


Lipoprotein(a)—60 Years Later—What Do We Know?


Abstract
:1. Introduction
2. Lipoprotein(a): Molecular Structure, Genetics, Production, and Catabolism
3. Pathogenicity of Lp(a)
4. Lipoprotein(a) and Cardiovascular Disease
5. Ethnicity and Non-Genetic Factors Influencing Lp(a) Concentrations
6. Lp(a) in Children and Youth
7. Measurement of Lipoprotein(a), Cut-Off Values and Risk Stratification
- 32–90 nmol/L—little chance of cardiovascular disease;
- 90–200 nmol/L—modest chance;
- 200–400 nmol/L—large chance;
- >400 nmol/L—very high chance.
8. Treatment of Increased Lp(a)
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Reyes-Soffer, G.; Ginsberg, H.N.; Berglund, L.; Duell, P.B.; Heffron, S.P.; Kamstrup, P.R.; Lloyd-Jones, D.M.; Marcovina, S.M.; Yeang, C.; Koschinsky, M.L. American Heart Association Council on Arteriosclerosis, Thrombosis and Vascular Biology; Council on Cardiovascular Radiology and Intervention; and Council on Peripheral Vascular Disease; Lipoprotein(a): A Genetically Determined, Causal, and Prevalent Risk Factor for Atherosclerotic Cardiovascular Disease: A Scientific Statement From the American Heart Association. Arterioscler. Thromb. Vasc. Biol. 2022, 42, e48–e60. [Google Scholar]
- Paré, G.; Çaku, A.; McQueen, M.; Anand, S.S.; Enas, E.; Clarke, R.; Boffa, M.B.; Koschinsky, M.; Wang, X.; Yusuf, S.; et al. Lipoprotein(a) Levels and the Risk of Myocardial Infarction Among 7 Ethnic Groups. Circulation 2019, 139, 1472–1482. [Google Scholar] [CrossRef]
- Langsted, A.; Kamstrup, P.R.; Nordestgaard, B.G. High lipoprotein(a) and high risk of mortality. Eur. Heart J. 2019, 40, 2760–2770. [Google Scholar] [CrossRef] [PubMed]
- Burgess, S.; Ference, B.A.; Staley, J.R.; Freitag, D.F.; Mason, A.M.; Nielsen, S.F.; Willeit, P.; Young, R.; Surendran, P.; Karthikeyan, S.; et al. Association of LPA variants with risk of coronary disease and the implicationsforlipoprotein(a)-lowering therapies: A Mendelian randomization analysis. JAMA Cardiol. 2018, 3, 619–627. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K.; Rodriguez, F. Lipoprotein(a) and Cardiovascular Disease Prevention across Diverse Populations. Cardiol. Ther. 2020, 9, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Noureen, A.; Kronenberg, F.; Utermann, G. Structure, function, and genetics of lipoprotein(a). J. Lipid Res. 2016, 57, 1339–1359. [Google Scholar] [CrossRef] [PubMed]
- Handhle, A.; Viljoen, A.; Wierzbicki, A.S. Elevated Lipoprotein(a): Background, Current Insights and Future Potential Therapies. Vasc. Health Risk Manag. 2021, 17, 527–542. [Google Scholar] [CrossRef]
- Kronenberg, F. Prediction of cardiovascular risk by Lp(a) concentrations or genetic variants within the LPA gene region. Clin. Res. Cardiol. Suppl. 2019, 14, 5–12. [Google Scholar] [CrossRef]
- Jawi, M.M.; Frohlich, J.; Chan, S.Y. Lipoprotein(a) the Insurgent: A New Insight into the Structure, Function, Metabolism, Pathogenicity, and Medications Affecting Lipoprotein(a) Molecule. J. Lipids 2020, 2020, 3491764. [Google Scholar] [CrossRef]
- Iannuzzo, G.; Tripaldella, M.; Mallardo, V.; Morgillo, M.; Vitelli, N.; Iannuzzi, A.; Aliberti, E.; Giallauria, F.; Tramontano, A.; Carluccio, R.; et al. Lipoprotein(a) Where Do We Stand? From the Physiopathology to Innovative Therapy. Biomedicines 2021, 9, 838. [Google Scholar] [CrossRef]
- Sally, P.; McCormick, A.; Schneider, W. Lipoprotein(a) catabolism: A case of multiple receptors. Pathology 2019, 51, 155–164. [Google Scholar]
- Berman, A.N.; Blankstein, R. Current and future role of lipoprotein(a) in preventive cardiology. Curr. Opin. Cardiol. 2019, 34, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Tada, H.; Takamura, M.; Kawashiri, M.-A. Lipoprotein(a) as an Old and New Causal Risk Factor of Atherosclerotic Cardiovascular Disease. J. Atheroscler. Thromb. 2019, 26, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; Bernard, S.; Baass, A. SLC22A3 is associated with lipoprotein (a) concentration and cardiovascular disease in familial hypercholesterolemia. Clin. Biochem. 2019, 66, 44–48. [Google Scholar] [CrossRef]
- Mach, F.; Baigent, C.; Catapano, A.L.; Koskinas, K.C.; Casula, M.; Badimon, L.; Chapman, M.J.; De Backer, G.G.; Delgado, V.; Ference, B.A.; et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: Lipid modification to reduce cardiovascular risk: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur. Heart J. 2020, 41, 111–188. [Google Scholar] [CrossRef]
- Cegla, J.; Neely, R.D.G.; France, M.; Ferns, G.; Byrne, C.D.; Halcox, J.; Datta, D.; Capps, N.; Shoulders, C.; Qureshi, N.; et al. HEART UK consensus statement on Lipoprotein(a): A call to action. Atherosclerosis 2019, 291, 62–70. [Google Scholar] [CrossRef]
- Koschinsky, M.L.; Kronenberg, F. The long journey of lipoprotein(a) from cardiovascular curiosity to therapeutic target. Atherosclerosis 2022, 349, 1–6. [Google Scholar] [CrossRef]
- Shah, N.P.; Pajidipati, N.J.; McGarrah, R.W.; Navar, A.M.; Vemulapalli, S.; Blazing, M.A.; Shah, S.H.; Hernandez, A.F.; Patel, M.R. Lipoprotein(a): An Update on a Marker of Residual Risk and Associated Clinical Manifestations. Am. J. Cardiol. 2020, 126, 94–102. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Petersen, K.S.; Kris-Etherton, P.M.; Berglund, L. Diet and Lp(a): Does dietary change modify residual cardiovascular risk conferred by Lp(a)? Nutrients 2020, 12, 2024. [Google Scholar] [CrossRef]
- Bhatia, H.S.; Wilkinson, M.J. Lipoprotein(a): Evidence for role as a Causal Risk Factor in cardiovascular Disease and Emerging Therapies. J. Clin. Med. 2022, 11, 6040. [Google Scholar] [CrossRef]
- Liu, H.; Fu, D.; Luo, Y.; Peng, D. Independent association of Lp(a) with platelet reactivity in subjects without statins or antiplatelet agents. Sci. Rep. 2022, 12, 16609. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Wang, M.; Pirruccello, J.P.; Ellinor, P.T.; Ng, K.; Kathiresan, S.; Khera, A.V. Lp(a) (Lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: New insights from a large National Biobank. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 465–474. [Google Scholar] [CrossRef] [PubMed]
- Maloberti, A.; Fabbri, S.; Colombo, V.; Gualini, E.; Monticelli, M.; Daus, F.; Busti, A.; Galasso, M.; De Censi, L.; Algeri, M.; et al. Lipoprotein(a): Cardiovascular Disease, Aortic Stenosis and New Therapeutic Option. Int. J. Mol. Sci. 2022, 24, 170. [Google Scholar] [CrossRef]
- Langsted, A.; Nordestgaard, B.G.; Kamstrup, P.R. Elevated Lipoprotein(a) and Risk of Ischemic Stroke. J. Am. Coll. Cardiol. 2019, 74, 54–66. [Google Scholar] [CrossRef] [PubMed]
- Larsson, S.C.; Gill, D.; Mason, A.M.; Jiang, T.; Bäck, M.; Butterworth, A.S.; Burgess, S. Lipoprotein(a) in Alzheimer, Atherosclerotic, Cerebrovascular, Thrombotic, and Valvular Disease. Circulation 2020, 141, 1826–1828. [Google Scholar] [CrossRef]
- Pavanello, C.; Pirazzi, C.; Bjorkman, K.; Sandstedt, J.; Tarlarini, C.; Mosca, L.; Romeo, S.; Calabresi, L.; Mancina, R.M. Individuals with familial hypercholesterolemia and cardiovascular events have higher circulating Lp(a) levels. J. Clin. Lipidol. 2019, 13, 778–787. [Google Scholar] [CrossRef]
- Swerdlow, D.I.; Rider, D.A.; Yavari, A.; Lindholm, M.W.; Campion, G.V.; Nissen, S.E. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: The emerging potential of RNA interference therapeutics. Cardiovasc. Res. 2022, 118, 1218–1231. [Google Scholar] [CrossRef]
- Vavuranakis, M.A.; Jones, S.R.; Cardoso, R.; Gerstenblith, G.; Leucker, T.M. The role of Lipoprotein(a) in cardiovascular disease: Current concepts and future perspectives. Hell. J. Cardiol. 2020, 61, 398–403. [Google Scholar] [CrossRef]
- Mohammadi-Shemirani, P.; Chong, M.; Narula, S.; Perrot, N.; Conen, D.; Roberts, J.; Thériault, S.; Bosséc, Y.; Lanktree, M.; Pigeyre, M.; et al. Elevated Lipoprotein(a) and Risk of Atrial Fibrillation: An Observational and Mendelian Randomization Study. J. Am. Coll. Cardiol. 2022, 79, 1579–1590. [Google Scholar] [CrossRef]
- Garg, P.K.; Guan, W.; Karger, A.B.; Steffen, B.T.; O’Neal, W.; Heckbert, S.R.; Michos, E.D.; Tsai, M.Y. Lp(a) (Lipoprotein [a]) and Risk for Incident Atrial Fibrillation: Multi-Ethnic Study of Atherosclerosis. Circ. Arrhythm. Electrophysiol. 2020, 13, e008401. [Google Scholar] [CrossRef]
- Xia, J.; Guo, C.; Liu, K.; Xie, Y.; Cao, H.; Peng, W.; Sun, Y.; Liu, X.; Li, B.; Zhang, L. Association of Lipoprotein (a) variants with risk of cardiovascular disease: A Mendelian randomization study. Lipids Health Dis. 2021, 20, 57. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Yang, X.; Qiu, Q.; Gao, F.; Chen, W.; Hu, L.; Xu, Y.; Yi, Y.; Hu, H.; Jiang, L. Low lipoprotein(a) concentration is associated with atrial fibrillation: A large retrospective cohort study. Lipids Health Dis. 2022, 21, 119. [Google Scholar] [CrossRef] [PubMed]
- Telyuk, P.; Austin, D.; Luvai, A.; Zaman, A. Lipoprotein(a): Insights for the Practicing Clinician. J. Clin. Med. 2022, 11, 3673. [Google Scholar] [CrossRef]
- Capoulade, R.; Yeang, C.; Chan, K.L.; Pibarot, P.; Tsimikas, S. Association of Mild to Moderate Aortic Valve Stenosis Progression with Higher Lipoprotein(a) and Oxidized Phospholipid Levels. JAMA Cardiol. 2018, 3, 1212. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, Y.; van der Toorn, J.E.; Singh, S.S.; Zheng, K.H.; Kavousi, M.; Sijbrands, E.J.G.; Stroes, E.S.G.; Vernooij, M.W.; de Rijke, Y.B.; Boekholdt, S.M.; et al. Lipoprotein(a) is associated with the onset but not the progression of aortic valve calcification. Eur. Heart J. 2022, 43, 3960–3967. [Google Scholar] [CrossRef]
- Verwer, M.C.; Waissi, F.; Mekke, J.M.; Dekker, M.; Stroes, E.S.; de Borst, G.J.; Kroon, J.; Hazenberg, C.E.; de Kleijn, D.P. High lipoprotein(a) is associated with major adverse limb events after femoral artery endarterectomy. Atherosclerosis 2022, 349, 196–203. [Google Scholar] [CrossRef]
- Sakata, K.; Kumakura, H.; Funada, R.; Matsuo, Y.; Nakashima, K.; Iwasaki, T.; Ichikawa, S. Lipoprotein(a) is a Promising Residual Risk Factor for Long-Term Clinical Prognosis in Peripheral Arterial Disease. Ann. Vasc. Dis. 2022, 15, 186–192. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Lombardi, R.; Paolini, E.; Macchi, C.; Corsini, A.; Sirtori, C.R.; Fracanzani, A.L.; Ruscica, M.; Dongiovanni, P. Low Lipoprotein(a) Levels Predict Hepatic Fibrosis in Patients with Nonalcoholic Fatty Liver Disease. Hepatol. Commun. 2022, 6, 535–549. [Google Scholar] [CrossRef]
- Mehta, A.; Jain, V.; Saeed, A.; Saseen, J.J.; Gulati, M.; Ballantyne, C.M.; Virani, S.S. Lipoprotein(a) and ethnicities. Atherosclerosis 2022, 349, 42–52. [Google Scholar] [CrossRef]
- Ebbeling, C.B.; Knapp, A.; Johnson, A.; Wong, J.M.; Greco, K.F.; Ma, K.; Mora, S.; Ludwig, D.S. Effects of a low-carbohydrate diet on insulin-resistant dyslipoproteinemia—A randomized controlled feeding trial. Am. J. Clin. Nutr. 2022, 115, 310. [Google Scholar] [CrossRef]
- Hadi, A.; Askarpour, M.; Ziaei, R.; Venkatakrishnan, K.; Ghaedi, E.; Ghavami, A. Impact of flaxseed supplementation on plasma lipoprotein(a) concentrations: A systematic review and meta-analysis of randomized controlled trials. Phytother. Res. 2020, 34, 1599–1608. [Google Scholar] [CrossRef] [PubMed]
- Enkhmaa, B.; Berglund, L. Non-genetic influences on lipoprotein(a) concentrations. Atherosclerosis 2022, 349, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Kaftan, A.N.; Naser, F.H.; Enaya, M.A. Changes of certain metabolic and cardiovascular markers Fructosamine, H-FABP and lipoprotein (a) in patients with hypothyroidism. Med. Arch. 2021, 75, 11–15. [Google Scholar] [CrossRef]
- Glynn, N.; Halsall, D.J.; Boran, G.; Cook, P.; McDermott, J.H.; Smith, D.; Tormey, W.; Thompson, C.J.; O’Gorman, D.; McKenna, M.J.; et al. Growth hormone replacement may influence the biological action of thyroid hormone on liver and bone tissue. Growth Horm. IGF Res. 2021, 57–58, 101393. [Google Scholar] [CrossRef]
- Hopewell, J.C.; Haynes, R.; Baigent, C. The role of lipoprotein(a) in chronic kidney disease. J. Lipid Res. 2018, 59, 577–585. [Google Scholar] [CrossRef]
- Gitto, S.; Cicero, A.F.G.; Loggi, E.; Giovannini, M.; Conti, F.; Grandini, E.; Guarneri, V.; Scuteri, A.; Vitale, G.; Cursaro, C.; et al. Worsening of serum lipid profile after direct acting antiviral treatment. Ann. Hepatol. 2018, 17, 64–75. [Google Scholar] [CrossRef]
- Kohn, B.; Ashraf, A.P.; Wilson, D.P. Should Lipoprotein(a) be Measured in Youth? J. Pediatr. 2021, 228, 285–289. [Google Scholar] [CrossRef]
- McNeal, C.J. Lipoprotein (a): Its relevance to the pediatric population. J. Clin. Lipidol. 2015, 9, 57–66. [Google Scholar] [CrossRef]
- Strandkjær, N.; Hansen, M.K.; Nielsen, S.T.; Frikke-Schmidt, R.; Tybjærg-Hansen, A.; Nordestgaard, B.G.; Tabor, A.; Bundgaard, H.; Iversen, K.; Kamstrup, P.R. Lipoprotein(a) levels at birth and in early childhood—The COMPARE study. J. Clin. Endocrinol. Metab. 2022, 107, 324–335. [Google Scholar] [CrossRef]
- De Boer, L.M.; Hof, M.H.; Wiegman, A.; Stroobants, A.K.; Kastelein, J.J.P.; Hutten, B.A. Lipoprotein(a) levels from childhood to adulthood: Data in nearly 3000 children who visited a pediatric lipid clinic. Atherosclerosis 2022, 349, 227–232. [Google Scholar] [CrossRef]
- Enkhmaa, B.; Anuurad, E.; Berglund, L. Lipoprotein(a): Impact by ethnicity and environmental and medical conditions. J. Lipid Res. 2016, 57, 1111–1125. [Google Scholar] [CrossRef] [PubMed]
- Boffa, M.B.; Koschinsky, M.L. Lipoprotein(a): Truly a direct prothrombotic factor in cardiovascular disease? J. Lipid Res. 2016, 57, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Zawacki, A.W.; Dodge, A.; Woo, K.M.; Ralphe, J.C.; Peterson, A.L. In pediatric familial hypercholesterolemia, lipoprotein(a) is more predictive than LDL-C for early onset of cardiovascular disease in family members. J. Clin. Lipidol. 2018, 12, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- de Ferranti, S.D.; Steinberger, J.; Ameduri, R.; Baker, A.; Gooding, H.; Kelly, A.S.; Mietus-Snyder, M.; Mitsnefes, M.M.; Peterson, A.L.; St-Pierre, J.; et al. Cardiovascular risk reduction in high-risk pediatric patients: A scientific statement from the American Heart Association. Circulation 2019, 139, e603–e634. [Google Scholar] [CrossRef] [PubMed]
- Foster, C.; Rahman, A.K.M.F.; Ashraf, A.P. Lipoprotein(a) Concentrations Correlate with LDL-C in Children with Type 1 and 2 Diabetes. J. Endocr. Soc. 2021, 5, bvab138. [Google Scholar] [CrossRef]
- Koutsogianni, A.; Liamis, G.; Liberopoulos, E.; Adamidis, P.S.; Florentin, M. Effects of Lipid-Modifying and Other Drugs on Lipoprotein(a) Levels—Potent Clinical Implications. Pharmaceuticals 2023, 16, 750. [Google Scholar] [CrossRef]
- Genovesi, S.; Giussani, M.; Lieti, G.; Orlando, A.; Patti, I.; Parati, G. Evidence and Uncertainties on Lipoprotein(a) as a Marker of Cardiovascular Health Risk in Children and Adolescents. Biomedicine 2023, 11, 1661. [Google Scholar] [CrossRef]
- Kronenberg, F. Lipoprotein(a) measurement issues: Are we making a mountain out of a Molehill? Atherosclerosis 2022, 349, 123–135. [Google Scholar] [CrossRef]
- Marcovina, S.M.; Viney, N.J.; Hughes, S.G.; Xia, S.; Witztum, J.L.; Tsimikas, S. Temporal variability in lipoprotein(a) levels in patients enrolled in the placebo arms of IONIS-APO(a)Rx and IONIS-APO(a)-LRx antisense oligonucleotide clinical trials. J. Clin. Lipidol. 2018, 12, 122–129.e2. [Google Scholar] [CrossRef]
- Grundy, S.M.; Stone, N.J.; Bailey, A.L.; Beam, C.; Birtcher, K.K.; Blumenthal, R.S.; Yeboah, J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/ PCNA guideline on the management of blood cholesterol. J. Am. Coll. Cardiol. 2019, 73, 3168–3209. [Google Scholar] [CrossRef]
- Pearson, G.J.; Thanassoulis, G.; Anderson, T.J.; Barry, A.R.; Couture, P.; Dayan, N.; Wray, W. 2021 Canadian Cardiovascular Society Guidelines for the Management of Dyslipidemia for the Prevention of Cardiovascular Disease in Adults. Can. J. Cardiol. 2021, 37, 1129–1150. [Google Scholar] [CrossRef]
- Wilson, D.P.; Jacobson, T.A.; Jones, P.H.; Koschinsky, M.L.; McNeal, C.J.; Nordestgaard, B.G.; Orringer, C.E. Use of Lipoprotein(a) in clinical practice: A biomarker whose time has come. A scientific statement from the National Lipid Association. J. Clin. Lipidol. 2019, 13, 374–392. [Google Scholar] [CrossRef]
- Stone, N.J.; Smith, S.C.; Orringer, C.E.; Rigotti, N.A.; Navar, A.M.; Khan, S.S.; Jones, D.W.; Goldberg, R.; Mora, S.; Blaha, M.; et al. Managing Atherosclerotic Cardiovascular Risk in Young Adults: JACC State-of-the-Art Review. J. Am. Coll. Cardiol. 2022, 79, 819–836. [Google Scholar] [CrossRef] [PubMed]
- Kronenberg, F.; Mora, S.; Stroes, E.S.G.; Ference, B.A.; Arsenault, B.J.; Berglund, L.; Dweck, M.R.; Koschinsky, M.; Lambert, G.; Mach, F.; et al. Lipoprotein(a) in atherosclerotic cardiovascular disease and aortic stenosis: A European Atherosclerosis Society consensus statement. Eur. Heart J. 2022, 43, 3925–3946. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, R.; Hoogevee, R.M.; Langste, A.; Stiekema, L.; Verweij, S.; Hoving, G.K.; Wareham, N.J.; Khav, K.; Boekholdt, S.M.; Nordestgaard, B.; et al. Cardiovascular disease risk associated with elevated lipoprotein(a) attenuates at low low-density lipoprotein cholesterol levels in a primary prevention setting. Eur. Heart J. 2018, 39, 2589–2596. [Google Scholar] [CrossRef]
- Madsen, C.M.; Kamstrup, P.R.; Langsted, A.; Varbo, A.; Nordestgaard, B.G. Lipoprotein(a)-Lowering by 50 mg/dL (105 nmol/L) May Be Needed to Reduce Cardiovascular Disease 20% in Secondary Prevention: A Population-Based Study. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, E.; Parhofer, K.G. Apheresis for severe hypercholesterolaemia and elevated lipoprotein(a). Pathology 2019, 51, 227–232. [Google Scholar] [CrossRef]
- Sachais, B.S.; Shaz, B.H. Apheresis to mitigate atherosclerotic vascular disease. Am. J. Hypertens. 2018, 31, 945–949. [Google Scholar] [CrossRef]
- Pokrovsky, S.N.; Afanasieva, O.I.; Ezhov, M.V. Therapeutic apheresis for management of Lp(a) hyperlipoproteinemia. Curr. Atheroscler. Rep. 2020, 22, 68. [Google Scholar] [CrossRef]
- Yahya, R.; Berk, K.; Verhoeven, A.; Bos, S.; van der Zee, L.; Touw, J.; Erhart, G.; Kronenberg, F.; Timman, R.; Sijbrands, E.; et al. Statin treatment increases lipoprotein(a) levels in subjects with low molecular weight apolipoprotein(a) phenotype. Atherosclerosis 2019, 289, 201–205. [Google Scholar] [CrossRef]
- Tsimikas, S.; Gordts, P.L.S.M.; Nora, C.; Yeang, C.; Witztum, J.L. Statin therapy increases lipoprotein(a) levels. Eur. Heart J. 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Parish, S.; Hopewell, J.C.; Hill, M.R.; Marcovina, S.; Valdes-Marquez, E.; Haynes, R.; Offer, A.; Pedersen, T.R.; Baigent, C.; Collins, R.; et al. Impact of Apolipoprotein(a) Isoform Size on Lipoprotein(a) Lowering in the HPS2-THRIVE Study. Circ. Genom. Precis. Med. 2018, 11, e001696. [Google Scholar] [CrossRef]
- Chemello, K.; Chan, D.C.; Lambert, G.; Watts, G.F. Recent advances in demystifying the metabolism of lipoprotein(a). Atherosclerosis 2022, 349, 82–91. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Fazio, S.; Giugliano, R.P.; Stroes, E.S.; Kanevsky, E.; Gouni-Berthold, I.; Im, K.; Pineda, A.L.; Wasserman, S.M.; Ceška, R.; et al. Lipoprotein(a), PCSK9 Inhibition, and Cardiovascular Risk insights from the FOURIER trial. Circulation 2019, 139, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Bittner, V.A.; Szarek, M.; Aylward, P.E.; Bhatt, D.L.; Diaz, R.; Edelberg, J.M.; Fras, Z.; Goodman, S.G.; Halvorsen, S.; Hanotin, C.; et al. Effect of Alirocumab on Lipoprotein(a) and Cardiovascular Risk After Acute Coronary Syndrome. J. Am. Coll. Cardiol. 2020, 75, 133–144. [Google Scholar] [CrossRef]
- Raal, F.J.; Kallend, D.; Ray, K.K.; Turner, T.; Koenig, W.; Wright, R.S.; Wijngaard, P.L.; Curcio, D.; Jaros, M.J.; Leiter, L.A.; et al. Inclisiran for the Treatment of Heterozygous Familial Hypercholesterolemia. N. Engl. J. Med. 2020, 382, 1520–1530. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Kastelein, J.J. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Macchi, C.; Sirtori, C.; Corsini, A.; Santos, R.; Watts, G.; Ruscica, M. A new dawn for managing dyslipidemias: The era of rna-based therapies. Pharmacol. Res. 2019, 150, 104413. [Google Scholar] [CrossRef]
- Tsimikas, S.; Karwatowska-Prokopczuk, E.; Gouni-Berthold, I.; Tardif, J.C.; Baum, S.J.; Steinhagen Thiessen, E.; Shapiro, M.D.; Stroes, E.S.; Moriarty, P.M.; Nordestgaard, B.G.; et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N. Engl. J. Med. 2020, 382, 244–255. [Google Scholar] [CrossRef]
- Stiekema, L.C.A.; Prange, K.H.M.; Hoogeveen, R.M.; Verweij, S.L.; Kroon, J.; Schnitzler, J.G.; Dzobo, K.E.; Cupido, A.J.; Tsimikas, S.; Stroes, E.S.G.; et al. Potent lipoprotein(a) lowering following apolipoprotein(a) antisense treatment reduces the pro-inflammatory activation of circulating monocytes in patients with elevated lipoprotein(a). Eur. Heart J. 2020, 41, 2262–2271. [Google Scholar] [CrossRef]
- Koren, M.J.; Moriarty, P.M.; Baum, S.J.; Neutel, J.; Hernandez-Illas, M.; Weintraub, H.S.; Florio, M.; Kassahun, H.; Melquist, S.; Varrieur, T.; et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat. Med. 2022, 28, 96–103. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; López, J.A.G.; Knusel, B.; Gencer, B.; Wang, H.; Wu, Y.; Kassahun, H.; Sabatine, M.S. Study design and rationale for the Olpasiran trials of Cardiovascular Events And lipoprotein(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am. Heart J. 2022, 251, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Nissen, S.E.; Wolski, K.; Balog, C.; Swerdlow, D.I.; Scrimgeour, A.C.; Rambaran, C.; Wilson, R.J.; Boyce, M.; Ray, K.K.; Cho, L.; et al. Single Ascending Dose Study of a Short Interfering RNA Targeting Lipoprotein(a) Production in Individuals with Elevated Plasma Lipoprotein(a) Levels. JAMA 2022, 327, 1679. [Google Scholar] [CrossRef] [PubMed]
- Milosavljevic, M.N.; Stefanovic, S.M.; Pejcic, A.V. Potential novel RNA -targeting agents for effective Lipoprotein (a) lowering: A systematic Assessment of the evidence from completed and ongoing developmental clinical trials. J. Cardiovasc. Pharmacol. 2023, 82, 1–12. [Google Scholar] [CrossRef]
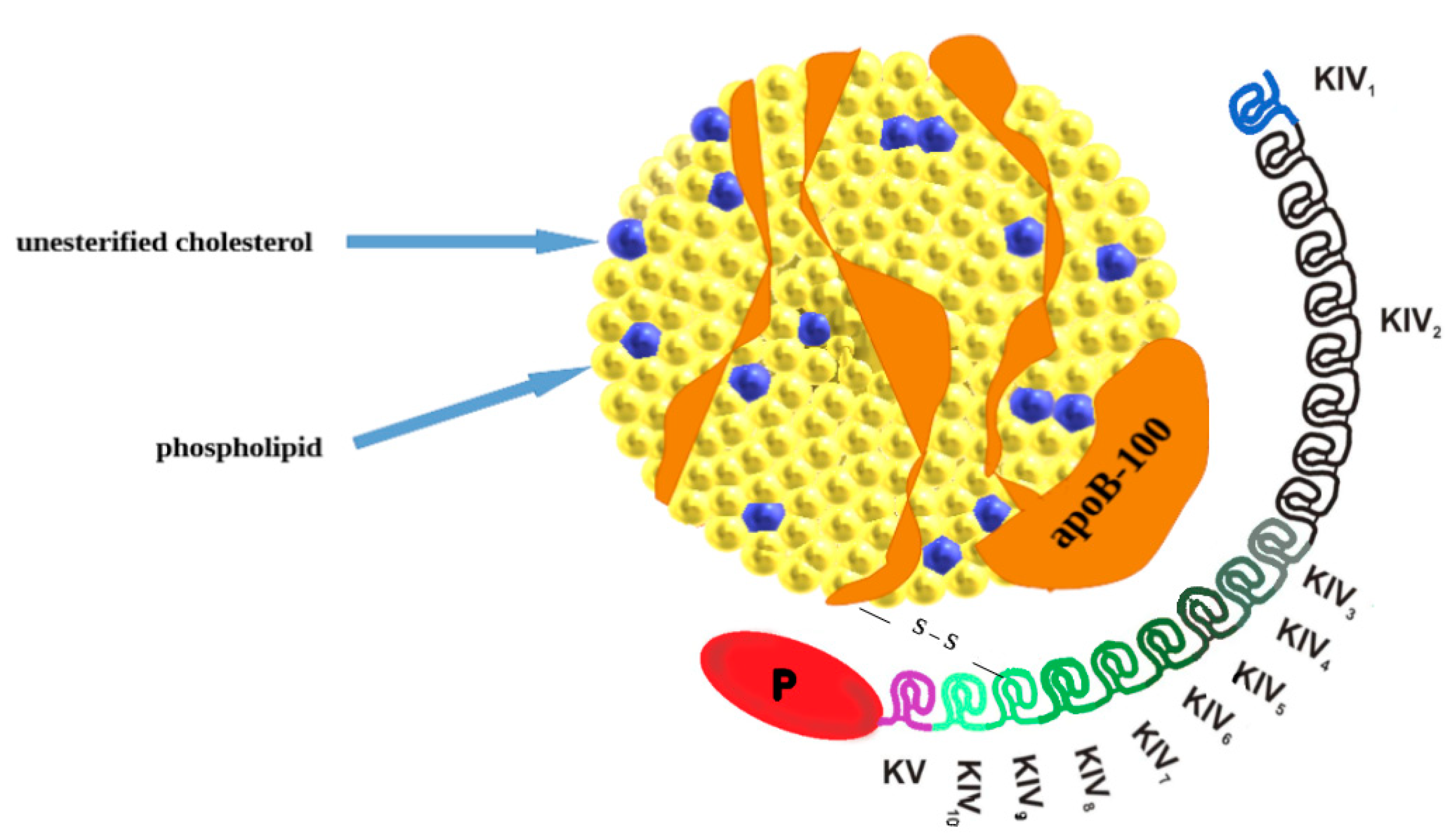

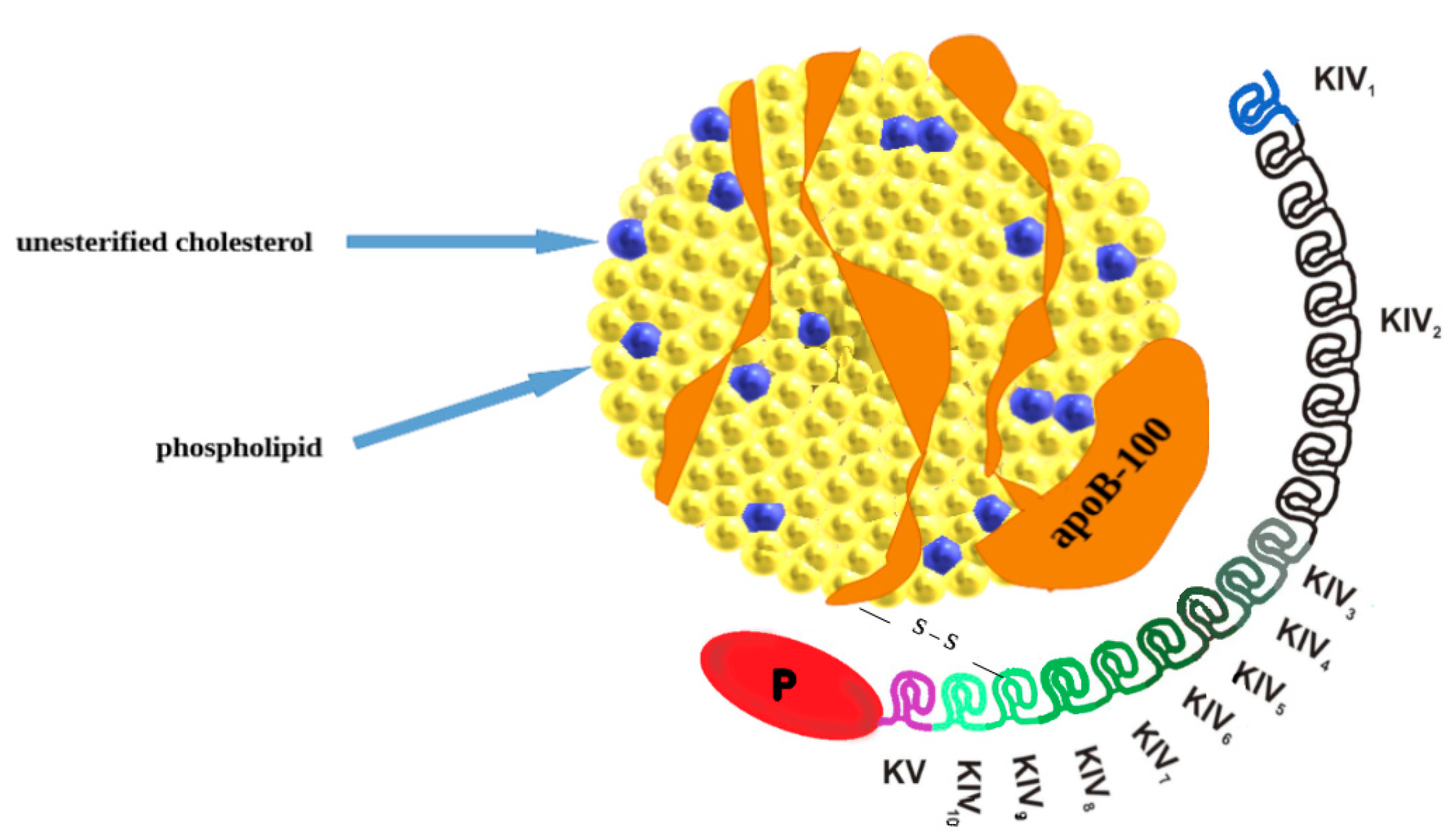{kind=link}
| Proatherogenic and Proinflammatory Properties of Lp(a) | Prothrombotic Properties of Lp(a) |
|---|---|
| ↑ Oxidized phospholipids ↑ Foam cell formation ↑ Endothelial dysfunction ↑ Smooth muscle cell proliferation ↑ Chemoattraction of monocytes ↑ Inflammation of the arterial wall (IL-8, monocyte chemotactic protein, TNF-α) | ↓ Plasminogen activation ↓ Fibrinolysis ↓ Tissue factor pathway inhibitor ↓ Clot permeability ↑ Platelet response ↑ Plasminogen activator inhibitor-1 (PAI-1) |
| Study | Group of Patients | Outcome | Implications |
|---|---|---|---|
| National Biobank [22] Lipoprotein(a) levels and the MI risk among different ethnicities, INTERHEART study [2] Lipoprotein(a) values vs. CV risk, meta-analysis [29] The Copenhagen City Heart study [24] The Copenhagen General Population study [24] | 460,506 12,943 126,634 10,813 49,699 | Prevalence of ASCVD Prevalence of MI Prevalence of CVA and CHD CV results based on register AVS prevalence | Elevated risk of ASCVD incidents Increased risk of MI Increased association of Lp(a) with CVA and CHD Elevated incidence of MI and AVS Three fold elevated AVS risk when Lp(a) > 90 mg/dL |
| Recommendation | Currently Applying Screening Guidelines |
|---|---|
| 2018 ACC/AHA (American College of Cardiology/American Heart Association) Cholesterol Guidelines [60] 2019 ESC/EAS (European Society of Cardiology /European Atherosclerosis Society) Dyslipidemia Guidelines [15] HEART UK Consensus Statement [16] 2021 Canadian Cardiovascular Society Guidelines [61] |
|
| Drug/Intervention | Mechanism of Action | Lp(a) Level Decrease | CV Risk Reduction |
|---|---|---|---|
| Non-specific therapies | |||
| Lipoprotein apheresis [67,68,69] | 2–3-h procedure; plasma exchange; removes LDL-c, VLDL, apoB containing particles (Lp(a)) | >50% | Yes |
| Statins [71,72] | Inhibition of HMG-CoA reductase enzyme | Conflicting results | Yes |
| Niacin [56,72,73] | Inhibits triglycerides synthesis | 20–40% | No |
| Evolocumab [23,74] | Anti-PCSK9 antibodies | 29.50% | Yes |
| Alirocumab [63,75] | Anti-PCSK9 antibodies | 23.50% | Yes |
| Inclisiran [76,77] | siRNA inhibiting PCSK9 | ≈20% | No |
| Specific (experimental) therapies | |||
| Pelacarsen [79,80] | ASO inhibiting apo(a) | ≈80% | Experimental phase (phase 3 ongoing) |
| Olpasiran [81,82] | siRNA inhibiting apo(a) | 80–98% | Experimental phase (phase 3 ongoing) |
| SLN360 [83] | siRNA inhibiting apo(a) | 98.00% | Experimental phase (phase 2 ongoing) |
| LY3819469 [84] | siRNA inhibiting apo(a) | unknown | Experimental phase (phase 2 registered) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pasławska, A.; Tomasik, P.J. Lipoprotein(a)—60 Years Later—What Do We Know? Cells 2023, 12, 2472. https://doi.org/10.3390/cells12202472
Pasławska A, Tomasik PJ. Lipoprotein(a)—60 Years Later—What Do We Know? Cells. 2023; 12(20):2472. https://doi.org/10.3390/cells12202472
Chicago/Turabian StylePasławska, Anna, and Przemysław J. Tomasik. 2023. "Lipoprotein(a)—60 Years Later—What Do We Know?" Cells 12, no. 20: 2472. https://doi.org/10.3390/cells12202472
APA StylePasławska, A., & Tomasik, P. J. (2023). Lipoprotein(a)—60 Years Later—What Do We Know? Cells, 12(20), 2472. https://doi.org/10.3390/cells12202472