

Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes
 , , ,
, , ,  ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ischemic Stroke Model (MCAO Model)
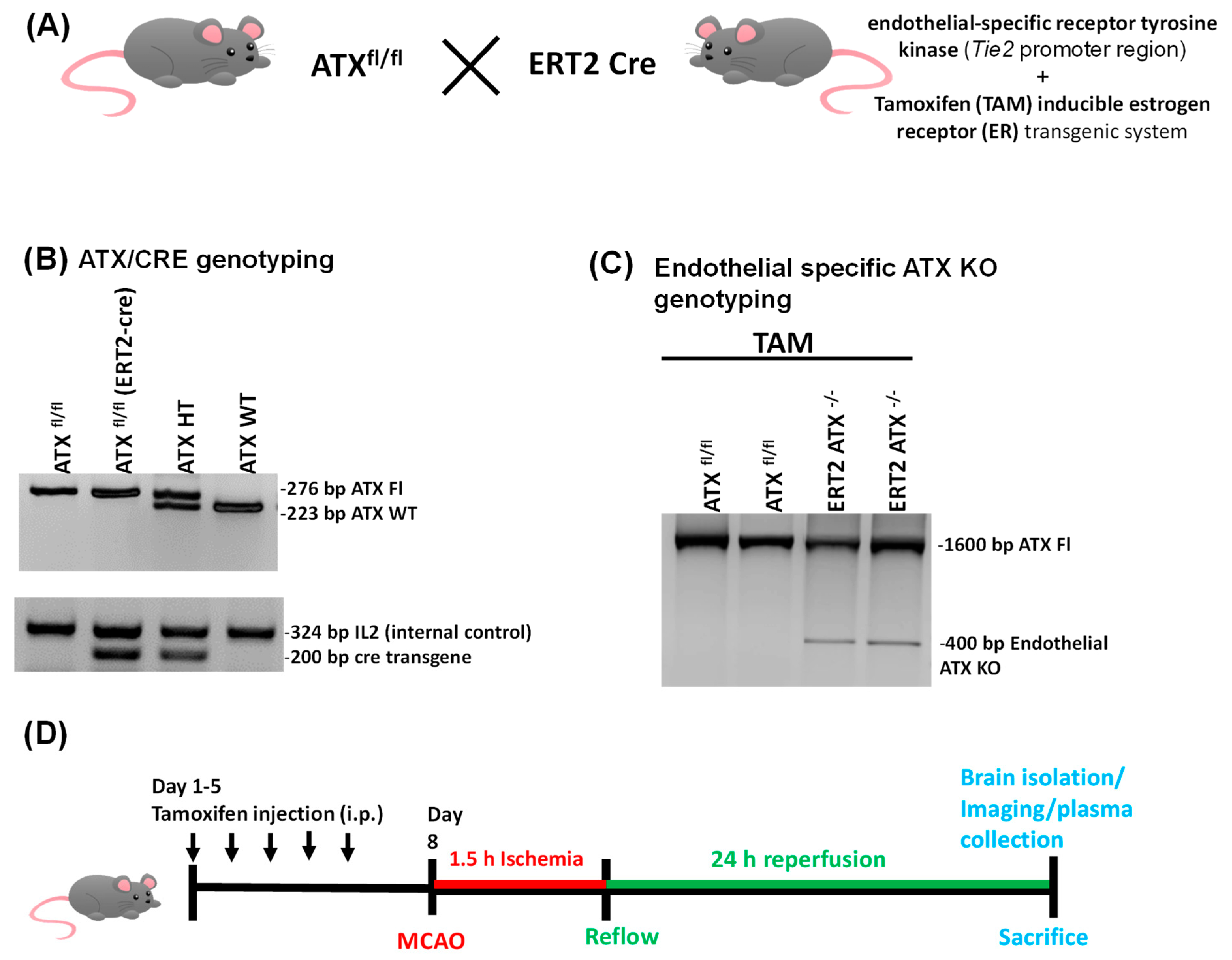
2.2. Generation of Inducible Endothelial-Specific ATX Knockout Mice (ERT2 ATX−/−)
2.3. Tamoxifen Treatment
2.4. ATX Activity Measurement
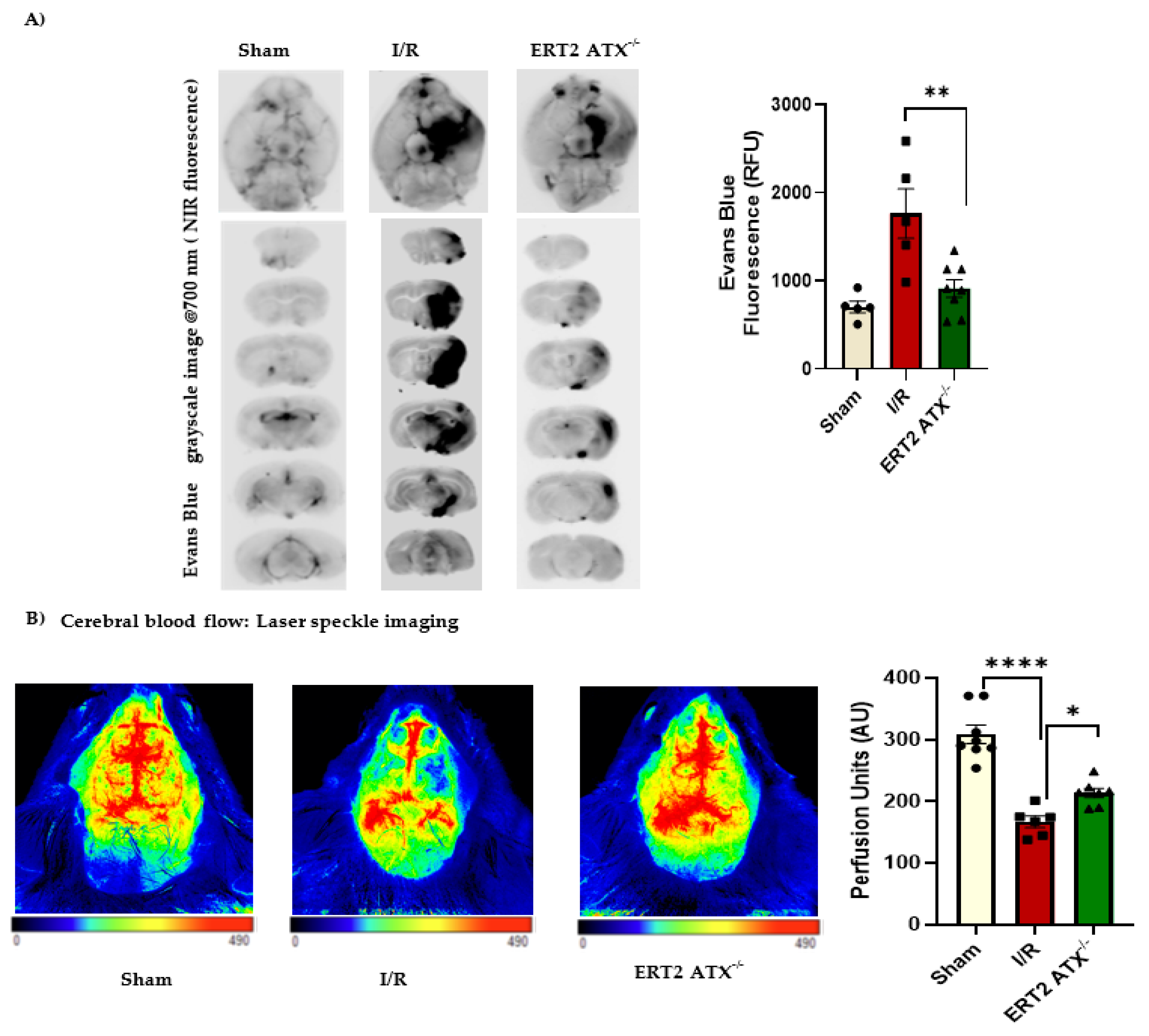
2.5. Evans Blue Permeability Measurement
2.6. Cresyl Violet Staining
2.7. PCR Genotyping
2.8. ELISA (Enzyme Linked Immunosorbent Assay)
2.9. Immunostaining
2.10. Laser Speckle Imaging
2.11. Statistical Analysis
3. Results
3.1. Generation and Genotyping of Inducible Endothelial ATX Knockout Mice (ERT2 ATX−/−)
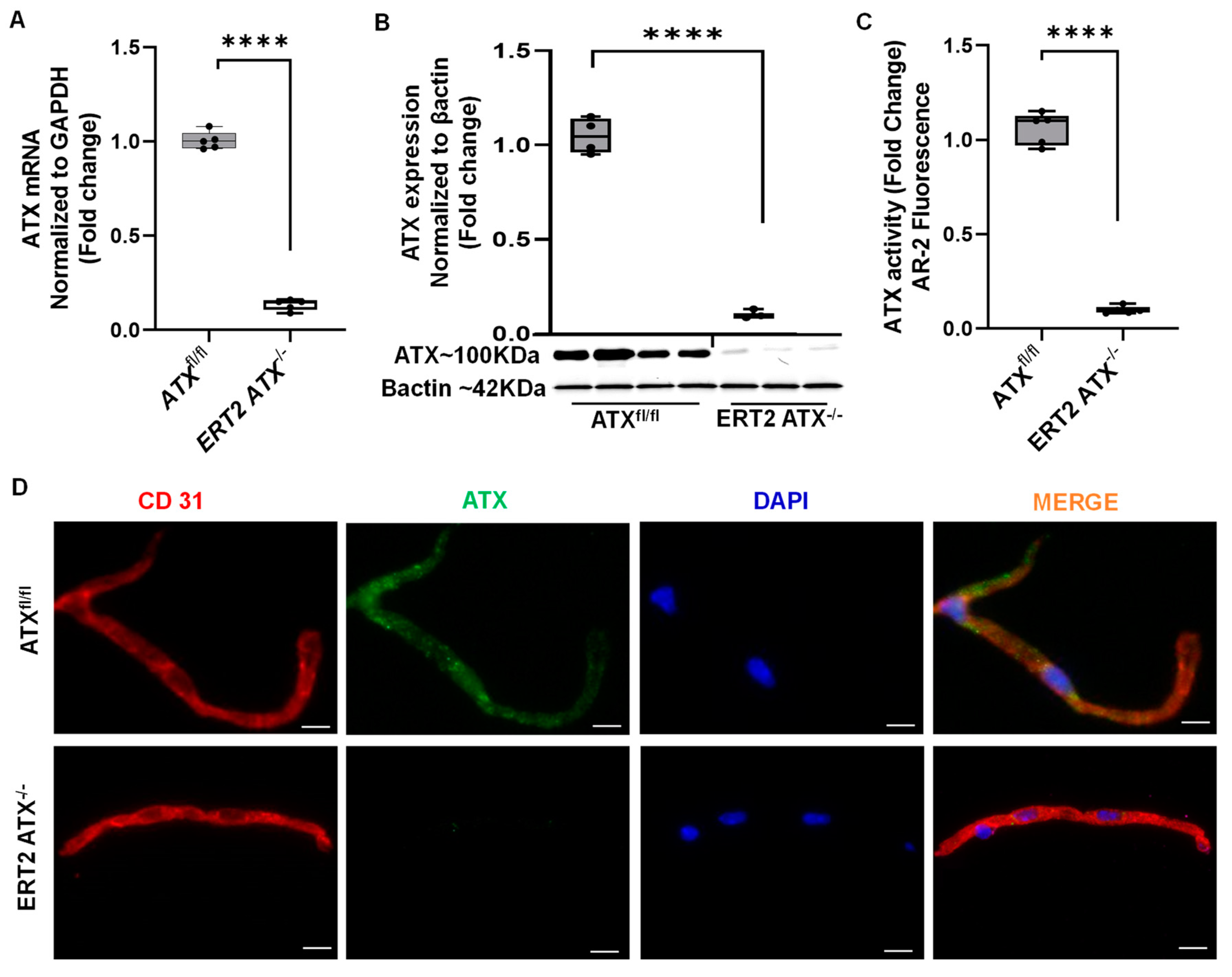
3.2. Inducible Inactivation of ATX in Adult Mice
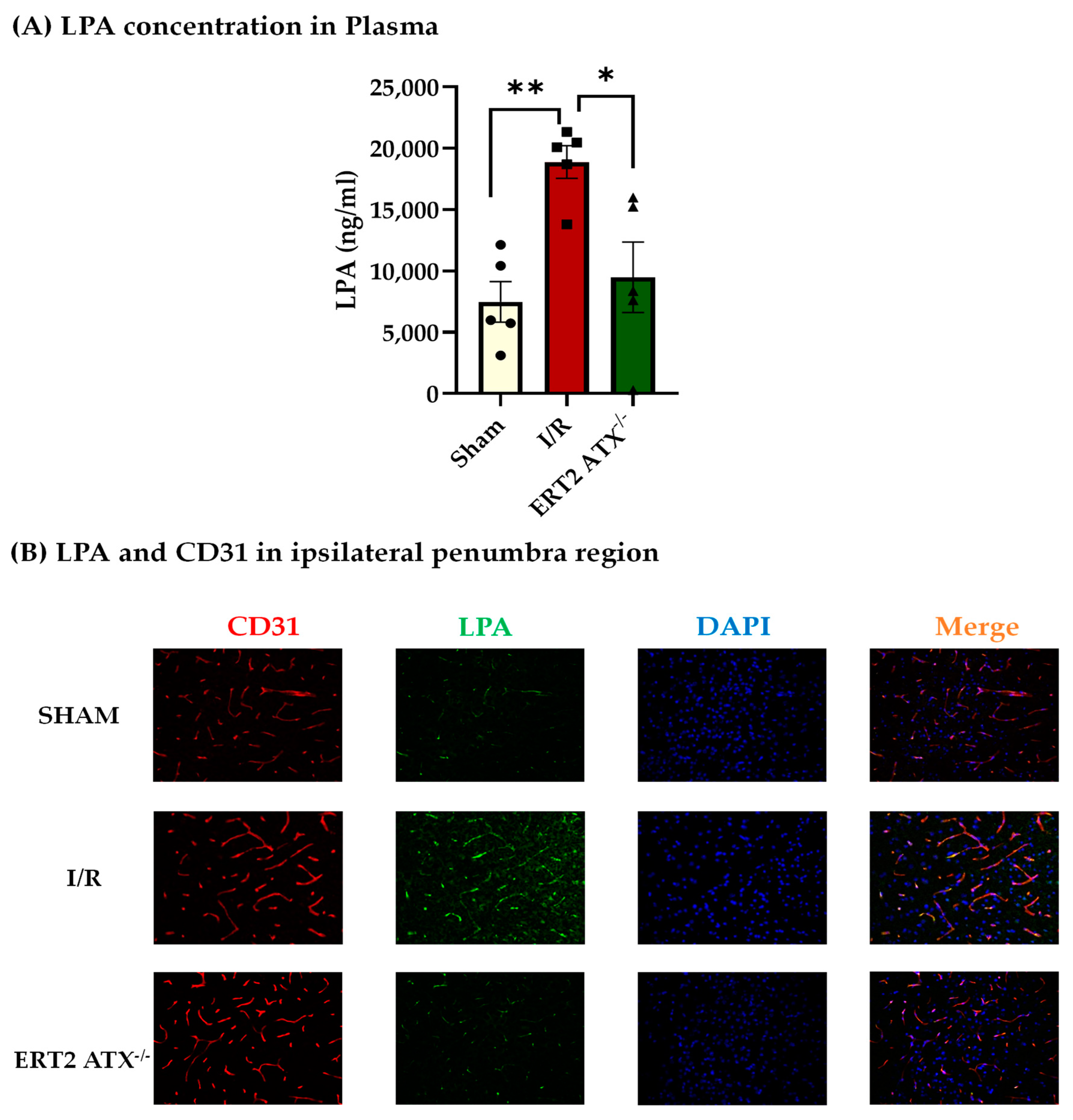
3.3. Reduction in LPA Concentration in ERT2 ATX−/− following I/R
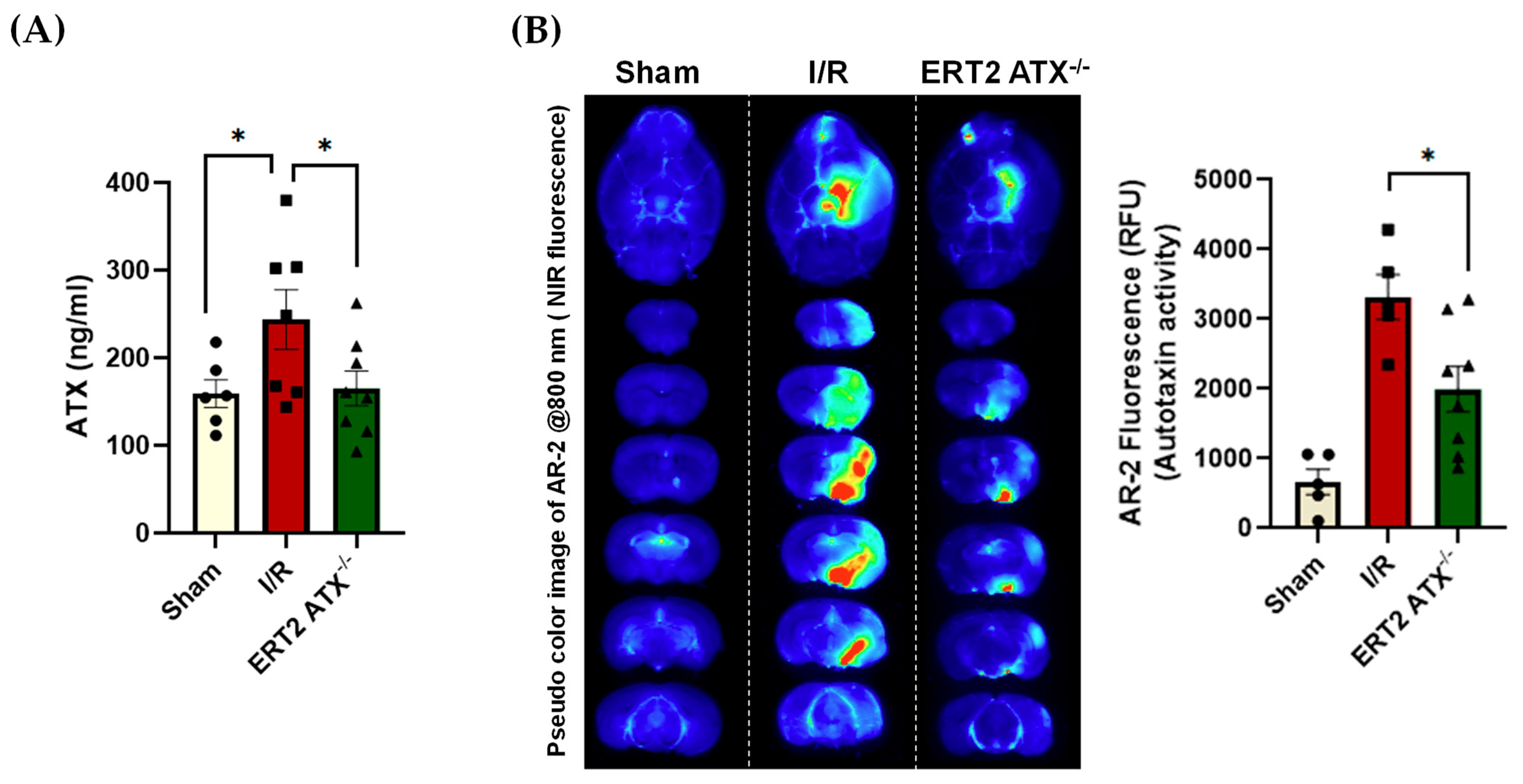
3.4. ATX Concentration and Activity Reduced in ERT2 ATX−/− following I/R
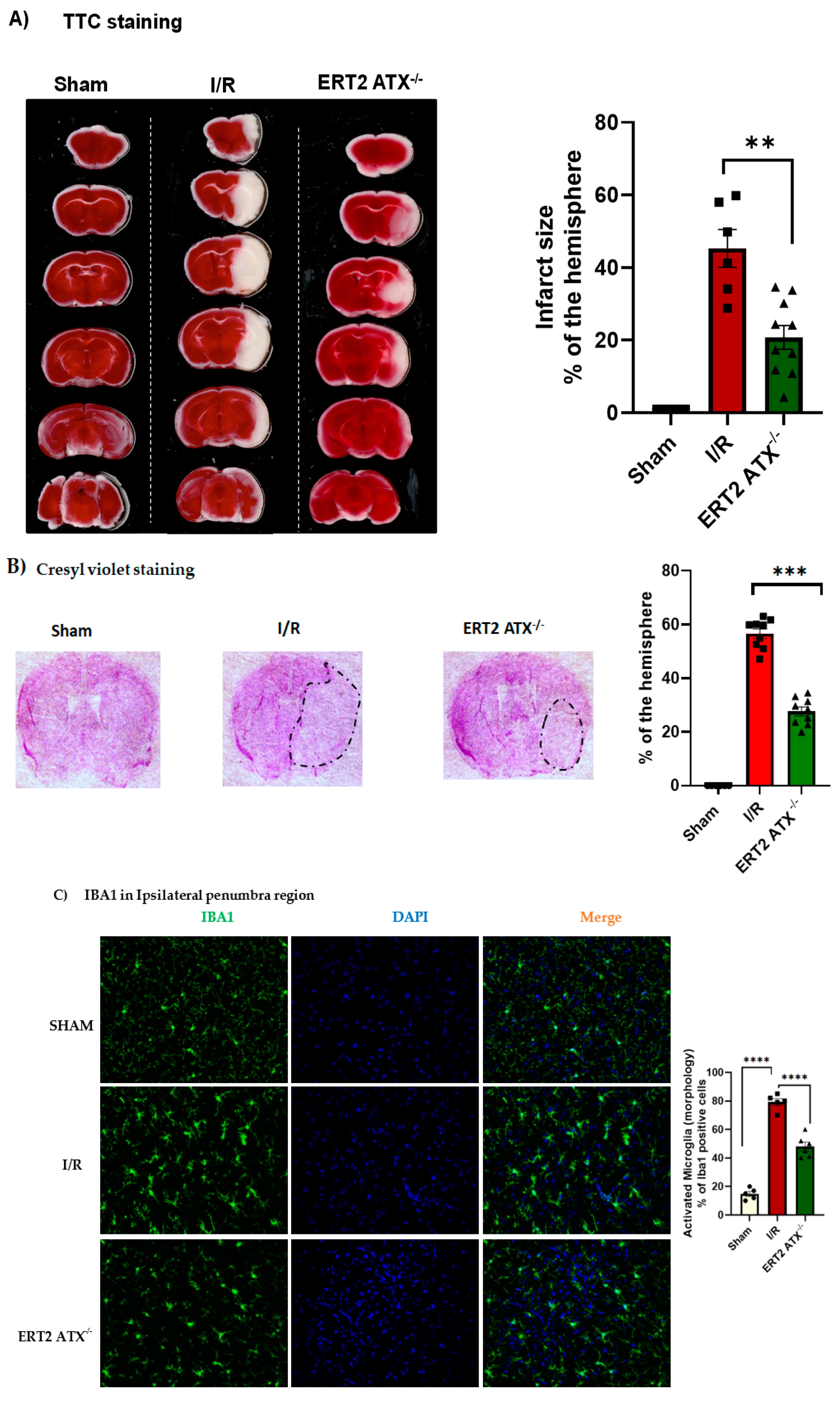
3.5. Infarct Size Reduced with Endothelial-Specific Deletion of ATX
3.6. Improved Stroke Functional Outcomes in ERT2 ATX−/− Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Meeteren, L.A.; Ruurs, P.; Stortelers, C.; Bouwman, P.; van Rooijen, M.A.; Pradère, J.P.; Pettit, T.R.; Wakelam, M.J.; Saulnier-Blache, J.S.; Mummery, C.L.; et al. Autotaxin, a secreted lysophospholipase D, is essential for blood vessel formation during development. Mol. Cell. Biol. 2006, 26, 5015–5022. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—An LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Fotopoulou, S.; Oikonomou, N.; Grigorieva, E.; Nikitopoulou, I.; Paparountas, T.; Thanassopoulou, A.; Zhao, Z.; Xu, Y.; Kontoyiannis, D.L.; Remboutsika, E.; et al. ATX expression and LPA signalling are vital for the development of the nervous system. Dev. Biol. 2010, 339, 451–464. [Google Scholar] [CrossRef]
- Geraldo, L.H.M.; Spohr, T.C.L.d.S.; Amaral, R.F.d.; Fonseca, A.C.C.d.; Garcia, C.; Mendes, F.d.A.; Freitas, C.; dosSantos, M.F.; Lima, F.R.S. Role of lysophosphatidic acid and its receptors in health and disease: Novel therapeutic strategies. Signal Transduct. Target. Ther. 2021, 6, 45. [Google Scholar] [CrossRef] [PubMed]
- Xiang, H.; Lu, Y.; Shao, M.; Wu, T. Lysophosphatidic Acid Receptors: Biochemical and Clinical Implications in Different Diseases. J. Cancer 2020, 11, 3519–3535. [Google Scholar] [CrossRef]
- Aoki, J. Mechanisms of lysophosphatidic acid production. Semin. Cell Dev. Biol. 2004, 15, 477–489. [Google Scholar] [CrossRef] [PubMed]
- van Meeteren, L.A.; Moolenaar, W.H. Regulation and biological activities of the autotaxin–LPA axis. Prog. Lipid Res. 2007, 46, 145–160. [Google Scholar] [CrossRef]
- Ninou, I.; Magkrioti, C.; Aidinis, V. Autotaxin in Pathophysiology and Pulmonary Fibrosis. Front. Med. 2018, 5, 180. [Google Scholar] [CrossRef]
- Gao, L.; Li, X.; Wang, H.; Liao, Y.; Zhou, Y.; Wang, K.; Hu, J.; Cheng, M.; Zeng, Z.; Wang, T.; et al. Autotaxin levels in serum and bronchoalveolar lavage fluid are associated with inflammatory and fibrotic biomarkers and the clinical outcome in patients with acute respiratory distress syndrome. J. Intensive Care 2021, 9, 44. [Google Scholar] [CrossRef]
- Jose, A.; Kienesberger, P.C. Autotaxin-LPA-LPP3 Axis in Energy Metabolism and Metabolic Disease. Int. J. Mol. Sci. 2021, 22, 9575. [Google Scholar] [CrossRef]
- Castelino, F.V.; Bain, G.; Pace, V.A.; Black, K.E.; George, L.; Probst, C.K.; Goulet, L.; Lafyatis, R.; Tager, A.M. An Autotaxin/Lysophosphatidic Acid/Interleukin-6 Amplification Loop Drives Scleroderma Fibrosis. Arthritis Rheumatol. 2016, 68, 2964–2974. [Google Scholar] [CrossRef]
- Zhang, G.; Cheng, Y.; Zhang, Q.; Li, X.; Zhou, J.; Wang, J.; Wei, L. ATX-LPA axis facilitates estrogen-induced endometrial cancer cell proliferation via MAPK/ERK signaling pathway. Mol. Med. Rep. 2018, 17, 4245–4252. [Google Scholar] [CrossRef]
- Tigyi, G.; Dacheux, M.A.; Lin, K.-H.; Yue, J.; Norman, D.; Benyó, Z.; Lee, S.C. Anti-cancer strategies targeting the autotaxin-lysophosphatidic acid receptor axis: Is there a path forward? Cancer Metastasis Rev. 2021, 40, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Bain, G.; Furuichi, K.; Iwata, Y.; Nakamura, M.; Hara, A.; Kitajima, S.; Sagara, A.; Miyake, T.; Toyama, T.; et al. The involvement of autotaxin in renal interstitial fibrosis through regulation of fibroblast functions and induction of vascular leakage. Sci. Rep. 2019, 9, 7414. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, S.; Sharma, S.; Ara, H.; Subedi, U.; Sun, G. Disrupted Blood-Brain Barrier and Mitochondrial Impairment by Autotaxin-Lysophosphatidic Acid Axis in Postischemic Stroke. J. Am. Heart Assoc. 2021, 10, e021511. [Google Scholar] [CrossRef] [PubMed]
- Umemura, K.; Yamashita, N.; Yu, X.; Arima, K.; Asada, T.; Makifuchi, T.; Murayama, S.; Saito, Y.; Kanamaru, K.; Goto, Y.; et al. Autotaxin expression is enhanced in frontal cortex of Alzheimer-type dementia patients. Neurosci. Lett. 2006, 400, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Smyth, S.S.; Mueller, P.; Yang, F.; Brandon, J.A.; Morris, A.J. Arguing the case for the autotaxin-lysophosphatidic acid-lipid phosphate phosphatase 3-signaling nexus in the development and complications of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 479–486. [Google Scholar] [CrossRef]
- Tripathi, H.; Al-Darraji, A.; Abo-Aly, M.; Peng, H.; Shokri, E.; Chelvarajan, L.; Donahue, R.R.; Levitan, B.M.; Gao, E.; Hernandez, G.; et al. Autotaxin inhibition reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction. J. Mol. Cell. Cardiol. 2020, 149, 95–114. [Google Scholar] [CrossRef]
- Bouchareb, R.; Mahmut, A.; Nsaibia, M.J.; Boulanger, M.C.; Dahou, A.; Lepine, J.L.; Laflamme, M.H.; Hadji, F.; Couture, C.; Trahan, S.; et al. Autotaxin Derived From Lipoprotein(a) and Valve Interstitial Cells Promotes Inflammation and Mineralization of the Aortic Valve. Circulation 2015, 132, 677–690. [Google Scholar] [CrossRef]
- Sun, S.; Wang, R.; Song, J.; Guan, M.; Li, N.; Zhang, X.; Zhao, Z.; Zhang, J. Blocking gp130 signaling suppresses autotaxin expression in adipocytes and improves insulin sensitivity in diet-induced obesity. J. Lipid Res. 2017, 58, 2102–2113. [Google Scholar] [CrossRef]
- Azare, J.; Doane, A.; Leslie, K.; Chang, Q.; Berishaj, M.; Nnoli, J.; Mark, K.; Al-Ahmadie, H.; Gerald, W.; Hassimi, M.; et al. Stat3 mediates expression of autotaxin in breast cancer. PLoS ONE 2011, 6, e27851. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, N.; Honjo, M. Crosstalk between transforming growth factor β-2 and Autotaxin in trabecular meshwork and different subtypes of glaucoma. J. Biomed. Sci. 2021, 28, 47. [Google Scholar] [CrossRef]
- Gierse, J.; Thorarensen, A.; Beltey, K.; Bradshaw-Pierce, E.; Cortes-Burgos, L.; Hall, T.; Johnston, A.; Murphy, M.; Nemirovskiy, O.; Ogawa, S.; et al. A Novel Autotaxin Inhibitor Reduces Lysophosphatidic Acid Levels in Plasma and the Site of Inflammation. J. Pharmacol. Exp. Ther. 2010, 334, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Albers, H.M.H.G.; Dong, A.; van Meeteren, L.A.; Egan, D.A.; Sunkara, M.; van Tilburg, E.W.; Schuurman, K.; van Tellingen, O.; Morris, A.J.; Smyth, S.S.; et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. USA 2010, 107, 7257–7262. [Google Scholar] [CrossRef] [PubMed]
- Jiang, G.; Madan, D.; Prestwich, G.D. Aromatic phosphonates inhibit the lysophospholipase D activity of autotaxin. Bioorg. Med. Chem. Lett. 2011, 21, 5098–5101. [Google Scholar] [CrossRef]
- Tang, X.; Wuest, M.; Benesch, M.G.K. Inhibition of Autotaxin with GLPG1690 Increases the Efficacy of Radiotherapy and Chemotherapy in a Mouse Model of Breast Cancer. Mol. Cancer Ther. 2020, 19, 63–74. [Google Scholar] [CrossRef]
- Hoshino, Y.; Okuno, T.; Saigusa, D.; Kano, K.; Yamamoto, S.; Shindou, H.; Aoki, J.; Uchida, K.; Yokomizo, T.; Ito, N. Lysophosphatidic acid receptor1/3 antagonist inhibits the activation of satellite glial cells and reduces acute nociceptive responses. FASEB J. 2022, 36, e22236. [Google Scholar] [CrossRef]
- Jiang, G.; Xu, Y.; Fujiwara, Y.; Tsukahara, T.; Tsukahara, R.; Gajewiak, J.; Tigyi, G.; Prestwich, G.D. α-Substituted Phosphonate Analogues of Lysophosphatidic Acid (LPA) Selectively Inhibit Production and Action of LPA. ChemMedChem 2007, 2, 679–690. [Google Scholar] [CrossRef]
- Valdés-Rives, S.A.; González-Arenas, A. Autotaxin-Lysophosphatidic Acid: From Inflammation to Cancer Development. Mediat. Inflamm. 2017, 2017, 9173090. [Google Scholar] [CrossRef]
- Bhattarai, S.; Sharma, S.; Subedi, U.; Ara, H.; Shum, A.; Milena, M.; Bhuiyan, M.S. The ATX-LPA Axis Regulates Vascular Permeability during Cerebral Ischemic-Reperfusion. Int. J. Mol. Sci. 2022, 23, 4138. [Google Scholar] [CrossRef] [PubMed]
- Longa, E.Z.; Weinstein, P.R.; Carlson, S.; Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 1989, 20, 84–91. [Google Scholar] [CrossRef]
- Fluri, F.; Schuhmann, M.K.; Kleinschnitz, C. Animal models of ischemic stroke and their application in clinical research. Drug Des. Dev. Ther. 2015, 9, 3445–3454. [Google Scholar] [CrossRef]
- Panchatcharam, M.; Salous, A.K.; Brandon, J.; Miriyala, S.; Wheeler, J.; Patil, P.; Sunkara, M.; Morris, A.J.; Escalante-Alcalde, D.; Smyth, S.S. Mice with targeted inactivation of ppap2b in endothelial and hematopoietic cells display enhanced vascular inflammation and permeability. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Madan, D.; Ferguson, C.G.; Lee, W.Y.; Prestwich, G.D.; Testa, C.A. Non-Invasive Imaging of Tumors by Monitoring Autotaxin Activity Using an Enzyme-Activated Near-Infrared Fluorogenic Substrate. PLoS ONE 2013, 8, e79065. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.W.; Lim, W.; Jo, D.; Kim, S.; Park, J.T.; Min, J.J.; Hyun, H.; Kim, H.S. Low-Dose Evans Blue Dye for Near-Infrared Fluorescence Imaging in Photothrombotic Stroke Model. Int. J. Med. Sci. 2018, 15, 696–702. [Google Scholar] [CrossRef]
- Kádár, A.; Wittmann, G.; Liposits, Z.; Fekete, C. Improved method for combination of immunocytochemistry and Nissl staining. J. Neurosci. Methods 2009, 184, 115–118. [Google Scholar] [CrossRef] [PubMed]
- Katsifa, A.; Kaffe, E.; Nikolaidou-Katsaridou, N.; Economides, A.N.; Newbigging, S.; McKerlie, C.; Aidinis, V. The Bulk of Autotaxin Activity Is Dispensable for Adult Mouse Life. PLoS ONE 2015, 10, e0143083. [Google Scholar] [CrossRef]
- Tokumura, A.; Majima, E.; Kariya, Y.; Tominaga, K.; Kogure, K.; Yasuda, K.; Fukuzawa, K. Identification of Human Plasma Lysophospholipase D, a Lysophosphatidic Acid-producing Enzyme, as Autotaxin, a Multifunctional Phosphodiesterase. J. Biol. Chem. 2002, 277, 39436–39442. [Google Scholar] [CrossRef]
- Schulze, C.; Smales, C.; Rubin, L.L.; Staddon, J.M. Lysophosphatidic acid increases tight junction permeability in cultured brain endothelial cells. J. Neurochem. 1997, 68, 991–1000. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.H.; Hu, D.E.; Fraser, P.A. Regulation of cerebromicrovascular permeability by lysophosphatidic acid. Microcirculation 2010, 17, 39–46. [Google Scholar] [CrossRef] [PubMed]
- van Nieuw Amerongen, G.P.; Vermeer, M.A.; van Hinsbergh, V.W. Role of RhoA and Rho kinase in lysophosphatidic acid-induced endothelial barrier dysfunction. Arterioscler. Thromb. Vasc. Biol. 2000, 20, e127–e133. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, G.A. Neurological diseases in relation to the blood-brain barrier. J. Cereb. Blood Flow Metab. 2012, 32, 1139–1151. [Google Scholar] [CrossRef]
- Ueda, H.; Neyama, H.; Sasaki, K.; Miyama, C.; Iwamoto, R. Lysophosphatidic acid LPA1 and LPA3 receptors play roles in the maintenance of late tissue plasminogen activator-induced central poststroke pain in mice. Neurobiol. Pain 2019, 5, 100020. [Google Scholar] [CrossRef] [PubMed]
- Guan, M.; Zhang, Z.; Li, S.; Liu, J.; Liu, L.; Yang, H.; Zhang, Y.; Wang, T.; Zhao, Z. Silver nanoparticles as matrix for MALDI FTICR MS profiling and imaging of diverse lipids in brain. Talanta 2018, 179, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Qin, J.; Liu, M.; Ruan, Q.; Li, Y.; Zhang, Z. Role of Rho kinase in lysophosphatidic acid-induced altering of blood-brain barrier permeability. Int. J. Mol. Med. 2014, 33, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Kufner, A.; Dell’Orco, A.; Rackoll, T.; Mekle, R.; Piper, S.K.; Fiebach, J.B.; Villringer, K.; Flöel, A.; Endres, M.; et al. Evolution of Blood-Brain Barrier Permeability in Subacute Ischemic Stroke and Associations With Serum Biomarkers and Functional Outcome. Front. Neurol. 2021, 12, 730923. [Google Scholar] [CrossRef]
- Merali, Z.; Huang, K.; Mikulis, D.; Silver, F.; Kassner, A. Evolution of blood-brain-barrier permeability after acute ischemic stroke. PLoS ONE 2017, 12, e0171558. [Google Scholar] [CrossRef]
- Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Blood-Brain Barrier Protection as a Therapeutic Strategy for Acute Ischemic Stroke. AAPS J. 2017, 19, 957–972. [Google Scholar] [CrossRef]
- Nikitopoulou, I.; Katsifa, A.; Kanellopoulou, P.; Jahaj, E. Autotaxin Has a Negative Role in Systemic Inflammation. Int. J. Mol. Sci. 2022, 23, 7920. [Google Scholar] [CrossRef]
- Brandon, J.A.; Kraemer, M.; Vandra, J.; Halder, S.; Ubele, M.; Morris, A.J.; Smyth, S.S. Adipose-derived autotaxin regulates inflammation and steatosis associated with diet-induced obesity. PLoS ONE 2019, 14, e0208099. [Google Scholar] [CrossRef]
- Isshiki, T.; Shimizu, H.; Sakamoto, S.; Yamasaki, A.; Miyoshi, S.; Nakamura, Y.; Homma, S.; Kishi, K. Serum autotaxin levels in chronic disease and acute exacerbation of fibrosing interstitial lung disease. ERJ Open Res. 2022, 8, 00683–02021. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Zhang, L.; Chen, X.; Li, Y.; Ha, F.; Liu, H.; Han, T. Autotaxin: An Early Warning Biomarker for Acute-on-chronic Liver Failure. J. Clin. Transl. Hepatol. 2020, 8, 240–245. [Google Scholar] [CrossRef]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. [Google Scholar] [CrossRef] [PubMed]
- Seo, E.J.; Kwon, Y.W.; Jang, I.H.; Kim, D.K.; Lee, S.I.; Choi, E.J.; Kim, K.-H.; Suh, D.-S.; Lee, J.H.; Choi, K.U.; et al. Autotaxin Regulates Maintenance of Ovarian Cancer Stem Cells through Lysophosphatidic Acid-Mediated Autocrine Mechanism. Stem Cells 2016, 34, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.K.; Conaghan, P.G.; Karim, Z.; Quinn, M.A.; Ikeda, K.; Peterfy, C.G.; Hensor, E.; Wakefield, R.J.; O’Connor, P.J.; Emery, P. An explanation for the apparent dissociation between clinical remission and continued structural deterioration in rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2958–2967. [Google Scholar] [CrossRef]
- Hoelzinger, D.B.; Nakada, M.; Demuth, T.; Rosensteel, T.; Reavie, L.B.; Berens, M.E. Autotaxin: A secreted autocrine/paracrine factor that promotes glioma invasion. J. Neuro-Oncol. 2008, 86, 297–309. [Google Scholar] [CrossRef]
- Karshovska, E.; Mohibullah, R.; Zhu, M.; Zahedi, F.; Thomas, D.; Magkrioti, C.; Geissler, C.; Megens, R.T.A.; Bianchini, M.; Nazari-Jahantigh, M.; et al. Endothelial ENPP2 (Ectonucleotide Pyrophosphatase/Phosphodiesterase 2) Increases Atherosclerosis in Female and Male Mice. Arterioscler. Thromb. Vasc. Biol. 2022, 42, 1023–1036. [Google Scholar] [CrossRef]
- Yaghi, S.; Willey, J.Z.; Cucchiara, B.; Goldstein, J.N.; Gonzales, N.R.; Khatri, P.; Kim, L.J.; Mayer, S.A.; Sheth, K.N.; Schwamm, L.H. Treatment and Outcome of Hemorrhagic Transformation After Intravenous Alteplase in Acute Ischemic Stroke: A Scientific Statement for Healthcare Professionals From the American Heart Association/American Stroke Association. Stroke 2017, 48, e343–e361. [Google Scholar] [CrossRef]
- Hollist, M.; Morgan, L.; Cabatbat, R.; Au, K.; Kirmani, M.F.; Kirmani, B.F. Acute Stroke Management: Overview and Recent Updates. Aging Dis. 2021, 12, 1000–1009. [Google Scholar] [CrossRef]
- Zheng, X.; Haupt, M.; Bähr, M.; Tatenhorst, L.; Doeppner, T.R. Treating Cerebral Ischemia: Novel Therapeutic Strategies from Experimental Stroke Research. In Cerebral Ischemia; Pluta, R., Ed.; Exon Publications: Brisbane, Australia, 2021. [Google Scholar]
- Salinet, A.S.; Panerai, R.B.; Robinson, T.G. The longitudinal evolution of cerebral blood flow regulation after acute ischaemic stroke. Cerebrovasc. Dis. Extra 2014, 4, 186–197. [Google Scholar] [CrossRef]
- Steiner, L.; Federspiel, A.; Jaros, J.; Slavova, N.; Wiest, R.; Steinlin, M.; Grunt, S.; Everts, R. Cerebral blood flow and cognitive outcome after pediatric stroke in the middle cerebral artery. Sci. Rep. 2021, 11, 19421. [Google Scholar] [CrossRef] [PubMed]
- Feigin, V.L.; Brainin, M. World Stroke Organization (WSO): Global Stroke Fact Sheet 2022. Int. J. Stroke 2022, 17, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Bitar, L.; Uphaus, T.; Thalman, C.; Muthuraman, M.; Gyr, L.; Ji, H.; Domingues, M.; Endle, H.; Groppa, S.; Steffen, F.; et al. Inhibition of the enzyme autotaxin reduces cortical excitability and ameliorates the outcome in stroke. Sci. Transl. Med. 2022, 14, eabk0135. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattarai, S.; Subedi, U.; Manikandan, S.; Sharma, S.; Sharma, P.; Miller, C.; Bhuiyan, M.S.; Kidambi, S.; Aidinis, V.; Sun, H.; et al. Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes. Cells 2023, 12, 511. https://doi.org/10.3390/cells12030511
Bhattarai S, Subedi U, Manikandan S, Sharma S, Sharma P, Miller C, Bhuiyan MS, Kidambi S, Aidinis V, Sun H, et al. Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes. Cells. 2023; 12(3):511. https://doi.org/10.3390/cells12030511
Chicago/Turabian StyleBhattarai, Susmita, Utsab Subedi, Shrivats Manikandan, Sudha Sharma, Papori Sharma, Chloe Miller, Md Shenuarin Bhuiyan, Srivatsan Kidambi, Vassilis Aidinis, Hong Sun, and et al. 2023. "Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes" Cells 12, no. 3: 511. https://doi.org/10.3390/cells12030511
APA StyleBhattarai, S., Subedi, U., Manikandan, S., Sharma, S., Sharma, P., Miller, C., Bhuiyan, M. S., Kidambi, S., Aidinis, V., Sun, H., Miriyala, S., & Panchatcharam, M. (2023). Endothelial Specific Deletion of Autotaxin Improves Stroke Outcomes. Cells, 12(3), 511. https://doi.org/10.3390/cells12030511