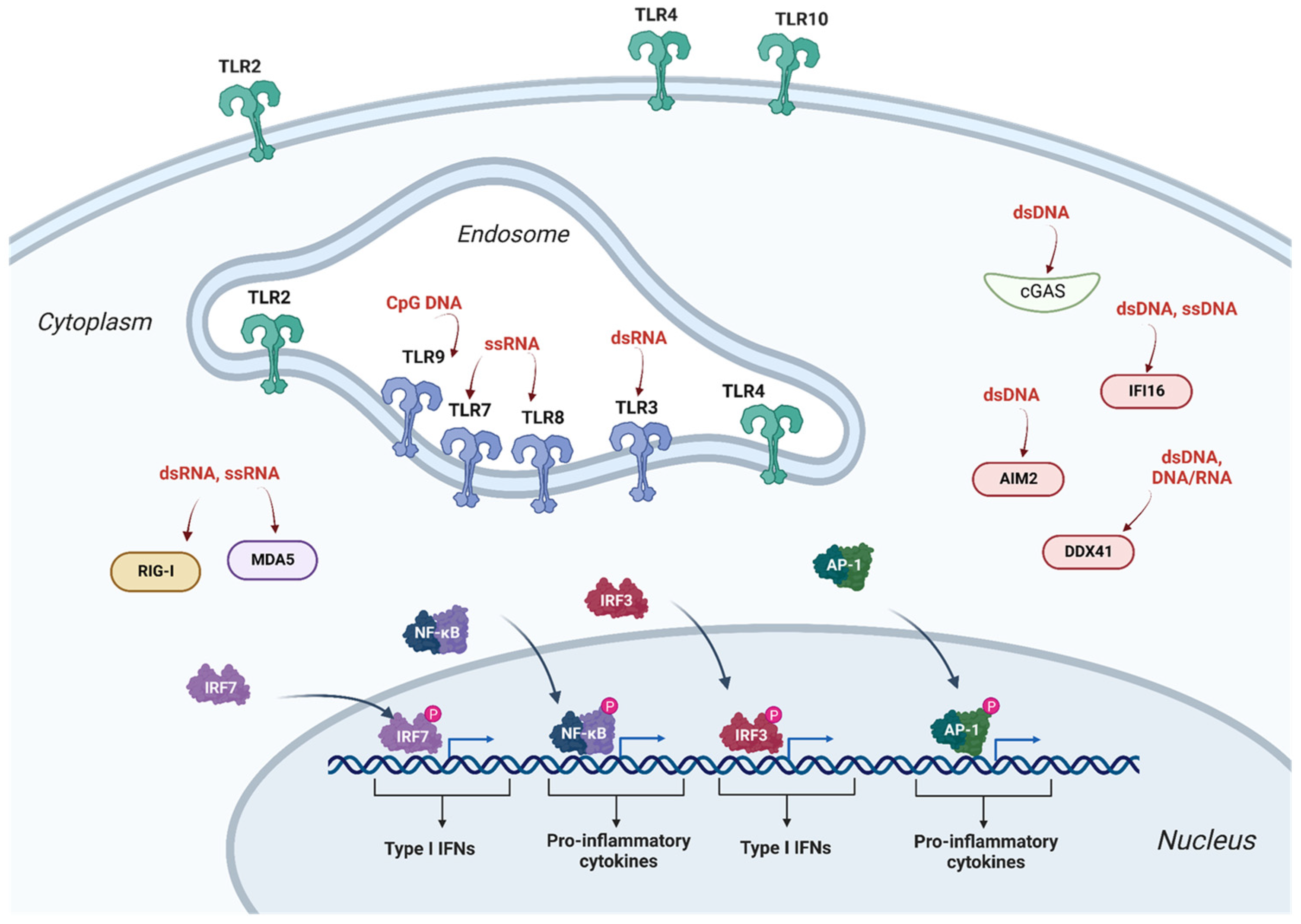
2.1. Innate Immunity and Antiviral Restriction Factors
2.1.1. Restriction Factors Sensing Lentiviral Vector RNA
2.1.2. Restriction Factors Sensing Lentiviral Vector Reverse Transcription Products
2.1.3. Restriction Factors Sensing Lentiviral Vector Proteins
2.1.4. Lentiviral Vector Intrinsic Restriction Factors

{kind=link}
{kind=link}
{kind=link}
| Viral Infection Phase | Sensor | Ligands | Adaptors | Effectors |
|---|---|---|---|---|
| Entry/ Uncoating | TLR2/4/10 | Viral proteins | MyD88 | Inflammatory cytokines/Type I IFNs |
| TLR3 | dsRNA | TRIF | Inflammatory cytokines/Type I IFNs | |
| TLR7 | ssRNA | MyD88 | Inflammatory cytokines/Type I IFNs | |
| TLR8 | ssRNA | MyD88 | Inflammatory cytokines/Type I IFNs | |
| TLR9 | CpG DNA Viral proteins | MyD88 | Inflammatory cytokines/Type I IFNs | |
| RIG-I/MDA5 | dsRNA, ppp-sRNA | MAVS | Inflammatory cytokines/Type I IFNs | |
| IFITMS | Viral envelope | d.a. | Abortive infection | |
| Uncoating/ Reverse Transcription | cGas | dsDNA | STING | Inflammatory cytokines/Type I IFNs |
| IFI16 | DNA/RNA | STING/ASC | Inflammatory cytokines/Type I IFNs Caspase-1 | |
| DDX41 | dsDNA, ssDNA | STING | IFNs | |
| TRIM5 | CA | d.a. | Abortive infection | |
| APOBEC3 | NC/ssRNA | d.a. | Abortive infection | |
| SAMDH1 | ssDNA, ssRNA, DNA/RNA | d.a. | Abortive infection | |
| Integration | cGas | DAMP response | STING | IFNs |
| Mx2 | CA | d.a. | Abortive infection | |
| Transcription/ Translation | DDX3X | Abortive viral RNA | MAVS | IFNs |
| Other | Tetherin | CA | d.a. | Abortive infection Inflammatory cytokines/Type I IFNs |
3. Adeno-Associated Vectors
3.1. Innate Immunity and Antiviral Restriction Factors
3.1.1. Restriction Factors Sensing AAV Vector DNA
3.1.2. Restriction Factors Sensing AAV Vector RNA Products
3.1.3. Restriction Factors Sensing AAV Vector Proteins
3.1.4. AAV Vector Intrinsic Restriction Factors
| Viral infection Phase | Sensor | Ligands | Adaptors | Effectors/Effect |
|---|---|---|---|---|
| Entry/ Uncoating | TLR2/4 | Capsid proteins | MyD88 | Inflammatory cytokines/Type I IFNs |
| TLR9 | CpG DNA dsDNA, DNA/RNA | MyD88 | Inflammatory cytokines/Type I IFNs | |
| cGas | dsDNA | STING | Inflammatory cytokines/Type I IFNs | |
| IFI16 | ssDNA, dsDNA | STING/ASC | Inflammatory cytokines/Type I IFNs Caspase-1 | |
| AIM2 | dsDNA, ssDNA, (DD) | ASC | Caspase-1 | |
| EGF Receptor tyrosine kinase (RTK) | Capsid | d.a. | Proteasomal degradation | |
| SUMO proteins | VP2, (DD) | d.a. | SUMOylation | |
| Nuclear entry/ dsDNA synthesis/Transcription | cGas | DAMP response | STING | IFNs |
| NBS1, Mre11, Rad50, Mdc1 | (DD) | STING | IFNs | |
| RIG-I/MDA5 | dsRNA, ssRNA | MAVS | Inflammatory cytokines/Type I IFNs | |
| FKBP52 | AAV genome | d.a. | Abortive double strand conversion | |
| PHF5A | AAV capsid | d.a. | Abortive double strand conversion |
4. Concluding Remarks and Future Directions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ma, C.-C.; Wang, Z.-L.; Xu, T.; He, Z.-Y.; Wei, Y.-Q. The approved gene therapy drugs worldwide: From 1998 to 2019. Biotechnol. Adv. 2020, 40, 107502. [Google Scholar] [CrossRef]
- Agency, E.M. Directive 2001/83/EC of the European Parliament and of the Council Of the European Parliament and of the Council of 6 November 2001 on the Community Code Relating to Medicinal Products for Human Use; Directive 2001/83/EC (Annex I Part IV); European Parliament: Strasbourg, France, 2001. [Google Scholar]
- Sauter, D.; Kirchhoff, F. Evolutionary conflicts and adverse effects of antiviral factors. Elife 2021, 10, e65243. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Fritsch, S.D.; Weichhart, T. Effects of Interferons and Viruses on Metabolism. Front. Immunol. 2016, 7, 630. [Google Scholar] [CrossRef]
- Yan, N.; Chen, Z.J. Intrinsic antiviral immunity. Nat. Immunol. 2012, 13, 214–222. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Z.J. Innate immune sensing and signaling of cytosolic nucleic acids. Annu. Rev. Immunol. 2014, 32, 461–488. [Google Scholar] [CrossRef]
- Hartmann, G.; Krieg, A.M. CpG DNA and LPS induce distinct patterns of activation in human monocytes. Gene Ther. 1999, 6, 893–903. [Google Scholar] [CrossRef]
- Hughes, T.S.; Langer, S.J.; Virtanen, S.I.; Chavez, R.A.; Watkins, L.R.; Milligan, E.D.; Leinwand, L.A. Immunogenicity of intrathecal plasmid gene delivery: Cytokine release and effects on transgene expression. J. Gene Med. 2009, 11, 782–790. [Google Scholar] [CrossRef]
- Li, S.; Wu, S.P.; Whitmore, M.; Loeffert, E.J.; Wang, L.; Watkins, S.C.; Pitt, B.R.; Huang, L. Effect of immune response on gene transfer to the lung via systemic administration of cationic lipidic vectors. Am. J. Physiol. 1999, 276, L796–L804. [Google Scholar] [CrossRef]
- Zhao, H.; Hemmi, H.; Akira, S.; Cheng, S.H.; Scheule, R.K.; Yew, N.S. Contribution of Toll-like receptor 9 signaling to the acute inflammatory response to nonviral vectors. Mol. Ther. 2004, 9, 241–248. [Google Scholar] [CrossRef] [PubMed]
- Gautam, A.; Densmore, C.L.; Golunski, E.; Xu, B.; Waldrep, J.C. Transgene Expression in Mouse Airway Epithelium by Aerosol Gene Therapy with PEI–DNA Complexes. Mol. Ther. 2001, 3, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Fausther-Bovendo, H.; Kobinger, G.P. Pre-existing immunity against Ad vectors: Humoral, cellular, and innate response, what’s important? Hum. Vaccin. Immunother. 2014, 10, 2875–2884. [Google Scholar] [CrossRef] [PubMed]
- Hendrickx, R.; Stichling, N.; Koelen, J.; Kuryk, L.; Lipiec, A.; Greber, U.F. Innate immunity to adenovirus. Hum. Gene Ther. 2014, 25, 265–284. [Google Scholar] [CrossRef]
- Monahan, P.E.; Négrier, C.; Tarantino, M.; Valentino, L.A.; Mingozzi, F. Emerging Immunogenicity and Genotoxicity Considerations of Adeno-Associated Virus Vector Gene Therapy for Hemophilia. J. Clin. Med. 2021, 10, 2471. [Google Scholar] [CrossRef]
- Muhuri, M.; Maeda, Y.; Ma, H.; Ram, S.; Fitzgerald, K.A.; Tai, P.W.; Gao, G. Overcoming innate immune barriers that impede AAV gene therapy vectors. J. Clin. Investig. 2021, 131, e143780. [Google Scholar] [CrossRef]
- Heine, A.; Juranek, S.; Brossart, P. Clinical and immunological effects of mRNA vaccines in malignant diseases. Mol. Cancer 2021, 20, 52. [Google Scholar] [CrossRef]
- Meng, Z.; Lu, M. RNA Interference-Induced Innate Immunity, Off-Target Effect, or Immune Adjuvant? Front. Immunol. 2017, 8, 331. [Google Scholar] [CrossRef]
- Minnaert, A.K.; Vanluchene, H.; Verbeke, R.; Lentacker, I.; De Smedt, S.C.; Raemdonck, K.; Sanders, N.N.; Remaut, K. Strategies for controlling the innate immune activity of conventional and self-amplifying mRNA therapeutics: Getting the message across. Adv. Drug Deliv. Rev. 2021, 176, 113900. [Google Scholar] [CrossRef]
- Coffin, J.; Hughes, S.; Varmus, H. Retroviruses; Cold Spring Harbor Laboratory Press: Long Island, NY, USA, 1997. [Google Scholar]
- Coroadinha, A.S. Cancer Gene Therapy: Development and Production of Lentiviral Vectors for Gene Therapy. Methods Mol. Biol. 2022, 2521, 297–315. [Google Scholar] [CrossRef]
- Finkelshtein, D.; Werman, A.; Novick, D.; Barak, S.; Rubinstein, M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc. Natl. Acad. Sci. USA 2013, 110, 7306–7311. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.M.; Buchholz, C.J. Surface-Engineered Lentiviral Vectors for Selective Gene Transfer into Subtypes of Lymphocytes. Mol. Ther.-Methods Clin. Dev. 2019, 12, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Guedan, S.; Calderon, H.; Posey, A.D., Jr.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Norberg, S.M.; Hinrichs, C.S. Engineered T cell therapy for viral and non-viral epithelial cancers. Cancer Cell 2022, 41, 58–69. [Google Scholar] [CrossRef]
- Liang, Q.; Catalano, F.; Vlaar, E.C.; Pijnenburg, J.M.; Stok, M.; van Helsdingen, Y.; Vulto, A.G.; van der Ploeg, A.T.; van Til, N.P.; Pijnappel, W. IGF2-tagging of GAA promotes full correction of murine Pompe disease at a clinically relevant dosage of lentiviral gene therapy. Mol. Ther. Methods Clin. Dev. 2022, 27, 109–130. [Google Scholar] [CrossRef]
- Cantore, A.; Naldini, L. WFH State-of-the-art paper 2020: In vivo lentiviral vector gene therapy for haemophilia. Haemophilia 2021, 27 (Suppl. S3), 122–125. [Google Scholar] [CrossRef]
- Lundberg, C.; Bjorklund, T.; Carlsson, T.; Jakobsson, J.; Hantraye, P.; Deglon, N.; Kirik, D. Applications of lentiviral vectors for biology and gene therapy of neurological disorders. Curr. Gene Ther. 2008, 8, 461–473. [Google Scholar] [CrossRef]
- van Montfoort, N.; Olagnier, D.; Hiscott, J. Unmasking immune sensing of retroviruses: Interplay between innate sensors and host effectors. Cytokine Growth Factor Rev. 2014, 25, 657–668. [Google Scholar] [CrossRef]
- Colomer-Lluch, M.; Ruiz, A.; Moris, A.; Prado, J.G. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front. Immunol. 2018, 9, 2876. [Google Scholar] [CrossRef]
- Kajaste-Rudnitski, A.; Naldini, L. Cellular innate immunity and restriction of viral infection: Implications for lentiviral gene therapy in human hematopoietic cells. Hum. Gene Ther. 2015, 26, 201–209. [Google Scholar] [CrossRef]
- Yin, X.; Langer, S.; Zhang, Z.; Herbert, K.M.; Yoh, S.; Konig, R.; Chanda, S.K. Sensor Sensibility-HIV-1 and the Innate Immune Response. Cells 2020, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef] [PubMed]
- Fonteneau, J.F.; Larsson, M.; Beignon, A.S.; McKenna, K.; Dasilva, I.; Amara, A.; Liu, Y.J.; Lifson, J.D.; Littman, D.R.; Bhardwaj, N. Human immunodeficiency virus type 1 activates plasmacytoid dendritic cells and concomitantly induces the bystander maturation of myeloid dendritic cells. J. Virol. 2004, 78, 5223–5232. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Case, L.K.; Wang, C.; Yurkovetskiy, L.; Dikiy, S.; Golovkina, T.V. Innate immune sensing of retroviral infection via Toll-like receptor 7 occurs upon viral entry. Immunity 2011, 35, 135–145. [Google Scholar] [CrossRef]
- Strebel, K. HIV accessory proteins versus host restriction factors. Curr. Opin. Virol. 2013, 3, 692–699. [Google Scholar] [CrossRef]
- Borsotti, C.; Borroni, E.; Follenzi, A. Lentiviral vector interactions with the host cell. Curr. Opin. Virol. 2016, 21, 102–108. [Google Scholar] [CrossRef]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef]
- Beignon, A.S.; McKenna, K.; Skoberne, M.; Manches, O.; DaSilva, I.; Kavanagh, D.G.; Larsson, M.; Gorelick, R.J.; Lifson, J.D.; Bhardwaj, N. Endocytosis of HIV-1 activates plasmacytoid dendritic cells via Toll-like receptor-viral RNA interactions. J. Clin. Investig. 2005, 115, 3265–3275. [Google Scholar] [CrossRef]
- Schlee, M.; Hartmann, G. The chase for the RIG-I ligand--recent advances. Mol. Ther. 2010, 18, 1254–1262. [Google Scholar] [CrossRef]
- Batool, M.; Kim, M.S.; Choi, S. Structural insights into the distinctive RNA recognition and therapeutic potentials of RIG-I-like receptors. Med. Res. Rev. 2022, 42, 399–425. [Google Scholar] [CrossRef]
- Lepelley, A.; Louis, S.; Sourisseau, M.; Law, H.K.; Pothlichet, J.; Schilte, C.; Chaperot, L.; Plumas, J.; Randall, R.E.; Si-Tahar, M.; et al. Innate sensing of HIV-infected cells. PLoS Pathog. 2011, 7, e1001284. [Google Scholar] [CrossRef] [PubMed]
- Solis, M.; Nakhaei, P.; Jalalirad, M.; Lacoste, J.; Douville, R.; Arguello, M.; Zhao, T.; Laughrea, M.; Wainberg, M.A.; Hiscott, J. RIG-I-mediated antiviral signaling is inhibited in HIV-1 infection by a protease-mediated sequestration of RIG-I. J. Virol. 2011, 85, 1224–1236. [Google Scholar] [CrossRef] [PubMed]
- Jakobsen, M.R.; Bak, R.O.; Andersen, A.; Berg, R.K.; Jensen, S.B.; Tengchuan, J.; Laustsen, A.; Hansen, K.; Ostergaard, L.; Fitzgerald, K.A.; et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proc. Natl. Acad. Sci. USA 2013, 110, E4571–E4580. [Google Scholar] [CrossRef] [PubMed]
- Stavrou, S.; Aguilera, A.N.; Blouch, K.; Ross, S.R. DDX41 Recognizes RNA/DNA Retroviral Reverse Transcripts and Is Critical for In Vivo Control of Murine Leukemia Virus Infection. mBio 2018, 9, e00923-18. [Google Scholar] [CrossRef]
- Gao, D.; Wu, J.; Wu, Y.T.; Du, F.; Aroh, C.; Yan, N.; Sun, L.; Chen, Z.J. Cyclic GMP-AMP synthase is an innate immune sensor of HIV and other retroviruses. Science 2013, 341, 903–906. [Google Scholar] [CrossRef]
- Nazli, A.; Kafka, J.K.; Ferreira, V.H.; Anipindi, V.; Mueller, K.; Osborne, B.J.; Dizzell, S.; Chauvin, S.; Mian, M.F.; Ouellet, M.; et al. HIV-1 gp120 induces TLR2- and TLR4-mediated innate immune activation in human female genital epithelium. J. Immunol. 2013, 191, 4246–4258. [Google Scholar] [CrossRef]
- Du, M.; Chen, Z.J. DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 2018, 361, 704–709. [Google Scholar] [CrossRef]
- Li, X.; Deng, M.; Petrucelli, A.S.; Zhu, C.; Mo, J.; Zhang, L.; Tam, J.W.; Ariel, P.; Zhao, B.; Zhang, S.; et al. Viral DNA Binding to NLRC3, an Inhibitory Nucleic Acid Sensor, Unleashes STING, a Cyclic Dinucleotide Receptor that Activates Type I Interferon. Immunity 2019, 50, 591–599.e596. [Google Scholar] [CrossRef]
- Chen, S.; Bonifati, S.; Qin, Z.; St Gelais, C.; Wu, L. SAMHD1 Suppression of Antiviral Immune Responses. Trends Microbiol. 2019, 27, 254–267. [Google Scholar] [CrossRef]
- Coquel, F.; Silva, M.J.; Técher, H.; Zadorozhny, K.; Sharma, S.; Nieminuszczy, J.; Mettling, C.; Dardillac, E.; Barthe, A.; Schmitz, A.L.; et al. SAMHD1 acts at stalled replication forks to prevent interferon induction. Nature 2018, 557, 57–61. [Google Scholar] [CrossRef]
- Ablasser, A.; Hemmerling, I.; Schmid-Burgk, J.L.; Behrendt, R.; Roers, A.; Hornung, V. TREX1 deficiency triggers cell-autonomous immunity in a cGAS-dependent manner. J. Immunol. 2014, 192, 5993–5997. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; van Kooyk, Y. DC-SIGN: A novel HIV receptor on DCs that mediates HIV-1 transmission. Curr. Top. Microbiol. Immunol. 2003, 276, 31–54. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Liu, L.; Wang, Y. Viral proteins recognized by different TLRs. J. Med. Virol. 2021, 93, 6116–6123. [Google Scholar] [CrossRef]
- Henrick, B.M.; Nag, K.; Yao, X.D.; Drannik, A.G.; Aldrovandi, G.M.; Rosenthal, K.L. Milk matters: Soluble Toll-like receptor 2 (sTLR2) in breast milk significantly inhibits HIV-1 infection and inflammation. PLoS ONE 2012, 7, e40138. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Drannik, A.G.; Abimiku, A.l.; Rosenthal, K.L.; INFANT Study Team. Soluble Toll-like receptor 2 is significantly elevated in HIV-1 infected breast milk and inhibits HIV-1 induced cellular activation, inflammation and infection. AIDS 2014, 28, 2023–2032. [Google Scholar] [CrossRef]
- Su, S.-B.; Tao, L.; Deng, Z.-P.; Chen, W.; Qin, S.-Y.; Jiang, H.-X. TLR10: Insights, controversies and potential utility as a therapeutic target. Scand. J. Immunol. 2021, 93, e12988. [Google Scholar] [CrossRef]
- Pichlmair, A.; Diebold, S.S.; Gschmeissner, S.; Takeuchi, Y.; Ikeda, Y.; Collins, M.K.; Reis e Sousa, C. Tubulovesicular structures within vesicular stomatitis virus G protein-pseudotyped lentiviral vector preparations carry DNA and stimulate antiviral responses via Toll-like receptor 9. J. Virol. 2007, 81, 539–547. [Google Scholar] [CrossRef]
- Bergantz, L.; Subra, F.; Deprez, E.; Delelis, O.; Richetta, C. Interplay between Intrinsic and Innate Immunity during HIV Infection. Cells 2019, 8, 922. [Google Scholar] [CrossRef]
- Siegrist, F.; Ebeling, M.; Certa, U. The small interferon-induced transmembrane genes and proteins. J. Interferon. Cytokine Res. 2011, 31, 183–197. [Google Scholar] [CrossRef] [PubMed]
- Chintala, K.; Mohareer, K.; Banerjee, S. Dodging the Host Interferon-Stimulated Gene Mediated Innate Immunity by HIV-1: A Brief Update on Intrinsic Mechanisms and Counter-Mechanisms. Front. Immunol. 2021, 12, 716927. [Google Scholar] [CrossRef] [PubMed]
- Brass, A.L.; Huang, I.C.; Benita, Y.; John, S.P.; Krishnan, M.N.; Feeley, E.M.; Ryan, B.J.; Weyer, J.L.; van der Weyden, L.; Fikrig, E.; et al. The IFITM Proteins Mediate Cellular Resistance to Influenza A H1N1 Virus, West Nile Virus, and Dengue Virus. Cell 2009, 139, 1243–1254. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Li, M.; Wilkins, J.; Ding, S.; Swartz, T.H.; Esposito, A.M.; Zheng, Y.-M.; Freed, E.O.; Liang, C.; Chen, B.K.; et al. IFITM Proteins Restrict HIV-1 Infection by Antagonizing the Envelope Glycoprotein. Cell Rep. 2015, 13, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Pan, Q.; Rong, L.; He, W.; Liu, S.L.; Liang, C. The IFITM proteins inhibit HIV-1 infection. J. Virol. 2011, 85, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.; Towers, G.J.; Qasim, W. Gene therapy strategies to exploit TRIM derived restriction factors against HIV-1. Viruses 2014, 6, 243–263. [Google Scholar] [CrossRef]
- Ribeiro, C.M.S.; Sarrami-Forooshani, R.; Setiawan, L.C.; Zijlstra-Willems, E.M.; van Hamme, J.L.; Tigchelaar, W.; van der Wel, N.N.; Kootstra, N.A.; Gringhuis, S.I.; Geijtenbeek, T.B.H. Receptor usage dictates HIV-1 restriction by human TRIM5α in dendritic cell subsets. Nature 2016, 540, 448–452. [Google Scholar] [CrossRef]
- Lascano, J.; Uchil, P.D.; Mothes, W.; Luban, J. TRIM5 Retroviral Restriction Activity Correlates with the Ability To Induce Innate Immune Signaling. J. Virol. 2016, 90, 308–316. [Google Scholar] [CrossRef]
- Evans, M.E.; Kumkhaek, C.; Hsieh, M.M.; Donahue, R.E.; Tisdale, J.F.; Uchida, N. TRIM5α Variations Influence Transduction Efficiency With Lentiviral Vectors in Both Human and Rhesus CD34+ Cells In Vitro and In Vivo. Mol. Ther. 2014, 22, 348–358. [Google Scholar] [CrossRef]
- Pertel, T.; Hausmann, S.; Morger, D.; Züger, S.; Guerra, J.; Lascano, J.; Reinhard, C.; Santoni, F.A.; Uchil, P.D.; Chatel, L.; et al. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 2011, 472, 361–365. [Google Scholar] [CrossRef]
- Asaoka, K.; Ikeda, K.; Hishinuma, T.; Horie-Inoue, K.; Takeda, S.; Inoue, S. A retrovirus restriction factor TRIM5α is transcriptionally regulated by interferons. Biochem. Biophys. Res. Commun. 2005, 338, 1950–1956. [Google Scholar] [CrossRef]
- Carthagena, L.; Bergamaschi, A.; Luna, J.M.; David, A.; Uchil, P.D.; Margottin-Goguet, F.; Mothes, W.; Hazan, U.; Transy, C.; Pancino, G.; et al. Human TRIM gene expression in response to interferons. PLoS ONE 2009, 4, e4894. [Google Scholar] [CrossRef]
- Desimmie, B.A.; Burdick, R.C.; Izumi, T.; Doi, H.; Shao, W.; Alvord, W.G.; Sato, K.; Koyanagi, Y.; Jones, S.; Wilson, E.; et al. APOBEC3 proteins can copackage and comutate HIV-1 genomes. Nucleic Acids Res. 2016, 44, 7848–7865. [Google Scholar] [CrossRef] [PubMed]
- Zennou, V.; Perez-Caballero, D.; Göttlinger, H.; Bieniasz, P.D. APOBEC3G incorporation into human immunodeficiency virus type 1 particles. J. Virol. 2004, 78, 12058–12061. [Google Scholar] [CrossRef] [PubMed]
- Hakata, Y.; Miyazawa, M. Deaminase-Independent Mode of Antiretroviral Action in Human and Mouse APOBEC3 Proteins. Microorganisms 2020, 8, 1976. [Google Scholar] [CrossRef] [PubMed]
- Trapp, S.; Derby, N.R.; Singer, R.; Shaw, A.; Williams, V.G.; Turville, S.G.; Bess, J.W., Jr.; Lifson, J.D.; Robbiani, M. Double-stranded RNA analog poly(I:C) inhibits human immunodeficiency virus amplification in dendritic cells via type I interferon-mediated activation of APOBEC3G. J. Virol. 2009, 83, 884–895. [Google Scholar] [CrossRef]
- Zhou, Y.; Wang, X.; Liu, M.; Hu, Q.; Song, L.; Ye, L.; Zhou, D.; Ho, W. A critical function of toll-like receptor-3 in the induction of anti-human immunodeficiency virus activities in macrophages. Immunology 2010, 131, 40–49. [Google Scholar] [CrossRef]
- Chang, M.O.; Suzuki, T.; Yamamoto, N.; Watanabe, M.; Takaku, H. HIV-1 Gag-virus-like particles inhibit HIV-1 replication in dendritic cells and T cells through IFN-α-dependent upregulation of APOBEC3G and 3F. J. Innate Immun. 2012, 4, 579–590. [Google Scholar] [CrossRef]
- Stopak, K.S.; Chiu, Y.L.; Kropp, J.; Grant, R.M.; Greene, W.C. Distinct patterns of cytokine regulation of APOBEC3G expression and activity in primary lymphocytes, macrophages, and dendritic cells. J. Biol. Chem. 2007, 282, 3539–3546. [Google Scholar] [CrossRef]
- Rennie, M.L.; McKelvie, S.A.; Bulloch, E.M.; Kingston, R.L. Transient dimerization of human MxA promotes GTP hydrolysis, resulting in a mechanical power stroke. Structure 2014, 22, 1433–1445. [Google Scholar] [CrossRef]
- Fricke, T.; White, T.E.; Schulte, B.; de Souza Aranha Vieira, D.A.; Dharan, A.; Campbell, E.M.; Brandariz-Nuñez, A.; Diaz-Griffero, F. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 2014, 11, 68. [Google Scholar] [CrossRef]
- Liu, Z.; Pan, Q.; Ding, S.; Qian, J.; Xu, F.; Zhou, J.; Cen, S.; Guo, F.; Liang, C. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 2013, 14, 398–410. [Google Scholar] [CrossRef]
- Schulte, B.; Buffone, C.; Opp, S.; Di Nunzio, F.; De Souza Aranha Vieira, D.A.; Brandariz-Nuñez, A.; Diaz-Griffero, F. Restriction of HIV-1 Requires the N-Terminal Region of MxB as a Capsid-Binding Motif but Not as a Nuclear Localization Signal. J. Virol. 2015, 89, 8599–8610. [Google Scholar] [CrossRef]
- Buffone, C.; Kutzner, J.; Opp, S.; Martinez-Lopez, A.; Selyutina, A.; Coggings, S.A.; Studdard, L.R.; Ding, L.; Kim, B.; Spearman, P.; et al. The ability of SAMHD1 to block HIV-1 but not SIV requires expression of MxB. Virology 2019, 531, 260–268. [Google Scholar] [CrossRef]
- Aebi, M.; Fäh, J.; Hurt, N.; Samuel, C.E.; Thomis, D.; Bazzigher, L.; Pavlovic, J.; Haller, O.; Staeheli, P. cDNA structures and regulation of two interferon-induced human Mx proteins. Mol. Cell Biol. 1989, 9, 5062–5072. [Google Scholar] [CrossRef] [PubMed]
- Beloglazova, N.; Flick, R.; Tchigvintsev, A.; Brown, G.; Popovic, A.; Nocek, B.; Yakunin, A.F. Nuclease activity of the human SAMHD1 protein implicated in the Aicardi-Goutieres syndrome and HIV-1 restriction. J. Biol. Chem. 2013, 288, 8101–8110. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ryoo, J.; Oh, C.; Hwang, S.; Ahn, K. SAMHD1 specifically restricts retroviruses through its RNase activity. Retrovirology 2015, 12, 46. [Google Scholar] [CrossRef] [PubMed]
- Seamon, K.J.; Sun, Z.; Shlyakhtenko, L.S.; Lyubchenko, Y.L.; Stivers, J.T. SAMHD1 is a single-stranded nucleic acid binding protein with no active site-associated nuclease activity. Nucleic Acids Res. 2015, 43, 6486–6499. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhan, Y.; Zhou, Y.; Jiang, Y.; Zheng, X.; Yu, L.; Tong, W.; Gao, F.; Li, L.; Huang, Q.; et al. Interferon regulatory factor 3 is a key regulation factor for inducing the expression of SAMHD1 in antiviral innate immunity. Sci. Rep. 2016, 6, 29665. [Google Scholar] [CrossRef]
- Zhu, M.; Lu, J.; Dong, X.; Zheng, N.; Li, T.; Chen, Z.; Pan, X.; Zhu, Y.; Yan, H.; Shen, Y.; et al. Interferon-stimulated gene factor 3 complex is required for the induction of sterile α motif and HD domain-containing protein 1 expression by interferon-α in SMMC-7721 cells. Mol. Med. Rep. 2015, 12, 7176–7180. [Google Scholar] [CrossRef]
- Baldauf, H.-M.; Pan, X.; Erikson, E.; Schmidt, S.; Daddacha, W.; Burggraf, M.; Schenkova, K.; Ambiel, I.; Wabnitz, G.; Gramberg, T.; et al. SAMHD1 restricts HIV-1 infection in resting CD4+ T cells. Nat. Med. 2012, 18, 1682–1688. [Google Scholar] [CrossRef]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. SAMHD1 restricts HIV-1 reverse transcription in quiescent CD4+T-cells. Retrovirology 2012, 9, 87. [Google Scholar] [CrossRef]
- Coiras, M.; Bermejo, M.; Descours, B.; Mateos, E.; García-Pérez, J.; López-Huertas, M.R.; Lederman, M.M.; Benkirane, M.; Alcamí, J. IL-7 Induces SAMHD1 Phosphorylation in CD4+ T Lymphocytes, Improving Early Steps of HIV-1 Life Cycle. Cell Rep. 2016, 14, 2100–2107. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Schenkova, K.; Adam, T.; Erikson, E.; Lehmann-Koch, J.; Sertel, S.; Verhasselt, B.; Fackler, O.T.; Lasitschka, F.; Keppler, O.T. SAMHD1’s protein expression profile in humans. J. Leukoc. Biol. 2015, 98, 5–14. [Google Scholar] [CrossRef]
- Li, D.; Schlaepfer, E.; Audigé, A.; Rochat, M.-A.; Ivic, S.; Knowlton, C.N.; Kim, B.; Keppler, O.T.; Speck, R.F. Vpx mediated degradation of SAMHD1 has only a very limited effect on lentiviral transduction rate in ex vivo cultured HSPCs. Stem Cell Res. 2015, 15, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Girard-Gagnepain, A.; Amirache, F.; Costa, C.; Lévy, C.; Frecha, C.; Fusil, F.; Nègre, D.; Lavillette, D.; Cosset, F.-L.; Verhoeyen, E. Baboon envelope pseudotyped LVs outperform VSV-G-LVs for gene transfer into early-cytokine-stimulated and resting HSCs. Blood 2014, 124, 1221–1231. [Google Scholar] [CrossRef]
- Li, S.X.; Barrett, B.S.; Heilman, K.J.; Messer, R.J.; Liberatore, R.A.; Bieniasz, P.D.; Kassiotis, G.; Hasenkrug, K.J.; Santiago, M.L. Tetherin promotes the innate and adaptive cell-mediated immune response against retrovirus infection in vivo. J. Immunol. 2014, 193, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Pujol, F.M.; Laketa, V.; Schmidt, F.; Mukenhirn, M.; Muller, B.; Boulant, S.; Grimm, D.; Keppler, O.T.; Fackler, O.T. HIV-1 Vpu Antagonizes CD317/Tetherin by Adaptor Protein-1-Mediated Exclusion from Virus Assembly Sites. J. Virol. 2016, 90, 6709–6723. [Google Scholar] [CrossRef]
- Amet, T.; Byrd, D.; Hu, N.; Sun, Q.; Li, F.; Zhao, Y.; Hu, S.; Grantham, A.; Yu, Q. BST-2 expression in human hepatocytes is inducible by all three types of interferons and restricts production of hepatitis C virus. Curr. Mol. Med. 2014, 14, 349–360. [Google Scholar] [CrossRef]
- Holmgren, A.M.; Miller, K.D.; Cavanaugh, S.E.; Rall, G.F. Bst2/Tetherin Is Induced in Neurons by Type I Interferon and Viral Infection but Is Dispensable for Protection against Neurotropic Viral Challenge. J. Virol. 2015, 89, 11011–11018. [Google Scholar] [CrossRef]
- Blanchet, F.P.; Stalder, R.; Czubala, M.; Lehmann, M.; Rio, L.; Mangeat, B.; Piguet, V. TLR-4 engagement of dendritic cells confers a BST-2/tetherin-mediated restriction of HIV-1 infection to CD4+ T cells across the virological synapse. Retrovirology 2013, 10, 6. [Google Scholar] [CrossRef]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef]
- Asokan, A.; Schaffer, D.V.; Jude Samulski, R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Mol. Ther. 2012, 20, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Sonntag, F.; Köther, K.; Schmidt, K.; Weghofer, M.; Raupp, C.; Nieto, K.; Kuck, A.; Gerlach, B.; Böttcher, B.; Müller, O.J.; et al. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 2011, 85, 12686–12697. [Google Scholar] [CrossRef] [PubMed]
- Kotin, R.M.; Siniscalco, M.; Samulski, R.J.; Zhu, X.D.; Hunter, L.; Laughlin, C.A.; McLaughlin, S.; Muzyczka, N.; Rocchi, M.; Berns, K.I. Site-specific integration by adeno-associated virus. Proc. Natl. Acad. Sci. USA 1990, 87, 2211–2215. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Samulski, R.J. Engineering adeno-associated virus vectors for gene therapy. Nat. Rev. Genet. 2020, 21, 255–272. [Google Scholar] [CrossRef]
- Kotterman, M.A.; Schaffer, D.V. Engineering adeno-associated viruses for clinical gene therapy. Nat. Rev. Genet. 2014, 15, 445–451. [Google Scholar] [CrossRef]
- Calcedo, R.; Vandenberghe, L.H.; Gao, G.; Lin, J.; Wilson, J.M. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 2009, 199, 381–390. [Google Scholar] [CrossRef]
- Erles, K.; Sebökovà, P.; Schlehofer, J.R. Update on the prevalence of serum antibodies (IgG and IgM) to adeno-associated virus (AAV). J. Med. Virol. 1999, 59, 406–411. [Google Scholar] [CrossRef]
- Popa-Wagner, R.; Porwal, M.; Kann, M.; Reuss, M.; Weimer, M.; Florin, L.; Kleinschmidt, J.A. Impact of VP1-specific protein sequence motifs on adeno-associated virus type 2 intracellular trafficking and nuclear entry. J. Virol. 2012, 86, 9163–9174. [Google Scholar] [CrossRef]
- Riyad, J.M.; Weber, T. Intracellular trafficking of adeno-associated virus (AAV) vectors: Challenges and future directions. Gene Ther. 2021, 28, 683–696. [Google Scholar] [CrossRef]
- Dudek, A.M.; Porteus, M.H. Answered and Unanswered Questions in Early-Stage Viral Vector Transduction Biology and Innate Primary Cell Toxicity for Ex-Vivo Gene Editing. Front. Immunol. 2021, 12, 660302. [Google Scholar] [CrossRef]
- Yan, Z.; Zak, R.; Luxton, G.W.; Ritchie, T.C.; Bantel-Schaal, U.; Engelhardt, J.F. Ubiquitination of both adeno-associated virus type 2 and 5 capsid proteins affects the transduction efficiency of recombinant vectors. J. Virol. 2002, 76, 2043–2053. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Huang, X.; Yang, Y. The TLR9-MyD88 pathway is critical for adaptive immune responses to adeno-associated virus gene therapy vectors in mice. J. Clin. Investig. 2009, 119, 2388–2398. [Google Scholar] [CrossRef] [PubMed]
- Martino, A.T.; Suzuki, M.; Markusic, D.M.; Zolotukhin, I.; Ryals, R.C.; Moghimi, B.; Ertl, H.C.; Muruve, D.A.; Lee, B.; Herzog, R.W. The genome of self-complementary adeno-associated viral vectors increases Toll-like receptor 9-dependent innate immune responses in the liver. Blood 2011, 117, 6459–6468. [Google Scholar] [CrossRef] [PubMed]
- Faust, S.M.; Bell, P.; Cutler, B.J.; Ashley, S.N.; Zhu, Y.; Rabinowitz, J.E.; Wilson, J.M. CpG-depleted adeno-associated virus vectors evade immune detection. J. Clin. Investig. 2013, 123, 2994–3001. [Google Scholar] [CrossRef]
- Chan, Y.K.; Wang, S.K.; Chu, C.J.; Copland, D.A.; Letizia, A.J.; Costa Verdera, H.; Chiang, J.J.; Sethi, M.; Wang, M.K.; Neidermyer, W.J., Jr.; et al. Engineering adeno-associated viral vectors to evade innate immune and inflammatory responses. Sci. Transl. Med. 2021, 13, eabd3438. [Google Scholar] [CrossRef]
- Zhang, C.; Shang, G.; Gui, X.; Zhang, X.; Bai, X.C.; Chen, Z.J. Structural basis of STING binding with and phosphorylation by TBK1. Nature 2019, 567, 394–398. [Google Scholar] [CrossRef]
- Chandler, L.C.; Barnard, A.R.; Caddy, S.L.; Patrício, M.I.; McClements, M.E.; Fu, H.; Rada, C.; MacLaren, R.E.; Xue, K. Enhancement of Adeno-Associated Virus-Mediated Gene Therapy Using Hydroxychloroquine in Murine and Human Tissues. Mol. Ther. Methods Clin. Dev. 2019, 14, 77–89. [Google Scholar] [CrossRef]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef]
- Jin, T.; Perry, A.; Jiang, J.; Smith, P.; Curry, J.A.; Unterholzner, L.; Jiang, Z.; Horvath, G.; Rathinam, V.A.; Johnstone, R.W.; et al. Structures of the HIN domain:DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity 2012, 36, 561–571. [Google Scholar] [CrossRef]
- Orzalli, M.H.; Conwell, S.E.; Berrios, C.; DeCaprio, J.A.; Knipe, D.M. Nuclear interferon-inducible protein 16 promotes silencing of herpesviral and transfected DNA. Proc. Natl. Acad. Sci. USA 2013, 110, E4492–E4501. [Google Scholar] [CrossRef]
- Nagai, Y.; Garrett, K.P.; Ohta, S.; Bahrun, U.; Kouro, T.; Akira, S.; Takatsu, K.; Kincade, P.W. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity 2006, 24, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Lentz, T.B.; Samulski, R.J. Insight into the mechanism of inhibition of adeno-associated virus by the Mre11/Rad50/Nbs1 complex. J. Virol. 2015, 89, 181–194. [Google Scholar] [CrossRef] [PubMed]
- Sanlioglu, S.; Benson, P.K.; Yang, J.; Atkinson, E.M.; Reynolds, T.; Engelhardt, J.F. Endocytosis and nuclear trafficking of adeno-associated virus type 2 are controlled by rac1 and phosphatidylinositol-3 kinase activation. J. Virol. 2000, 74, 9184–9196. [Google Scholar] [CrossRef] [PubMed]
- Alexander, I.E.; Russell, D.W.; Miller, A.D. DNA-damaging agents greatly increase the transduction of nondividing cells by adeno-associated virus vectors. J. Virol. 1994, 68, 8282–8287. [Google Scholar] [CrossRef]
- Nidetz, N.F.; McGee, M.C.; Tse, L.V.; Li, C.; Cong, L.; Li, Y.; Huang, W. Adeno-associated viral vector-mediated immune responses: Understanding barriers to gene delivery. Pharmacol. Ther. 2020, 207, 107453. [Google Scholar] [CrossRef]
- Shao, W.; Earley, L.F.; Chai, Z.; Chen, X.; Sun, J.; He, T.; Deng, M.; Hirsch, M.L.; Ting, J.; Samulski, R.J.; et al. Double-stranded RNA innate immune response activation from long-term adeno-associated virus vector transduction. JCI Insight 2018, 3, e120474. [Google Scholar] [CrossRef]
- Hösel, M.; Broxtermann, M.; Janicki, H.; Esser, K.; Arzberger, S.; Hartmann, P.; Gillen, S.; Kleeff, J.; Stabenow, D.; Odenthal, M.; et al. Toll-like receptor 2–mediated innate immune response in human nonparenchymal liver cells toward adeno-associated viral vectors. Hepatology 2012, 55, 287–297. [Google Scholar] [CrossRef]
- Balakrishnan, B.; Sen, D.; Hareendran, S.; Roshini, V.; David, S.; Srivastava, A.; Jayandharan, G.R. Activation of the cellular unfolded protein response by recombinant adeno-associated virus vectors. PLoS ONE 2013, 8, e53845. [Google Scholar] [CrossRef]
- Mano, M.; Ippodrino, R.; Zentilin, L.; Zacchigna, S.; Giacca, M. Genome-wide RNAi screening identifies host restriction factors critical for in vivo AAV transduction. Proc. Natl. Acad. Sci. USA 2015, 112, 11276–11281. [Google Scholar] [CrossRef]
- Zhao, W.; Zhong, L.; Wu, J.; Chen, L.; Qing, K.; Weigel-Kelley, K.A.; Larsen, S.H.; Shou, W.; Warrington, K.H., Jr.; Srivastava, A. Role of cellular FKBP52 protein in intracellular trafficking of recombinant adeno-associated virus 2 vectors. Virology 2006, 353, 283–293. [Google Scholar] [CrossRef]
- Qing, K.; Li, W.; Zhong, L.; Tan, M.; Hansen, J.; Weigel-Kelley, K.A.; Chen, L.; Yoder, M.C.; Srivastava, A. Adeno-associated virus type 2-mediated gene transfer: Role of cellular T-cell protein tyrosine phosphatase in transgene expression in established cell lines in vitro and transgenic mice in vivo. J. Virol. 2003, 77, 2741–2746. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Chen, L.; Li, Y.; Qing, K.; Weigel-Kelley, K.A.; Chan, R.J.; Yoder, M.C.; Srivastava, A. Self-complementary adeno-associated virus 2 (AAV)-T cell protein tyrosine phosphatase vectors as helper viruses to improve transduction efficiency of conventional single-stranded AAV vectors in vitro and in vivo. Mol. Ther. 2004, 10, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, W.; Yang, Z.; Qing, K.; Tan, M.; Hansen, J.; Li, Y.; Chen, L.; Chan, R.J.; Bischof, D.; et al. Impaired nuclear transport and uncoating limit recombinant adeno-associated virus 2 vector-mediated transduction of primary murine hematopoietic cells. Hum. Gene Ther. 2004, 15, 1207–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhong, L.; Li, W.; Yang, Z.; Chen, L.; Li, Y.; Qing, K.; Weigel-Kelley, K.A.; Yoder, M.C.; Shou, W.; Srivastava, A. Improved transduction of primary murine hepatocytes by recombinant adeno-associated virus 2 vectors in vivo. Gene Ther. 2004, 11, 1165–1169. [Google Scholar] [CrossRef]
- Czar, M.J.; Owens-Grillo, J.K.; Dittmar, K.D.; Hutchison, K.A.; Zacharek, A.M.; Leach, K.L.; Deibel, M.R., Jr.; Pratt, W.B. Characterization of the protein-protein interactions determining the heat shock protein (hsp90.hsp70.hsp56) heterocomplex. J. Biol. Chem. 1994, 269, 11155–11161. [Google Scholar] [CrossRef]
- Czar, M.J.; Owens-Grillo, J.K.; Yem, A.W.; Leach, K.L.; Deibel, M.R., Jr.; Welsh, M.J.; Pratt, W.B. The hsp56 immunophilin component of untransformed steroid receptor complexes is localized both to microtubules in the cytoplasm and to the same nonrandom regions within the nucleus as the steroid receptor. Mol. Endocrinol. 1994, 8, 1731–1741. [Google Scholar] [CrossRef]
- Schreiber, C.A.; Sakuma, T.; Izumiya, Y.; Holditch, S.J.; Hickey, R.D.; Bressin, R.K.; Basu, U.; Koide, K.; Asokan, A.; Ikeda, Y. An siRNA Screen Identifies the U2 snRNP Spliceosome as a Host Restriction Factor for Recombinant Adeno-associated Viruses. PLoS Pathog. 2015, 11, e1005082. [Google Scholar] [CrossRef]
- Mitchell, A.M.; Samulski, R.J. Mechanistic insights into the enhancement of adeno-associated virus transduction by proteasome inhibitors. J. Virol. 2013, 87, 13035–13041. [Google Scholar] [CrossRef]
- Zhong, L.; Li, B.; Mah, C.S.; Govindasamy, L.; Agbandje-McKenna, M.; Cooper, M.; Herzog, R.W.; Zolotukhin, I.; Warrington, K.H., Jr.; Weigel-Van Aken, K.A.; et al. Next generation of adeno-associated virus 2 vectors: Point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc. Natl. Acad. Sci. USA 2008, 105, 7827–7832. [Google Scholar] [CrossRef]
- Zhong, L.; Zhao, W.; Wu, J.; Li, B.; Zolotukhin, S.; Govindasamy, L.; Agbandje-McKenna, M.; Srivastava, A. A dual role of EGFR protein tyrosine kinase signaling in ubiquitination of AAV2 capsids and viral second-strand DNA synthesis. Mol. Ther. 2007, 15, 1323–1330. [Google Scholar] [CrossRef]
- Aslanidi, G.V.; Rivers, A.E.; Ortiz, L.; Song, L.; Ling, C.; Govindasamy, L.; Van Vliet, K.; Tan, M.; Agbandje-McKenna, M.; Srivastava, A. Optimization of the capsid of recombinant adeno-associated virus 2 (AAV2) vectors: The final threshold? PLoS ONE 2013, 8, e59142. [Google Scholar] [CrossRef] [PubMed]
- Büning, H.; Srivastava, A. Capsid Modifications for Targeting and Improving the Efficacy of AAV Vectors. Mol. Ther. Methods Clin. Dev. 2019, 12, 248–265. [Google Scholar] [CrossRef] [PubMed]
- Hölscher, C.; Sonntag, F.; Henrich, K.; Chen, Q.; Beneke, J.; Matula, P.; Rohr, K.; Kaderali, L.; Beil, N.; Erfle, H.; et al. The SUMOylation Pathway Restricts Gene Transduction by Adeno-Associated Viruses. PLoS Pathog. 2015, 11, e1005281. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Njenga, R.; Leuchs, B.; Chiocca, S.; Kleinschmidt, J.; Müller, M. SUMOylation Targets Adeno-associated Virus Capsids but Mainly Restricts Transduction by Cellular Mechanisms. J. Virol. 2020, 94, e00871-20. [Google Scholar] [CrossRef] [PubMed]
- Everett, R.D.; Boutell, C.; Hale, B.G. Interplay between viruses and host sumoylation pathways. Nat. Rev. Microbiol. 2013, 11, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, S.; Wodrich, H. Virion factors that target Daxx to overcome intrinsic immunity. J. Virol. 2013, 87, 10412–10422. [Google Scholar] [CrossRef] [PubMed]
- Madigan, V.J.; Tyson, T.O.; Yuziuk, J.A.; Pillai, M.; Moller-Tank, S.; Asokan, A. A CRISPR Screen Identifies the Cell Polarity Determinant Crumbs 3 as an Adeno-associated Virus Restriction Factor in Hepatocytes. J. Virol. 2019, 93, e00943-19. [Google Scholar] [CrossRef]
- Meisen, W.H.; Nejad, Z.B.; Hardy, M.; Zhao, H.; Oliverio, O.; Wang, S.; Hale, C.; Ollmann, M.M.; Collins, P.J. Pooled Screens Identify GPR108 and TM9SF2 as Host Cell Factors Critical for AAV Transduction. Mol. Ther. Methods Clin. Dev. 2020, 17, 601–611. [Google Scholar] [CrossRef]
- Verdera, H.C.; Kuranda, K.; Mingozzi, F. AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Mol. Ther. 2020, 28, 723–746. [Google Scholar] [CrossRef]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11 (Suppl. S1), S10–S17. [Google Scholar] [CrossRef]
- Li, X.; Wei, X.; Lin, J.; Ou, L. A versatile toolkit for overcoming AAV immunity. Front. Immunol. 2022, 13, 991832. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, C.; Thorne, L.G.; Unali, G.; Schiroli, G.; Giordano, A.M.S.; Piras, F.; Cuccovillo, I.; Petit, S.J.; Ahsan, F.; Noursadeghi, M.; et al. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing In Human Hematopoietic Stem Cells. Cell Stem Cell 2018, 23, 820–832. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coroadinha, A.S. Host Cell Restriction Factors Blocking Efficient Vector Transduction: Challenges in Lentiviral and Adeno-Associated Vector Based Gene Therapies. Cells 2023, 12, 732. https://doi.org/10.3390/cells12050732
Coroadinha AS. Host Cell Restriction Factors Blocking Efficient Vector Transduction: Challenges in Lentiviral and Adeno-Associated Vector Based Gene Therapies. Cells. 2023; 12(5):732. https://doi.org/10.3390/cells12050732
Chicago/Turabian StyleCoroadinha, Ana Sofia. 2023. "Host Cell Restriction Factors Blocking Efficient Vector Transduction: Challenges in Lentiviral and Adeno-Associated Vector Based Gene Therapies" Cells 12, no. 5: 732. https://doi.org/10.3390/cells12050732
APA StyleCoroadinha, A. S. (2023). Host Cell Restriction Factors Blocking Efficient Vector Transduction: Challenges in Lentiviral and Adeno-Associated Vector Based Gene Therapies. Cells, 12(5), 732. https://doi.org/10.3390/cells12050732