

Direct Cell Reprogramming and Phenotypic Conversion: An Analysis of Experimental Attempts to Transform Astrocytes into Neurons in Adult Animals

 ,
, 
Abstract
1. Introduction
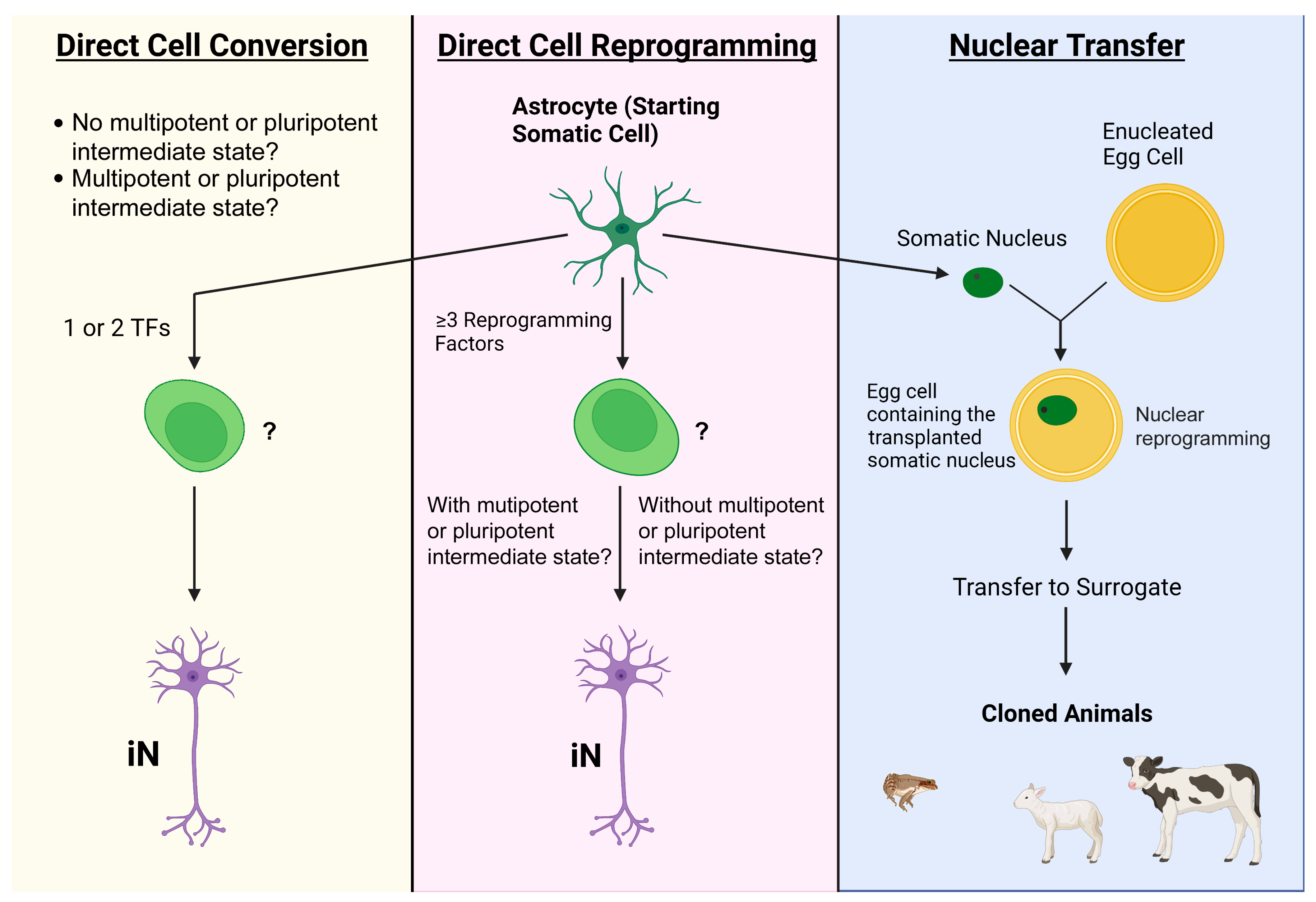
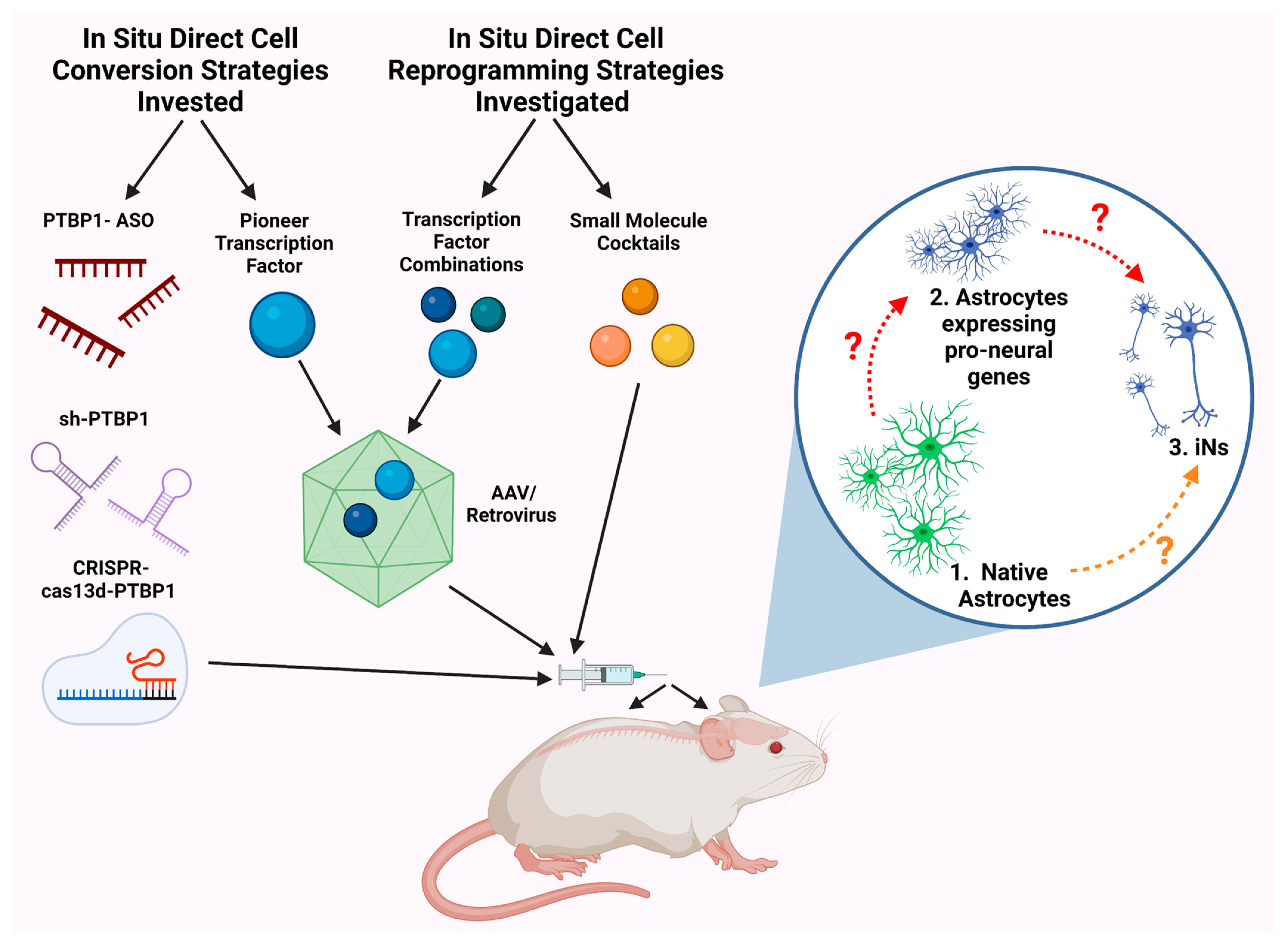
2. Common Bench Approaches to Directly Attaining Neurons from Astrocytes
2.1. Transcription Factors

{kind=link}
{kind=link}
{kind=link}
| Direct Cell Conversion Strategy | Induction Factor | In Vitro/In Vivo | Starting Cell Type/Animal Model | Vector/Delivery System | Induction Efficiency (%) | Phenotype |
|---|---|---|---|---|---|---|
| Common Bench Approaches to AtN Conversion | ||||||
| Proneural factors/ pioneer transcription factors | Ascl1 (Mash1) [29] | In vitro | Dorsal midbrain astrocytes, WT mice (P5–P7) | Lentivirus | 76.8 ± 6.4 | Glutamatergic (19.4%), GABAergic (8/38 cells) |
| In vivo | Dorsal midbrain astrocytes, WT mice (P60), M + F | AAV micropipette injection | 92.1 ± 1.5 | GABAergic (11.7 ± 4.0%), Glutamatergic (6.3 ± 1.3%) | ||
| NeuroD1 | In vivo | Cortical astrocytes, stab injury mouse model (P90–180), M + F [23] | AAV needle injection | 90.6 ± 5.2 | Glutamatergic, GABAergic | |
| Ischemic stroke model, GFAP-Cre × Rosa-YFP mice (adult), M [30] | Lentivirus stereotaxic injection | ~66 | Glutamatergic (~80%) | |||
| Contusive SCI model T10 acute phase, WT mice (P60–P120), M + F [22] | Cre-FLEX AAV needle injection | ~55 | Glutamatergic | |||
| Contusive SCI model T11-T12 chronic phase, WT mice (P60-P120), M + F [22] | Cre-FLEX AAV needle injection | >95 | Glutamatergic | |||
| Neurog2 (Ngn2) | In vivo [24] | Dorsal midbrain astrocytes, WT mice (adult) | AAV stereotaxic needle injection | 96.3 ± 1.7 | Glutamatergic (64.97 ± 8.04%), GABAergic (2.26 ± 2.07%) | |
| Dorsal horn T8–T10, WT mice (adult) | 80.11 ± 5.42 | Glutamatergic (50/9%), GABAergic (38.5%) | ||||
| Complete transection SCI model T8–T10, WT mice (adult) | AAV injection at L1–L2 dorsal surface | 41.62 ± 22.82 | Data not provided | |||
| Dlx2 | In vivo [31] | Striatal astrocytes, stab injury model, WT C57BL/6J mice (P60–P150), M + F | Retrovirus needle injection | ~20 (30 dpi) | DCX+ immature neurons | |
| Striatal astrocytes in stab injury model, Aldh1l1-CreERT2 mice, (P60–P150), M + F | AAV9 needle injection | ~70 (60 dpi) | MSN | |||
| NeuroD1 + Dlx2 | In vivo [27] | Striatal astrocytes, WT mice (P60–P140), M + F | rAAV2/5 stereotaxic injection | 72.7 | GABAergic (~85.0%), MSN (55.7%), interneurons (9.6%) | |
| Striatal astrocytes, R6/2 transgenic Huntington’s disease mouse model (P60–P150), M + F | rAAV2/5 stereotaxic injection | 78.6 | GABAergic, MSN, interneurons | |||
| Striatum, YAC128 transgenic Huntington’s disease mouse model (middle aged, 15 months), M + F | rAAV2/5 stereotaxic injection | 50.0 | GABAergic, MSN, interneurons | |||
| Ascl1 + Dlx2 | In vivo [32] | Hippocampus, mesial temporal lobe epilepsy model, C57BL/6J mice (2–3 months), M | Retrovirus stereotaxic injection | 70 | GABAergic interneurons (~75%) | |
| PTBP1 knockout | PTBP1 | In vivo | Dentate gyrus, adult GFAP-CreERT2CAG-lox-stop-lox-tdTomato mice (5 months), M + F [33] | ASO-PTBP1 CSF injection | 15 (2 mpi) | Granule cell layer neurons |
| Dentate gyrus, aged GFAP-CreERT2CAG-lox-stop-lox-tdTomato mice (1 year), M + F [33] | ASO-PTBP1 CSF injection | 5 (2 mpi) | Granule cell layer neurons | |||
| Midbrain astrocytes, 6-OHDA Parkinson’s disease mouse model GFAP-Cre transgenic mouse [34] | AAV-shPTBP1 | 30–35 (12 wpi) | Dopaminergic | |||
| Striatum of adult C57BL/6 mice (~P70), M [35] | AAV-GFAP-CasRx-Ptbp1 with gRNAs 5 + 6 targeting Ptbp1 stereotaxic injection | 48.0 ± 10.0 | Glutamatergic (~50%) | |||
| Common Bench Approaches to AtN Reprogramming | ||||||
| Transcription factor and other reprogramming factor in combinations | NeAL218 * [26] | In vitro | Human midbrain astrocytes | Lentivirus carrying rtTA | 16.48 ±8.6 | Dopaminergic (100%) |
| In vivo | Ipsilateral striatum, transgenic (GFAP-tTA)110Pop/J mice (adult, P60–P180) | Tet-regulated lentivirus/stereotaxic injection | 14.63 ± 8.5 cells/section | Dopaminergic | ||
| Small molecules | SLDC * [36] | In vitro | Human cortical astrocytes | Direct application to culture medium | 71 | Glutamatergic (78%), GABAergic (2%), dopaminergic (1%) |
| DFICBY [14] | In vitro | TauEGFP reporter murine astrocytes | Direct application to FCBG* culture medium | 89.2 ± 1.4 (TuJ1+, 16 dpi) 77.8 ± 11.1 (NeuN+, 30 dpi) | Glutamatergic, GABAergic | |
| In vivo | Striatum, mGfap-Cre/Rosa26-tdTomato/TauEGFP mice (P56) | Osmotic minipump for 2 weeks at a constant rate | >350 NeuN/tdTomato+ cells (8 wpi)/127 ± 24 tdTomato+/NEUN+ cells per slice at injection core | Data not provided | ||
| MCMs * [37] | In vitro | Human cortical astrocytes | Applied to culture medium in step-wise manner | 68.7 ± 4.2 | Glutamatergic (88.3 ± 4%), GABAergic (8.2 ± 1.5%) | |
2.2. Gene Delivery Vehicles
2.2.1. Viral Vectors
2.2.2. Other Vectors
2.3. PTBP1 Knockdown
2.4. Small Molecules
2.5. Other Tactics
2.5.1. Micro-RNAs (miRs)
2.5.2. DNA Binding Domains
2.5.3. CRISPR
3. Major Challenges in the Field of Direct AtN Reprogramming and Conversion Research
3.1. Transcriptional Mechanisms and Quality Control
| Induction Factor(s) | Vector/ Delivery System | Cell Type/ Anatomical Target | Induction Efficiency (%) | Criteria | iN Phenotype/ Criteria | iN Features | |
|---|---|---|---|---|---|---|---|
| In vitro astrocyte to neuron reprogramming | |||||||
| NeAL218 + MP * [26] | Lentivirus carrying rtTA [26] | ATCC (SVGp12, cat. n CRL86-21), midbrain (hIAs) | 30.97 ± 5.3 | TH+, MAP2+ (84.6 ± 1.9%), TUBB3+ | Dopaminergic (100% of iN)—DDC, SLC6A3, FOXA2, EN1, and SLC18A | Simple neuron-like morphologies and lack emDAs membrane properties | |
| NeAL218 + RTMP * [26] | ATCC (SVGp12, cat. n CRL86-21), midbrain (hIAs) | 16.48 ± 8.6 | TH+, TUBB3+, MAP2+, SYN+ | Dopaminergic (100% of iN)—DDC, SLC6A3, ALDH1A1, and KCNJ6 | Ca2+ response upon depolarization (55 mM KCl), generate AP, sEA + AP at 13–17 days, current clamp recordings show different firing properties upon current injection (none, single AP, multiple AP), and 2/7 (≈29%) generate multiple AP | ||
| Lonza (normal human astrocytes, cat. n CC-2565), hPAs | 12.4 ± 2.7 | TH+, MAP2+, RBFOX3+ | Dopaminergic (100% of iN)—DDC, SLC6A3, ALDH1A1, KCNJ6, and PBX1 | ||||
| In vitro astrocyte to neuron conversion | |||||||
| Ascl1 (Mash1) [29,88] | Lentivirus FUGW [29] | Isolated from P5–P7 mice, postnatal dorsal midbrain | 76.8 ± 6.4 | Tuj1+ MAP2+, and Synapsin I+ | Glutamatergic (19.4%)—blocked by CNQX, GABAergic (8/38, ≈21%)—blocked by Bicuculline | Produce AP and sPSC in 85.3% | |
| Retroviral VSV-G [88] | C57BL/6 mice P5–P7, pNCC | 37 ± 11% and 14 ± 2% Tuj1+, and >40% TuJ1- | TuJ1+ | No TuJ1+/Tbr1+, no clear nuclear staining for Ascl1 (Mash1) | Intrinsic excitability, generate typical neuronal AP, and virtual absence of spontaneous synaptic input | ||
| Neurog2 (Ngn2) [88,89] | Retroviral VSV-G [88] | C57BL/6 mice P5–P7, pNCC | >85%, 71 ± 16%, and 16 ± 18% clones TuJ1+, and ~10% clones TuJ1- | TuJ1+ | Glutamatergic (≈33%)—TuJ1+/Tbr1+, blocked by CNQX GABA (polysynaptic, UD)— >5 ms delay, blocked by both CNQX and Bicuculline | Fire repetitive AP, ↑ negative resting mV, ↓ IR, ↑ AP amp over time, functional but ↓ PS response, and not generate SR from neighboring neurons | |
| pCAG-IRES-DsRed (self- silencing, long-acting) [89] | C57BL/6J or GLAST::CreERT2/Z/EG mice P5–P7, pNCC | 70.2 ± 6.3%, | BIII tubulin+, GFAP- | Glutamatergic (58.3%)—BIII tubulin+/vGlut1+ puncta (85.4 ± 5.0%) GABA (0%) | MAP2 in 2–3 weeks and Ca2+ transients (63.8%) | ||
| In neurosphere [89] | 91.4 ± 2.2% | MAP2+ | Glutamatergic—MAP2+/vGlut1+, AC/SC (9/21, ≈43%), CNQX-sensitive sSC (8/30, ≈27%) | Low IR | |||
| Dlx2 [89] | pCAG-IRES-DsRed (self- silencing, long-acting) [89] | C57BL/6J or GLAST::CreERT2/Z/EG mice P5–P7, pNCC | 35.9 ± 13.0% | BIII tubulin+, MAP2+ | GABAergic—Autapses, vGlut1-, BIII tubulin/vGaT+ (33.7 ± 3.6%), sSC with slow decay time (9/33, ≈27%), AR blocked by Bicuculline | Neuron morphology, fire AP, distinct firing patterns (regular, stuttering, and low-threshold), 7/9 (≈78%) immature firing pattern, and 2/9 (≈22%) mature interneuron-like firing pattern | |
| In neurosphere [89] | 94.7 ± 0.3% | MAP2+ | GABAergic—MAP2+/vGaT+ puncta, slow decay time (9/10, ≈90%, UD) | ↓ IR and no Ca2+ transients | |||
| Induction Factor(s) | Vector/ Delivery System | Animal Model/Sex | Anatomical Target | Direct Reprogramming Efficiency (%) | Criteria | iN Phenotype/ Criteria | iN Features |
| In vivo astrocyte to neuron reprogramming | |||||||
| ALN * [90] | Cre-inducible AAV5/injection | Adult GFAP-Cre mice (P84–P112) | Striatum | 46.8 ± 2.9 | NeuN+ | Glutamatergic—vGlut1+ (16%) GABAergic—GAD65/67+ (68%) | rMP (−61.4 ± 9.7 mV), AP mean amp (33.5 ± 2.29 mV), and AP threshold (25 ± 7.19 pA) |
| NeAl218 * [26] | Tet-regulated NeAL218 lentiviruses/stereotactic needle injection | Adult Tg(GFAP-tTA)110Pop/J mice (P60–P180) | Ipsilateral striatum | 14.63 ± 8.5 | TH+ | Dopaminergic—TH+/SLC6A3+, RBFOX3+, NR4A2+, and PBX1+ | TH+/SLC6A3+ iNs produced Ih |
| In vivo astrocyte to neuron conversion | |||||||
| Ascl1 [29] | AAV/ micropipette injection [29] | Adolescent WT mice (P12–P15), M + F | Dorsal midbrain | 93.1 ± 1.7 | NeuN+ | GABAergic—NeuN+/Gad1+ (13.2 ± 4.2%) Glutamatergic—NeuN+/VGLUT2+ (6.5 ± 2.2%) | Producing AP, sPSC observed, IOC in VCM, MΩ (177.3 ± 16.6), and ↓ RMP (−61.9 ± 1.0) |
| Adult WT mice (P60), M + F | 92.1 ± 1.5 | NeuN+ | GABAergic—NeuN+/Gad1+ (11.7 ± 4.0%) Glutamatergic—NeuN+/VGLUT2+ (6.3 ± 1.3%) | Producing AP, sPSC observed, IOC in VCM, MΩ (240.0 ± 81.9), and ↓ RMP (−61.0 ± 1.2) | |||
| Striatum | 64.4 ± 3.4 | NeuN+ | GABAergic (according to electrophysiological test performed) | Fire APs in CCM (13/16, ≈81%), sEPSC and sIPSCs (12/16, ≈75%), and IOC in VCM (15/16, ≈94%) | |||
| Somatosensory cortex | 93.9 ± 1.2 | NeuN+ | Glutamatergic or GABAergic (according to electrophysiological verification) | Record 163.3 ± 35.9 MΩ, dMP (−67± 2.2 mV), APs, IOC, sEPSC, and sIPSCs | |||
| AAV-FLEX/ micropipettes injection [29] | Adult Aldh1l1–Cre transgenic mice (P60), M + F | Dorsal midbrain | 90.1 ± 2.1 | NeuN+ | GABAergic (according to electrophysiological verification) | Exhibit firing patterns identical to midbrain endogenous GABAergic neurons | |
| AAV/needle injection [29] | Injured dorsal midbrain | 54.2 ± 6.9 | NeuN+ | Glutamatergic or GABAergic (according to electrophysiological verification) | 424.7 ± 88.7 MΩ, rMP (−61.2 ± 1.6 mV), IOC in VCM, rAPs fired in CCM, sEPSC, and sIPSCs | ||
| NeuroD1 [21,22,23] | AAV/stereotactic needle injection and infusion pump [21] | Adult Macaca mulatta (9–21 years old), M | Cortex | 94.4 ± 5.5 | NeuN+/ Tbr1+ | Glutamatergic—Tbr1+, projection neurons | ↑ SV2 and significantly recovered MAP2 |
| AAV9/ stereotactic needle injection [23] | Adult WT mice (P90–P180), M + F | Cortex | 90.6 ± 5.2 | NeuN+ | Glutamatergic—vGlutT1+ GABAergic—GAD67+ | ↑ SMI32, ↑ vGluT1 and GAD67, large Na+/K+ currents (13/15, ≈87%), rAPs (7/10, ≈70%), glutamatergic SE (10/13, ≈77%), and GABAergic SE (9/13, ≈69%) | |
| Cre-FLEX AAV/needle injection [22] | Adult WT mice (P60–P120), M + F | Stab-injured dorsal horn T10 | ~95.0 | NeuN+ | Glutamatergic—NeuN+/Tlx3+ (62.6 ± 3.3%) GABAergic—NeuN+/Pax2+ (8.8 ± 1.3%) | rAPs, large Na+/K+ current, robust spontaneous EPSCs, and no difference in Na+ current and sEPSCs compared with neighboring native neurons | |
| Contusive SCI model T10 acute phase | ~55.0 | NeuN+ | Glutamatergic—Neu+/Tlx3+ in dorsal horn | ↑ SV2 | |||
| Contusive SCI model T11–T12 chronic phase | >95.0 | NeuN+ | Glutamatergic—Neu+/Tlx3+ in dorsal horn | ↑ SV2 | |||
| Neurog2 [24] | AAV/stereotactic needle injection | Adult WT mice | Dorsal midbrain | 96.3 ± 1.7 | NeuN+ | Glutamatergic—NeuN+/VGLUT2+ (64. 97 ± 8.04%) GABAergic—NeuN+/Gad1+ (2.26 ± 2.07%) | Multiple APs, IOC in VCM, EPSC, MC and the IR of iN are largely comparable with local neurons, and neuronal profile |
| Dorsal horn T8–T10 | 80.11 ± 5.42 | NeuN+ | Glutamatergic—Tlx3+ (50.9 ± 8.8%) GABAergic—Pax2+ (38.5 ± 8.3%) | Produce IOC in VCM, multiple APs (9/11, ≈82%; ↓ AP amp), and MC and iR comparable to native neurons | |||
| AAV/injection from L1–L2 dorsal surface | Transected SC T8–T10 | 41.62 ± 22.82 | NeuN+ | Data not provided | Data not provided | ||
| Ptbp1 knockout [35] | AAV-GFAP-CasRx-Ptbp1 with gRNAs 5 + 6 for Ptbp/stereotactic injection | Adult C57BL/6 mice (~P70) | Striatum | 48.00 ± 10.00 | NeuN+ | Glutamatergic—50% iNs glutaminase+ | Data not provided |
| Ipsilateral striatum/PD model | 32.00 ± 7.00 | TH+ | Dopaminergic—TH+/DAT+ (31 ± 7%), ~15% TH+/DDC+, ~37% TH+/FOXA2+ iNs were ALDH1A1+, GIRK2+, and CB– | rAPs (20/22, ≈91%) in response to depolarizing current injection in the CCM, sPSC observed in VCM (Vc = −70 mV), delayed voltage rectification induced by Ih (4/10, 40%), and majority iNs were VMAT2+ | |||
| NeuroD1 + Dlx2 [22,27] | rAAV2/5/stereotactic bilateral needle injection [27] | Adult WT mice (P60–P140), M + F | Striatum | 72.7 | NeuN+ | MSN—NeuN+/DARPP32+ (55.7%) GABAergic—NeuN+/GAD67+ (83.9%) GABAergic—NeuN+/GABA+ (85.0%) Interneurons—NeuN+/PV+ (9.6%) NeuN+/SST+ or NPY+ or CalR+ (<5%) | Data not provided |
| Adult R6/2 transgenic mice (P60–P150), M + F | 78.6 | NeuN+ | MSN, GABAergic, and interneuron; additional expression: DARPP32 (56.6%), GAD67 (82.4%), GABA (88.7%), PV (8.4%), and <5% (SST, NPY, CalR) | iNs rAPs (17/18, ≈94%), 72.2% firing at <80 Hz, 22.2% firing at >80 Hz, detected sEPSCs and sIPSCs in all iN, and ↑ iR, ↓ cC, ↓ RMP, and ↓ AP amp compared with control | |||
| Middle-aged YAC128 transgenic mice (15 months), M + F | 50.0 | NeuN+ | MSN, GABAergic, and interneuron; additional expression: DARPP32 (29.8%), GABA (half), and PV (3.9%) | Data not provided | |||
| Cre-FLEX AAV/needle injection [22] | Adult WT mice (P60–P120), M + F | Stab-injured dorsal horn T11–T12 | N/A | Tlx3+ Pax2+ | Glutamatergic—Tlx3+ (56.2 ± 3.4%) GABAergic—Pax2+ (32.5 ± 2.1%) | Data not provided | |
| Ascl1 + Nurr1 [41] | FLEX-switch AAV/ microinjection | Adult mGFAP-Cre mice (P60–90), M + F | Injury cortex model | 40.0 (24 dpi) 70.0 (72 dpi) | NeuN+ | iNs variable morphology | * Ascl alone served as a control and was shown to have a conversation efficiency of ≈20.0% (NeuN+) |
| Neurog2 + Nurr1 [41] | 53.0 (24 dpi) 80.0 (72 dpi) | NeuN+ | NeuN+/CUX1+ iNs in upper layer, NeuN+/CUX+ iNs in deeper layer; both displayed stereotypical pyramidal-shaped cell soma; single and combinatorial labeling for CUX1, SATB2, and BRN2+ iNs in upper layers FOXP2+, CTIP2+, TLE4+, and TBR1+ iNs in lower layer | rMP, iR, APs comparable to endogenous neurons, and E/I input blocked by NBQX. * Nurr1 alone served as a control and was shown to have a conversation efficiency of ≈20.0% (NeuN+) | |||
3.2. Specific Epigenetic Mechanisms
3.3. Metabolic Transition
3.4. Other Potential Therapeutic Effects Derived from the Process of AtN Conversion
4. Common Issues concerning Cell Phenotype Reprogramming and Conversion Protocols
4.1. Control of Specific Subtypes of iNs
4.2. Age of Starting Cells
4.3. Astrocytic Regional Identity
5. Cell Lineage Tracing
6. Future Directions
6.1. Micro-3D Cell Culture Systems
6.2. Spatial Biology
6.3. Other Delivery Methods
7. Additional Discussions and Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Aaku-Saraste, E.; Hellwig, A.; Huttner, W.B. Loss of occluding and functional tight junctions, but not ZO-1, during neural tube closure-remodelling of the neuroepithelium prior to neurogenesis. Dev. Biol. 1996, 180, 664–679. [Google Scholar] [CrossRef]
- Hamilton, A. The division of differentiated cells in the central nervous system of the white rat. J. Comp. Neurol. 1901, 11, 297–320. [Google Scholar] [CrossRef]
- Kjell, J.; Fischer-Sternjak, J.; Thompson, A.J.; Friess, C.; Sticco, M.J.; Salinas, F.; Cox, J.; Martinelli, D.C.; Ninkovic, J.; Franze, K.; et al. Defining the Adult Neural Stem Cell Niche Proteome Identifies Key Regulators of Adult Neurogenesis. Cell Stem Cell 2020, 26, 277–293. [Google Scholar] [CrossRef] [PubMed]
- Kase, Y.; Shimazaki, T.; Okano, H. Current understanding of adult neurogenesis in the mammalian brain: How does adult neurogenesis decrease with age? Inflamm. Regen. 2020, 40, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, J.W. The Struggle to Make CNS Axons Regenerate: Why Has It Been so Difficult? Neurochem. Res. 2020, 45, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Karova, K.; Wainwright, J.V.; Machova-Urdzikova, L.; Pisal, R.V.; Schmidt, M.; Jendelova, P.; Jhanwar-Uniyal, M. Transplantation of neural precursors generated from spinal progenitor cells reduces inflammation in spinal cord injury via NF-κB pathway inhibition. J. Neuroinflamm. 2019, 16, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Vonderwalde, I.; Azimi, A.; Rolvink, G.; Ahlfors, J.E.; Shoichet, M.S.; Morshead, C.M. Transplantation of Directly Reprogrammed Human Neural Precursor Cells Following Stroke Promotes Synaptogenesis and Functional Recovery. Transl. Stroke Res. 2020, 11, 93–107. [Google Scholar] [CrossRef]
- Gurdon, J.B. The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpoles. J. Embryol. Exp. Morphol. 1962, 10, 622–640. [Google Scholar] [CrossRef]
- Weintraub, H.; Tapscott, S.J.; Davis, R.L.; Thayer, M.J.; Adam, M.A.; Lassar, A.B.; Miller, A.D. Activation of muscle-specific genes in pigment, nerve, fat, liver, and fibroblast cell lines by forced expression of MyoD. Proc. Natl. Acad. Sci. USA 1989, 86, 5434–5438. [Google Scholar] [CrossRef]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef]
- Gurdon, J.B. Chapter Twenty Four-Cell Fate Determination by Transcription Factors. Essays Dev. Biol. Part A 2016, 116, 445–454. [Google Scholar]
- Grath, A.; Dai, G. Direct cell reprogramming for tissue engineering and regenerative medicine. J. Biol. Eng. 2019, 13, 14–29. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gon, X. Organ-on-a-chip: Recent breakthroughs and future prospects. BioMed. Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef]
- Ma, Y.; Xie, H.; Du, X.; Wang, L.; Jin, X.; Zhang, Q.; Han, Y.; Sun, S.; Wang, L.; Li, X.; et al. In vivo chemical reprogramming of astrocytes into neurons. Cell Discov. 2021, 7, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Brown, J.; Kanarek, A.; Rajagopal, J.; Melton, D.A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 2008, 455, 627–632. [Google Scholar] [CrossRef]
- Jeon, J.; Correa-Medina, M.; Ricordi, C.; Edlund, H.; Diez, J.A. Endocrine cell clustering during human pancreas development. J. Histochem. Cytochem. 2009, 57, 811–824. [Google Scholar] [CrossRef]
- Pavlou, M.A.S.; Grandbarbe, L.; Buckley, N.J.; Niclou, S.P.; Michelucci, A. Transcriptional and epigenetic mechanisms underlying astrocyte identity. Prog. Neurobiol. 2019, 174, 36–52. [Google Scholar] [CrossRef]
- Eguchi, R.; Hamano, M.; Iwata, M.; Nakamura, T.; Oki, S.; Yamanishi, Y. TRANSDIRE: Data-driven direct reprogramming by a pioneer factor-guided trans-omics approach. Bioinformatics 2022, 38, 2839–2846. [Google Scholar] [CrossRef]
- Park, N.I.; Guilhamon, P.; Desai, K.; McAdam, R.F.; Langille, E.; O’Connor, M.; Lan, X.; Whetstone, H.; Coutinho, F.J.; Vanner, R.J.; et al. ASCL1 Reorganizes Chromatin to Direct Neuronal Fate and Suppress Tumorigenicity of Glioblastoma Stem Cells. Cell Stem Cell 2017, 21, 209–224. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Ma, N.X.; Pei, Z.F.; Wu, Z.; Do-Monte, F.H.; Keefe, S.; Yellin, E.; Chen, M.S.; Yin, J.C.; Lee, G.; et al. A NeuroD1 AAV-Based Gene Therapy for Functional Brain Repair after Ischemic Injury through In Vivo Astrocyte-to-Neuron Conversion. Mol. Ther. 2020, 28, 217–234. [Google Scholar] [CrossRef]
- Ge, L.J.; Yang, F.H.; Li, W.; Wang, T.; Lin, Y.; Feng, J.; Chen, N.H.; Jiang, M.; Wang, J.H.; Hu, X.T.; et al. In Vivo Neuroregeneration to Treat Ischemic Stroke through NeuroD1 AAV-Based Gene Therapy in Adult Non-human Primates. Front. Cell Dev. Biol. 2020, 8, 590008. [Google Scholar] [CrossRef]
- Puls, B.; Ding, Y.; Zhang, F.; Pan, M.; Lei, Z.; Pei, Z.; Jiang, M.; Bai, Y.; Forsyth, C.; Metzger, M.; et al. Regeneration of Functional Neurons After Spinal Cord Injury via in situ NeuroD1-Mediated Astrocyte-to-Neuron Conversion. In Front. Cell Dev. Biol. 2020, 8, 1595. [Google Scholar] [CrossRef]
- Zhang, L.; Lei, Z.; Guo, Z.; Pei, Z.; Chen, Y.; Zhang, F.; Cai, A.; Mok, G.; Lee, G.; Swaminathan, V.; et al. Development of Neuroregenerative Gene Therapy to Reverse Glial Scar Tissue Back to Neuron-Enriched Tissue. Front. Cell. Neurosci. 2020, 14, 594170. [Google Scholar] [CrossRef]
- Liu, F.; Zhang, Y.; Chen, F.; Yuan, J.; Li, S.; Han, S.; Lu, D.; Geng, J.; Rao, Z.; Sun, L.; et al. Neurog2 directly converts astrocytes into functional neurons in midbrain and spinal cord. Cell Death Dis. 2021, 12, 225. [Google Scholar] [CrossRef]
- Niu, W.; Zang, T.; Zou, Y.; Fang, S.; Smith, D.K.; Bachoo, R.; Zhang, C.L. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 2013, 15, 1164–1175. [Google Scholar] [CrossRef]
- Rivetti di Val Cervo, P.; Romanov, R.A.; Spigolon, G.; Masini, D.; Martín-Montañez, E.; Toledo, E.M.; La Manno, G.; Feyder, M.; Pifl, C.; Ng, Y.H.; et al. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017, 35, 444–452. [Google Scholar] [CrossRef]
- Wu, Z.; Parry, M.; Hou, X.Y.; Liu, M.H.; Wang, H.; Cain, R.; Pei, Z.F.; Chen, Y.C.; Guo, Z.Y.; Abhijeet, S.; et al. Gene therapy conversion of striatal astrocytes into GABAergic neurons in mouse models of Huntington’s disease. Nat. Commun. 2020, 11, 1105–1123. [Google Scholar] [CrossRef]
- Hu, Q.; Chen, R.; Teesalu, T.; Ruoslahti, E.; Clegg, D.O. Reprogramming human retinal pigmented epithelial cells to neurons using recombinant proteins. Stem Cells Transl. Med. 2014, 3, 1526–1534. [Google Scholar] [CrossRef]
- Liu, Y.; Miao, Q.; Yuan, J.; Han, S.; Zhang, P.; Li, S.; Rao, Z.; Zhao, W.; Ye, Q.; Geng, J.; et al. Ascl1 Converts Dorsal Midbrain Astrocytes into Functional Neurons In Vivo. J. Neurosci. 2015, 35, 9336–9355. [Google Scholar] [CrossRef]
- Jiang, M.Q.; Yu, S.P.; Wei, Z.Z.; Zhong, W.; Cao, W.; Gu, X.; Wu, A.; McCrary, M.R.; Berglund, K.; Wei, L. Conversion of Reactive Astrocytes to Induced Neurons Enhances Neuronal Repair and Functional Recovery After Ischemic Stroke. Front. Aging Neurosci. 2021, 13, 612856. [Google Scholar] [CrossRef]
- Liu, M.H.; Xu, Y.G.; Bai, X.N.; Lin, J.H.; Xiang, Z.Q.; Wang, T.; Xu, L.; Li, W.; Chen, G. Efficient Dlx2-mediated astrocyte-to-neuron conversion and inhibition of neuroinflammation by NeuroD1. bioRxiv 2022. [Google Scholar] [CrossRef]
- Lentini, C.; d’Orange, M.; Marichal, N.; Trottmann, M.M.; Vignoles, R.; Foucault, L.; Verrier, C.; Massera, C.; Raineteau, O.; Conzelmann, K.K.; et al. Reprogramming reactive glia into interneurons reduces chronic seizure activity in a mouse model of mesial temporal lobe epilepsy. Cell Stem Cell 2021, 28, 2104–2121. [Google Scholar] [CrossRef]
- Maimon, R.; Chillon-Marinas, C.; Snethlage, C.E.; Singhal, S.M.; McAlonis-Downes, M.; Ling, K.; Rigo, F.; Bennett, C.F.; Da Cruz, S.; Hnasko, T.S.; et al. Therapeutically viable generation of neurons with antisense oligonucleotide suppression of PTB. Nat. Neurosci. 2021, 24, 1089–1099. [Google Scholar] [CrossRef]
- Qian, H.; Kang, X.; Hu, J.; Zhang, D.; Liang, Z.; Meng, F.; Zhang, X.; Xue, Y.; Maimon, R.; Dowdy, S.F.; et al. Reversing a model of Parkinson’s disease with in situ converted nigral neurons. Nature 2020, 582, 550–556. [Google Scholar] [CrossRef]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603. [Google Scholar] [CrossRef]
- Yin, J.C.; Zhang, L.; Ma, N.X.; Wang, Y.; Lee, G.; Hou, X.Y.; Lei, Z.F.; Zhang, F.Y.; Dong, F.P.; Wu, G.Y.; et al. Chemical Conversion of Human Fetal Astrocytes into Neurons through Modulation of Multiple Signalling Pathways. Stem Cell Rep. 2019, 12, 488–501. [Google Scholar] [CrossRef]
- Zhang, L.; Yin, J.C.; Yeh, H.; Ma, N.X.; Lee, G.; Chen, X.A.; Wang, Y.; Lin, L.; Chen, L.; Jin, P.; et al. Small Molecules Efficiently Reprogram Human Astroglial Cells into Functional Neurons. Cell Stem Cell 2015, 17, 735–747. [Google Scholar] [CrossRef]
- Zaiss, A.K.; Liu, Q.; Bowen, G.P.; Wong, N.C.W.; Bartlett, J.S.; Muruve, D.A. Differential activation of innate immune responses by adenovirus and adeno-associated virus vectors. J. Virol. 2002, 76, 4580–4590. [Google Scholar] [CrossRef]
- Chan, K.Y.; Jang, M.J.; Yoo, B.B.; Greenbaum, A.; Ravi, N.; Wu, W.L.; Sánchez-Guardado, L.; Lois, C.; Mazmanian, S.K.; Deverman, B.E.; et al. Engineered AAVs for efficient non-invasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017, 20, 1172–1179. [Google Scholar] [CrossRef]
- Xiang, Z.; Xu, L.; Liu, M.; Wang, Q.; Li, W.; Lei, W.; Chen, G. Lineage tracing of direct astrocyte-to-neuron conversion in the mouse cortex. Neural Regen. Res. 2021, 16, 750–756. [Google Scholar]
- Mattugini, N.; Bocchi, R.; Scheuss, V.; Russo, G.L.; Torper, O.; Lao, C.L.; Götz, M. Inducing Different Neuronal Subtypes from Astrocytes in the Injured Mouse Cerebral Cortex. Neuron 2019, 103, 1086–1095. [Google Scholar] [CrossRef]
- Lim, D.A.; Alvarez-Buylla, A. Adult neural stem cells stake their ground. Trends Neurosci. 2014, 37, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Gresita, A.; Glavan, D.; Udristoiu, I.; Catalin, B.; Hermann, D.M.; Popa-Wagner, A. Very Low Efficiency of Direct Reprogramming of Astrocytes into Neurons in the Brains of Young and Aged Mice After Cerebral Ischemia. Front. Aging Neurosci. 2019, 11, 334–341. [Google Scholar] [CrossRef]
- Brommel, C.M.; Cooney, A.L.; Sinn, P.L. Adeno-Associated Virus-Based Gene Therapy for Lifelong Correction of Genetic Disease. Hum. Gene Ther. 2020, 31, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Thakor, D.K.; Teng, Y.D.; Tabata, Y. Neuronal gene delivery by negatively charged pullulan-spermine/DNA anioplexes. Biomaterials 2009, 30, 1815–1826. [Google Scholar] [CrossRef] [PubMed]
- Thakor, D.K.; Teng, Y.D.; Obata, H.; Nagane, K.; Saito, S.; Tabata, Y. Nontoxic genetic engineering of mesenchymal stem cells using serum-compatible pullulan-spermine/DNA anioplexes. Tissue Eng. Part C Methods 2011, 17, 131–144. [Google Scholar] [CrossRef] [PubMed]
- Ye, H.; Wang, Q. Efficient Generation of Non-Integration and Feeder-Free Induced Pluripotent Stem Cells from Human Peripheral Blood Cells by Sendai Virus. Cell. Physiol. Biochem. 2018, 50, 1318–1331. [Google Scholar] [CrossRef]
- Fusaki, N.; Ban, H.; Nishiyama, A.; Saeki, K.; Hasegawa, M. Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc. Jpn. Acad. 2009, 85, 348–362. [Google Scholar] [CrossRef]
- Seki, T.; Yuasa, S.; Oda, M.; Egashira, T.; Yae, K.; Kusumoto, D.; Nakata, H.; Tohyama, S.; Hashimoto, H.; Kodaira, M.; et al. Generation of induced pluripotent stem cells from human terminally differentiated circulating T cells. Cell Stem Cell 2010, 7, 11–14. [Google Scholar] [CrossRef]
- Ban, H.; Nishishita, N.; Fusaki, N.; Tabata, T.; Saeki, K.; Shikamura, M.; Takada, N.; Inoue, M.; Hasegawa, M.; Kawamata, S.; et al. Efficient generation of transgene-free human induced pluripotent stem cells (iPSCs) by temperature-sensitive Sendai virus vectors. Proc. Natl. Acad. Sci. USA 2011, 108, 14234–14239. [Google Scholar] [CrossRef] [PubMed]
- Chakritbudsabong, W.; Sariya, L.; Jantahiran, P.; Chaisilp, N.; Chaiwattanarungruengpaisan, S.; Rungsiwiwut, R.; Ferreira, J.N.; Rungarunlert, S. Generation of Porcine Induced Neural Stem Cells Using the Sendai Virus. Front. Vet. Sci. 2021, 8, 806785. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, K.; Akiyama, M.; Tamura, F.; Isomi, M.; Yamakawa, H.; Sadahiro, T.; Muraoka, N.; Kojima, H.; Haginiwa, S.; Kurotsu, S.; et al. Direct In Vivo Reprogramming with Sendai Virus Vectors Improves Cardiac Function after Myocardial Infarction. Cell Stem Cell 2018, 22, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Zu, H.; Gao, D. Non-viral Vectors in Gene Therapy: Recent Development, Challenges, and Prospects. AAPS J. 2021, 23, 78–90. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Keskin, D.; Shi, L. Poly(β-Amino Esters): Synthesis, Formulations, and Their Biomedical Applications. Adv. Healthc. Mater. 2019, 8, 1801359. [Google Scholar] [CrossRef]
- Qi, R.; Mullen, D.G.; Baker, J.R.J.; Banaszak Holl, M.M. The Mechanism of Polyplex Internalization into Cells: Testing the GM1/Caveolin-1 Lipid Raft Mediated Endocytosis Pathway. Mol. Pharm. 2010, 7, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.F.; Grigsby, C.L.; Kulangara, K.; Wang, H.; Yasuda, R.; Leong, K.W. Nonviral direct conversion of primary mouse embryonic fibroblasts to neuronal cells. Mol. Ther. 2012, 1, 32–43. [Google Scholar] [CrossRef]
- Mattiassi, S.; Rizwan, M.; Grigsby, C.; Zaw, A.M.; Leong, K.; Yim, E. Enhanced efficiency of nonviral direct neuronal reprogramming on topographical patterns. Biomater. Sci. 2021, 9, 5175–5191. [Google Scholar] [CrossRef]
- Li, X.; Kozielski, K.; Cheng, Y.H.; Liu, H.; Zamboni, C.G.; Green, J.; Mao, H.Q. Nanoparticle-mediated conversion of primary human astrocytes into neurons and oligodendrocytes. Biomater. Sci. 2016, 4, 1100–1112. [Google Scholar] [CrossRef]
- Niu, W.; Zang, T.; Smith, D.K.; Vue, T.Y.; Zou, Y.; Bachoo, R.; Johnson, J.E.; Zhang, C.L. SOX2 Reprograms Resident Astrocytes into Neural Progenitors in the Adult Brain. Stem Cell Rep. 2015, 4, 780–794. [Google Scholar] [CrossRef]
- Xue, Y.; Qian, H.; Hu, J.; Zhou, B.; Zhou, Y.; Hu, X.; Karakhanyan, A.; Pang, Z.; Fu, X.D. Sequential regulatory loops as key gatekeepers for neuronal reprogramming in human cells. Nat. Neurosci. 2016, 19, 807–815. [Google Scholar] [CrossRef]
- Xue, Y.; Ouyang, K.; Huang, J.; Zhou, Y.; Ouyang, H.; Li, H.; Wang, G.; Wu, Q.; Wei, C.; Bi, Y.; et al. Direct conversion of fibroblasts to neurons by reprogramming PTB-regulated microRNA circuits. Cell 2013, 152, 82–96. [Google Scholar] [CrossRef]
- Feng, Y.; Bai, S.; Li, G.; Nie, H.; Chen, S.; Pan, C.; Zhang, P.; Tang, Y.; Liu, N.; Tang, Z. Reprogramming rat astrocytes into neurons using small molecules for cell replacement following intracerebral haemorrhage. Brain Sci. Adv. 2021, 7, 184–198. [Google Scholar] [CrossRef]
- Ma, N.X.; Yin, J.C.; Chen, G. Transcriptome Analysis of Small Molecule–Mediated Astrocyte-to-Neuron Reprogramming. In Front. Cell Dev. Biol. 2019, 7, 82. [Google Scholar] [CrossRef]
- Xu, J.; Hao, X.; Yin, M.X.; Lu, Y.; Jin, Y.; Xu, J.; Ge, L.; Wu, W.; Ho, M.; Yang, Y.; et al. Prevention of medulla neuron dedifferentiation by Nerfin-1 requires inhibition of Notch activity. Development 2017, 144, 1510–1517. [Google Scholar]
- Dauth, S.; Grevesse, T.; Pantazopoulos, H.; Campbell, P.H.; Maoz, B.M.; Berretta, S.; Parker, K.K. Extracellular matrix protein expression is brain region dependent. J. Comp. Neurol. 2016, 524, 1309–1336. [Google Scholar] [CrossRef]
- Reginensi, D.; Ortiz, D.; Pravia, A.; Burillo, A.; Morales, F.; Morgan, C.; Jimenez, L.; Dave, K.R.; Perez-Pinzon, M.A.; Gittens, R.A. Role of Region-Specific Brain Decellularized Extracellular Matrix on In Vitro Neuronal Maturation. Tissue Eng. 2020, 26, 964–978. [Google Scholar] [CrossRef]
- Cho, A.N.; Jin, Y.; An, Y.; Kim, J.; Choi, Y.S.; Kim, J.; Choi, W.; Koo, D.J.; Yu, W.; Chang, G.E.; et al. Microfluidic device with brain extracellular matrix promotes structural and functional maturation of human brain organoids. Nat. Commun. 2021, 12, 4730–4754. [Google Scholar] [CrossRef]
- Yoo, A.S.; Staahl, B.T.; Chen, L.; Crabtree, G.R. MicroRNA-mediated switching of chromatin-remodelling complexes in neural development. Nature 2009, 460, 642–646. [Google Scholar] [CrossRef]
- Abernathy, D.G.; Kim, W.K.; McCoy, M.J.; Lake, A.M.; Ouwenga, R.; Lee, S.W.; Xing, X.; Li, D.; Lee, H.J.; Heuckeroth, R.O.; et al. MicroRNAs Induce a Permissive Chromatin Environment that Enables Neuronal Subtype-Specific Reprogramming of Adult Human Fibroblasts. Cell Stem Cell 2017, 21, 332–348. [Google Scholar] [CrossRef]
- Papadimitriou, E.; Koutsoudaki, P.N.; Karamitros, T.; Karagkouni, D.; Chroni-Tzartou, D.; Gkemisis, C.; Margariti, M.; Xingi, E.; Thanou, I.; Tzartos, S.J.; et al. A miR-124-mediated post-transcriptional mechanism controlling the cell fate switch of astrocytes to induced-neurons. bioRxiv 2021. [Google Scholar] [CrossRef]
- Cates, K.; McCoy, M.J.; Kwon, J.S.; Liu, Y.; Abernathy, D.G.; Zhang, B.; Liu, S.; Gontarz, P.; Kim, W.K.; Chen, S.; et al. Deconstructing Stepwise Fate Conversion of Human Fibroblasts to Neurons by MicroRNAs. Cell Stem Cell 2021, 28, 127–140. [Google Scholar] [CrossRef]
- Church, V.A.; Cates, K.; Capano, L.; Aryal, S.; Kim, W.K.; Yoo, A.S. Generation of Human Neurons by microRNA-Mediated Direct Conversion of Dermal Fibroblasts. Methods Mol. Biol. 2021, 2239, 77–100. [Google Scholar]
- Vardjan, N.; Horvat, A.; Anderson, J.E.; Yu, D.; Croom, D.; Zeng, X.; Lužnik, Z.; Kreft, M.; Teng, Y.D.; Kirov, S.A.; et al. Adrenergic activation attenuates astrocyte swelling induced by hypotonicity and neurotrauma. Glia 2016, 64, 1034–1049. [Google Scholar] [CrossRef]
- Al-Naama, N.; Mackeh, R.; Kino, T. C2H2-Type Zinc Finger Proteins in Brain Development, Neurodevelopmental, and Other Neuropsychiatric Disorders: Systematic Literature-Based Analysis. Front. Neurol. 2020, 11, 32. [Google Scholar] [CrossRef]
- Williams, A.J.; Khachigian, L.M.; Shows, T.; Collins, T. Isolation and characterization of a novel zinc-finger protein with transcription repressor activity. J. Biol. Chem. 1995, 270, 22143–22152. [Google Scholar] [CrossRef]
- Nowick, K.; Gernat, T.; Almaas, E.; Stubbs, L. Differences in human and chimpanzee gene expression patterns define an evolving network of transcription factors in brain. Proc. Natl. Acad. Sci. USA 2009, 106, 22358–22363. [Google Scholar] [CrossRef]
- Kwak, S.; Kim, T.W.; Kang, B.H.; Kim, J.H.; Lee, J.S.; Lee, H.T.; Hwang, I.Y.; Shin, J.; Lee, J.H.; Cho, E.J.; et al. Zinc finger proteins orchestrate active gene silencing during embryonic stem cell differentiation. Nucleic Acids Res. 2018, 46, 6592–6607. [Google Scholar] [CrossRef]
- Gersbach, C.A.; Perez-Pinera, P. Activating human genes with zinc finger proteins, transcription activator-like effectors and CRISPR/Cas9 for gene therapy and regenerative medicine. Expert Opin. Ther. Targets 2014, 18, 835–839. [Google Scholar] [CrossRef]
- Zarei-Kheirabadi, M.; Hesaraki, M.; Kiani, S.; Baharvand, H. In vivo conversion of rat astrocytes into neuronal cells through neural stem cells in injured spinal cord with a single zinc-finger transcription factor. Stem Cell Res. Ther. 2019, 10, 380–399. [Google Scholar] [CrossRef]
- Baumann, V.; Wiesbeck, M.; Breunig, C.T.; Braun, J.M.; Köferle, A.; Ninkovic, J.; Götz, M.; Stricker, S.H. Targeted removal of epigenetic barriers during transcriptional reprogramming. Nat. Commun. 2019, 10, 2119–2131. [Google Scholar] [CrossRef]
- Matharu, N.; Rattanasopha, S.; Tamura, S.; Maliskova, L.; Wang, Y.; Bernard, A.; Hardin, A.; Eckalbar, W.L.; Vaisse, C.; Ahituv, N. CRISPR-mediated activation of a promoter or enhancer rescues obesity caused by haploinsufficiency. Science 2019, 363, eaau0629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, J.; Zhou, C.; Gao, N.; Rao, Z.; Li, H.; Hu, X.; Li, C.; Yao, X.; Shen, X.; et al. In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR–dCas9-activator transgenic mice. Nat. Neurosci. 2018, 21, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Luginbühl, J.; Kouno, T.; Nakano, R.; Chater, T.E.; Sivaraman, D.M.; Kishima, M.; Roudnicky, F.; Carninci, P.; Plessy, C.; Shin, J.W. Decoding Neuronal Diversification by Multiplexed Single-cell RNA-Seq. Stem Cell Rep. 2021, 16, 810–824. [Google Scholar] [CrossRef] [PubMed]
- Wapinski, O.L.; Vierbuchen, T.; Qu, K.; Lee, Q.Y.; Chanda, S.; Fuentes, D.R.; Giresi, P.G.; Ng, Y.H.; Marro, S.; Neff, N.F.; et al. Hierarchical mechanisms for direct reprogramming of fibroblasts to neurons. Cell 2013, 155, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Kareta, M.S.; Chanda, S.; Ahlenius, H.; Perotti, N.; Zhou, B.; Grieder, S.D.; Ge, X.; Drake, S.; Euong Ang, C.; et al. Myt1l safeguards neuronal identity by actively repressing many non-neuronal fates. Nature 2017, 544, 245–249. [Google Scholar] [CrossRef]
- Rao, Z.; Wang, R.; Li, S.; Shi, Y.; Mo, L.; Han, S.; Yuan, J.; Jing, N.; Cheng, L. Molecular Mechanisms Underlying Ascl1-Mediated Astrocyte-to-Neuron Conversion. Stem Cell Rep. 2021, 16, 534–547. [Google Scholar] [CrossRef]
- Ma, N.X.; Puls, B.; Chen, G. Transcriptomic analyses of NeuroD1-mediated astrocyte-to-neuron conversion. Dev. Neurobiol. 2022, 82, 375–391. [Google Scholar] [CrossRef]
- Berninger, B.; Costa, M.R.; Koch, U.; Schroeder, T.; Sutor, B.; Grothe, B.; Götz, M. Functional Properties of Neurons Derived from in Vitro Reprogrammed Postnatal Astroglia. J. Neurosci. 2007, 27, 8654–8664. [Google Scholar] [CrossRef]
- Heinrich, C.; Blum, R.; Gascón, S.; Masserdotti, G.; Tripathi, P.; Sánchez, R.; Tiedt, S.; Schroeder, T.; Götz, M.; Berninger, B. Directing Astroglia from the Cerebral Cortex into Subtype Specific Functional Neurons. PLoS Biol. 2010, 8, e1000373. [Google Scholar] [CrossRef]
- Torper, O.; Ottosson, D.R.; Pereira, M.; Lau, S.; Cardoso, T.; Grealish, S.; Parmar, M. In Vivo Reprogramming of Striatal NG2 Glia into Functional Neurons That Integrate into Local Host Circuitry. Cell Rep. 2015, 12, 474–481. [Google Scholar] [CrossRef]
- Luo, C.; Lee, Q.Y.; Wapinski, O.; Castanon, R.; Nery, J.R.; Mall, M.; Kareta, M.S.; Cullen, S.M.; Goodell, M.A.; Chang, H.Y.; et al. Global DNA methylation remodelling during direct reprogramming of fibroblasts to neurons. eLife 2019, 8, e40197. [Google Scholar] [CrossRef] [PubMed]
- Cruz, C.; Della Rosa, M.; Krueger, C.; Gao, Q.; Horkai, D.; King, M.; Field, L.; Houseley, J. Tri-methylation of histone H3 lysine 4 facilitates gene expression in ageing cells. eLife 2018, 7, e34081. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Zheng, M.; Wang, L.; Zhou, Y.; Yin, C.; Liu, J.; Qian, L. Re-patterning of H3K27me3, H3K4me3 and DNA methylation during fibroblast conversion into induced cardiomyocytes. Stem Cell Res. 2016, 16, 507–518. [Google Scholar] [CrossRef] [PubMed]
- Barbagiovanni, G.; Germain, P.L.; Zech, M.; Atashpaz, S.; Lo Riso, P.; D’Antonio-Chronowska, A.; Tenderini, E.; Caiazzo, M.; Boesch, S.; Jech, R.; et al. KMT2B Is Selectively Required for Neuronal Transdifferentiation and Its Loss Exposes Dystonia Candidate Genes. Cell Rep. 2018, 25, 988–1001. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Mathelier, A.; Zhang, X. Super-enhancers are transcriptionally more active and cell type-specific than stretch enhancers. Epigenetics 2018, 13, 910–922. [Google Scholar] [CrossRef]
- Hu, Y.; Wilson, G.S. A temporary local energy pool coupled to neuronal activity: Fluctuations of extracellular lactate levels in rat brain monitored with rapid-response enzyme-based sensor. J. Neurochem. 1997, 69, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Gascón, S.; Murenu, E.; Masserdotti, G.; Ortega, F.; Russo, G.L.; Petrik, D.; Deshpande, A.; Heinrich, C.; Karow, M.; Robertson, S.P.; et al. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell 2016, 18, 396–409. [Google Scholar] [CrossRef]
- Zheng, X.; Boyer, L.; Jin, M.; Mertens, J.; Kim, Y.; Ma, L.; Ma, L.; Hamm, M.; Gage, F.H.; Hunter, T. Metabolic reprogramming during neuronal differentiation from aerobic glycolysis to neuronal oxidative phosphorylation. eLife 2016, 5, e13374. [Google Scholar] [CrossRef]
- Yu, D.; Neeley, W.L.; Pritchard, C.D.; Slotkin, J.R.; Woodard, E.J.; Langer, R.; Teng, Y.D. Blockade of peroxynitrite-induced neural stem cell death in the acutely injured spinal cord by drug-releasing polymer. Stem Cells 2009, 27, 1212–1222. [Google Scholar] [CrossRef]
- Russo, G.L.; Sonsalla, G.; Natarajan, P.; Breunig, C.T.; Bulli, G.; Merl-Pham, J.; Schmitt, S.; Giehrl-Schwab, J.; Giesert, F.; Jastroch, M.; et al. CRISPR-Mediated Induction of Neuron-Enriched Mitochondrial Proteins Boosts Direct Glia-to-Neuron Conversion. Cell Stem Cell 2021, 28, 524–534. [Google Scholar] [CrossRef]
- Fecher, C.; Trovò, L.; Müller, S.A.; Snaidero, N.; Wettmarshausen, J.; Heink, S.; Ortiz, O.; Wagner, I.; Kühn, R.; Hartmann, J.; et al. Cell-type-specific profiling of brain mitochondria reveals functional and molecular diversity. Nat. Neurosci. 2019, 22, 1731–1742. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Li, K.; Li, J.; Zheng, J.; Qin, S. Reactive Astrocytes in Neurodegenerative Diseases. Aging Dis. 2019, 10, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Du, S.S.; Zhai, X.; Poczos, B.; Singh, A. Gradient Descent Provably Optimizes Over-Parameterized Neural Networks. arXiv 2019, arXiv:1810.02054. [Google Scholar]
- Zhang, L.; Lei, Z.; Guo, Z.; Pei, Z.; Chen, Y.; Zhang, F.; Cai, A.; Mok, Y.K.; Lee, G.; Swaminnathan, V.; et al. Reversing Glial Scar Back to Neural Tissue through NeuroD1-Mediated Astrocyte-to-Neuron Conversion. bioRxiv 2018. [Google Scholar] [CrossRef]
- Tai, W.; Wu, W.; Wang, L.L.; Ni, H.; Chen, C.; Yang, J.; Zang, T.; Zou, Y.; Xu, X.M.; Zhang, C.L. In vivo reprogramming of NG2 glia enables adult neurogenesis and functional recovery following spinal cord injury. Cell Stem Cell 2021, 28, 923–937. [Google Scholar] [CrossRef]
- Burda, J.E.; Bernstein, A.M.; Sofroniew, M.V. Astrocyte roles in traumatic brain injury. Exp. Neurol. 2016, 275, 305–315. [Google Scholar] [CrossRef]
- Endo, M.; Ubulkasim, G.; Kobayashi, C.; Onishi, R.; Aiba, A.; Minami, Y. Critical role of Ror2 receptor tyrosine kinase in regulating cell cycle progression of reactive astrocytes following brain injury. Glia 2017, 65, 182–197. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Sun, Q.; Fan, J.; Jiang, Y.; Yang, W.; Cui, Y.; Yu, Z.; Jiang, H.; Li, B. Role of Astrocytes in Post-traumatic Epilepsy. Front. Neurol. 2019, 10, 1149. [Google Scholar] [CrossRef]
- Teng, Y.D. Functional Multipotency of Stem Cells and Recovery Neurobiology of Injured Spinal Cords. Cell Transplant. 2019, 28, 451–459. [Google Scholar] [CrossRef]
- Teng, Y.D.; Lavik, E.B.; Qu, X.; Park, K.I.; Ourednik, J.; Zurakowski, D.; Langer, R.; Snyder, E.Y. Functional recovery following traumatic spinal cord injury mediated by a unique polymer scaffold seeded with neural stem cells. Proc. Natl. Acad. Sci. USA 2002, 99, 3024–3029. [Google Scholar] [CrossRef]
- Ropper, A.E.; Thakor, D.K.; Han, I.; Yu, D.; Zeng, X.; Anderson, J.E.; Aljuboori, Z.; Kim, S.W.; Wang, H.; Sidman, R.L.; et al. Defining recovery neurobiology of injured spinal cord by synthetic matrix-assisted hMSC implantation. Proc. Natl. Acad. Sci. USA 2017, 114, 820–829. [Google Scholar] [CrossRef]
- An, H.; Lee, H.L.; Cho, D.W.; Hong, J.; Lee, H.Y.; Lee, J.M.; Woo, J.; Lee, J.; Park, M.; Yang, Y.S.; et al. TRANsCre-DIONE transdifferentiates scar-forming reactive astrocytes into functional motor neurons. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fu, X.; Zhu, J.; Duan, Y.; Li, G.; Cai, H.; Zheng, L.; Qian, H.; Zhang, C.; Jin, Z.; Fu, X.D.; et al. Visual function restoration in genetically blind mice via endogenous cellular reprogramming. bioRxiv 2020. [Google Scholar] [CrossRef]
- Khouri-Farah, N.; Guo, Q.; Morgan, K.; Shin, J.; Li, J.Y.H. Integrated single-cell transcriptomic and epigenetic study of cell state transition and lineage commitment in embryonic mouse cerebellum. Sci. Adv. 2022, 8, eabl9156. [Google Scholar] [CrossRef]
- Trokovic, R.; Weltner, J.; Noisa, P.; Raivio, T.; Otonkoski, T. Combined negative effect of donor age and time in culture on the reprogramming efficiency into induced pluripotent stem cells. Stem Cell Res. 2015, 15, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Lo Sardo, V.; Ferguson, W.; Erikson, G.A.; Topol, E.J.; Baldwin, K.K.; Torkamani, A. Influence of donor age on induced pluripotent stem cells. Nat. Biotechnol. 2017, 35, 69–74. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef]
- Skaansar, O.; Tverdal, C.; Rønning, P.A.; Skogen, K.; Brommeland, T.; Røise, O.; Aarhus, M.; Andelic, N.; Helseth, E. Traumatic brain injury—The effects of patient age on treatment intensity and mortality. BMC Neurol. 2020, 20, 376–386. [Google Scholar] [CrossRef]
- Yoo, J.W.; Hong, B.Y.; Jo, L.; Kim, J.S.; Park, J.G.; Shin, B.K.; Lim, S.H. Effects of Age on Long-Term Functional Recovery in Patients with Stroke. Medicina 2020, 56, 451. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Zhang, L.; Wu, Z.; Chen, Y.; Wang, F.; Chen, G. In Vivo Direct Reprogramming of Reactive Glial Cells into Functional Neurons after Brain Injury and in an Alzheimer’s Disease Model. Cell Stem Cell 2014, 14, 188–202. [Google Scholar] [CrossRef] [PubMed]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.M.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-dependent instability of mature neuronal fate in induced neurons from Alzheimer’s patients. Cell Stem Cell 2021, 28, 1533–1548. [Google Scholar] [CrossRef]
- Traxler, L.; Edenhofer, F.; Mertens, J. Next-generation disease modelling with direct conversion: A new path to old neurons. FEBS Lett. 2019, 593, 3316–3337. [Google Scholar] [CrossRef]
- Koen, J.D. Age-related neural dedifferentiation for individual stimuli: An across-participant pattern similarity analysis. Neuropsychol. Dev. Cognition. Sect. B Aging Neuropsychol. Cogn. 2022, 29, 552–576. [Google Scholar] [CrossRef] [PubMed]
- Preininger, M.K.; Kaufer, D. Blood-Brain Barrier Dysfunction and Astrocyte Senescence as Reciprocal Drivers of Neuropathology in Aging. Int. J. Mol. Sci. 2022, 23, 6217. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and health span. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef] [PubMed]
- Lanjakornsiripan, D.; Pior, B.J.; Kawaguchi, D.; Furutachi, S.; Tahara, T.; Katsuyama, Y.; Suzuki, Y.; Fukazawa, Y.; Gotoh, Y. Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat. Commun. 2018, 9, 1623–1638. [Google Scholar] [CrossRef] [PubMed]
- Kronschläger, M.T.; Siegert, A.S.M.; Resch, F.J.; Rajendran, P.S.; Khakh, B.S.; Sandkühler, J. Lamina-specific properties of spinal astrocytes. Glia 2021, 69, 1749–1766. [Google Scholar] [CrossRef]
- Herrero-Navarro, Á.; Puche-Aroca, L.; Moreno-Juan, V.; Sempere-Ferràndez, A.; Espinosa, A.; Susín, R.; Torres-Masjoan, L.; Leyva-Díaz, E.; Karow, M.; Figueres-Oñate, M.; et al. Astrocytes and neurons share region-specific transcriptional signatures that confer regional identity to neuronal reprogramming. Sci. Adv. 2021, 7, eabe8978. [Google Scholar] [CrossRef]
- Iwasa, S.N.; Babona-Pilipos, R.; Morshead, C.M. Environmental Factors That Influence Stem Cell Migration: An “Electric Field”. Stem Cells Int. 2017, 2017, 4276927. [Google Scholar] [CrossRef]
- d’Alessandro, J.; Barbier-Chebbah, A.; Cellerin, V.; Benichou, O.; Mège, R.M.; Voituriez, R.; Ladoux, B. Cell migration guided by long-lived spatial memory. Nat. Commun. 2021, 12, 4118. [Google Scholar] [CrossRef] [PubMed]
- Blackshaw, S.; Hoang, T.; Kim, D.W.; Appel, H.; Pannullo, N.; Ozawa, M.; Zheng, S.; Yu, M.; Peachey, N.; Kim, J. Ptbp1 Deletion Does Not Induce Glia-to-Neuron Conversion in Adult Mouse Retina and Brain. bioRxiv 2021. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, Q.; Huang, Q.; Ma, S.; Li, M. Repressing PTBP1 fails to convert reactive astrocytes to dopaminergic neurons in a 6-hydroxydopamine mouse model of Parkinson’s disease. eLife 2022, 11, e75636. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Yan, Z.; Wu, X.; Zhang, M.; Xu, C.; Shi, L.; Yang, H.; Fang, K. Ptbp1 knockdown in mouse striatum did not induce astrocyte-to-neuron conversion using HA-tagged labelling system. bioRxiv 2022. [Google Scholar] [CrossRef]
- Hoang, T.; Kim, D.W.; Appel, H.; Pannullo, N.A.; Leavey, P.; Ozawa, M.; Zheng, S.; Yu, M.; Peachey, N.S.; Blackshaw, S. Genetic loss of function of Ptbp1 does not induce glia-to-neuron conversion in retina. Cell Rep. 2022, 39, 110849. [Google Scholar] [CrossRef]
- Wang, L.L.; Serrano, C.; Zhong, X.; Ma, S.; Zou, Y.; Zhang, C.L. Revisiting astrocyte to neuron conversion with lineage tracing in vivo. Cell 2021, 184, 5465–5481. [Google Scholar] [CrossRef]
- Wang, L.L.; Garcia, C.S.; Zhong, X.; Ma, S.; Zhang, C.L. Rapid and efficient astrocyte-to-neuron conversion with regional identity and connectivity? bioRxiv 2020. [Google Scholar] [CrossRef]
- Koh, I.; Kim, P. In Vitro Reconstruction of Brain Tumor Microenvironment. BioChip J. 2019, 13, 1–7. [Google Scholar] [CrossRef]
- Boussommier-Calleja, A.; Li, R.; Chen, M.B.; Wong, S.C.; Kamm, R.D. Microfluidics: A new tool for modelling cancer-immune interactions. Trends Cancer 2016, 2, 6–19. [Google Scholar] [CrossRef]
- Kim, H.N.; Choi, N. Consideration of the Mechanical Properties of Hydrogels for Brain Tissue Engineering and Brain-on-a-chip. BioChip J. 2019, 13, 8–19. [Google Scholar] [CrossRef]
- Jahromi, M.A.; Abdoli, A.; Rahmanian, M.; Bardania, H.; Bayandori, M.; Moosavi Basri, S.M.; Kalbasi, A.; Aref, A.R.; Karimi, M.; Hamblin, M.R. Microfluidic Brain-on-a-Chip: Perspectives for Mimicking Neural System Disorders. Mol. Neurobiol. 2019, 56, 8489–8512. [Google Scholar] [CrossRef]
- Koo, B.; Choi, B.; Park, H.; Yoon, K.J. Past, Present, and Future of Brain Organoid Technology. Mol. Cells 2019, 42, 617–627. [Google Scholar] [PubMed]
- Shou, Y.; Liang, F.; Xu, S.; Li, X. The Application of Brain Organoids: From Neuronal Development to Neurological Diseases. Front. Cell Dev. Biol. 2020, 8, 579659. [Google Scholar] [CrossRef] [PubMed]
- Mansour, A.A.; Schafer, S.T.; Gage, F.H. Cellular complexity in brain organoids: Current progress and unsolved issues. Semin. Cell Dev. Biol. 2021, 111, 32–39. [Google Scholar] [CrossRef]
- Sun, N.; Meng, X.; Liu, Y.; Song, D.; Jiang, C.; Cai, J. Applications of brain organoids in neurodevelopment and neurological diseases. J. Biomed. Sci. 2021, 28, 30–46. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.M.; de Haan, P.; Ronaldson-Bouchard, K.; Kim, G.A.; Ko, J.; Rho, H.S.; Chen, Z.; Habibovic, P.; Jeon, N.L.; Takayama, S.; et al. A guide to the organ-on-a-chip. Nat. Rev. Methods Prim. 2022, 2, 33. [Google Scholar] [CrossRef]
- Hofer, M.; Lutolf, M.P. Engineering organoids. Nat. Rev. Mater. 2021, 6, 402–420. [Google Scholar] [CrossRef]
- Shiri, Z.; Simorgh, S.; Naderi, S.; Baharvand, H. Optogenetics in the Era of Cerebral Organoids. Trends Biotechnol. 2019, 37, 1282–1294. [Google Scholar] [CrossRef]
- Fiorenzano, A.; Sozzi, E.; Birtele, M.; Kajtez, J.; Giacomoni, J.; Nilsson, F.; Bruzelius, A.; Sharma, Y.; Zhang, Y.; Mattsson, B.; et al. Single-cell transcriptomics captures features of human midbrain development and dopamine neuron diversity in brain organoids. Nat. Commun. 2021, 12, 7302. [Google Scholar] [CrossRef]
- Xiaoshuai, L.; Qiushi, W.; Rui, W. Advantages of CRISPR-Cas9 combined organoid model in the study of congenital nervous system malformations. Front. Bioeng. Biotechnol. 2022, 10, 932936. [Google Scholar] [CrossRef]
- Yi, H.G.; Jeong, Y.H.; Kim, Y.; Choi, Y.J.; Moon, H.; Park, S.; Kang, K.; Bae, M.; Jang, J.; Youn, H.; et al. A bioprinted human-glioblastoma-on-a-chip for the identification of patient-specific responses to chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Nascimento, J.M.; Saia-Cereda, V.M.; Sartore, R.C.; da Costa, R.M.; Schitine, C.S.; Freitas, H.R.; Murgu, M.; de Melo Reis, R.A.; Rehen, S.K.; Martins-de-Souza, D. Human Cerebral Organoids and Fetal Brain Tissue Share Proteomic Similarities. Front. Cell Dev. Biol. 2019, 7, 303. [Google Scholar] [CrossRef] [PubMed]
- Doss, M.X.; Sachinidis, A. Current Challenges of iPSC-Based Disease Modelling and Therapeutic Implications. Cells 2019, 8, 403. [Google Scholar] [CrossRef]
- Al-Ghraiyba, N.F.; Wang, J.; Alkhalifa, A.E.; Roberts, A.B.; Raj, R.; Yang, E.; Kaddoumi, A. Glial Cell-Mediated Neuroinflammation in Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 10572. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Gunduz, M.A.; Semeano, A.T.; Yılmaz, E.C.; Alanazi, F.A.H.; Imir, O.B.; Yener, U.; Arbelaez, C.A.; Usuga, E.; Teng, Y.D. Coexistence of chronic hyperalgesia and multilevel neuroinflammatory responses after experimental SCI: A systematic approach to profiling neuropathic pain. J. Neuroinflamm. 2022, 19, 264. [Google Scholar] [CrossRef] [PubMed]
- Abreu, C.M.; Gama, L.; Krasemann, S.; Chesnut, M.; Odwin-Dacosta, S.; Hogberg, H.T.; Hartung, T.; Pamies, D. Microglia Increase Inflammatory Responses in iPSC-Derived Human BrainSpheres. Front. Microbiol. 2018, 9, 2766. [Google Scholar] [CrossRef]
- Ormel, P.R.; Vieira de Sá, R.; Van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.; Johansen, L.E.; van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef]
- Bodnar, B.; Zhang, Y.; Liu, J.; Lin, Y.; Wang, P.; Wei, Z.; Saribas, S.; Zhu, Y.; Li, F.; Wang, X.; et al. Novel scalable and simplified system to generate microglia-containing cerebral organoids from human induced pluripotent stem cells. Front. Cell. Neurosci. 2021, 15, 682272. [Google Scholar] [CrossRef]
- Xu, R.; Boreland, A.J.; Li, X.; Erickson, C.; Jin, M.; Atkins, C.; Pang, Z.P.; Daniels, B.P.; Jiang, P. Developing human pluripotent stem cell-based cerebral organoids with a controllable microglia ratio for modelling brain development and pathology. Stem Cell Rep. 2021, 16, 1923–1937. [Google Scholar] [CrossRef]
- Sharf, T.; van der Molen, T.; Guzman, E.; Glasauer, S.M.; Luna, G.; Cheng, Z.; Audouard, M.; Ranasinghe, K.G.; Kudo, K.; Nagarajan, S.; et al. Intrinsic network activity in human brain organoids. SSRN Electron. J. 2021, 3797268, 1–47. [Google Scholar] [CrossRef]
- Hogberg, H.; Smirnova, L. The Future of 3D Brain Cultures in Developmental Neurotoxicity Testing. Front. Toxicol. 2022, 4, 808620. [Google Scholar] [CrossRef]
- Makrygianni, E.A.; Chrousos, G.P. From Brain Organoids to Networking Assembloids: Implications for Neuroendocrinology and Stress Medicine. Front. Physiol. 2021, 12, 621970. [Google Scholar] [CrossRef] [PubMed]
- Vallmitjana, A.; Gu, J.; La, K.; Xu, Q.; Flores, J.; Zimak, J.; Shiu, J.; Hosohama, L.; Wu, J.; Douglas, C.; et al. Spatial transcriptomics using combinatorial fluorescence spectral and lifetime encoding, imaging and analysis. Nat. Commun. 2022, 13, 169. [Google Scholar]
- Tang, Q.; Liu, L.; Guo, Y.; Zhang, X.; Zhang, S.; Jia, Y.; Du, Y.; Cheng, B.; Yang, L.; Huang, Y.; et al. Optical Cell Tagging for Spatially Resolved Single-Cell RNA Sequencing. Angew. Chem. Int. Ed. 2022, 61, e202113929. [Google Scholar]
- Curreli, S.; Bonato, J.; Romanzi, S.; Panzeri, S.; Fellin, T. Complementary encoding of spatial information in hippocampal astrocytes. PLOS Biol. 2022, 20, e3001530. [Google Scholar] [CrossRef] [PubMed]
- Strebhardt, K.; Ullrich, A. Paul Ehrlich’s magic bullet concept: 100 years of progress. Nat. Rev. Cancer 2008, 8, 473–480. [Google Scholar] [CrossRef]
- Pei, D.; Buyanova, M. Overcoming Endosomal Entrapment in Drug Delivery. Bioconjug. Chem. 2019, 30, 273–283. [Google Scholar] [CrossRef]
- Yang, Y.; Li, Q.; Guo, X.; Tu, J.; Zhang, D. Mechanisms underlying sonoporation: Interaction between microbubbles and cells. Ultrason. Sonochem. 2020, 67, 105096. [Google Scholar] [CrossRef]
- Leite, P.E.C.; Pereira, M.R.; Harris, G.; Pamies, D.; Dos Santos, L.; Granjeiro, J.; Hogberg, H.; Hartung, T.; Smirnova, L. Suitability of 3D human brain spheroid models to distinguish toxic effects of gold and poly-lactic acid nanoparticles to assess biocompatibility for brain drug delivery. Part Fibre Toxicol. 2019, 16, 22–42. [Google Scholar] [CrossRef]
- Sokolova, V.; Mekky, G.; van der Meer, S.B.; Seeds, M.C.; Atala, A.J.; Epple, M. Transport of ultrasmall gold nanoparticles (2 nm) across the blood–brain barrier in a six-cell brain spheroid model. Sci. Rep. 2020, 10, 18033. [Google Scholar] [CrossRef]
- Darrigues, E.; Nima, Z.A.; Griffin, R.J.; Anderson, J.M.; Biris, A.S.; Rodriguez, A. 3D cultures for modelling nanomaterial-based photothermal therapy. Nanoscale Horiz. 2020, 5, 400–430. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-penetrating peptides: From basic research to clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- Kardani, K.; Milani, A.H.; Shabani, S.; Bolhassani, A. Cell penetrating peptides: The potent multi-cargo intracellular carriers. Expert Opin. Drug Deliv. 2019, 16, 1227–1258. [Google Scholar] [CrossRef] [PubMed]
- Corbett, J.L.; Tosh, D. Conversion of one cell type into another: Implications for understanding organ development, pathogenesis of cancer and generating cells for therapy. Biochem. Soc. Trans. 2014, 42, 609–616. [Google Scholar] [CrossRef]
- Song, I.Y.; Balmain, A. Cellular reprogramming in skin cancer. Semin. Cancer Biol. 2015, 32, 32–39. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dennison, R.; Usuga, E.; Chen, H.; Paul, J.Z.; Arbelaez, C.A.; Teng, Y.D. Direct Cell Reprogramming and Phenotypic Conversion: An Analysis of Experimental Attempts to Transform Astrocytes into Neurons in Adult Animals. Cells 2023, 12, 618. https://doi.org/10.3390/cells12040618
Dennison R, Usuga E, Chen H, Paul JZ, Arbelaez CA, Teng YD. Direct Cell Reprogramming and Phenotypic Conversion: An Analysis of Experimental Attempts to Transform Astrocytes into Neurons in Adult Animals. Cells. 2023; 12(4):618. https://doi.org/10.3390/cells12040618
Chicago/Turabian StyleDennison, Rachel, Esteban Usuga, Harriet Chen, Jacob Z. Paul, Christian A. Arbelaez, and Yang D. Teng. 2023. "Direct Cell Reprogramming and Phenotypic Conversion: An Analysis of Experimental Attempts to Transform Astrocytes into Neurons in Adult Animals" Cells 12, no. 4: 618. https://doi.org/10.3390/cells12040618
APA StyleDennison, R., Usuga, E., Chen, H., Paul, J. Z., Arbelaez, C. A., & Teng, Y. D. (2023). Direct Cell Reprogramming and Phenotypic Conversion: An Analysis of Experimental Attempts to Transform Astrocytes into Neurons in Adult Animals. Cells, 12(4), 618. https://doi.org/10.3390/cells12040618