
Innate Immune Pathways in Atherosclerosis—From Signaling to Long-Term Epigenetic Reprogramming



Abstract
:
1. Introduction
2. Cellular Innate Immune Responses in Atherosclerosis
3. Heterogeneity and Function of Plaque Macrophages—Then and Now
3.1. The Simplified View
3.2. Macrophage Heterogeneity as Revealed by Emerging Techniques
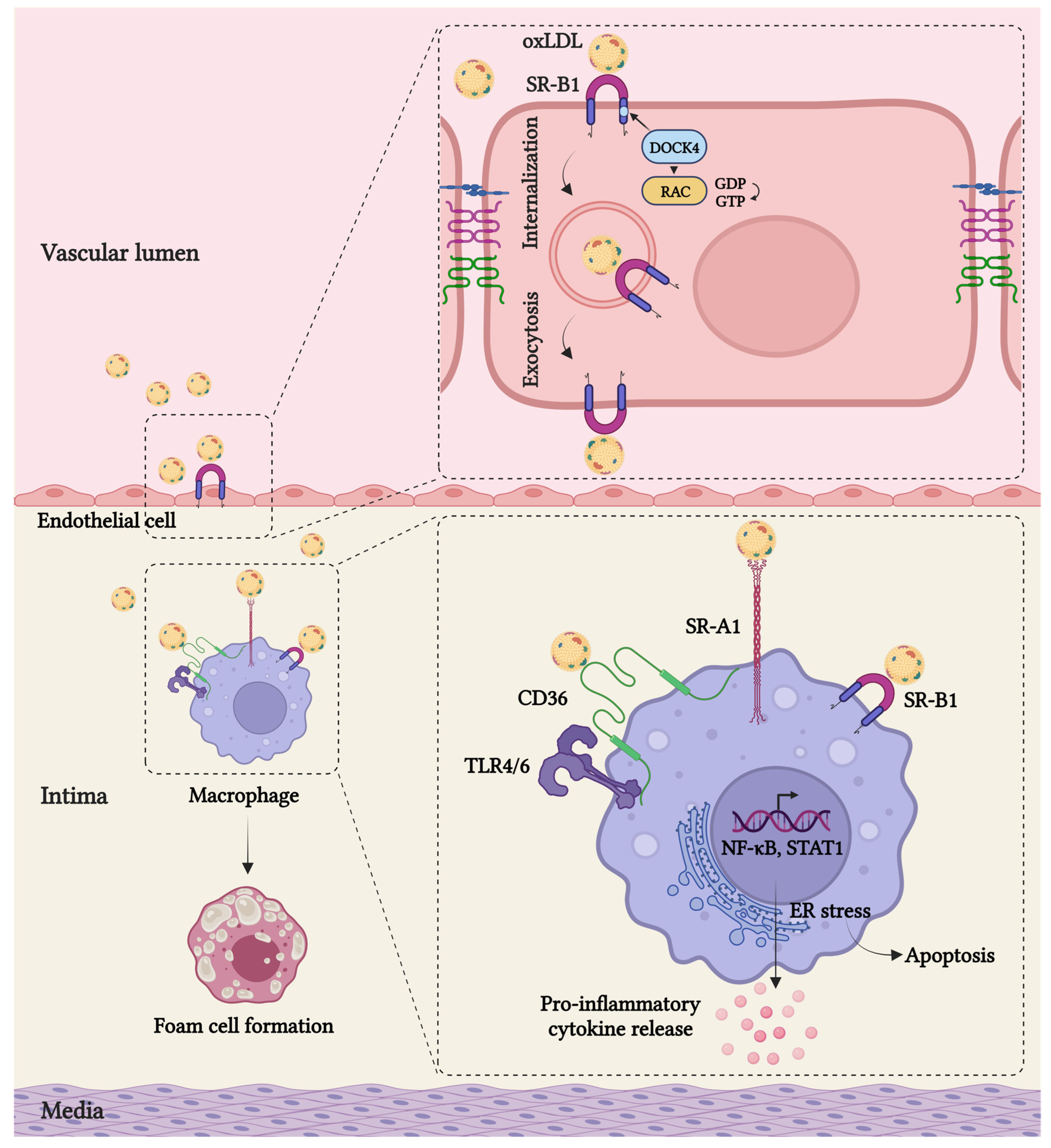
4. Scavenger Receptors in Arterial Inflammation
Classes, Structure, and Complexity

{kind=link}
{kind=link}
{kind=link}
| Class | Receptor Name(s) | Ligands | Expression Profile |
|---|---|---|---|
| A | SR-A1/SR-A | AcLDL, oxLDL, maleylated or glycated BSA, β-amyloid, heat shock proteins and hepatitis C virus [60]; poly G and poly I, polysaccharides, including LTA and LPS, of Gram-positive and Gram-negative bacteria. | Macrophages, monocytes, mast and dendritic cells, endothelial, smooth muscle cells [60]. |
| SR-A6/MARCO | AcLDL, oxLDL [60], unopsonized environmental particles (TIO2, FE2O3, silica, and nanoparticles) [61]. | Alveolar macrophages, macrophages of lymph nodes sinuses, thymus, spleen, and intestine [62], Kupffer cells in the liver [63]. | |
| B | SR-B1 | HDL [64], LDL, oxLDL [17], apoptotic cells, hepatitis C virus [65]. | Monocytes/macrophages, dendritic cells, endothelial cells, hepatocytes, and adipocytes [17,65,66]. |
| SR-B2/CD-36 | AcLDL, oxLDL [67], HDL, LDL, VLDL, apoptotic cells [68], β-amyloid [69]. | Macrophages, platelets, adipocytes, epithelial and endothelial cells [68]. | |
| C | SR-C1 | Gram-positive and Gram-negative bacteria [70]. | Drosophila melanogaster macrophages [70]. |
| D | SR-D1/CD68 | OxLDL [66], apoptotic cells. | Monocytes/macrophages [66]. |
| E | SR-E1/LOX-1 | OxLDL [71], C-reactive protein [72], AGE, HSP60 and HSP70 [73], apoptotic cells, activated platelets, bacteria. | Endothelial [71] and smooth muscle cells [74], macrophages, and platelets. |
| F | SR-F1/SCARF1 | AcLDL [75], apoptotic cells via C1q [58], calreticulin [76], HSP70, HSP90, HSP110 [77]. | Endothelial, dendritic cells, and macrophages [58]. |
| G | SR-G1/CXCL16/ SR-PSOX | Phosphatidylserine, oxLDL [56], [78]. | Endothelial cells, dendritic cells, macrophages [79], smooth muscle cells. |
| H | SR-H1/STAB1 | AcLDL, oxLDL [80], AGEs [81], extracellular protein SPARC, TGFBi, Periostin, Reelin [82]. | Monocytes/macrophages, endothelial cells [80]. |
| SR-H2/STAB2 | AcLDL, oxLDL [80], AGEs [81], TGFBi, Periostin, Reelin [82], hyaluronan [83]. | ||
| I | SR-I1/CD163 | Haptoglobin–hemoglobin complexes [84]. | Monocyte/macrophages [84,85]. |
| J | SR-J1/RAGE | HMGB1, β-amyloid, phosphatidylserine [86]. | Endothelial cells, hepatocytes, smooth muscle cells, macrophages [86]. |
| K | SR-K1/CD44 | HA [87]. | Monocytes/macrophages [87]. |
| L | SR-L1/LRP1 | VLDL [88], defensin, HSP70, HSP90. | Dendritic cells, monocytes/macrophages. |
5. Scavenger Receptor-Mediated Lipid Uptake in Endothelial Cells and Macrophages
6. Downstream Signaling Events Mediated by Scavenger Receptors
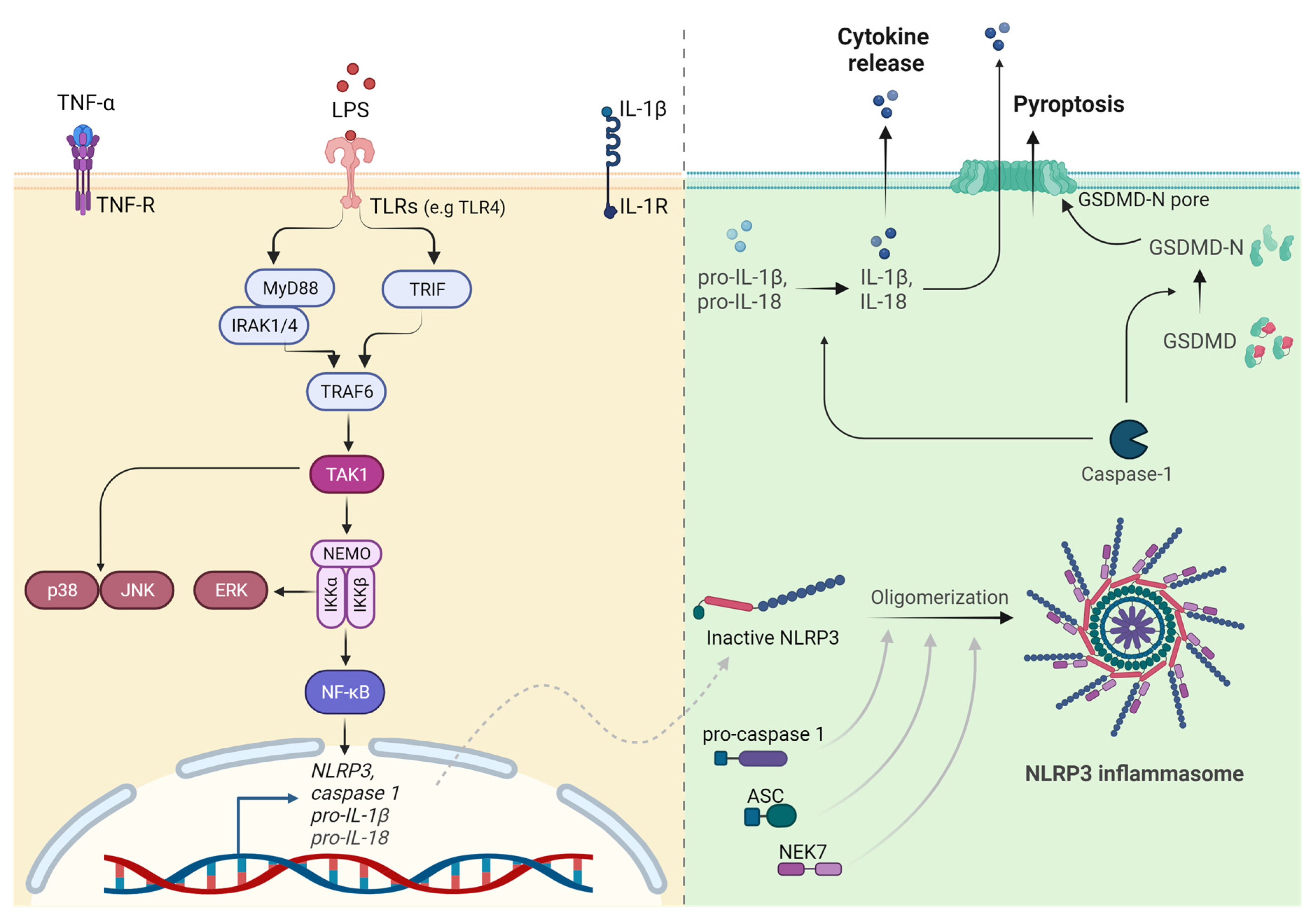
7. TLR and NLR Signaling in Arterial Inflammation
8. NLRP3 Inflammasome Activation in the Pathogenesis of Atherosclerosis
9. Crosstalk between Scavenger Receptors and TLRs in Chronic Vascular Inflammation
10. Reprogramming of Immune Cells in Atherosclerosis
11. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mensah, G.A.; Roth, G.A.; Fuster, V. The Global Burden of Cardiovascular Diseases and Risk Factors: 2020 and Beyond. J. Am. Coll. Cardiol. 2019, 74, 2529–2532. [Google Scholar] [CrossRef] [PubMed]
- Soehnlein, O.; Libby, P. Targeting inflammation in atherosclerosis—From experimental insights to the clinic. Nat. Rev. Drug Discov. 2021, 20, 589–610. [Google Scholar] [CrossRef] [PubMed]
- Roy, P.; Orecchioni, M.; Ley, K. How the immune system shapes atherosclerosis: Roles of innate and adaptive immunity. Nat. Rev. Immunol. 2022, 22, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Weber, C.; Noels, H. Atherosclerosis: Current pathogenesis and therapeutic options. Nat. Med. 2011, 17, 1410–1422. [Google Scholar] [CrossRef]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef]
- Pryshchep, O.; Ma-Krupa, W.; Younge, B.R.; Goronzy, J.J.; Weyand, C.M. Vessel-specific Toll-like receptor profiles in human medium and large arteries. Circulation 2008, 118, 1276–1284. [Google Scholar] [CrossRef]
- Cole, J.E.; Georgiou, E.; Monaco, C. The expression and functions of toll-like receptors in atherosclerosis. Mediat. Inflamm. 2010, 2010, 393946. [Google Scholar] [CrossRef]
- Zhou, Y.; Little, P.J.; Downey, L.; Afroz, R.; Wu, Y.; Ta, H.T.; Xu, S.; Kamato, D. The Role of Toll-like Receptors in Atherothrombotic Cardiovascular Disease. ACS Pharmacol. Transl. Sci. 2020, 3, 457–471. [Google Scholar] [CrossRef]
- Ohashi, K.; Burkart, V.; Flohe, S.; Kolb, H. Cutting edge: Heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J. Immunol. 2000, 164, 558–561. [Google Scholar] [CrossRef]
- Yu, L.; Wang, L.; Chen, S. Endogenous toll-like receptor ligands and their biological significance. J. Cell Mol. Med. 2010, 14, 2592–2603. [Google Scholar] [CrossRef]
- Chen, S.; Shimada, K.; Crother, T.R.; Erbay, E.; Shah, P.K.; Arditi, M. Chlamydia and Lipids Engage a Common Signaling Pathway That Promotes Atherogenesis. J. Am. Coll. Cardiol. 2018, 71, 1553–1570. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Cui, Z.Y.; Huang, X.F.; Zhang, D.D.; Guo, R.J.; Han, M. Inflammation and atherosclerosis: Signaling pathways and therapeutic intervention. Signal Transduct. Target. Ther. 2022, 7, 131. [Google Scholar] [CrossRef] [PubMed]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circ. Res. 2018, 122, 1722–1740. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Bornfeldt, K.E. Inflammasomes and Atherosclerosis: A Mixed Picture. Circ. Res. 2023, 132, 1505–1520. [Google Scholar] [CrossRef]
- Park, I.; Goddard, M.E.; Cole, J.E.; Zanin, N.; Lyytikainen, L.P.; Lehtimaki, T.; Andreakos, E.; Feldmann, M.; Udalova, I.; Drozdov, I.; et al. C-type lectin receptor CLEC4A2 promotes tissue adaptation of macrophages and protects against atherosclerosis. Nat. Commun. 2022, 13, 215. [Google Scholar] [CrossRef]
- Huang, L.; Chambliss, K.L.; Gao, X.; Yuhanna, I.S.; Behling-Kelly, E.; Bergaya, S.; Ahmed, M.; Michaely, P.; Luby-Phelps, K.; Darehshouri, A.; et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature 2019, 569, 565–569. [Google Scholar] [CrossRef]
- Febbraio, M.; Podrez, E.A.; Smith, J.D.; Hajjar, D.P.; Hazen, S.L.; Hoff, H.F.; Sharma, K.; Silverstein, R.L. Targeted disruption of the class B scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Investig. 2000, 105, 1049–1056. [Google Scholar] [CrossRef]
- Geijtenbeek, T.B.; Gringhuis, S.I. Signalling through C-type lectin receptors: Shaping immune responses. Nat. Rev. Immunol. 2009, 9, 465–479. [Google Scholar] [CrossRef]
- Maskrey, B.H.; Megson, I.L.; Whitfield, P.D.; Rossi, A.G. Mechanisms of resolution of inflammation: A focus on cardiovascular disease. Arter. Thromb. Vasc. Biol. 2011, 31, 1001–1006. [Google Scholar] [CrossRef]
- Butcher, E.C. Leukocyte-endothelial cell recognition: Three (or more) steps to specificity and diversity. Cell 1991, 67, 1033–1036. [Google Scholar] [CrossRef]
- Serhan, C.N.; Brain, S.D.; Buckley, C.D.; Gilroy, D.W.; Haslett, C.; O’Neill, L.A.; Perretti, M.; Rossi, A.G.; Wallace, J.L. Resolution of inflammation: State of the art, definitions and terms. FASEB J. 2007, 21, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Back, M.; Yurdagul, A., Jr.; Tabas, I.; Oorni, K.; Kovanen, P.T. Inflammation and its resolution in atherosclerosis: Mediators and therapeutic opportunities. Nat. Rev. Cardiol. 2019, 16, 389–406. [Google Scholar] [CrossRef] [PubMed]
- Engelen, S.E.; Robinson, A.J.B.; Zurke, Y.X.; Monaco, C. Therapeutic strategies targeting inflammation and immunity in atherosclerosis: How to proceed? Nat. Rev. Cardiol. 2022, 19, 522–542. [Google Scholar] [CrossRef] [PubMed]
- Getz, G.S.; Reardon, C.A. Natural killer T cells in atherosclerosis. Nat. Rev. Cardiol. 2017, 14, 304–314. [Google Scholar] [CrossRef]
- Klingenberg, R.; Gerdes, N.; Badeau, R.M.; Gistera, A.; Strodthoff, D.; Ketelhuth, D.F.; Lundberg, A.M.; Rudling, M.; Nilsson, S.K.; Olivecrona, G.; et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J. Clin. Investig. 2013, 123, 1323–1334. [Google Scholar] [CrossRef]
- Williams, J.W.; Zaitsev, K.; Kim, K.W.; Ivanov, S.; Saunders, B.T.; Schrank, P.R.; Kim, K.; Elvington, A.; Kim, S.H.; Tucker, C.G.; et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat. Immunol. 2020, 21, 1194–1204. [Google Scholar] [CrossRef]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef]
- Stremmel, C.; Stark, K.; Schulz, C. Heterogeneity of Macrophages in Atherosclerosis. Thromb. Haemost. 2019, 119, 1237–1246. [Google Scholar] [CrossRef]
- Barrett, T.J. Macrophages in Atherosclerosis Regression. Arter. Thromb. Vasc. Biol. 2020, 40, 20–33. [Google Scholar] [CrossRef]
- Susser, L.I.; Rayner, K.J. Through the layers: How macrophages drive atherosclerosis across the vessel wall. J. Clin. Investig. 2022, 132. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.J.; Tabas, I. Macrophages in the pathogenesis of atherosclerosis. Cell 2011, 145, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Willemsen, L.; de Winther, M.P. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J. Pathol. 2020, 250, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Blagov, A.V.; Markin, A.M.; Bogatyreva, A.I.; Tolstik, T.V.; Sukhorukov, V.N.; Orekhov, A.N. The Role of Macrophages in the Pathogenesis of Atherosclerosis. Cells 2023, 12, 522. [Google Scholar] [CrossRef]
- Khallou-Laschet, J.; Varthaman, A.; Fornasa, G.; Compain, C.; Gaston, A.T.; Clement, M.; Dussiot, M.; Levillain, O.; Graff-Dubois, S.; Nicoletti, A.; et al. Macrophage plasticity in experimental atherosclerosis. PLoS ONE 2010, 5, e8852. [Google Scholar] [CrossRef]
- Bouhlel, M.A.; Derudas, B.; Rigamonti, E.; Dievart, R.; Brozek, J.; Haulon, S.; Zawadzki, C.; Jude, B.; Torpier, G.; Marx, N.; et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007, 6, 137–143. [Google Scholar] [CrossRef]
- Moore, K.J.; Sheedy, F.J.; Fisher, E.A. Macrophages in atherosclerosis: A dynamic balance. Nat. Rev. Immunol. 2013, 13, 709–721. [Google Scholar] [CrossRef]
- Kadl, A.; Meher, A.K.; Sharma, P.R.; Lee, M.Y.; Doran, A.C.; Johnstone, S.R.; Elliott, M.R.; Gruber, F.; Han, J.; Chen, W.; et al. Identification of a novel macrophage phenotype that develops in response to atherogenic phospholipids via Nrf2. Circ. Res. 2010, 107, 737–746. [Google Scholar] [CrossRef]
- Gleissner, C.A.; Shaked, I.; Erbel, C.; Bockler, D.; Katus, H.A.; Ley, K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ. Res. 2010, 106, 203–211. [Google Scholar] [CrossRef]
- Boyle, J.J.; Johns, M.; Lo, J.; Chiodini, A.; Ambrose, N.; Evans, P.C.; Mason, J.C.; Haskard, D.O. Heme induces heme oxygenase 1 via Nrf2: Role in the homeostatic macrophage response to intraplaque hemorrhage. Arter. Thromb. Vasc. Biol. 2011, 31, 2685–2691. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; van Rooijen, N.; MacDonald, A.S.; Allen, J.E. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed]
- Gundra, U.M.; Girgis, N.M.; Ruckerl, D.; Jenkins, S.; Ward, L.N.; Kurtz, Z.D.; Wiens, K.E.; Tang, M.S.; Basu-Roy, U.; Mansukhani, A.; et al. Alternatively activated macrophages derived from monocytes and tissue macrophages are phenotypically and functionally distinct. Blood 2014, 123, e110–e122. [Google Scholar] [CrossRef] [PubMed]
- Cochain, C.; Vafadarnejad, E.; Arampatzi, P.; Pelisek, J.; Winkels, H.; Ley, K.; Wolf, D.; Saliba, A.E.; Zernecke, A. Single-Cell RNA-Seq Reveals the Transcriptional Landscape and Heterogeneity of Aortic Macrophages in Murine Atherosclerosis. Circ. Res. 2018, 122, 1661–1674. [Google Scholar] [CrossRef] [PubMed]
- Zernecke, A.; Winkels, H.; Cochain, C.; Williams, J.W.; Wolf, D.; Soehnlein, O.; Robbins, C.S.; Monaco, C.; Park, I.; McNamara, C.A.; et al. Meta-Analysis of Leukocyte Diversity in Atherosclerotic Mouse Aortas. Circ. Res. 2020, 127, 402–426. [Google Scholar] [CrossRef] [PubMed]
- Winkels, H.; Ehinger, E.; Vassallo, M.; Buscher, K.; Dinh, H.Q.; Kobiyama, K.; Hamers, A.A.J.; Cochain, C.; Vafadarnejad, E.; Saliba, A.E.; et al. Atlas of the Immune Cell Repertoire in Mouse Atherosclerosis Defined by Single-Cell RNA-Sequencing and Mass Cytometry. Circ. Res. 2018, 122, 1675–1688. [Google Scholar] [CrossRef]
- Lin, J.D.; Nishi, H.; Poles, J.; Niu, X.; McCauley, C.; Rahman, K.; Brown, E.J.; Yeung, S.T.; Vozhilla, N.; Weinstock, A.; et al. Single-cell analysis of fate-mapped macrophages reveals heterogeneity, including stem-like properties, during atherosclerosis progression and regression. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Kim, K.; Shim, D.; Lee, J.S.; Zaitsev, K.; Williams, J.W.; Kim, K.W.; Jang, M.Y.; Seok Jang, H.; Yun, T.J.; Lee, S.H.; et al. Transcriptome Analysis Reveals Nonfoamy Rather Than Foamy Plaque Macrophages Are Proinflammatory in Atherosclerotic Murine Models. Circ. Res. 2018, 123, 1127–1142. [Google Scholar] [CrossRef]
- McArdle, S.; Buscher, K.; Ghosheh, Y.; Pramod, A.B.; Miller, J.; Winkels, H.; Wolf, D.; Ley, K. Migratory and Dancing Macrophage Subsets in Atherosclerotic Lesions. Circ. Res. 2019, 125, 1038–1051. [Google Scholar] [CrossRef]
- Ensan, S.; Li, A.; Besla, R.; Degousee, N.; Cosme, J.; Roufaiel, M.; Shikatani, E.A.; El-Maklizi, M.; Williams, J.W.; Robins, L.; et al. Self-renewing resident arterial macrophages arise from embryonic CX3CR1(+) precursors and circulating monocytes immediately after birth. Nat. Immunol. 2016, 17, 159–168. [Google Scholar] [CrossRef]
- Lim, H.Y.; Lim, S.Y.; Tan, C.K.; Thiam, C.H.; Goh, C.C.; Carbajo, D.; Chew, S.H.S.; See, P.; Chakarov, S.; Wang, X.N.; et al. Hyaluronan Receptor LYVE-1-Expressing Macrophages Maintain Arterial Tone through Hyaluronan-Mediated Regulation of Smooth Muscle Cell Collagen. Immunity 2018, 49, 1191. [Google Scholar] [CrossRef]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Macrophage biology in development, homeostasis and disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.E. Cellular signaling in macrophage migration and chemotaxis. J. Leukoc. Biol. 2000, 68, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.M.; Febbraio, M.; Silverstein, R.L. CD36 modulates migration of mouse and human macrophages in response to oxidized LDL and may contribute to macrophage trapping in the arterial intima. J. Clin. Investig. 2009, 119, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y.; Weissman, I.L.; Leeper, N.J. The Role of Efferocytosis in Atherosclerosis. Circulation 2017, 135, 476–489. [Google Scholar] [CrossRef]
- PrabhuDas, M.R.; Baldwin, C.L.; Bollyky, P.L.; Bowdish, D.M.E.; Drickamer, K.; Febbraio, M.; Herz, J.; Kobzik, L.; Krieger, M.; Loike, J.; et al. A Consensus Definitive Classification of Scavenger Receptors and Their Roles in Health and Disease. J. Immunol. 2017, 198, 3775–3789. [Google Scholar] [CrossRef]
- Sheikine, Y.; Sirsjö, A. CXCL16/SR-PSOX—A friend or a foe in atherosclerosis? Atherosclerosis 2008, 197, 487–495. [Google Scholar] [CrossRef]
- Canton, J.; Neculai, D.; Grinstein, S. Scavenger receptors in homeostasis and immunity. Nat. Rev. Immunol. 2013, 13, 621–634. [Google Scholar] [CrossRef]
- Ramirez-Ortiz, Z.G.; Pendergraft, W.F.; Prasad, A.; Byrne, M.H.; Iram, T.; Blanchette, C.J.; Luster, A.D.; Hacohen, N.; Khoury, J.E.; Means, T.K. The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nat. Immunol. 2013, 14, 917–926. [Google Scholar] [CrossRef]
- Suzuki, H.; Kurihara, Y.; Takeya, M.; Kamada, N.; Kataoka, M.; Jishage, K.; Ueda, O.; Sakaguchi, H.; Higashi, T.; Suzuki, T.; et al. A role for macrophage scavenger receptors in atherosclerosis and susceptibility to infection. Nature 1997, 386, 292–296. [Google Scholar] [CrossRef]
- Plüddemann, A.; Neyen, C.; Gordon, S. Macrophage scavenger receptors and host-derived ligands. Methods 2007, 43, 207–217. [Google Scholar] [CrossRef]
- Hirano, S.; Fujitani, Y.; Furuyama, A.; Kanno, S. Macrophage receptor with collagenous structure (MARCO) is a dynamic adhesive molecule that enhances uptake of carbon nanotubes by CHO-K1 Cells. Toxicol. Appl. Pharmacol. 2012, 259, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Palecanda, A.; Paulauskis, J.; Al-Mutairi, E.; Imrich, A.; Qin, G.; Suzuki, H.; Kodama, T.; Tryggvason, K.; Koziel, H.; Kobzik, L. Role of the Scavenger Receptor MARCO in Alveolar Macrophage Binding of Unopsonized Environmental Particles. J. Exp. Med. 1999, 189, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- van der Laan, L.J.; Döpp, E.A.; Haworth, R.; Pikkarainen, T.; Kangas, M.; Elomaa, O.; Dijkstra, C.D.; Gordon, S.; Tryggvason, K.; Kraal, G. Regulation and functional involvement of macrophage scavenger receptor MARCO in clearance of bacteria in vivo. J. Immunol. 1999, 162, 939–947. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of Scavenger Receptor SR-BI as a High Density Lipoprotein Receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef]
- Barth, H.; Schnober, E.K.; Neumann-Haefelin, C.; Thumann, C.; Zeisel, M.B.; Diepolder, H.M.; Hu, Z.; Liang, T.J.; Blum, H.E.; Thimme, R.; et al. Scavenger Receptor Class B Is Required for Hepatitis C Virus Uptake and Cross-Presentation by Human Dendritic Cells. J. Virol. 2008, 82, 3466–3479. [Google Scholar] [CrossRef]
- Ramprasad, M.P.; Terpstra, V.; Kondratenko, N.; Quehenberger, O.; Steinberg, D. Cell surface expression of mouse macrosialin and human CD68 and their role as macrophage receptors for oxidized low density lipoprotein. Proc. Natl. Acad. Sci. USA 1996, 93, 14833–14838. [Google Scholar] [CrossRef]
- Navazo, M.D.P.; Daviet, L.; Ninio, E.; McGregor, J.L. Identification on Human CD36 of a Domain (155-183) Implicated in Binding Oxidized Low-Density Lipoproteins (Ox-LDL). Arter. Thromb. Vasc. Biol. 1996, 16, 1033–1039. [Google Scholar] [CrossRef]
- Greenberg, M.E.; Sun, M.; Zhang, R.; Febbraio, M.; Silverstein, R.; Hazen, S.L. Oxidized phosphatidylserine–CD36 interactions play an essential role in macrophage-dependent phagocytosis of apoptotic cells. J. Exp. Med. 2006, 203, 2613–2625. [Google Scholar] [CrossRef]
- El Khoury, J.B.; Moore, K.J.; Means, T.K.; Leung, J.; Terada, K.; Toft, M.; Freeman, M.W.; Luster, A.D. CD36 Mediates the Innate Host Response to β-Amyloid. J. Exp. Med. 2003, 197, 1657–1666. [Google Scholar] [CrossRef]
- Rämet, M.; Pearson, A.; Manfruelli, P.; Li, X.; Koziel, H.; Göbel, V.; Chung, E.; Krieger, M.; Ezekowitz, R.A. Drosophila scavenger receptor CI is a pattern recognition receptor for bacteria. Immunity 2001, 15, 1027–1038. [Google Scholar] [CrossRef]
- Sawamura, T.; Kume, N.; Aoyama, T.; Moriwaki, H.; Hoshikawa, H.; Aiba, Y.; Tanaka, T.; Miwa, S.; Katsura, Y.; Kita, T.; et al. An endothelial receptor for oxidized low-density lipoprotein. Nature 1997, 386, 73–77. [Google Scholar] [CrossRef]
- Shih, H.H.; Zhang, S.; Cao, W.; Hahn, A.; Wang, J.; Paulsen, J.E.; Harnish, D.C. CRP is a novel ligand for the oxidized LDL receptor LOX-1. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H1643–H1650. [Google Scholar] [CrossRef] [PubMed]
- Delneste, Y.; Magistrelli, G.; Gauchat, J.; Haeuw, J.; Aubry, J.; Nakamura, K.; Kawakami-Honda, N.; Goetsch, L.; Sawamura, T.; Bonnefoy, J.; et al. Involvement of LOX-1 in dendritic cell-mediated antigen cross-presentation. Immunity 2002, 17, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, H.; Kume, N.; Miyamoto, S.; Minami, M.; Morimoto, M.; Hayashida, K.; Hashimoto, N.; Kita, T. Oxidized LDL modulates Bax/Bcl-2 through the lectinlike Ox-LDL receptor-1 in vascular smooth muscle cells. Arter. Thromb. Vasc. Biol. 2001, 21, 955–960. [Google Scholar] [CrossRef]
- Adachi, H.; Tsujimoto, M.; Arai, H.; Inoue, K. Expression cloning of a novel scavenger receptor from human endothelial cells. J. Biol. Chem. 1997, 272, 31217–31220. [Google Scholar] [CrossRef] [PubMed]
- Berwin, B.; Delneste, Y.; Lovingood, R.V.; Post, S.R.; Pizzo, S.V. SREC-I, a type F scavenger receptor, is an endocytic receptor for calreticulin. J. Biol. Chem. 2004, 279, 51250–51257. [Google Scholar] [CrossRef]
- Murshid, A.; Gong, J.; Calderwood, S.K. Heat shock protein 90 mediates efficient antigen cross presentation through the scavenger receptor expressed by endothelial cells-I. J. Immunol. 2010, 185, 2903–2917. [Google Scholar] [CrossRef]
- Shimaoka, T.; Kume, N.; Minami, M.; Hayashida, K.; Kataoka, H.; Kita, T.; Yonehara, S. Molecular Cloning of a Novel Scavenger Receptor for Oxidized Low Density Lipoprotein, SR-PSOX, on Macrophages. J. Biol. Chem. 2000, 275, 40663–40666. [Google Scholar] [CrossRef]
- Tabata, S.; Kadowaki, N.; Kitawaki, T.; Shimaoka, T.; Yonehara, S.; Yoshie, O.; Uchiyama, T. Distribution and kinetics of SR-PSOX/CXCL16 and CXCR6 expression on human dendritic cell subsets and CD4+ T cells. J. Leukoc. Biol. 2005, 77, 777–786. [Google Scholar] [CrossRef]
- Li, R.; Oteiza, A.; Sørensen, K.K.; McCourt, P.; Olsen, R.; Smedsrød, B.; Svistounov, D. Role of liver sinusoidal endothelial cells and stabilins in elimination of oxidized low-density lipoproteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, G71–G81. [Google Scholar] [CrossRef]
- Tamura, Y.; Adachi, H.; Osuga, J.-i.; Ohashi, K.; Yahagi, N.; Sekiya, M.; Okazaki, H.; Tomita, S.; Iizuka, Y.; Shimano, H.; et al. FEEL-1 and FEEL-2 are endocytic receptors for advanced glycation end products. J. Biol. Chem. 2003, 278, 12613–12617. [Google Scholar] [CrossRef]
- Manta, C.-P.; Leibing, T.; Friedrich, M.; Nolte, H.; Adrian, M.; Schledzewski, K.; Krzistetzko, J.; Kirkamm, C.; David Schmid, C.; Xi, Y.; et al. Targeting of Scavenger Receptors Stabilin-1 and Stabilin-2 Ameliorates Atherosclerosis by a Plasma Proteome Switch Mediating Monocyte/Macrophage Suppression. Circulation 2022, 146, 1783–1799. [Google Scholar] [CrossRef]
- Hansen, B.; Longati, P.; Elvevold, K.; Nedredal, G.-I.; Schledzewski, K.; Olsen, R.; Falkowski, M.; Kzhyshkowska, J.; Carlsson, F.; Johansson, S.; et al. Stabilin-1 and stabilin-2 are both directed into the early endocytic pathway in hepatic sinusoidal endothelium via interactions with clathrin/AP-2, independent of ligand binding. Exp. Cell Res. 2005, 303, 160–173. [Google Scholar] [CrossRef]
- Kristiansen, M.; Graversen, J.H.; Jacobsen, C.; Sonne, O.; Hoffman, H.J.; Law, S.K.; Moestrup, S.K. Identification of the haemoglobin scavenger receptor. Nature 2001, 409, 198–201. [Google Scholar] [CrossRef]
- Van Gorp, H.; Delputte, P.L.; Nauwynck, H.J. Scavenger receptor CD163, a Jack-of-all-trades and potential target for cell-directed therapy. Mol. Immunol. 2010, 47, 1650–1660. [Google Scholar] [CrossRef]
- He, M.; Kubo, H.; Morimoto, K.; Fujino, N.; Suzuki, T.; Takahasi, T.; Yamada, M.; Yamaya, M.; Maekawa, T.; Yamamoto, Y.; et al. Receptor for advanced glycation end products binds to phosphatidylserine and assists in the clearance of apoptotic cells. EMBO Rep. 2011, 12, 358–364. [Google Scholar] [CrossRef]
- Kawana, H.; Karaki, H.; Higashi, M.; Miyazaki, M.; Hilberg, F.; Kitagawa, M.; Harigaya, K. CD44 Suppresses TLR-Mediated Inflammation1. J. Immunol. 2008, 180, 4235–4245. [Google Scholar] [CrossRef]
- Potere, N.; Del Buono, M.G.; Mauro, A.G.; Abbate, A.; Toldo, S. Low Density Lipoprotein Receptor-Related Protein-1 in Cardiac Inflammation and Infarct Healing. Front. Cardiovasc. Med. 2019, 6, 1–18. [Google Scholar] [CrossRef]
- Van Berkel, T.J.C.; Out, R.; Hoekstra, M.; Kuiper, J.; Biessen, E.; Van Eck, M. Scavenger receptors: Friend or foe in atherosclerosis? Curr. Opin. Lipidol. 2005, 16, 525–535. [Google Scholar] [CrossRef]
- Kunjathoor, V.V.; Febbraio, M.; Podrez, E.A.; Moore, K.J.; Andersson, L.; Koehn, S.; Rhee, J.S.; Silverstein, R.; Hoff, H.F.; Freeman, M.W. Scavenger receptors class AI/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002, 277, 49982–49988. [Google Scholar] [CrossRef]
- Babaev, V.R.; Gleaves, L.A.; Carter, K.J.; Suzuki, H.; Kodama, T.; Fazio, S.; Linton, M.F. Reduced atherosclerotic lesions in mice deficient for total or macrophage-specific expression of scavenger receptor-A. Arter. Thromb. Vasc. Biol. 2000, 20, 2593–2599. [Google Scholar] [CrossRef]
- Moore, K.J.; Kunjathoor, V.V.; Koehn, S.L.; Manning, J.J.; Tseng, A.A.; Silver, J.M.; McKee, M.; Freeman, M.W. Loss of receptor-mediated lipid uptake via scavenger receptor A or CD36 pathways does not ameliorate atherosclerosis in hyperlipidemic mice. J. Clin. Investig. 2005, 115, 2192–2201. [Google Scholar] [CrossRef]
- Guy, E.; Kuchibhotla, S.; Silverstein, R.; Febbraio, M. Continued inhibition of atherosclerotic lesion development in long term Western diet fed CD36°/apoE° mice. Atherosclerosis 2007, 192, 123–130. [Google Scholar] [CrossRef]
- Kuchibhotla, S.; Vanegas, D.; Kennedy, D.J.; Guy, E.; Nimako, G.; Morton, R.E.; Febbraio, M. Absence of CD36 protects against atherosclerosis in ApoE knock-out mice with no additional protection provided by absence of scavenger receptor A I/II. Cardiovasc. Res. 2008, 78, 185–196. [Google Scholar] [CrossRef]
- Marleau, S.; Harb, D.; Bujold, K.; Avallone, R.; Iken, K.; Wang, Y.; Demers, A.; Sirois, M.G.; Febbraio, M.; Silverstein, R.L.; et al. EP 80317, a ligand of the CD36 scavenger receptor, protects apolipoprotein E-deficient mice from developing atherosclerotic lesions. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2005, 19, 1869–1871. [Google Scholar] [CrossRef]
- Guo, M.; Härtlova, A.; Gierliński, M.; Prescott, A.; Castellvi, J.; Losa, J.H.; Petersen, S.K.; Wenzel, U.A.; Dill, B.D.; Emmerich, C.H.; et al. Triggering MSR1 promotes JNK-mediated inflammation in IL-4-activated macrophages. EMBO J. 2019, 38, e100299. [Google Scholar] [CrossRef]
- Govaere, O.; Petersen, S.K.; Martinez-Lopez, N.; Wouters, J.; Van Haele, M.; Mancina, R.M.; Jamialahmadi, O.; Bilkei-Gorzo, O.; Lassen, P.B.; Darlay, R.; et al. Macrophage scavenger receptor 1 mediates lipid-induced inflammation in non-alcoholic fatty liver disease. J. Hepatol. 2022, 76, 1001–1012. [Google Scholar] [CrossRef]
- Rahaman, S.O.; Lennon, D.J.; Febbraio, M.; Podrez, E.A.; Hazen, S.L.; Silverstein, R.L. A CD36-dependent signaling cascade is necessary for macrophage foam cell formation. Cell Metab. 2006, 4, 211–221. [Google Scholar] [CrossRef]
- Li, X.-Y.; Kong, L.-X.; Li, J.; He, H.-X.; Zhou, Y.-D. Kaempferol suppresses lipid accumulation in macrophages through the downregulation of cluster of differentiation 36 and the upregulation of scavenger receptor class B type I and ATP-binding cassette transporters A1 and G1. Int. J. Mol. Med. 2013, 31, 331–338. [Google Scholar] [CrossRef]
- Agrawal, S.; Febbraio, M.; Podrez, E.; Cathcart, M.K.; Stark, G.R.; Chisolm, G.M. Signal transducer and activator of transcription 1 is required for optimal foam cell formation and atherosclerotic lesion development. Circulation 2007, 115, 2939–2947. [Google Scholar] [CrossRef]
- Hong, D.; Bai, Y.-P.; Gao, H.-C.; Wang, X.; Li, L.-F.; Zhang, G.-G.; Hu, C.-P. Ox-LDL induces endothelial cell apoptosis via the LOX-1-dependent endoplasmic reticulum stress pathway. Atherosclerosis 2014, 235, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Ryoo, S.; Bhunia, A.; Chang, F.; Shoukas, A.; Berkowitz, D.E.; Romer, L.H. OxLDL-dependent activation of arginase II is dependent on the LOX-1 receptor and downstream RhoA signaling. Atherosclerosis 2011, 214, 279–287. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors-redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen recognition and innate immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef]
- Duan, T.; Du, Y.; Xing, C.; Wang, H.Y.; Wang, R.F. Toll-Like Receptor Signaling and Its Role in Cell-Mediated Immunity. Front. Immunol. 2022, 13, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.C.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Medzhitov, R.; Preston-hurlburt, P.; Jr, C.A.J. Widow of Nepali shot by PC seeks compensation. South China Morning Post 2012, 388, 6–9. [Google Scholar]
- Edfeldt, K.; Swedenborg, J.; Hansson, G.K.; Yan, Z.Q. Expression of toll-like receptors in human atherosclerotic lesions: A possible pathway for plaque activation. Circulation 2002, 105, 1158–1161. [Google Scholar] [CrossRef]
- Michelsen, K.S.; Wong, M.H.; Shah, P.K.; Zhang, W.; Yano, J.; Doherty, T.M.; Akira, S.; Rajavashisth, T.B.; Arditi, M. Lack of toll-like receptor 4 or myeloid differentiation factor 88 reduces atherosclerosis and alters plaque phenotype in mice deficient in apolipoprotein E. Proc. Natl. Acad. Sci. USA 2004, 101, 10679–10684. [Google Scholar] [CrossRef]
- Li, H.; Sun, B. Toll-like receptor 4 in atherosclerosis. J. Cell. Mol. Med. 2007, 11, 88–95. [Google Scholar] [CrossRef] [PubMed]
- Zeuke, S.; Ulmer, A.J.; Kusumoto, S.; Katus, H.A.; Heine, H. TLR4-mediated inflammatory activation of human coronary artery endothelial cells by LPS. Cardiovasc. Res. 2002, 56, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Smiley, S.T.; King, J.A.; Hancock, W.W. Fibrinogen Stimulates Macrophage Chemokine Secretion Through Toll-Like Receptor 4. J. Immunol. 2001, 167, 2887–2894. [Google Scholar] [CrossRef] [PubMed]
- Higashimori, M.; Tatro, J.B.; Moore, K.J.; Mendelsohn, M.E.; Galper, J.B.; Beasley, D. Role of toll-like receptor 4 in intimal foam cell accumulation in apolipoprotein E-deficient mice. Arter. Thromb. Vasc. Biol. 2011, 31, 50–57. [Google Scholar] [CrossRef]
- Chou, W.C.; Jha, S.; Linhoff, M.W.; Ting, J.P.Y. The NLR gene family: From discovery to present day. Nat. Rev. Immunol. 2023, 23, 635–654. [Google Scholar] [CrossRef]
- Latz, E. The inflammasomes: Mechanisms of activation and function. Curr. Opin. Immunol. 2010, 22, 28–33. [Google Scholar] [CrossRef]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 36. [Google Scholar] [CrossRef]
- Man, S.M.; Karki, R.; Kanneganti, T.D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol. Rev. 2017, 277, 61–75. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef]
- Westerterp, M.; Fotakis, P.; Ouimet, M.; Bochem, A.E.; Zhang, H.; Molusky, M.M.; Wang, W.; Abramowicz, S.; la Bastide-van Gemert, S.; Wang, N.; et al. Cholesterol Efflux Pathways Suppress Inflammasome Activation, NETosis, and Atherogenesis. Circulation 2018, 138, 898–912. [Google Scholar] [CrossRef] [PubMed]
- Evavold, C.L.; Ruan, J.; Tan, Y.; Xia, S.; Wu, H.; Kagan, J.C. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 2018, 48, 35–44.e36. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Günther, P.; Lauterbach, M.A.R.; Duewell, P.; Biswas, D.; Pelka, K.; Scholz, C.J.; Oosting, M.; Haendler, K.; Baßler, K.; et al. Western Diet Triggers NLRP3-Dependent Innate Immune Reprogramming HHS Public Access. Cell 2018, 172, 162–175. [Google Scholar] [CrossRef] [PubMed]
- Asare, Y.; Shnipova, M.; Zivkovic, L.; Schlegl, C.; Tosato, F.; Aronova, A.; Brandhofer, M.; Strohm, L.; Beaufort, N.; Malik, R.; et al. IKKbeta binds NLRP3 providing a shortcut to inflammasome activation for rapid immune responses. Signal Transduct. Target. Ther. 2022, 7, 355. [Google Scholar] [CrossRef]
- Schmacke, N.A.; O’Duill, F.; Gaidt, M.M.; Szymanska, I.; Kamper, J.M.; Schmid-Burgk, J.L.; Madler, S.C.; Mackens-Kiani, T.; Kozaki, T.; Chauhan, D.; et al. IKKbeta primes inflammasome formation by recruiting NLRP3 to the trans-Golgi network. Immunity 2022, 55, 2271–2284.e7. [Google Scholar] [CrossRef]
- Nanda, S.K.; Prescott, A.R.; Figueras-Vadillo, C.; Cohen, P. IKKbeta is required for the formation of the NLRP3 inflammasome. EMBO Rep. 2021, 22, e50743. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Paramel Varghese, G.; Folkersen, L.; Strawbridge, R.J.; Halvorsen, B.; Yndestad, A.; Ranheim, T.; Krohg-Sørensen, K.; Skjelland, M.; Espevik, T.; Aukrust, P.; et al. NLRP3 Inflammasome Expression and Activation in Human Atherosclerosis. J. Am. Heart Assoc. 2016, 5, e003031. [Google Scholar] [CrossRef]
- Zhuang, T.; Liu, J.; Chen, X.; Zhang, L.; Pi, J.; Sun, H.; Li, L.; Bauer, R.; Wang, H.; Yu, Z.; et al. Endothelial Foxp1 Suppresses Atherosclerosis via Modulation of Nlrp3 Inflammasome Activation. Circ. Res. 2019, 125, 590–605. [Google Scholar] [CrossRef]
- Wang, R.; Wu, W.; Li, W.; Huang, S.; Li, Z.; Liu, R.; Shan, Z.; Zhang, C.; Li, W.; Wang, S. Activation of NLRP3 Inflammasome Promotes Foam Cell Formation in Vascular Smooth Muscle Cells and Atherogenesis Via HMGB1. J. Am. Heart Assoc. 2018, 7, e008596. [Google Scholar] [CrossRef]
- Coll, R.C.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Croker, D.E.; et al. Potential Therapeutic for Inflammatory Diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Van Hout, G.P.J.; Bosch, L.; Ellenbroek, G.H.J.M.; De Haan, J.J.; Van Solinge, W.W.; Cooper, M.A.; Arslan, F.; De Jager, S.C.A.; Robertson, A.A.B.; Pasterkamp, G.; et al. The selective NLRP3-inflammasome inhibitor MCC950 reduces infarct size and preserves cardiac function in a pig model of myocardial infarction. Eur. Heart J. 2017, 38, 828–836. [Google Scholar] [CrossRef] [PubMed]
- Van Der Heijden, T.; Kritikou, E.; Venema, W.; Van Duijn, J.; Van Santbrink, P.J.; Slütter, B.; Foks, A.C.; Bot, I.; Kuiper, J. NLRP3 Inflammasome Inhibition by MCC950 Reduces Atherosclerotic Lesion Development in Apolipoprotein E-Deficient Mice—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1457–1461. [Google Scholar] [CrossRef]
- Stewart, C.R.; Stuart, L.M.; Wilkinson, K.; van Gils, J.M.; Deng, J.; Halle, A.; Rayner, K.J.; Boyer, L.; Zhong, R.; Frazier, W.A.; et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat. Immunol. 2010, 11, 155–161. [Google Scholar] [CrossRef]
- Murshid, A.; Gong, J.; Prince, T.; Borges, T.J.; Calderwood, S.K. Scavenger receptor SREC-I mediated entry of TLR4 into lipid microdomains and triggered inflammatory cytokine release in RAW 2647 cells upon LPS activation. PLoS ONE 2015, 10, e0122529. [Google Scholar] [CrossRef] [PubMed]
- Murshid, A.; Gong, J.; Ahmad, R.; Borges, T.J.; Calderwood, S.K. Scavenger receptor SREC-I promotes double stranded RNA-mediated TLR3 activation in human monocytes. Immunobiology 2015, 220, 823–832. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Varin, A.; Chen, Y.; Liu, B.; Tryggvason, K.; Gordon, S. SR-A/MARCO-mediated ligand delivery enhances intracellular TLR and NLR function, but ligand scavenging from cell surface limits TLR4 response to pathogens. Blood 2011, 117, 1319–1328. [Google Scholar] [CrossRef]
- Netea, M.G.; van der Meer, J.W.M. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 2017, 21, 297–300. [Google Scholar] [CrossRef]
- Quintin, J.; Saeed, S.; Martens, J.H.; Giamarellos-Bourboulis, E.J.; Ifrim, D.C.; Logie, C.; Jacobs, L.; Jansen, T.; Kullberg, B.-J.; Wijmenga, C.; et al. Candida albicans Infection Affords Protection against Reinfection via Functional Reprogramming of Monocytes. Cell Host Microbe 2012, 12, 223–232. [Google Scholar] [CrossRef]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guérin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef]
- Bekkering, S.; Quintin, J.; Joosten, L.A.B.; van der Meer, J.W.M.; Netea, M.G.; Riksen, N.P. Oxidized Low-Density Lipoprotein Induces Long-Term Proinflammatory Cytokine Production and Foam Cell Formation via Epigenetic Reprogramming of Monocytes. Arter. Thromb. Vasc. Biol. 2014, 34, 1731–1738. [Google Scholar] [CrossRef] [PubMed]
- van der Valk, F.M.; Bekkering, S.; Kroon, J.; Yeang, C.; Van den Bossche, J.; van Buul, J.D.; Ravandi, A.; Nederveen, A.J.; Verberne, H.J.; Scipione, C.; et al. Oxidized Phospholipids on Lipoprotein(a) Elicit Arterial Wall Inflammation and an Inflammatory Monocyte Response in Humans. Circulation 2016, 134, 611–624. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Bekkering, S.; Latz, E.; Riksen, N.P. Long-term activation of the innate immune system in atherosclerosis. Semin. Immunol. 2016, 28, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Klemm, S.L.; Shipony, Z.; Greenleaf, W.J. Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet. 2019, 20, 207–220. [Google Scholar] [CrossRef]
- Bekkering, S.; van den Munckhof, I.; Nielen, T.; Lamfers, E.; Dinarello, C.; Rutten, J.; de Graaf, J.; Joosten, L.A.B.; Netea, M.G.; Gomes, M.E.R.; et al. Innate immune cell activation and epigenetic remodeling in symptomatic and asymptomatic atherosclerosis in humans in vivo. Atherosclerosis 2016, 254, 228–236. [Google Scholar] [CrossRef]
- Bekkering, S.; Arts, R.J.W.; Novakovic, B.; Kourtzelis, I.; van der Heijden, C.D.C.C.; Li, Y.; Popa, C.D.; ter Horst, R.; van Tuijl, J.; Netea-Maier, R.T.; et al. Metabolic Induction of Trained Immunity through the Mevalonate Pathway. Cell 2018, 172, 135–146.e139. [Google Scholar] [CrossRef]
- Bekkering, S.; Stiekema, L.C.A.; Bernelot Moens, S.; Verweij, S.L.; Novakovic, B.; Prange, K.; Versloot, M.; Roeters van Lennep, J.E.; Stunnenberg, H.; de Winther, M.; et al. Treatment with Statins Does Not Revert Trained Immunity in Patients with Familial Hypercholesterolemia. Cell Metab. 2019, 30, 1–2. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsen, K.; van der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Greißel, A.; Culmes, M.; Burgkart, R.; Zimmermann, A.; Eckstein, H.-H.; Zernecke, A.; Pelisek, J. Histone acetylation and methylation significantly change with severity of atherosclerosis in human carotid plaques. Cardiovasc. Pathol. 2016, 25, 79–86. [Google Scholar] [CrossRef]
- Arts, R.J.W.; Moorlag, S.J.C.F.M.; Novakovic, B.; Li, Y.; Wang, S.-Y.; Oosting, M.; Kumar, V.; Xavier, R.J.; Wijmenga, C.; Joosten, L.A.B.; et al. BCG Vaccination Protects against Experimental Viral Infection in Humans through the Induction of Cytokines Associated with Trained Immunity. Cell Host Microbe 2018, 23, 89–100.e105. [Google Scholar] [CrossRef]
- Ishii, M.; Wen, H.; Corsa, C.A.S.; Liu, T.; Coelho, A.L.; Allen, R.M.; Carson, W.F.; Cavassani, K.A.; Li, X.; Lukacs, N.W.; et al. Epigenetic regulation of the alternatively activated macrophage phenotype. Blood 2009, 114, 3244–3254. [Google Scholar] [CrossRef] [PubMed]
- Takeuch, O.; Akira, S. Epigenetic control of macrophage polarization. Eur. J. Immunol. 2011, 41, 2490–2493. [Google Scholar] [CrossRef] [PubMed]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F.; Mukherjee, S.; Jolanta, G.; Tomasz, C.; et al. Cytokine Induced Phenotypic and Epigenetic Signatures Are Key to Establishing Specific Macrophage Phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Ding, N.; Zhang, H.; Liu, K.; Xiong, J.; Ma, S.; Ai, Y.; Zhang, H.; Jiang, Y. SNF5 promotes IL-1β expression via H3K4me1 in atherosclerosis induced by homocysteine. Int. J. Biochem. Cell Biol. 2021, 135, 105974. [Google Scholar] [CrossRef]
- Bai, L.; Li, Z.; Li, Q.; Guan, H.; Zhao, S.; Liu, R.; Wang, R.; Zhang, J.; Jia, Y.; Fan, J.; et al. Mediator 1 Is Atherosclerosis Protective by Regulating Macrophage Polarization. Arter. Thromb. Vasc. Biol. 2017, 37, 1470–1481. [Google Scholar] [CrossRef]
- Guo, Z.; Wang, L.; Liu, H.; Xie, Y. Innate Immune Memory in Monocytes and Macrophages: The Potential Therapeutic Strategies for Atherosclerosis. Cells 2022, 11, 4072. [Google Scholar] [CrossRef]
- Riksen, N.P.; Bekkering, S.; Mulder, W.J.M.; Netea, M.G. Trained immunity in atherosclerotic cardiovascular disease. Nat. Rev. Cardiol. 2023. [Google Scholar] [CrossRef]
- Kuznetsova, T.; Prange, K.H.M.; Glass, C.K.; de Winther, M.P.J. Transcriptional and epigenetic regulation of macrophages in atherosclerosis. Nat. Rev. Cardiol. 2020, 17, 216–228. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aronova, A.; Tosato, F.; Naser, N.; Asare, Y. Innate Immune Pathways in Atherosclerosis—From Signaling to Long-Term Epigenetic Reprogramming. Cells 2023, 12, 2359. https://doi.org/10.3390/cells12192359
Aronova A, Tosato F, Naser N, Asare Y. Innate Immune Pathways in Atherosclerosis—From Signaling to Long-Term Epigenetic Reprogramming. Cells. 2023; 12(19):2359. https://doi.org/10.3390/cells12192359
Chicago/Turabian StyleAronova, Arailym, Federica Tosato, Nawraa Naser, and Yaw Asare. 2023. "Innate Immune Pathways in Atherosclerosis—From Signaling to Long-Term Epigenetic Reprogramming" Cells 12, no. 19: 2359. https://doi.org/10.3390/cells12192359
APA StyleAronova, A., Tosato, F., Naser, N., & Asare, Y. (2023). Innate Immune Pathways in Atherosclerosis—From Signaling to Long-Term Epigenetic Reprogramming. Cells, 12(19), 2359. https://doi.org/10.3390/cells12192359