
Erythrocyte Plasma Membrane Lipid Composition Mirrors That of Neurons and Glial Cells in Murine Experimental In Vitro and In Vivo Inflammation

 ,
,  ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Study Design
2.2. Histology and Immunofluorescence
2.3. Lipid Analysis
2.4. Macrophage (RAW 264.7) Cell Line and In Vitro Inflammation
2.5. Primary Cultures and In Vitro Challenges
2.6. Lipid Probes and Microscopy
2.7. RBC Scan Electron Microscopy
2.8. Fluorescent Microscopy and Postprocess Imaging
2.9. Statistical Analysis
3. Results
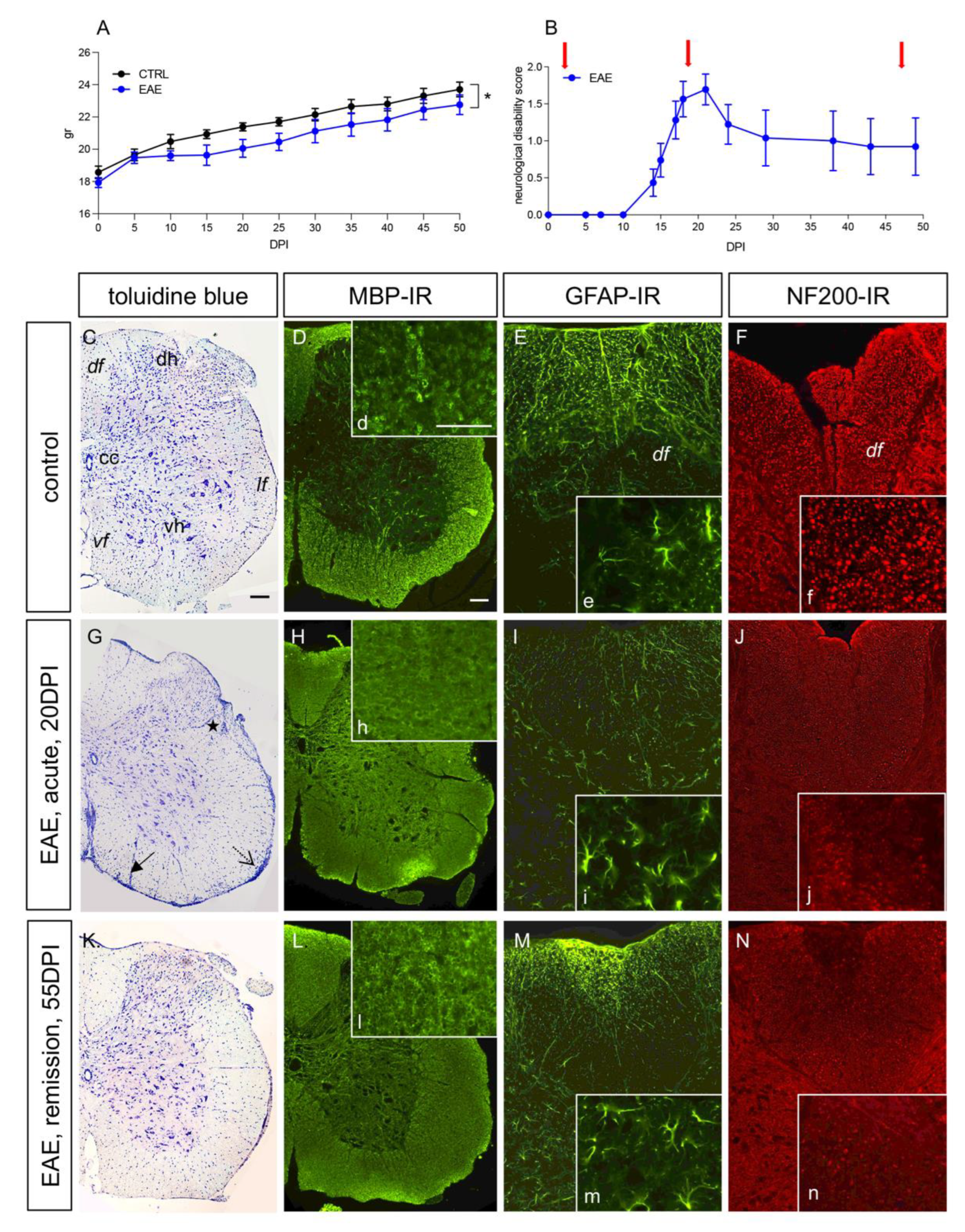
3.1. The EAE Mouse Model Reflects Remittent Neuroinflammation and Demyelination
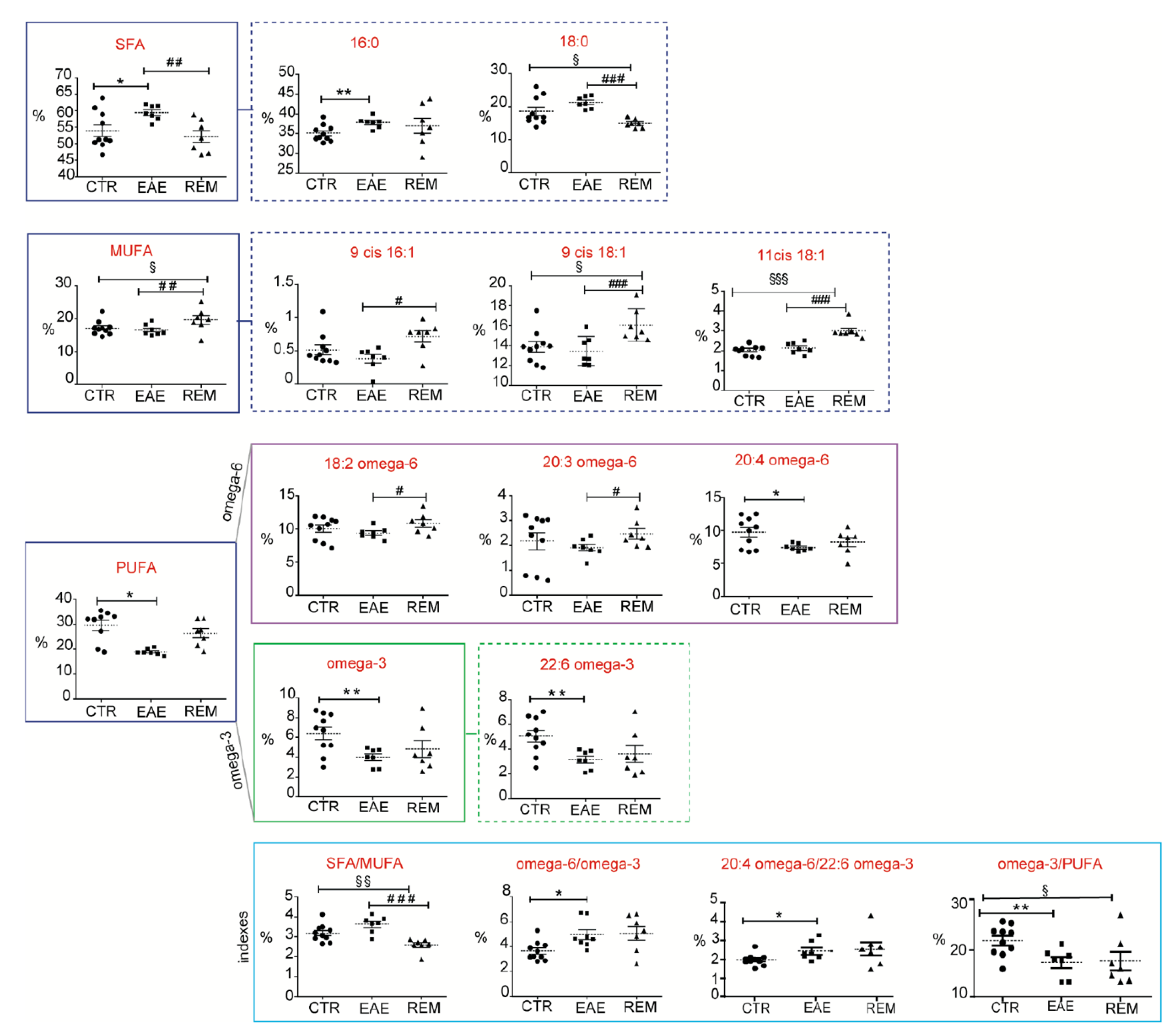
3.2. Fatty-Acid-Based Membrane Lipidome Analysis of RBCs and Spinal Cords
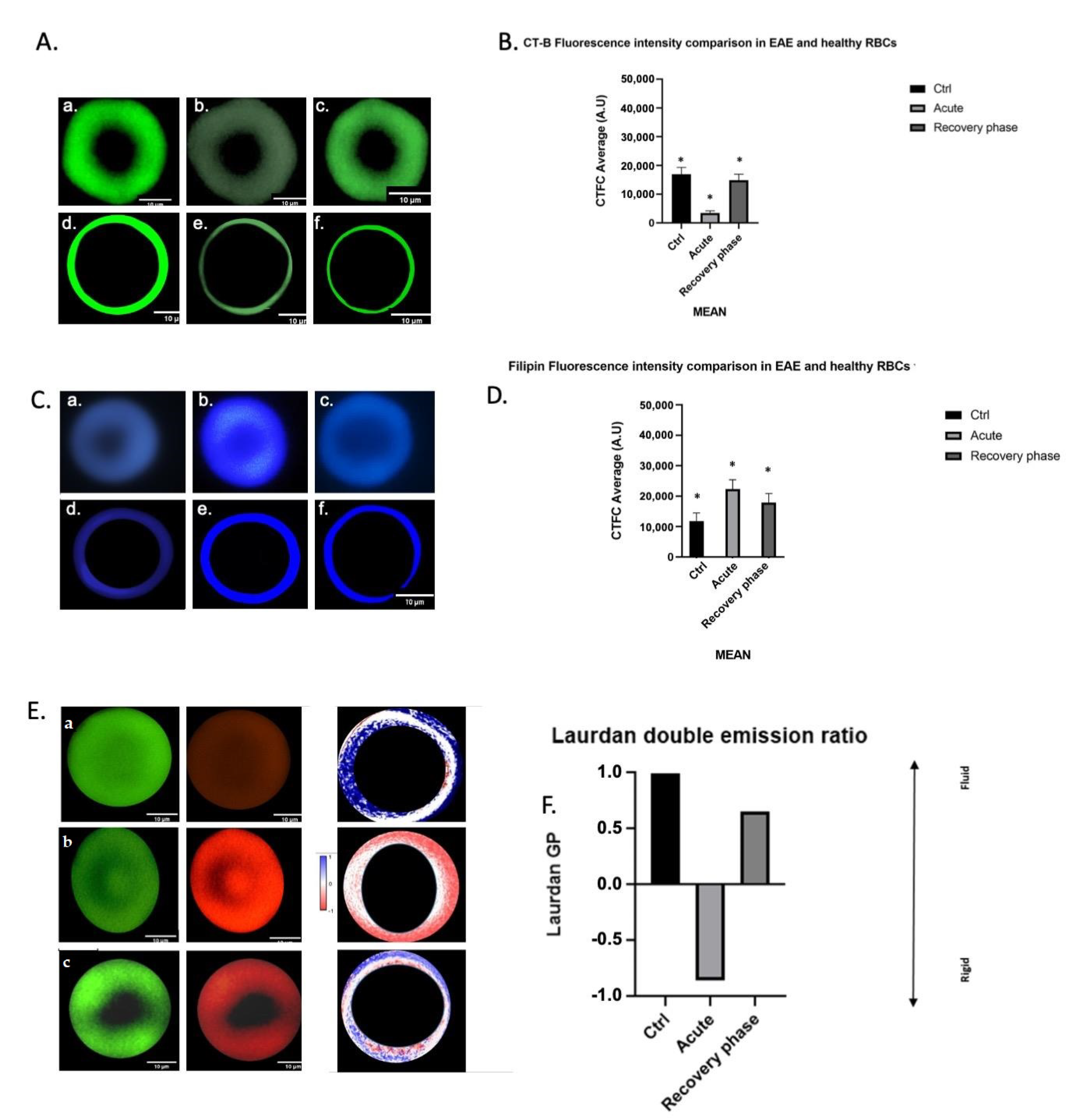
3.3. Erythrocyte Morphology and Membrane Lipid Probes during EAE
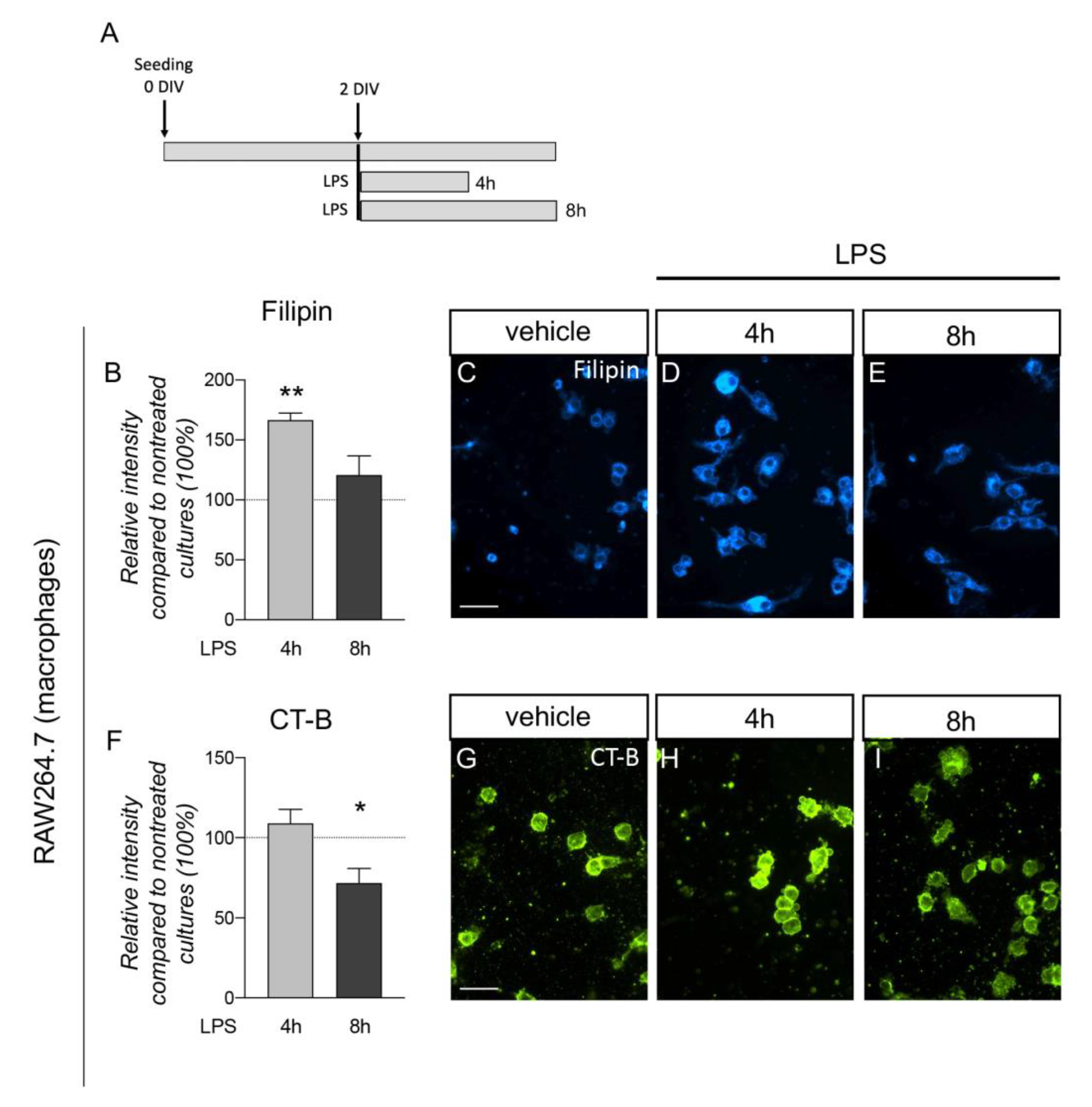
3.4. Plasma Membrane Lipids of Macrophages Dynamically Respond to Inflammatory Stimuli
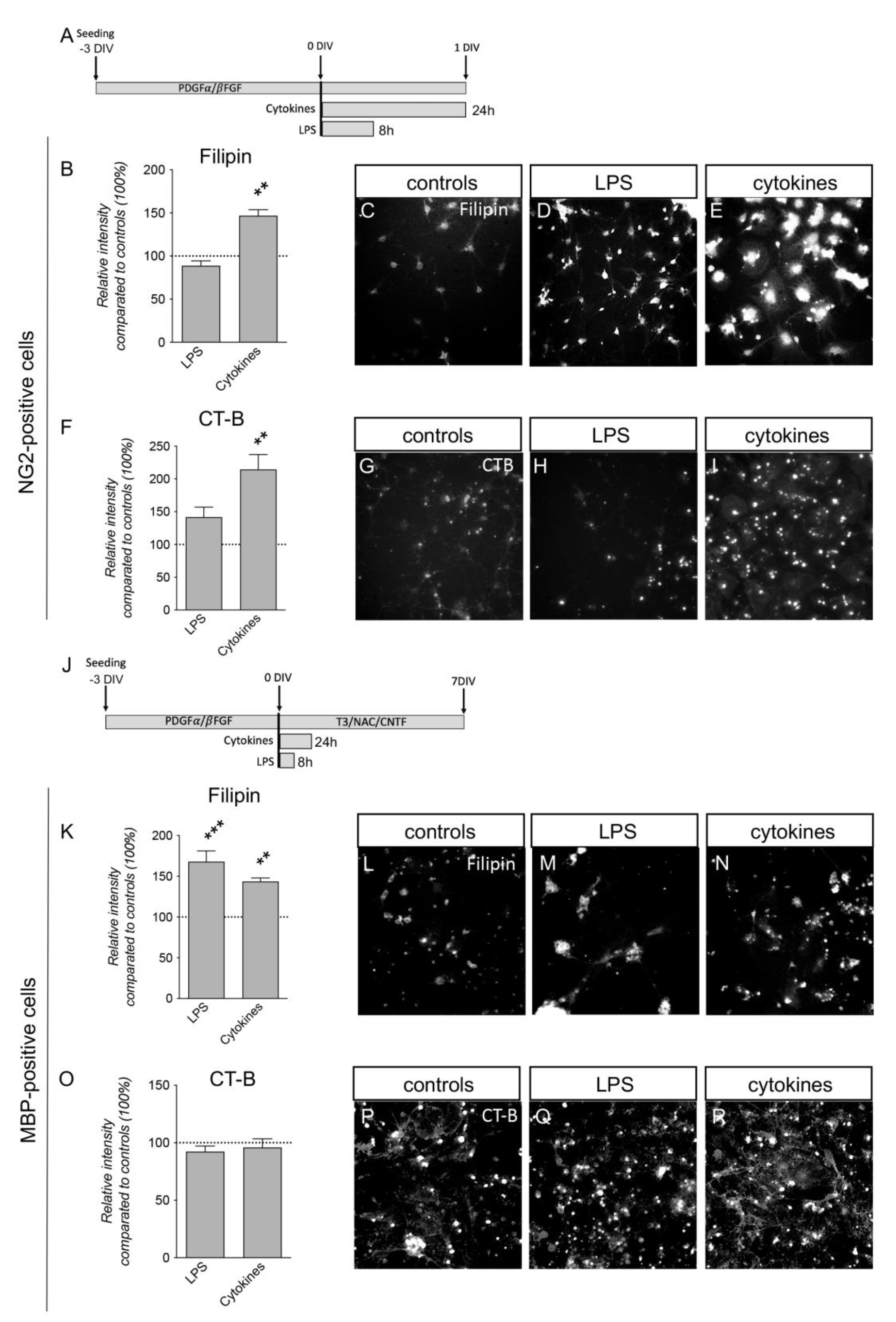
3.5. Inflammatory Challenges Acutely Affect Plasma Membrane Lipid Composition in Primary Cortical Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ARA | arachidonic acid |
| CNS | central nervous system |
| DHA | docosahexaenoic acid |
| EAE | experimental allergic encephalomyelitis |
| EPA | eicosapentaenoic acid |
| FA | fatty acid |
| GM1 | monosialotetrahexosylganglioside |
| LPS | lipopolysaccharide |
| MBP | myelin basic protein |
| MS | multiple sclerosis |
| MUFA | monounsaturated fatty acid |
| OL | oligodendrocyte |
| OPC | oligodendrocyte precursor cell |
| PUFA | polyunsaturated FA |
| RBC | red blood cell |
References
- Aureli, M.; Grassi, S.; Prioni, S.; Sonnino, S.; Prinetti, A. Lipid membrane domains in the brain. Biochim. Biophys. Acta 2015, 1851, 1006–1016. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.A.; Deber, C.M.; Klrschner, O.A. The Structure and Function of Central Nervous System Myelin. Crit. Rev. Clin. Lab. Sci. 1993, 30, 29–64. [Google Scholar] [CrossRef] [PubMed]
- Poitelon, Y.; Kopec, A.M.; Belin, S. Myelin Fat Facts: An Overview of Lipids and Fatty Acid Metabolism. Cells 2020, 9, 812. [Google Scholar] [CrossRef] [PubMed]
- Roy, D.; Tedeschi, A. The Role of Lipids, Lipid Metabolism and Ectopic Lipid Accumulation in Axon Growth, Regeneration and Repair after CNS Injury and Disease. Cells 2021, 10, 1078. [Google Scholar] [CrossRef]
- Montani, L. Lipids in regulating oligodendrocyte structure and function. Semin. Cell Dev. Biol. 2020, 112, 114–122. [Google Scholar] [CrossRef]
- Meschkat, M.; Steyer, A.M.; Weil, M.-T.; Kusch, K.; Jahn, O.; Piepkorn, L.; Agüi-Gonzalez, P.; Phan, N.T.N.; Ruhwedel, T.; Sadowski, B.; et al. White matter integrity in mice requires continuous myelin synthesis at the inner tongue. Nat. Commun. 2022, 13, 1163. [Google Scholar] [CrossRef]
- Chrast, R.; Saher, G.; Nave, K.-A.; Verheijen, M.H. Lipid metabolism in myelinating glial cells: Lessons from human inherited disorders and mouse models. J. Lipid Res. 2011, 52, 419–434. [Google Scholar] [CrossRef]
- Werner, H.; Buscham, T.; Eichel, M.; Siems, S. Turning to myelin turnover. Neural Regen. Res. 2019, 14, 2063–2066. [Google Scholar] [CrossRef]
- Williamson, J.M.; Lyons, D.A. Myelin Dynamics Throughout Life: An Ever-Changing Landscape? Front. Cell. Neurosci. 2018, 12, 424. [Google Scholar] [CrossRef]
- Ferrer, I. Proteomics and lipidomics in the human brain. Handb. Clin. Neurol. 2018, 150, 285–302. [Google Scholar] [CrossRef]
- Castellanos, D.B.; Martín-Jiménez, C.A.; Rojas-Rodríguez, F.; Barreto, G.E.; González, J. Brain lipidomics as a rising field in neurodegenerative contexts: Perspectives with Machine Learning approaches. Front. Neuroendocr. 2021, 61, 100899. [Google Scholar] [CrossRef]
- Tredinnick, A.R.; Probst, Y.C. Evaluating the Effects of Dietary Interventions on Disease Progression and Symptoms of Adults with Multiple Sclerosis: An Umbrella Review. Adv. Nutr. Int. Rev. J. 2020, 11, 1603–1615. [Google Scholar] [CrossRef]
- Huang, H.-T.; Tsai, S.-F.; Wu, H.-T.; Huang, H.-Y.; Hsieh, H.-H.; Kuo, Y.-M.; Chen, P.-S.; Yang, C.-S.; Tzeng, S.-F. Chronic exposure to high fat diet triggers myelin disruption and interleukin-33 upregulation in hypothalamus. BMC Neurosci. 2019, 20, 33. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nat. Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Bogie, J.F.; Haidar, M.; Kooij, G.; Hendriks, J.J. Fatty acid metabolism in the progression and resolution of CNS disorders. Adv. Drug Deliv. Rev. 2020, 159, 198–213. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Parks, N.E.; Jackson-Tarlton, C.S.; Vacchi, L.; Merdad, R.; Johnston, B.C. Dietary interventions for multiple sclerosis-related outcomes. Cochrane Database Syst. Rev. 2020, 2020, CD004192. [Google Scholar] [CrossRef]
- Ferreri, C.; Mennella, M.F.; Formisano, C.; Landi, L.; Chatgilialoglu, C. Arachidonate geometrical isomers generated by thiyl radicals: The relationship with trans lipids detected in biological samples. Free Radic. Biol. Med. 2002, 33, 1516–1526. [Google Scholar] [CrossRef]
- Sansone, A.; Melchiorre, M.; Chatgilialoglu, C.; Ferreri, C. Hexadecenoic Fatty Acid Isomers: A Chemical Biology Approach for Human Plasma Biomarker Development. Chem. Res. Toxicol. 2013, 26, 1703–1709. [Google Scholar] [CrossRef]
- Menounou, G.; Giacometti, G.; Scanferlato, R.; Dambruoso, P.; Sansone, A.; Tueros, I.; Amézaga, J.; Chatgilialoglu, C.; Ferreri, C. Trans Lipid Library: Synthesis of Docosahexaenoic Acid (DHA) Monotrans Isomers and Regioisomer Identification in DHA-Containing Supplements. Chem. Res. Toxicol. 2018, 31, 191–200. [Google Scholar] [CrossRef]
- Küçüksayan, E.; Sansone, A.; Chatgilialoglu, C.; Ozben, T.; Tekeli, D.; Talibova, G.; Ferreri, C. Sapienic Acid Metabolism Influences Membrane Plasticity and Protein Signaling in Breast Cancer Cell Lines. Cells 2022, 11, 225. [Google Scholar] [CrossRef]
- Ferreri, C.; Sansone, A.; Buratta, S.; Urbanelli, L.; Costanzi, E.; Emiliani, C.; Chatgilialoglu, C. The n-10 Fatty Acids Family in the Lipidome of Human Prostatic Adenocarcinoma Cell Membranes and Extracellular Vesicles. Cancers 2020, 12, 900. [Google Scholar] [CrossRef] [PubMed]
- Crisi, P.E.; Luciani, A.; Di Tommaso, M.; Prasinou, P.; De Santis, F.; Chatgilialoglu, C.; Pietra, M.; Procoli, F.; Sansone, A.; Giordano, M.V.; et al. The Fatty Acid-Based Erythrocyte Membrane Lipidome in Dogs with Chronic Enteropathy. Animals 2021, 11, 2604. [Google Scholar] [CrossRef] [PubMed]
- Prasinou, P.; Crisi, P.E.; Chatgilialoglu, C.; Di Tommaso, M.; Sansone, A.; Gramenzi, A.; Belà, B.; De Santis, F.; Boari, A.; Ferreri, C. The Erythrocyte Membrane Lipidome of Healthy Dogs: Creating a Benchmark of Fatty Acid Distribution and Interval Values. Front. Veter. Sci. 2020, 7, 502. [Google Scholar] [CrossRef] [PubMed]
- Carillon, J.; Saby, M.; Barial, S.; Sansone, A.; Scanferlato, R.; Gayrard, N.; Lajoix, A.-D.; Jover, B.; Chatgilialoglu, C.; Ferreri, C. Melon juice concentrate supplementation in an animal model of obesity: Involvement of relaxin and fatty acid pathways. J. Funct. Foods 2019, 59, 92–100. [Google Scholar] [CrossRef]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [CrossRef]
- Dolci, L.; Perone, R.; Di Gesù, R.; Kurakula, M.; Gualandi, C.; Zironi, E.; Gazzotti, T.; Tondo, M.; Pagliuca, G.; Gostynska, N.; et al. Design and In Vitro Study of a Dual Drug-Loaded Delivery System Produced by Electrospinning for the Treatment of Acute Injuries of the Central Nervous System. Pharmaceutics 2021, 13, 848. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calzà, L. Vulnerability to oxygen-glucose deprivation of primary neurons derived from Tg2576 Alzheimer mice: Role of intraneuronal Abeta accumulation and astrocytes. Dis. Model. Mech. 2017, 10, 671–678. [Google Scholar] [CrossRef]
- Baldassarro, V.A. High-Content Screening Differentiation and Maturation Analysis of Fetal and Adult Neural Stem Cell-Derived Oligodendrocyte Precursor Cell Cultures. J. Vis. Exp. 2021, 169, e61988. [Google Scholar] [CrossRef]
- Borjini, N.; Fernández, M.; Giardino, L.; Calzà, L. Cytokine and chemokine alterations in tissue, CSF, and plasma in early presymptomatic phase of experimental allergic encephalomyelitis (EAE), in a rat model of multiple sclerosis. J. Neuroinflamm. 2016, 13, 1–16. [Google Scholar] [CrossRef]
- Fernández, M.; Baldassarro, V.A.; Sivilia, S.; Giardino, L.; Calzà, L. Inflammation severely alters thyroid hormone signaling in the central nervous system during experimental allergic encephalomyelitis in rat: Direct impact on OPCs differentiation failure. Glia 2016, 64, 1573–1589. [Google Scholar] [CrossRef]
- Wilhelm, L.P.; Voilquin, L.; Kobayashi, T.; Tomasetto, C.; Alpy, F. Intracellular and Plasma Membrane Cholesterol Labeling and Quantification Using Filipin and GFP-D4. Intracell. Lipid Transp. Methods Protoc. 2019, 1949, 137–152. [Google Scholar] [CrossRef]
- Wenzel, M.; Vischer, N.O.E.; Strahl, H.; Hamoen, L.W. Assessing Membrane Fluidity and Visualizing Fluid Membrane Domains in Bacteria Using Fluorescent PMembrane Dyes. Bio. Protoc. 2018, 8, e3063. [Google Scholar] [CrossRef]
- Owen, D.M.; Sauer, M.; Gaus, K. Fluorescence localization microscopy. Commun. Integr. Biol. 2012, 5, 345–349. [Google Scholar] [CrossRef]
- Calzà, L.; Giardino, L.; Pozza, M.; Micera, A.; Aloe, L. Time-course changes of nerve growth factor, corticotropin-releasing hormone, and nitric oxide synthase isoforms and their possible role in the development of inflammatory response in experimental allergic encephalomyelitis. Proc. Natl. Acad. Sci. USA 1997, 94, 3368–3373. [Google Scholar] [CrossRef]
- Pyka-Fosciak, G.; Stasiolek, M.; Litwin, J.A. Immunohistochemical analysis of spinal cord components in mouse model of experimental autoimmune encephalomyelitis. Folia Histochem. Cytobiol. 2018, 56, 151–158. [Google Scholar] [CrossRef]
- Valerii, M.C.; Turroni, S.; Ferreri, C.; Zaro, M.; Sansone, A.; Dalpiaz, A.; Botti, G.; Ferraro, L.; Spigarelli, R.; Bellocchio, I.; et al. Effect of a Fiber D-Limonene-Enriched Food Supplement on Intestinal Microbiota and Metabolic Parameters of Mice on a High-Fat Diet. Pharmaceutics 2021, 13, 1753. [Google Scholar] [CrossRef]
- Fragopoulou, A.F.; Polyzos, A.; Papadopoulou, M.-D.; Sansone, A.; Manta, A.K.; Balafas, E.; Kostomitsopoulos, N.; Skouroliakou, A.; Chatgilialoglu, C.; Georgakilas, A.; et al. Hippocampal lipidome and transcriptome profile alterations triggered by acute exposure of mice to GSM 1800 MHz mobile phone radiation: An exploratory study. Brain Behav. 2018, 8, e01001. [Google Scholar] [CrossRef]
- Rossol, M.; Heine, H.; Meusch, U.; Quandt, D.; Klein, C.; Sweet, M.J.; Hauschildt, S. LPS-induced Cytokine Production in Human Monocytes and Macrophages. Crit. Rev. Immunol. 2011, 31, 379–446. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calzà, L. Differential effects of glucose deprivation on the survival of fetal versus adult neural stem cells-derived oligodendrocyte precursor cells. Glia 2019, 68, 898–917. [Google Scholar] [CrossRef]
- Baldassarro, V.A.; Marchesini, A.; Giardino, L.; Calzà, L. PARP activity and inhibition in fetal and adult oligodendrocyte precursor cells: Effect on cell survival and differentiation. Stem Cell Res. 2017, 22, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Cermenati, G.; Mitro, N.; Audano, M.; Melcangi, R.C.; Crestani, M.; De Fabiani, E.; Caruso, D. Lipids in the nervous system: From biochemistry and molecular biology to patho-physiology. Biochim. Biophys. Acta 2015, 1851, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Turri, M.; Marchi, C.; Adorni, M.P.; Calabresi, L.; Zimetti, F. Emerging role of HDL in brain cholesterol metabolism and neurodegenerative disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159123. [Google Scholar] [CrossRef] [PubMed]
- Guest, J.; Garg, M.; Bilgin, A.; Grant, R. Relationship between central and peripheral fatty acids in humans. Lipids Health Dis. 2013, 12, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Bai, S.; Hao, Y.; Guan, Y. Fatty acids role in multiple sclerosis as “metabokines”. J. Neuroinflamm. 2022, 19, 1–21. [Google Scholar] [CrossRef]
- Ariyama, H.; Kono, N.; Matsuda, S.; Inoue, T.; Arai, H. Decrease in Membrane Phospholipid Unsaturation Induces Unfolded Protein Response. J. Biol. Chem. 2010, 285, 22027–22035. [Google Scholar] [CrossRef]
- Draycott, S.A.V.; Elmes, M.J.; Muhlhausler, B.S.; Langley-Evans, S. Omega-6: Omega-3 Fatty Acid Ratio and Total Fat Content of the Maternal Diet Alter Offspring Growth and Fat Deposition in the Rat. Nutrients 2020, 12, 2505. [Google Scholar] [CrossRef]
- Abbott, S.K.; Else, P.L.; Atkins, T.A.; Hulbert, A.J. Fatty Acid Composition of Membrane Bilayers: Importance of Diet Polyunsaturated Fat Balance. Biochim. Biophys. Acta 2012, 1818, 1309–1317. [Google Scholar] [CrossRef]
- Salvati, S.; Di Biase, A.; Attorri, L.; Di Benedetto, R.; Sanchez, M.; Lorenzini, L.; Alessandri, M.; Calzà, L. Ethyl-eicosapentaenoic acid ameliorates the clinical course of experimental allergic encephalomyelitis induced in dark agouti rats. J. Nutr. Biochem. 2013, 24, 1645–1654. [Google Scholar] [CrossRef]
- Maulucci, G.; Cohen, O.; Daniel, B.; Sansone, A.; Petropoulou, P.I.; Filou, S.; Spyridonidis, A.; Pani, G.; De Spirito, M.; Chatgilialoglu, C.; et al. Fatty acid-related modulations of membrane fluidity in cells: Detection and implications. Free Radic. Res. 2016, 50 (Suppl. S1), S40–S50. [Google Scholar] [CrossRef]
- Ávila-Martín, G.; Galán-Arriero, I.; Gómez-Soriano, J.; Taylor, J. Treatment of Rat Spinal Cord Injury with the Neurotrophic Factor Albumin-Oleic Acid: Translational Application for Paralysis, Spasticity and Pain. PLoS ONE 2011, 6, e26107. [Google Scholar] [CrossRef]
- Hon, G.; Hassan, M.; van Rensburg, S.; Abel, S.; Marais, D.; van Jaarsveld, P.; Smuts, C.; Henning, F.; Erasmus, R.; Matsha, T. Erythrocyte membrane fatty acids in patients with multiple sclerosis. Mult. Scler. J. 2009, 15, 759–762. [Google Scholar] [CrossRef]
- Koch, M.; Ramsaransing, G.S.; Fokkema, M.R.; Heersema, D.J.; De Keyser, J. Erythrocyte membrane fatty acids in benign and progressive forms of multiple sclerosis. J. Neurol. Sci. 2006, 244, 123–126. [Google Scholar] [CrossRef]
- Ferreri, C.; Chatgilialoglu, C. Membrane Lipidomics for Personalized Health; Wiley and Sons: Chichester, UK, 2015. [Google Scholar]
- Jauregibeitia, I.; Portune, K.; Rica, I.; Tueros, I.; Velasco, O.; Grau, G.; Castaño, L.; Di Nolfo, F.; Ferreri, C.; Arranz, S. Potential of Erythrocyte Membrane Lipid Profile as a Novel Inflammatory Biomarker to Distinguish Metabolically Healthy Obesity in Children. J. Pers. Med. 2021, 11, 337. [Google Scholar] [CrossRef]
- Li, H.; Lykotrafitis, G. Erythrocyte Membrane Model with Explicit Description of the Lipid Bilayer and the Spectrin Network. Biophys. J. 2014, 107, 642–653. [Google Scholar] [CrossRef]
- Mohandas, N.; Gallagher, P.G. Red cell membrane: Past, present, and future. Blood 2008, 112, 3939–3948. [Google Scholar] [CrossRef]
- Mikirova, N.; Riordan, H.D.; A Jackson, J.; Wong, K.; Miranda-Massari, J.R.; Gonzalez, M.J. Erythrocyte membrane fatty acid composition in cancer patients. Puerto Rico Health Sci. J. 2004, 23, 107–113. [Google Scholar]
- Acar, N.; Berdeaux, O.; Grégoire, S.; Cabaret, S.; Martine, L.; Gain, P.; Thuret, G.; Creuzot-Garcher, C.P.; Bron, A.M.; Bretillon, L. Lipid Composition of the Human Eye: Are Red Blood Cells a Good Mirror of Retinal and Optic Nerve Fatty Acids? PLoS ONE 2012, 7, e35102. [Google Scholar] [CrossRef]
- Fernández-Pérez, E.; Sepúlveda, F.; Peoples, R.; Aguayo, L. Role of membrane GM1 on early neuronal membrane actions of Aβ during onset of Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 3105–3116. [Google Scholar] [CrossRef]
- Klymchenko, A.S.; Kreder, R. Fluorescent Probes for Lipid Rafts: From Model Membranes to Living Cells. Chem. Biol. 2014, 21, 97–113. [Google Scholar] [CrossRef]
- Goossens, P.; Rodriguez-Vita, J.; Etzerodt, A.; Masse, M.; Rastoin, O.; Gouirand, V.; Ulas, T.; Papantonopoulou, O.; Van Eck, M.; Auphan-Anezin, N.; et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019, 29, 1376–1389.e4. [Google Scholar] [CrossRef] [PubMed]
- Abourbeh, G.; Thézé, B.; Maroy, R.; Dubois, A.; Brulon, V.; Fontyn, Y.; Dollé, F.; Tavitian, B.; Boisgard, R. Imaging Microglial/Macrophage Activation in Spinal Cords of Experimental Autoimmune Encephalomyelitis Rats by Positron Emission Tomography Using the Mitochondrial 18 kDa Translocator Protein Radioligand [18F]DPA-714. J. Neurosci. 2012, 32, 5728–5736. [Google Scholar] [CrossRef] [PubMed]
- Schumann, J. It is all about fluidity: Fatty acids and macrophage phagocytosis. Eur. J. Pharmacol. 2016, 785, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Schoeniger, A.; Adolph, S.; Fuhrmann, H.; Schumann, J. The Impact of Membrane Lipid Composition on Macrophage Activation in the Immune Defense against Rhodococcus equi and Pseudomonas aeruginosa. Int. J. Mol. Sci. 2011, 12, 7510. [Google Scholar] [CrossRef] [PubMed]
- Cammarota, E.; Soriani, C.; Taub, R.; Morgan, F.; Sakai, J.; Veatch, S.L.; Bryant, C.E.; Cicuta, P. Criticality of plasma membrane lipids reflects activation state of macrophage cells. J. R. Soc. Interface 2020, 17, 20190803. [Google Scholar] [CrossRef]
- Camargo, N.; Goudriaan, A.; Van Deijk, A.-L.F.; Otte, W.; Brouwers, J.F.; Lodder, H.; Gutmann, D.; Nave, K.-A.; Dijkhuizen, R.M.; Mansvelder, H.; et al. Oligodendroglial myelination requires astrocyte-derived lipids. PLOS Biol. 2017, 15, e1002605. [Google Scholar] [CrossRef]
- Marangon, D.; Boccazzi, M.; Lecca, D.; Fumagalli, M. Regulation of Oligodendrocyte Functions: Targeting Lipid Metabolism and Extracellular Matrix for Myelin Repair. J. Clin. Med. 2020, 9, 470. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Sun, T.; Hosang, L.; Depp, C.; Sasmita, A.O.; Vasileva, M.H.; Scholz, P.; Zhao, Y.; Krueger-Burg, D.; et al. Neuronal cholesterol synthesis is essential for repair of chronically demyelinated lesions in mice. Cell Rep. 2021, 37, 109889. [Google Scholar] [CrossRef]
- Berghoff, S.A.; Spieth, L.; Saher, G. Local cholesterol metabolism orchestrates remyelination. Trends Neurosci. 2022, 45, 272–283. [Google Scholar] [CrossRef]
- Mocchetti, I. Exogenous gangliosides, neuronal plasticity and repair, and the neurotrophins. Cell. Mol. Life Sci. CMLS 2005, 62, 2283–2294. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Lunghi, G.; Di Biase, E.; Fazzari, M.; Sonnino, S.; Mauri, L. GM1 Ganglioside Is A Key Factor in Maintaining the Mammalian Neuronal Functions Avoiding Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 868. [Google Scholar] [CrossRef]
- Tessitore, A.; Martin, M.D.P.; Sano, R.; Ma, Y.; Mann, L.; Ingrassia, A.; Laywell, E.D.; Steindler, D.A.; Hendershot, L.M.; D’Azzo, A. GM1-Ganglioside-Mediated Activation of the Unfolded Protein Response Causes Neuronal Death in a Neurodegenerative Gangliosidosis. Mol. Cell 2004, 15, 753–766. [Google Scholar] [CrossRef]
- Pan, X.; De Aragão, C.B.P.; Velasco-Martin, J.P.; Priestman, D.A.; Wu, H.Y.; Takahashi, K.; Yamaguchi, K.; Sturiale, L.; Garozzo, D.; Platt, F.M.; et al. Neuraminidases 3 and 4 regulate neuronal function by catabolizing brain gangliosides. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2017, 31, 3467–3483. [Google Scholar] [CrossRef]
- Sipione, S.; Monyror, J.; Galleguillos, D.; Steinberg, N.; Kadam, V. Gangliosides in the Brain: Physiology, Pathophysiology and Therapeutic Applications. Front. Neurosci. 2020, 14, 572965. [Google Scholar] [CrossRef]
- Galleguillos, D.; Wang, Q.; Steinberg, N.; Zaidi, A.; Shrivastava, G.; Dhami, K.; Daskhan, G.C.; Schmidt, E.N.; Dworsky-Fried, Z.; Giuliani, F.; et al. Anti-inflammatory role of GM1 and other gangliosides on microglia. J. Neuroinflamm. 2022, 19, 9. [Google Scholar] [CrossRef]
- Zhang, Y.; Huang, Q.; Zhao, C.; Tang, J.; Wang, Y. GM1 improves neurofascin155 association with lipid rafts and prevents rat brain myelin injury after hypoxia-ischemia. Braz. J. Med. Biol. Res. 2011, 44, 553–561. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spinal Cord CTRL (n = 3) | Spinal Cord EAE (n = 3) | |
|---|---|---|
| FAME 1 (% rel quant) | mean ± sd | mean ± sd |
| 14:0 | 0.27 ± 0.03 | 0.40 ± 0.18 |
| 16:0 | 16.35 ± 0.36 | 17.00 ± 0.94 |
| 16:1 trans | 0.01 ± 0.00 | 0.00 ± 0.00 |
| (6 + 7) 16:1 cis | 0.16 ± 0.01 | 0.43 ± 0.18 |
| 9 cis 16:1 | 0.50 ± 0.03 | 0.52 ± 0.13 |
| 18:0 | 15.86 ± 0.31 | 15.63 ± 0.19 |
| 9 trans 18:1 | 0.04 ± 0.05 | 0.04 ± 0.01 |
| 8 cis 18:1 | 0.02 ± 0.01 | 0.06 ± 0.03 |
| 9 cis 18:1 | 28.44 ± 0.92 | 32.35 ± 1.99 * |
| 11 cis 18:1 | 5.66 ± 0.06 | 5.04 ± 0.24 * |
| 5 cis, 8 cis 18:2 | Traces | Traces |
| mtrans 18:2 | 0.01 ± 0.00 | 0.03 ± 0.03 |
| 18:2 omega-6 | 0.98 ± 0.17 | 0.71 ± 0.06 |
| 18:3 omega-6 | 0.11 ± 0.01 | 0.11 ± 0.02 |
| 18:3 omega-3 | 0.11 ± 0.03 | 0.07 ± 0.03 |
| 20:0 | 1.73 ± 0.04 | 1.67 ± 0.30 |
| 11 cis 20:1 | 7.54 ± 0.47 | 6.15 ± 1.12 |
| 13 cis 20:1 | 2.06 ± 0.04 | 1.71 ± 0.41 |
| 20:2 omega-6 | 1.25 ± 0.06 | 1.17 ± 0.37 |
| 20:3 omega-6 | 0.58 ± 0.04 | 0.58 ± 0.08 |
| 20:4 omega-6 | 6.47 ± 0.18 | 6.22 ± 0.16 |
| mtrans 20:4 | 0.03 ± 0.01 | 0.04 ± 0.00 |
| 20:5 omega-3 | 0.04 ± 0.01 | 0.03 ± 0.00 |
| 13 cis 22:1 | 0.74 ± 0.03 | 0.69 ± 0.10 |
| 22:5 omega-3 | 0.31 ± 0.04 | 0.27 ± 0.00 |
| 22:6 omega-3 | 9.47 ± 0.42 | 8.08 ± 0.63 * |
| 24:0 | 0.42 ± 0.08 | 1.07 ± 0.22 ** |
| 15 cis 24:1 | 0.26 ± 0.00 | 0.25 ± 0.03 |
| SFA | 34.63 ± 0.73 | 35.77 ± 0.88 |
| MUFA | 44.39 ± 1.14 | 46.25 ± 0.53 |
| PUFA | 19.32 ± 0.55 | 17.25 ± 1.01 |
| omega-6 | 9.38 ± 0.14 | 8.80 ± 0.43 |
| omega-3 | 9.93 ± 0.41 | 8.45 ± 0.65 * |
| omega-6/omega-3 | 0.95 ± 0.03 | 1.04 ± 0.05 * |
| SFA/MUFA | 0.78 ± 0.03 | 0.77 ± 0.01 |
| ARA/EPA | 166.59 ± 4.64 | 251 ± 32.99 * |
| ARA/DHA | 0.68 ± 0.02 | 0.77 ± 0.4 * |
| omega-3/PUFA*100 | 51 ± 1 | 49 ± 1 |
| tot trans | 0.08 ± 0.04 | 0.11 ± 0.04 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanzani, A.; Sansone, A.; Brenna, C.; Baldassarro, V.A.; Alastra, G.; Lorenzini, L.; Chatgilialoglu, C.; Laface, I.; Ferreri, C.; Neri, L.M.; et al. Erythrocyte Plasma Membrane Lipid Composition Mirrors That of Neurons and Glial Cells in Murine Experimental In Vitro and In Vivo Inflammation. Cells 2023, 12, 561. https://doi.org/10.3390/cells12040561
Stanzani A, Sansone A, Brenna C, Baldassarro VA, Alastra G, Lorenzini L, Chatgilialoglu C, Laface I, Ferreri C, Neri LM, et al. Erythrocyte Plasma Membrane Lipid Composition Mirrors That of Neurons and Glial Cells in Murine Experimental In Vitro and In Vivo Inflammation. Cells. 2023; 12(4):561. https://doi.org/10.3390/cells12040561
Chicago/Turabian StyleStanzani, Agnese, Anna Sansone, Cinzia Brenna, Vito Antonio Baldassarro, Giuseppe Alastra, Luca Lorenzini, Chryssostomos Chatgilialoglu, Ilaria Laface, Carla Ferreri, Luca Maria Neri, and et al. 2023. "Erythrocyte Plasma Membrane Lipid Composition Mirrors That of Neurons and Glial Cells in Murine Experimental In Vitro and In Vivo Inflammation" Cells 12, no. 4: 561. https://doi.org/10.3390/cells12040561
APA StyleStanzani, A., Sansone, A., Brenna, C., Baldassarro, V. A., Alastra, G., Lorenzini, L., Chatgilialoglu, C., Laface, I., Ferreri, C., Neri, L. M., & Calzà, L. (2023). Erythrocyte Plasma Membrane Lipid Composition Mirrors That of Neurons and Glial Cells in Murine Experimental In Vitro and In Vivo Inflammation. Cells, 12(4), 561. https://doi.org/10.3390/cells12040561