
Aurora B SUMOylation Is Restricted to Centromeres in Early Mitosis and Requires RANBP2

 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Synchronisation and Treatments
2.2. Plasmids and Mutagenesis
2.3. Generation of Stable Inducible Cell Lines
2.4. RNA Interference
2.5. Immunofluorescence (IF)
2.6. Proximity Ligation Assay (PLA)
2.7. Microscopy
2.8. Western Immunoblotting
2.9. Statistical Analysis
3. Results
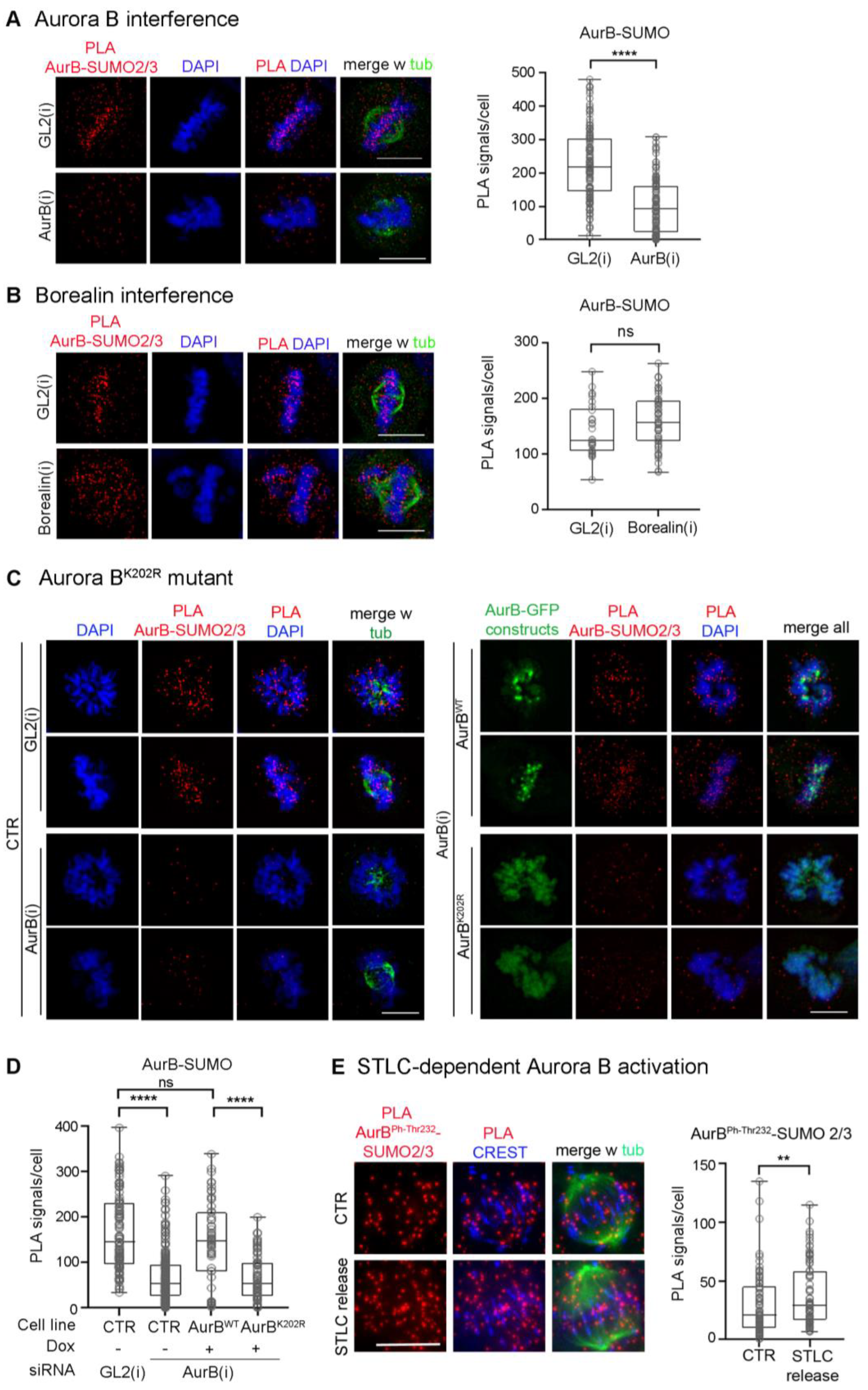
3.1. Aurora B-SUMO2/3 Ligation Products Can Be Visualised by PLA at Centromeres/Kinetochores in Intact Human Cells
3.2. SUMOylated Aurora B Is Restricted to the G2-to-Metaphase Window
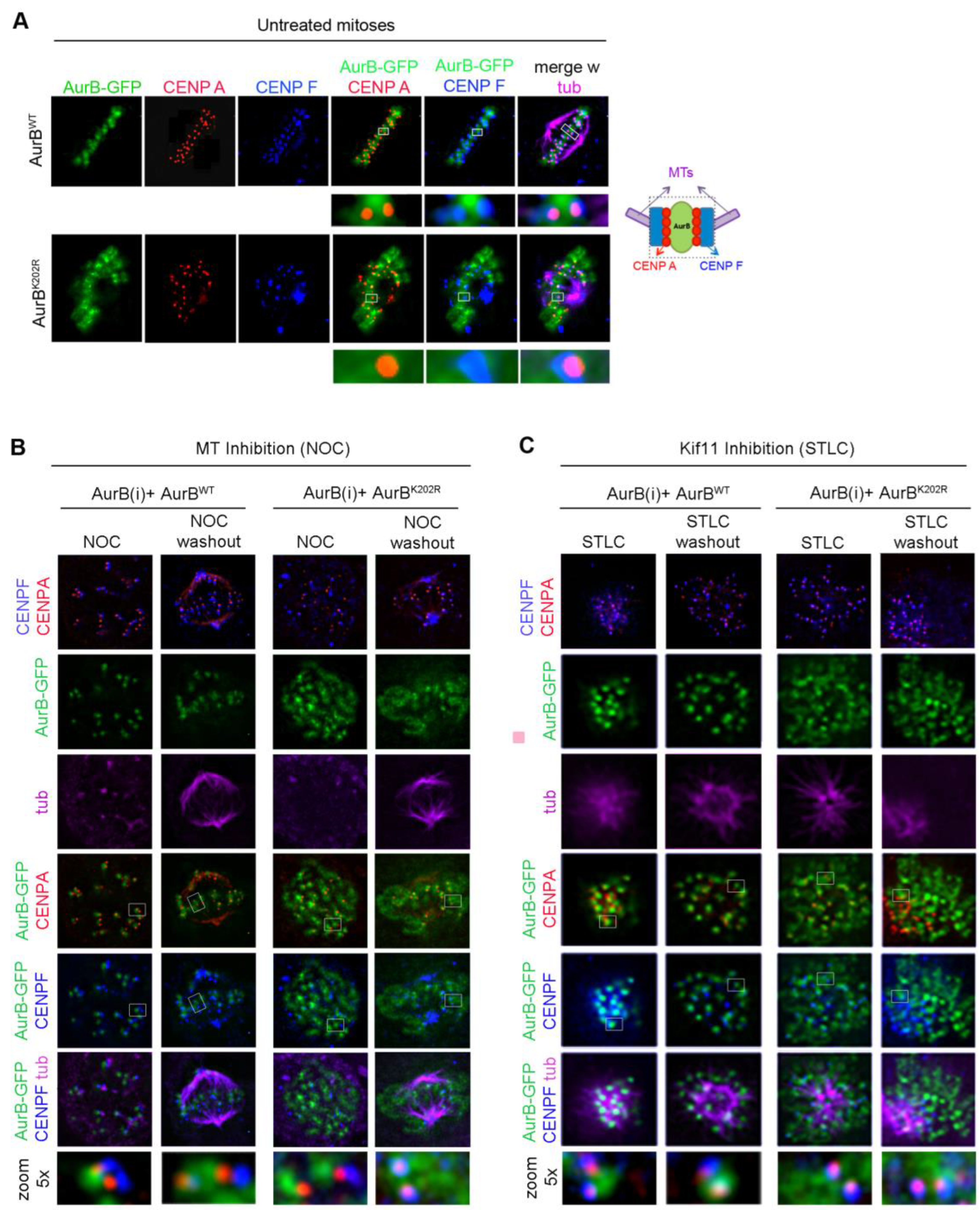
3.3. Mutation of Lysine K202 Impairs Multiple Aspects of Aurora B Function
3.4. RANBP2 Contributes to the SUMOylated State of Aurora B
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Flotho, A.; Melchior, F. Sumoylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Varejão, N.; Lascorz, J.; Li, Y.; Reverter, D. Molecular mechanisms in SUMO conjugation. Biochem. Soc. Trans. 2020, 48, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.O. Signalling mechanisms and cellular functions of SUMO. Nat. Rev. Mol. Cell Biol. 2022, 23, 715–731. [Google Scholar] [CrossRef] [PubMed]
- Raman, N.; Nayak, A.; Muller, S. The SUMO system: A master organizer of nuclear protein assemblies. Chromosoma 2013, 122, 475–485. [Google Scholar] [CrossRef]
- Wotton, D.; Pemberton, L.F.; Merrill-Schools, J. SUMO and Chromatin Remodeling. Adv. Exp. Med. Biol. 2017, 963, 35–50. [Google Scholar]
- Zhao, X. SUMO-Mediated Regulation of Nuclear Functions and Signaling Processes. Mol. Cell 2018, 71, 409–418. [Google Scholar] [CrossRef]
- Celen, A.B.; Sahin, U. Sumoylation on its 25th anniversary: Mechanisms, pathology, and emerging concepts. FEBS J. 2020, 287, 3110–3140. [Google Scholar] [CrossRef] [PubMed]
- Talamillo, A.; Barroso-Gomila, O.; Giordano, I.; Ajuria, L.; Grillo, M.; Mayor, U.; Barrio, R. The role of SUMOylation during development. Biochem. Soc. Trans. 2020, 48, 463–478. [Google Scholar] [CrossRef]
- Cubeñas-Potts, C.; Goeres, J.D.; Matunis, M.J. SENP1 and SENP2 affect spatial and temporal control of sumoylation in mitosis. Mol. Biol. Cell 2013, 24, 3483–3495. [Google Scholar] [CrossRef] [PubMed]
- Cubeñas-Potts, C.; Srikumar, T.; Lee, C.; Osula, O.; Subramonian, D.; Zhang, X.D.; Cotter, R.J.; Raught, B.; Matunis, M.J. Identification of SUMO-2/3-modified proteins associated with mitotic chromosomes. Proteomics 2015, 15, 763–772. [Google Scholar] [CrossRef]
- Wei, B.; Huang, C.; Liu, B.; Wang, Y.; Xia, N.; Fan, Q.; Chen, G.Q.; Cheng, J. Mitotic Phosphorylation of SENP3 Regulates DeSUMOylation of Chromosome-Associated Proteins and Chromosome Stability. Cancer Res. 2018, 78, 2171–2178. [Google Scholar] [CrossRef] [PubMed]
- Hassebroek, V.A.; Park, H.; Pandey, N.; Lerbakken, B.T.; Aksenova, V.; Arnaoutov, A.; Dasso, M.; Azuma, Y. PICH regulates the abundance and localization of SUMOylated proteins on mitotic chromosomes. Mol. Biol. Cell 2020, 31, 2537–2556. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Subramonian, D.; Zhang, X.-D. SUMOylation in control of accurate chromosome segregation during mitosis. Curr. Protein Pept. Sci. 2012, 13, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Cuijpers, S.A.G.; Vertegaal, A.C.O. Guiding Mitotic Progression by Crosstalk between Post-translational Modifications. Trends Biochem. Sci. 2018, 43, 251–268. [Google Scholar] [CrossRef]
- Abrieu, A.; Liakopoulos, D. How Does SUMO Participate in Spindle Organization? Cells 2019, 8, 801. [Google Scholar] [CrossRef] [PubMed]
- Pichler, A.; Gast, A.; Seeler, J.S.; Dejean, A.; Melchior, F. The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 2002, 108, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Matunis, M.J.; Coutavas, E.; Blobel, G. A novel ubiquitin-like modification modulates the partitioning of the Ran-GTPase-activating protein RanGAP1 between the cytosol and the nuclear pore complex. J. Cell Biol. 1996, 135, 1457–1470. [Google Scholar] [CrossRef]
- Mahajan, R.; Delphin, C.; Guan, T.; Gerace, L.; Melchior, F. A small ubiquitin-related polypeptide involved in targeting RanGAP1 to nuclear pore complex protein RanBP2. Cell 1997, 88, 97–107. [Google Scholar] [CrossRef]
- Matunis, M.J.; Wu, J.; Blobel, G. SUMO-1 modification and its role in targeting the Ran GTPase-activating protein, RanGAP1, to the nuclear pore complex. J. Cell Biol. 1998, 140, 499–509. [Google Scholar] [CrossRef]
- Lee, G.W.; Melchior, F.; Matunis, M.J.; Mahajan, R.; Tian, Q.; Anderson, P. Modification of Ran GTPase-activating protein by the small ubiquitin-related modifier SUMO-1 requires Ubc9, an E2-type ubiquitin-conjugating enzyme homologue. J. Biol. Chem. 1998, 273, 6503–6507. [Google Scholar] [CrossRef]
- Bernier-Villamor, V.; Sampson, D.A.; Matunis, M.J.; Lima, C.D. Structural basis for E2-mediated SUMO conjugation revealed by a complex between ubiquitin-conjugating enzyme Ubc9 and RanGAP1. Cell 2002, 108, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Reverter, D.; Lima, C.D. Insights into E3 ligase activity revealed by a SUMO-RanGAP1-Ubc9-Nup358 complex. Nature 2005, 435, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Werner, A.; Flotho, A.; Melchior, F. The RanBP2/RanGAP1*SUMO1/Ubc9 complex is a multisubunit SUMO E3 ligase. Mol. Cell 2012, 46, 287–298. [Google Scholar] [CrossRef] [PubMed]
- Ritterhoff, T.; Das, H.; Hofhaus, G.; Schröder, R.R.; Flotho, A.; Melchior, F. The RanBP2/RanGAP1*SUMO1/Ubc9 SUMO E3 ligase is a disassembly machine for Crm1-dependent nuclear export complexes. Nat. Commun. 2016, 7, 11482. [Google Scholar] [CrossRef]
- Ptak, C.; Wozniak, R.W. SUMO and Nucleocytoplasmic Transport. Adv. Exp. Med. Biol. 2017, 963, 111–126. [Google Scholar]
- Joseph, J.; Liu, S.T.; Jablonski, S.A.; Yen, T.J.; Dasso, M. The RanGAP1-RanBP2 complex is essential for microtubule-kinetochore interactions in vivo. Curr. Biol. 2004, 14, 611–617. [Google Scholar] [CrossRef]
- Roscioli, E.; Di Francesco, L.; Bolognesi, A.; Giubettini, M.; Orlando, S.; Harel, A.; Schininà, M.E.; Lavia, P. Importin-β negatively regulates multiple aspects of mitosis including RANGAP1 recruitment to kinetochores. J. Cell Biol. 2012, 196, 435–450. [Google Scholar] [CrossRef]
- Gilistro, E.; de Turris, V.; Damizia, M.; Verrico, A.; Moroni, S.; De Santis, R.; Rosa, A.; Lavia, P. Importin-β and CRM1 control a RANBP2 spatiotemporal switch essential for mitotic kinetochore function. J. Cell Sci. 2017, 130, 2564–2578. [Google Scholar] [CrossRef]
- Salina, D.; Enarson, P.; Rattner, J.B.; Burke, B. Nup358 integrates nuclear envelope breakdown with kinetochore assembly. J. Cell Biol. 2003, 162, 991–1001. [Google Scholar] [CrossRef]
- Hamada, M.; Haeger, A.; Jeganathan, K.B.; van Ree, J.H.; Malureanu, L.; Wälde, S.; Joseph, J.; Kehlenbach, R.H.; van Deursen, J.M. Ran-dependent docking of importin-beta to RanBP2/Nup358 filaments is essential for protein import and cell viability. J. Cell Biol. 2011, 194, 597–612. [Google Scholar] [CrossRef]
- Hashizume, C.; Kobayashi, A.; Wong, R.W. Down-modulation of nucleoporin RanBP2/Nup358 impaired chromosomal alignment and induced mitotic catastrophe. Cell Death Dis. 2013, 4, e854. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, M.M.; Malureanu, L.; Jeganathan, K.B.; Kao, E.; Sustmann, C.; Tahk, S.; Shuai, K.; Grosschedl, R.; van Deursen, J.M. Resolution of sister centromeres requires RanBP2-mediated SUMOylation of topoisomerase IIalpha. Cell 2008, 133, 103–115. [Google Scholar] [CrossRef]
- Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell Biol. 2003, 163, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Martínez, L.A.; Giménez-Abián, J.F.; Azuma, Y.; Guacci, V.; Giménez-Martín, G.; Lanier, L.M.; Clarke, D.J. PIASgamma is required for faithful chromosome segregation in human cells. PLoS ONE 2006, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Arnaoutov, A.; Anan, T.; Dasso, M. PIASy mediates SUMO-2 conjugation of Topoisomerase-II on mitotic chromosomes. EMBO J. 2005, 24, 2172–2182. [Google Scholar] [CrossRef]
- Ryu, H.; Furuta, M.; Kirkpatrick, D.; Gygi, S.P.; Azuma, Y. PIASy-dependent SUMOylation regulates DNA topoisomerase IIalpha activity. J. Cell Biol. 2010, 191, 783–784. [Google Scholar] [CrossRef]
- Klein, U.R.; Haindl, M.; Nigg, E.A.; Muller, S. RanBP2 and SENP3 function in a mitotic SUMO2/3 conjugation-deconjugation cycle on Borealin. Mol. Biol. Cell 2009, 20, 410–418. [Google Scholar] [CrossRef]
- Fernández-Miranda, G.; Pérez De Castro, I.; Carmena, M.; Aguirre-Portolés, C.; Ruchaud, S.; Fant, X.; Montoya, G.; Earnshaw, W.C.; Malumbres, M. SUMOylation modulates the function of Aurora-B kinase. J. Cell Sci. 2010, 123, 2823–2833. [Google Scholar] [CrossRef]
- Ban, R.; Nishida, T.; Urano, T. Mitotic kinase Aurora-B is regulated by SUMO-2/3 conjugation/deconjugation during mitosis. Genes Cells 2011, 16, 652–669. [Google Scholar] [CrossRef]
- Pelisch, F.; Sonneville, R.; Pourkarimi, E.; Agostinho, A.; Blow, J.J.; Gartner, A.; Hay, R.T. Dynamic SUMO modification regulates mitotic chromosome assembly and cell cycle progression in Caenorhabditis elegans. Nat. Commun. 2014, 5, 5485. [Google Scholar] [CrossRef]
- Van Der Horst, A.; Lens, S.M.A. Cell division: Control of the chromosomal passenger complex in time and space. Chromosoma 2014, 123, 25–42. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Musacchio, A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front. Oncol. 2015, 5, 225. [Google Scholar] [CrossRef] [PubMed]
- Broad, A.J.; DeLuca, J.G. The right place at the right time: Aurora B kinase localization to centromeres and kinetochores. Essays Biochem. 2020, 64, 299–311. [Google Scholar]
- Di Fiore, B.; Pines, J. How cyclin A destruction escapes the spindle assembly checkpoint. J. Cell Biol. 2010, 190, 501–509. [Google Scholar] [CrossRef]
- Santaguida, S.; Vernieri, C.; Villa, F.; Ciliberto, A.; Musacchio, A. Evidence that Aurora B is implicated in spindle checkpoint signalling independently of error correction. EMBO J. 2011, 30, 1508–1519. [Google Scholar] [CrossRef] [PubMed]
- Ulrich, A.; Andersen, K.R.; Schwartz, T.U. Exponential megapriming PCR (EMP) cloning--seamless DNA insertion into any target plasmid without sequence constraints. PLoS ONE 2012, 7, e5336. [Google Scholar] [CrossRef]
- Ditchfield, C.; Johnson, V.L.; Tighe, A.; Ellston, R.; Haworth, C.; Johnson, T.; Mortlock, A.; Keen, N.; Taylor, S.S. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J. Cell Biol. 2003, 161, 267–280. [Google Scholar] [CrossRef]
- Sessa, F.; Mapelli, M.; Ciferri, C.; Tarricone, C.; Areces, L.B.; Schneider, T.R.; Stukenberg, P.T.; Musacchio, A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol. Cell 2005, 18, 379–391. [Google Scholar] [CrossRef]
- Gassmann, R.; Carvalho, A.; Henzing, A.J.; Ruchaud, S.; Hudson, D.F.; Honda, R.; Nigg, E.A.; Gerloff, D.L.; Earnshaw, W.C. Borealin: A novel chromosomal passenger required for stability of the bipolar mitotic spindle. J. Cell Biol. 2004, 166, 179–191. [Google Scholar] [CrossRef]
- Di Francesco, L.; Verrico, A.; Asteriti, I.A.; Rovella, P.; Cirigliano, P.; Guarguaglini, G.; Schininà, M.E.; Lavia, P. Visualization of human karyopherin beta-1/importin beta-1 interactions with protein partners in mitotic cells by co-immunoprecipitation and proximity ligation assays. Sci. Rep. 2018, 8, 1850. [Google Scholar] [CrossRef]
- Verrico, A.; Rovella, P.; Di Francesco, L.; Damizia, M.; Staid, D.S.; Le Pera, L.; Schininà, M.E.; Lavia, P. Importin-β/karyopherin-β1 modulates mitotic microtubule function and taxane sensitivity in cancer cells via its nucleoporin-binding region. Oncogene 2020, 39, 454–468. [Google Scholar] [CrossRef] [PubMed]
- Lampson, M.A.; Renduchitala, K.; Khodjakov, A.; Kapoor, T.M. Correcting improper chromosome-spindle attachments during cell division. Nat. Cell Biol. 2004, 6, 232–237. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; McCollum, D. A role for metaphase spindle elongation forces in correction of merotelic kinetochore attachments. Curr. Biol. 2012, 22, 225–230. [Google Scholar] [CrossRef] [PubMed]
- Zuccolo, M.; Alves, A.; Galy, V.; Bolhy, S.; Formstecher, E.; Racine, V.; Sibarita, J.B.; Fukagawa, T.; Shiekhattar, R.; Yen, T.; et al. The human Nup107-160 nuclear pore subcomplex contributes to proper kinetochore functions. EMBO J. 2007, 26, 1853–1864. [Google Scholar] [CrossRef]
- Belgareh, N.; Rabut, G.; Baï, S.W.; Van Overbeek, M.; Beaudouin, J.; Daigle, N.; Zatsepina, O.V.; Pasteau, F.; Labas, V.; Fromont-Racine, M.; et al. An evolutionarily conserved NPC subcomplex, which redistributes in part to kinetochores in mammalian cells. J. Cell Biol. 2001, 154, 1147–1160. [Google Scholar] [CrossRef]
- Loïodice, I.; Alves, A.; Rabut, G.; Van Overbeek, M.; Ellenberg, J.; Sibarita, J.B.; Doye, V. The entire Nup107-160 complex, including three new members, is targeted as one entity to kinetochores in mitosis. Mol. Biol. Cell 2004, 15, 3333–3344. [Google Scholar] [CrossRef]
- Goeres, J.; Chan, P.K.; Mukhopadhyay, D.; Zhang, H.; Raught, B.; Matunis, M.J. The SUMO-specific isopeptidase SENP2 associates dynamically with nuclear pore complexes through interactions with karyopherins and the Nup107-160 nucleoporin subcomplex. Mol. Biol. Cell 2011, 22, 4868–4882. [Google Scholar] [CrossRef]
- Chow, K.H.; Elgort, S.; Dasso, M.; Powers, M.A.; Ullman, K.S. The SUMO proteases SENP1 and SENP2 play a critical role in nucleoporin homeostasis and nuclear pore complex function. Mol. Biol. Cell 2014, 25, 160–168. [Google Scholar] [CrossRef]
- Platani, M.; Santarella-Mellwig, R.; Posch, M.; Walczak, R.; Swedlow, J.R.; Mattaj, I.W. The Nup107-160 nucleoporin complex promotes mitotic events via control of the localization state of the chromosome passenger complex. Mol. Biol. Cell 2009, 20, 5260–5275. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Cesare, E.; Moroni, S.; Bartoli, J.; Damizia, M.; Giubettini, M.; Koerner, C.; Krenn, V.; Musacchio, A.; Lavia, P. Aurora B SUMOylation Is Restricted to Centromeres in Early Mitosis and Requires RANBP2. Cells 2023, 12, 372. https://doi.org/10.3390/cells12030372
Di Cesare E, Moroni S, Bartoli J, Damizia M, Giubettini M, Koerner C, Krenn V, Musacchio A, Lavia P. Aurora B SUMOylation Is Restricted to Centromeres in Early Mitosis and Requires RANBP2. Cells. 2023; 12(3):372. https://doi.org/10.3390/cells12030372
Chicago/Turabian StyleDi Cesare, Erica, Sara Moroni, Jessica Bartoli, Michela Damizia, Maria Giubettini, Carolin Koerner, Veronica Krenn, Andrea Musacchio, and Patrizia Lavia. 2023. "Aurora B SUMOylation Is Restricted to Centromeres in Early Mitosis and Requires RANBP2" Cells 12, no. 3: 372. https://doi.org/10.3390/cells12030372
APA StyleDi Cesare, E., Moroni, S., Bartoli, J., Damizia, M., Giubettini, M., Koerner, C., Krenn, V., Musacchio, A., & Lavia, P. (2023). Aurora B SUMOylation Is Restricted to Centromeres in Early Mitosis and Requires RANBP2. Cells, 12(3), 372. https://doi.org/10.3390/cells12030372