
Orai3 Calcium Channel Contributes to Oral/Oropharyngeal Cancer Stemness through the Elevation of ID1 Expression

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Laser-Captured Microdissection (LCM)
2.3. Quantitative Real-Time PCR (qPCR)
2.4. Western Blotting
2.5. Tumor Sphere Formation Assay
2.6. Migration Assay
2.7. Ectopic Expression of Orai3
2.8. SOCE Assay (Single-Cell Ca2+ Imaging)
2.9. Cell Proliferation Assay
2.10. Anchorage-Independent Growth
2.11. In Vivo Xenograft Tumor Assay
2.12. ALDH1 Assay
2.13. Small Interfering RNA (siRNA) Transfection
2.14. Mouse Models
2.15. Statistical Analysis
3. Results
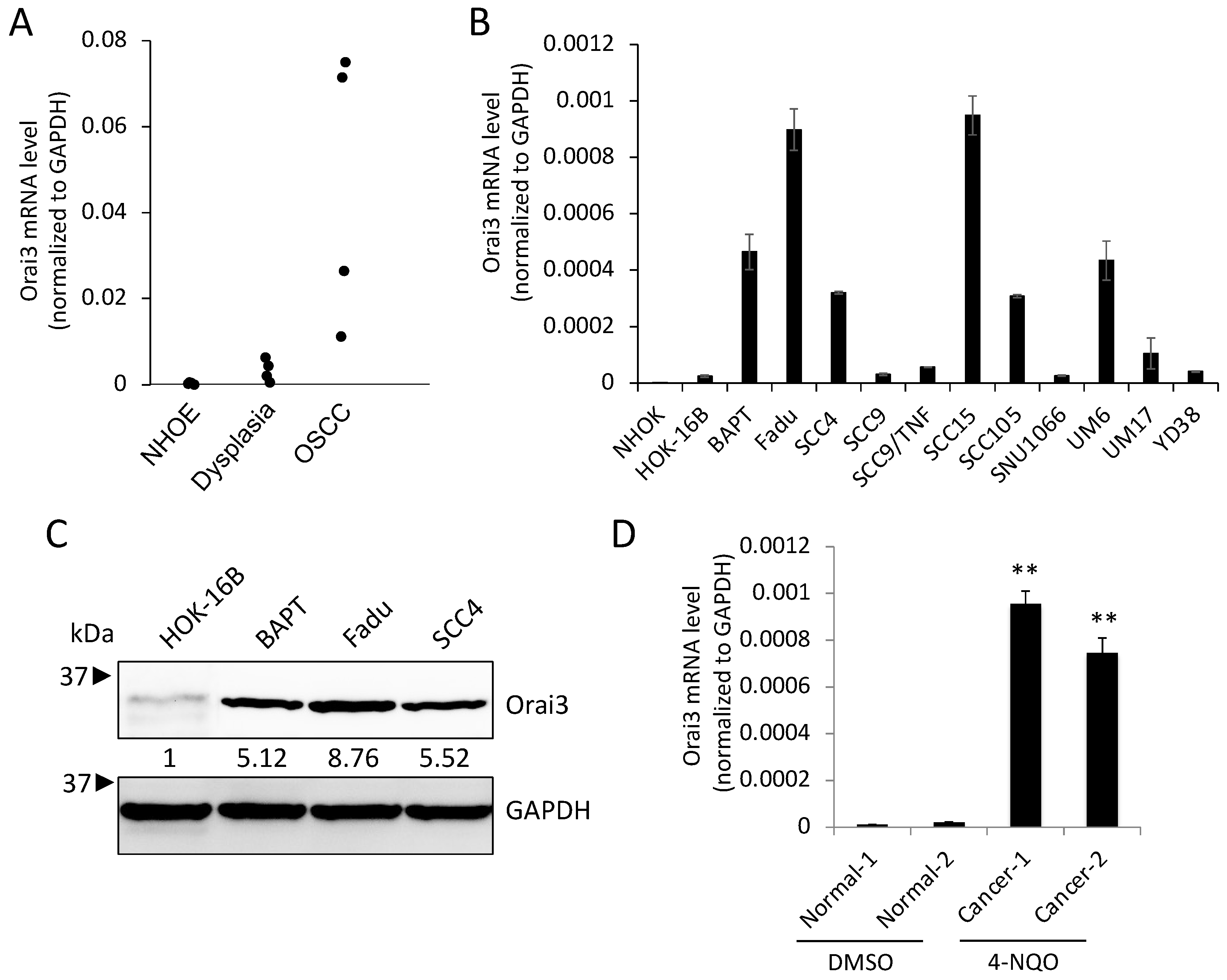
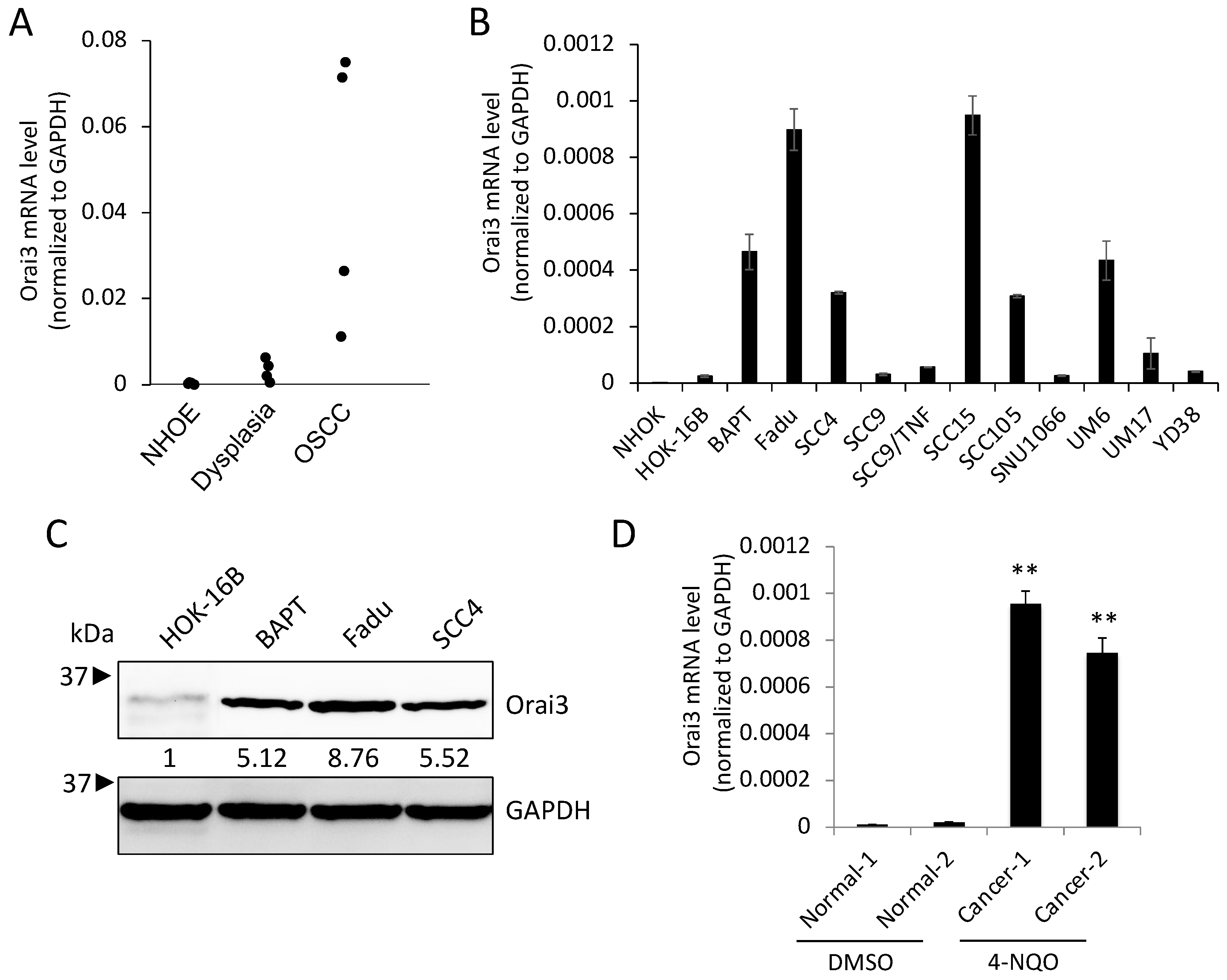
3.1. Stepwise Increase in Orai3 Expression during Oral Carcinogenesis
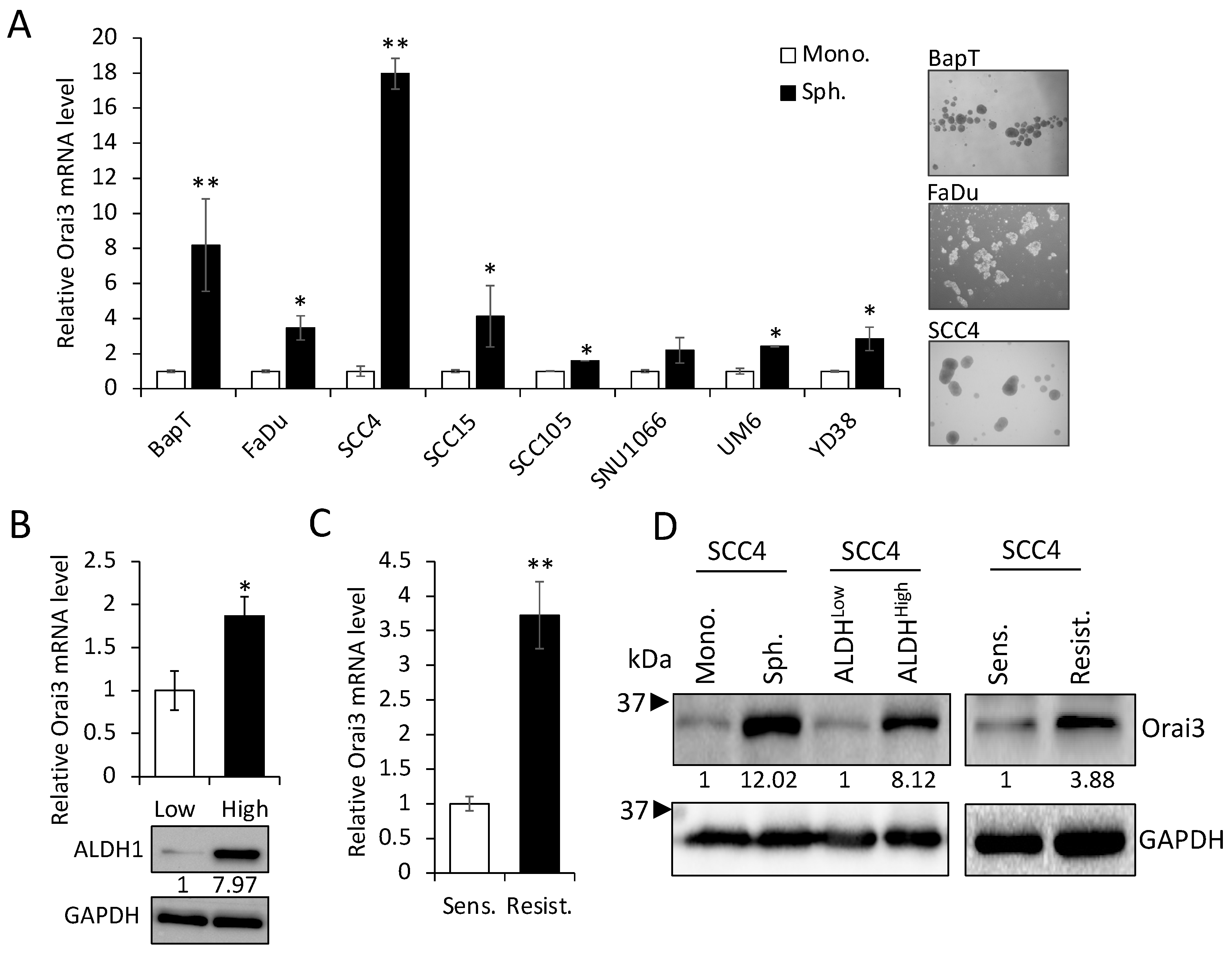
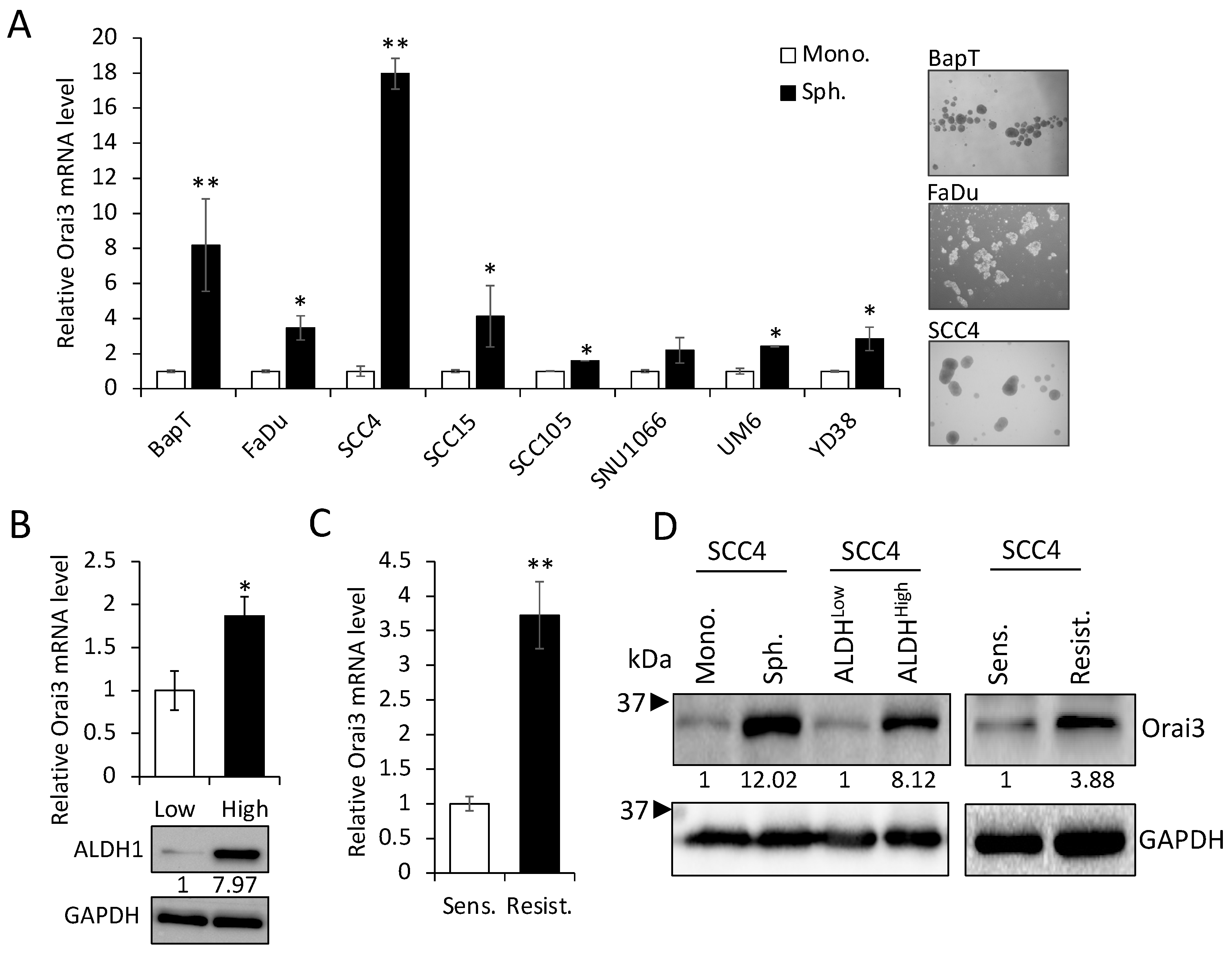
3.2. Orai3 Is Enriched in CSC Populations Derived from OSCC
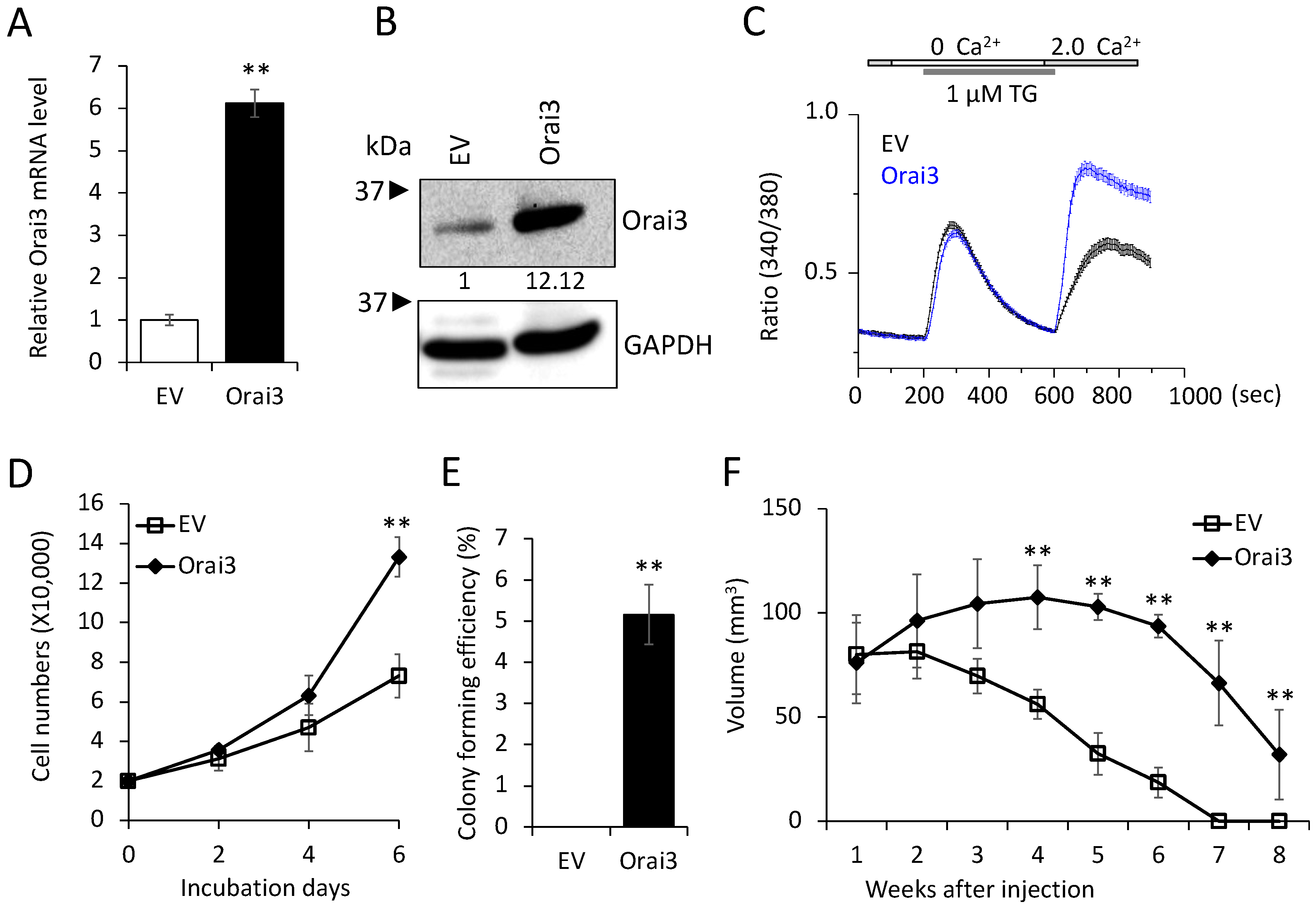
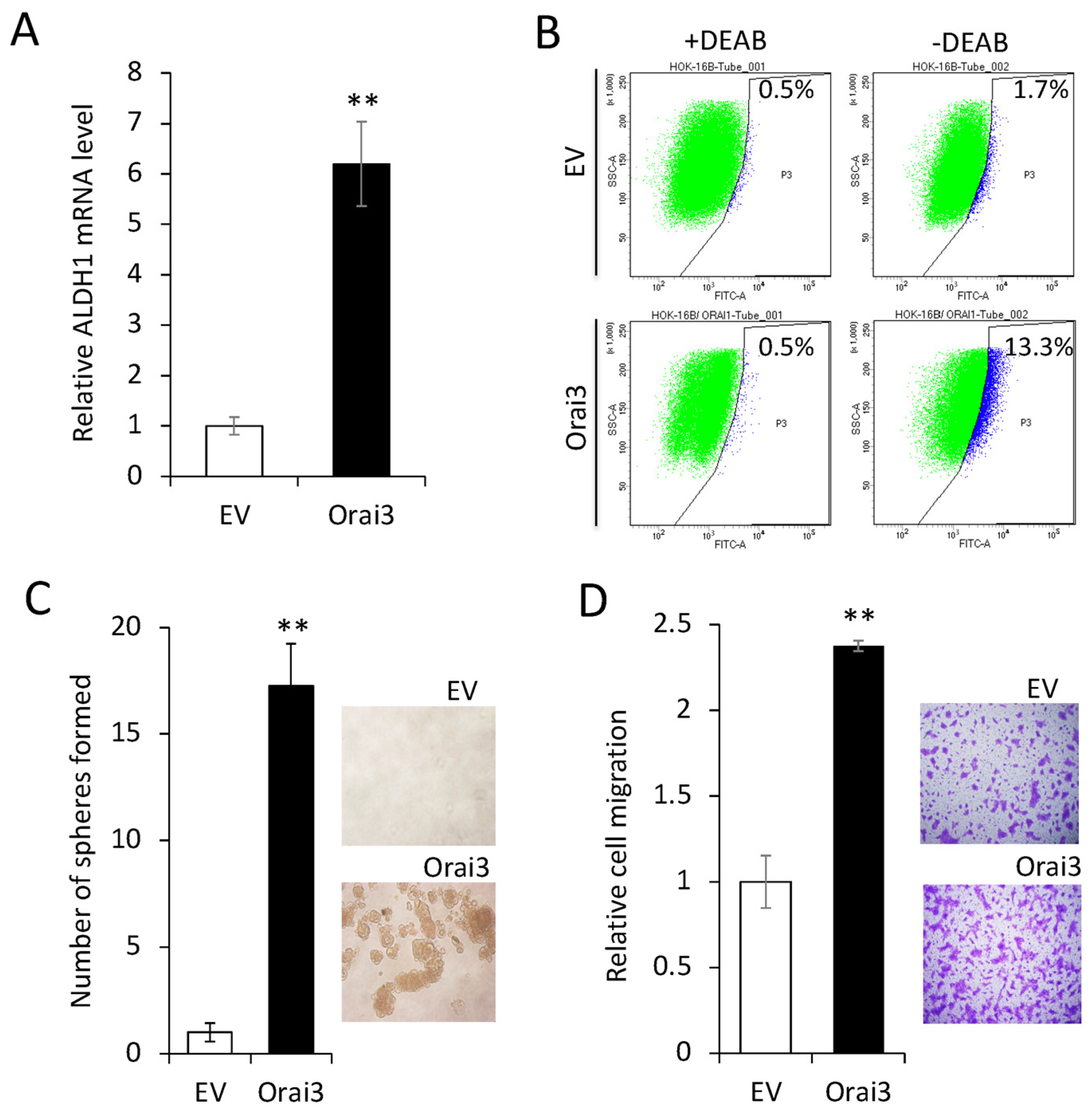
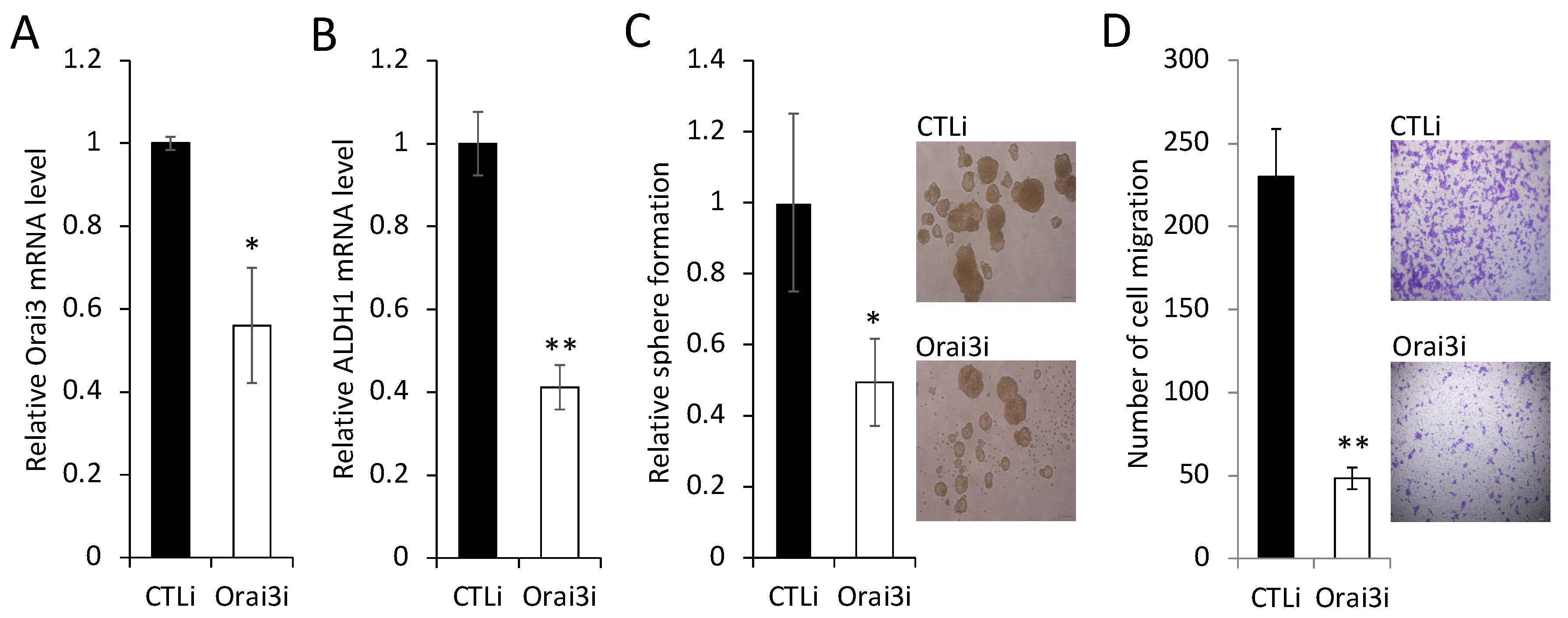
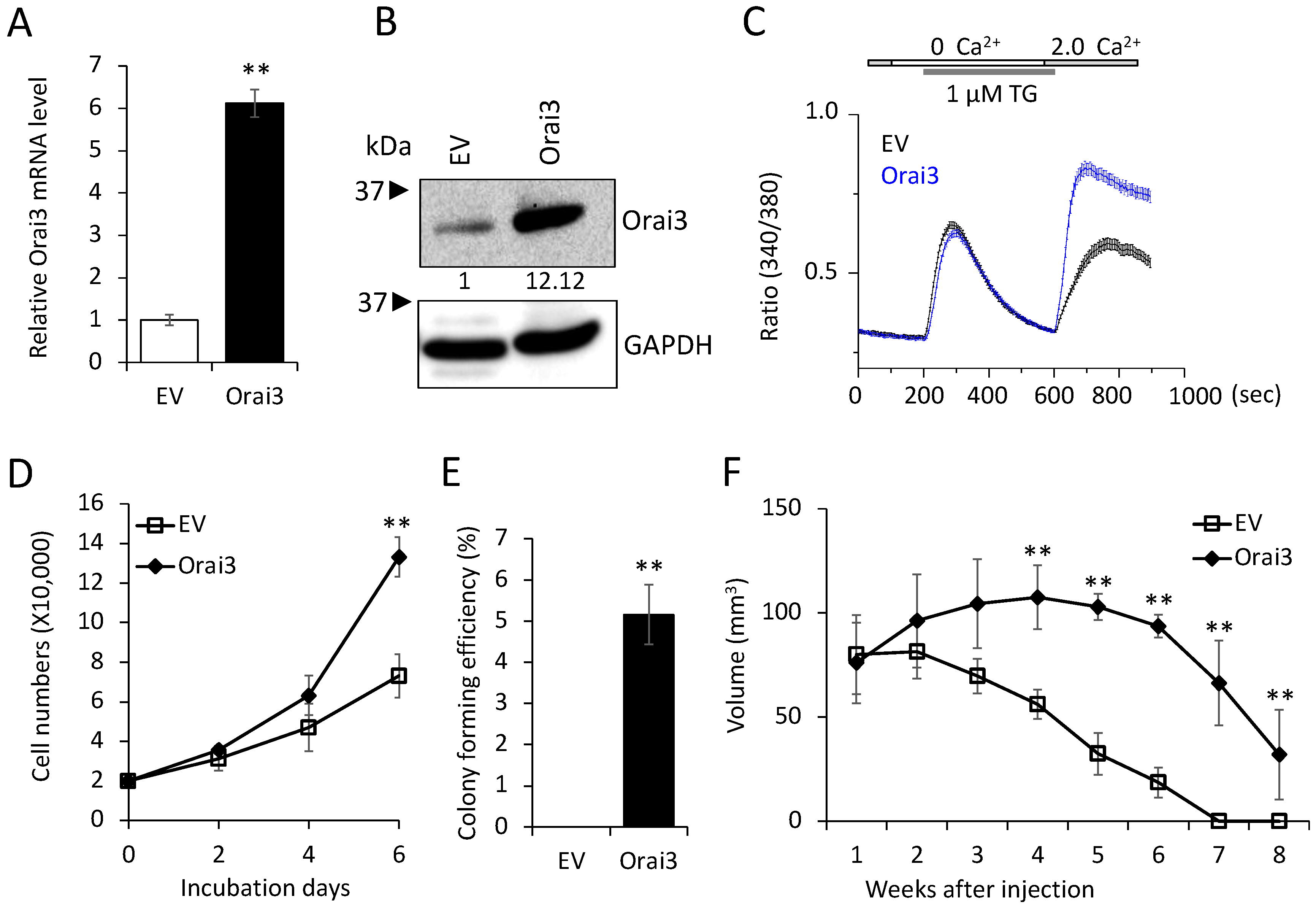
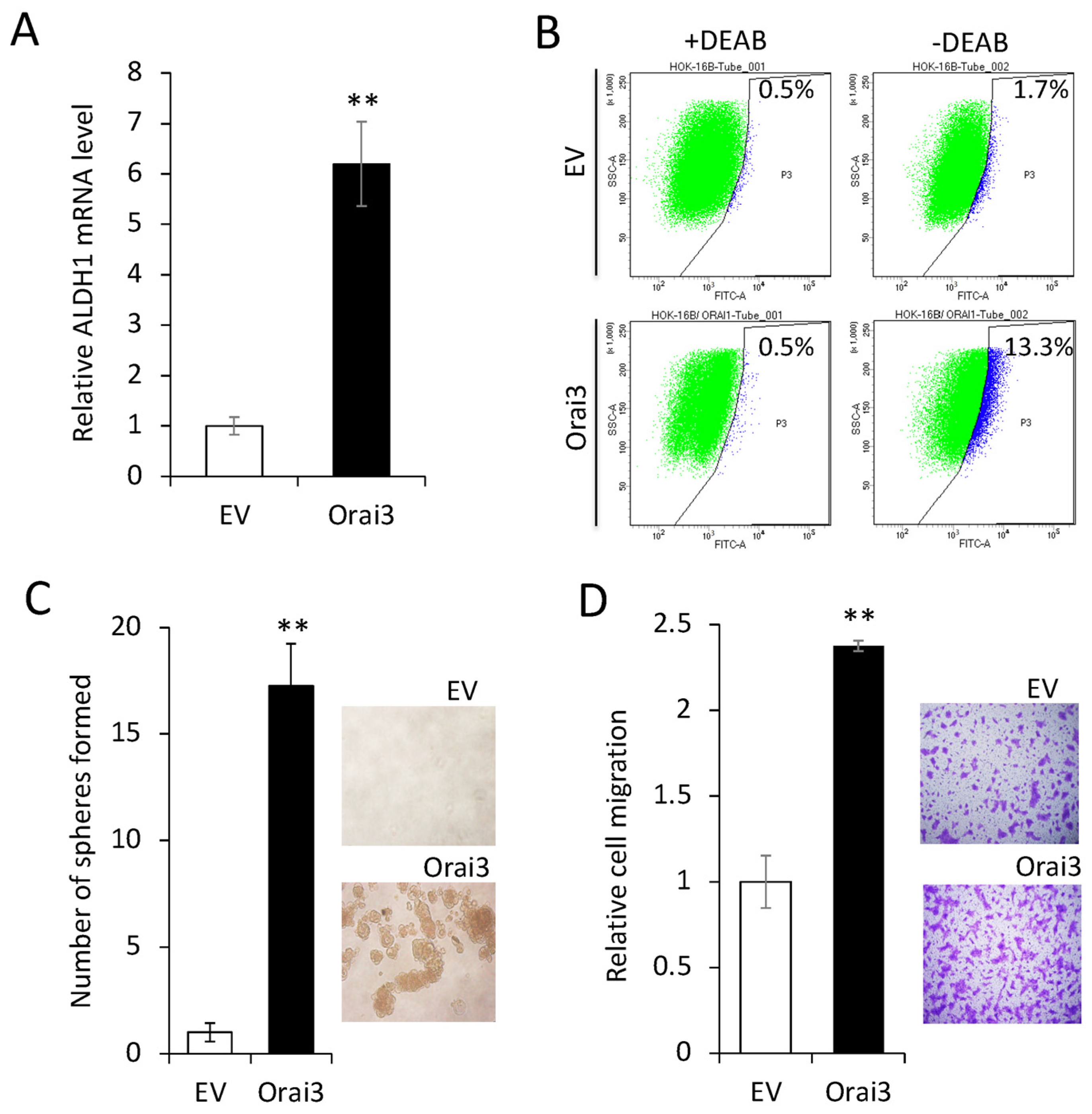
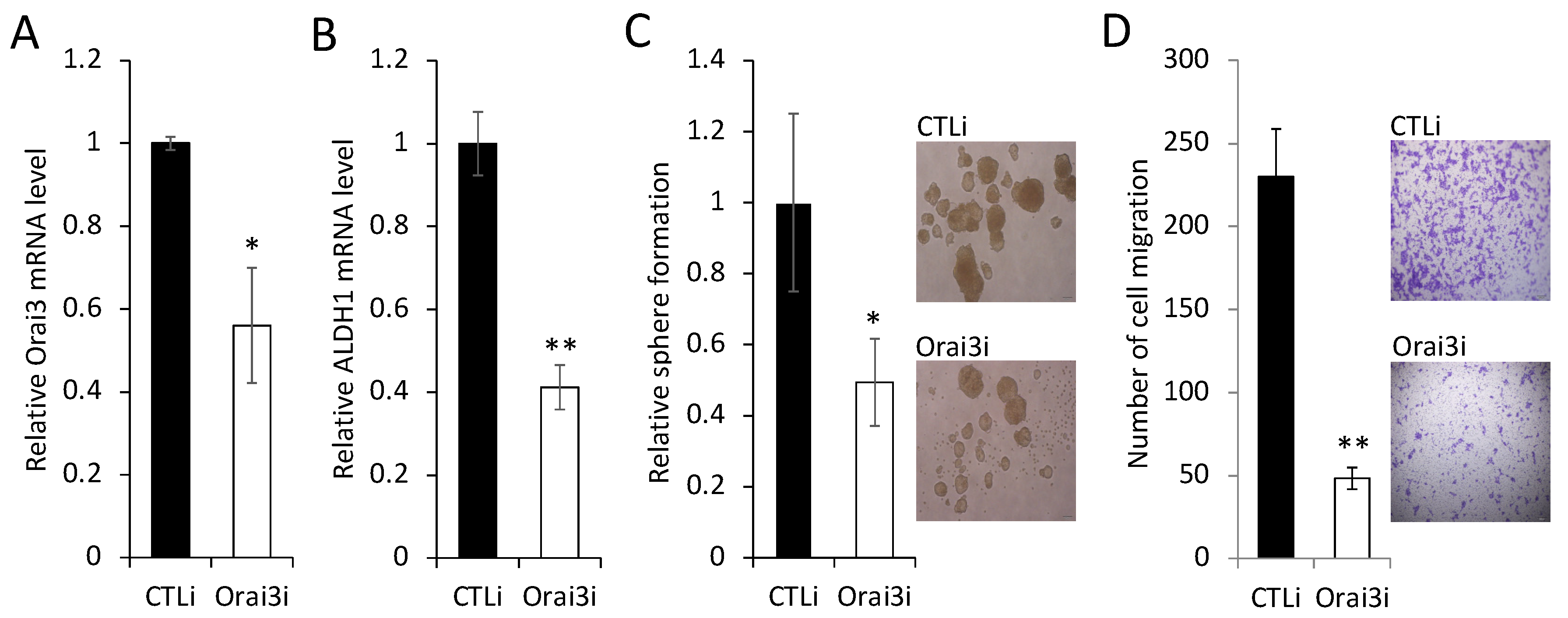
3.3. Elevated Orai3 Supports CSC Phenotype
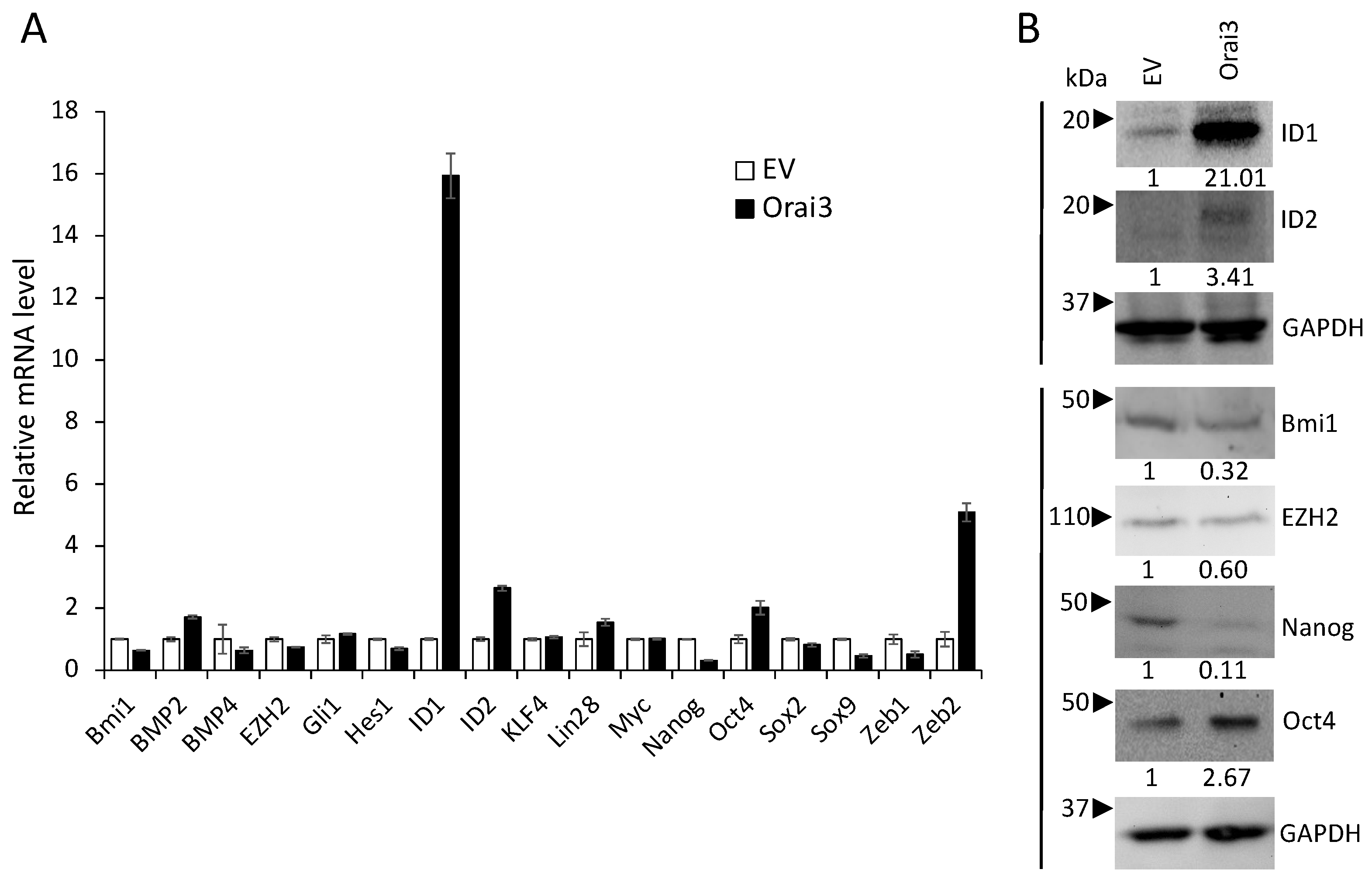
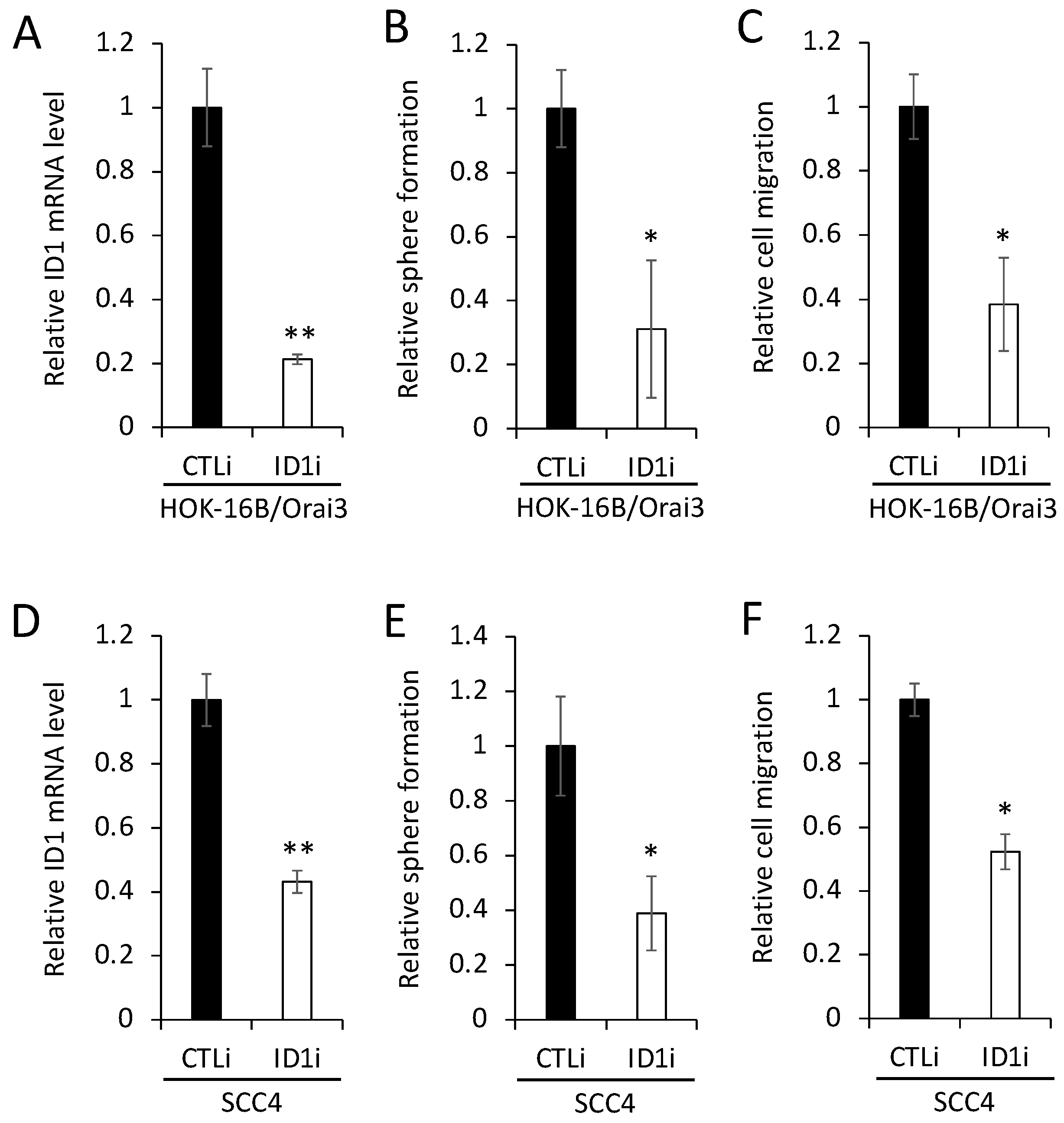
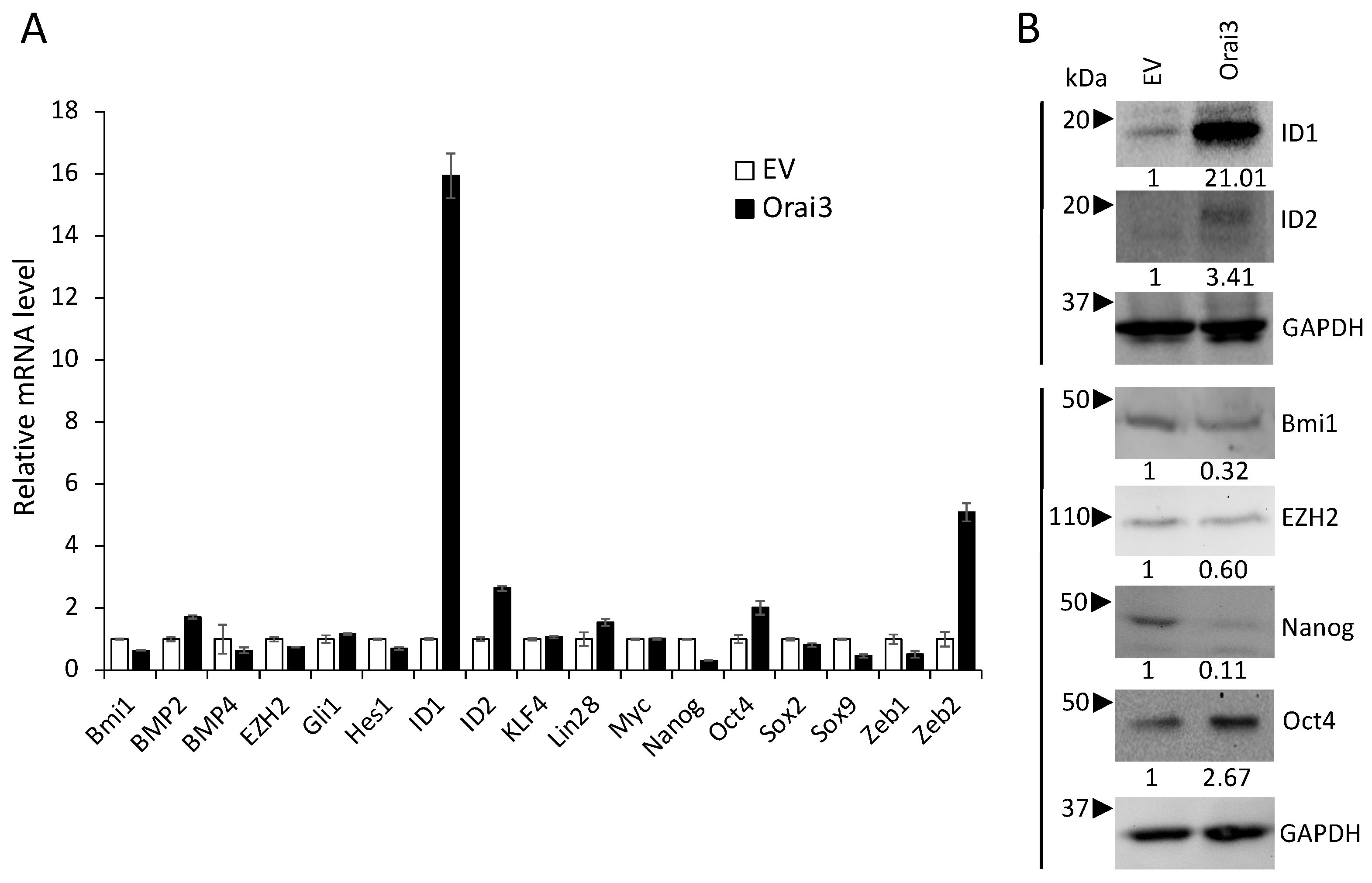
3.4. ID1 Is Required for Orai3-Induced CSC Phenotype
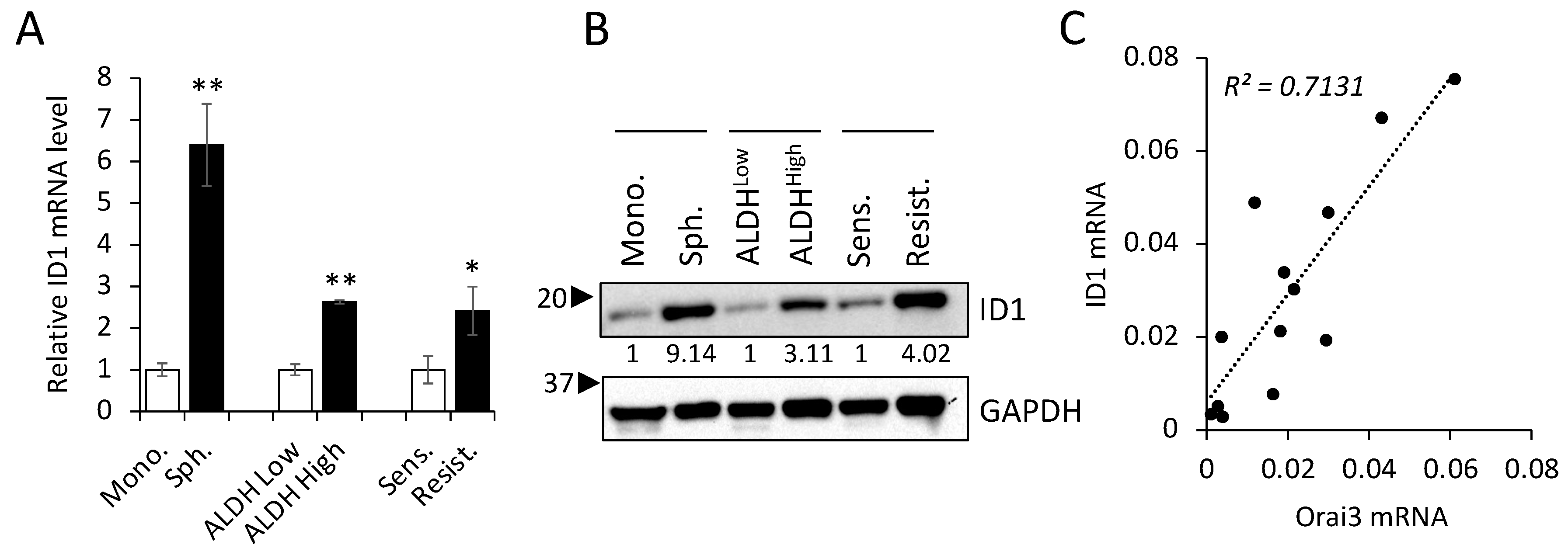
3.5. Orai3 and ID1 Are Highly Expressed in CSC-Enriched Populations, and Their Expression Levels Are Positively Correlated in OSCC Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nassar, D.; Blanpain, C. Cancer Stem Cells: Basic Concepts and Therapeutic Implications. Annu. Rev. Pathol. 2016, 11, 47–76. [Google Scholar] [CrossRef] [PubMed]
- Shin, K.-H.; Kim, R.H. An Updated Review of Oral Cancer Stem Cells and Their Stemness Regulation. Crit. Rev. Oncog. 2018, 23, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Reibel, J.; Bouquot, J.; Dabelsteen, E. Oral epithelial dysplasia classification systems: Predictive value, utility, weaknesses and scope for improvement. J. Oral. Pathol. Med. 2008, 37, 127–133. [Google Scholar] [CrossRef]
- Brennan, M.; Migliorati, C.A.; Lockhart, P.B.; Wray, D.; Al-Hashimi, I.; Axell, T.; Bruce, A.J.; Carpenter, W.; Eisenberg, E.; Epstein, J.B.; et al. Management of oral epithelial dysplasia: A review. Oral. Surg. Oral. Med. Oral. Pathol. Oral. Radiol. Endod. 2007, 103, e11–e12. [Google Scholar] [CrossRef] [PubMed]
- Marcial, V.A.; Pajak, T.F.; Mohiuddin, M.; Cooper, J.S.; al Sarraf, M.; Mowry, P.A.; Curran, W.; Crissman, J.; Rodriguez, M.; Velez-Garcia, E. Concomitant cisplatin chemotherapy and radiotherapy in advanced mucosal squamous cell carcinoma of the head and neck. Long-term results of the Radiation Therapy Oncology Group study 81-17. Cancer 1990, 66, 1861–1868. [Google Scholar] [CrossRef]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Monteith, G.R.; McAndrew, D.; Faddy, H.M.; Roberts-Thomson, S.J. Calcium and cancer: Targeting Ca2+ transport. Nat. Rev. Cancer 2007, 7, 519–530. [Google Scholar] [CrossRef]
- Roderick, H.L.; Cook, S.J. Ca2+ signalling checkpoints in cancer: Remodelling Ca2+ for cancer cell proliferation and survival. Nat. Rev. Cancer 2008, 8, 361–375. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Skryma, R.; Shuba, Y. Calcium in tumour metastasis: New roles for known actors. Nat. Rev. Cancer 2011, 11, 609–618. [Google Scholar] [CrossRef]
- Flourakis, M.; Lehen’kyi, V.; Beck, B.; Raphael, M.; Vandenberghe, M.; Abeele, F.V.; Roudbaraki, M.; Lepage, G.; Mauroy, B.; Romanin, C.; et al. Orai1 contributes to the establishment of an apoptosis-resistant phenotype in prostate cancer cells. Cell Death Dis. 2010, 1, e75. [Google Scholar] [CrossRef]
- Yang, S.; Zhang, J.J.; Huang, X.Y. Orai1 and STIM1 are critical for breast tumor cell migration and metastasis. Cancer Cell 2009, 15, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Hughes, J.D.; Rollins, S.; Chen, B.; Perkins, E. Calcium entry via ORAI1 regulates glioblastoma cell proliferation and apoptosis. Exp. Mol. Pathol. 2011, 91, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lkhagvadorj, S.; Lee, M.R.; Hwang, K.H.; Chung, H.C.; Jung, J.H.; Cha, S.K.; Eom, M. Orai1 and STIM1 are critical for cell migration and proliferation of clear cell renal cell carcinoma. Biochem. Biophys. Res. Commun. 2014, 448, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Rigas, N.K.; Lee, C.R.; Bang, A.; Srikanth, S.; Gwack, Y.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. Orai1 promotes tumor progression by enhancing cancer stemness via NFAT signaling in oral/oropharyngeal squamous cell carcinoma. Oncotarget 2016, 7, 43239–43255. [Google Scholar] [CrossRef]
- Wang, L.; Hao, J.; Zhang, Y.; Yang, Z.; Cao, Y.; Lu, W.; Shu, Y.; Jiang, L.; Hu, Y.; Lv, W.; et al. Orai1 mediates tumor-promoting store-operated Ca2+ entry in human gastrointestinal stromal tumors via c-KIT and the extracellular signal-regulated kinase pathway. Tumour Biol. 2017, 39, 1010428317691426. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Huang, H.; Sun, L.; Dong, S.; Huang, N.; Shi, M.; Bin, J.; Liao, Y.; Liao, W. Elevated Orai1 and STIM1 expressions upregulate MACC1 expression to promote tumor cell proliferation, metabolism, migration, and invasion in human gastric cancer. Cancer Lett. 2016, 381, 31–40. [Google Scholar] [CrossRef]
- Deng, W.; Wang, J.; Zhang, J.; Cai, J.; Bai, Z.; Zhang, Z. Orai1, a Direct Target of microRNA-519, Promotes Progression of Colorectal Cancer via Akt/GSK3beta Signaling Pathway. Dig. Dis. Sci. 2016, 61, 1553–1560. [Google Scholar] [CrossRef]
- Zhan, Z.Y.; Zhong, L.X.; Feng, M.; Wang, J.F.; Liu, D.B.; Xiong, J.P. Over-expression of Orai1 mediates cell proliferation and associates with poor prognosis in human non-small cell lung carcinoma. Int. J. Clin. Exp. Pathol. 2015, 8, 5080–5088. [Google Scholar]
- Motiani, R.K.; Abdullaev, I.F.; Trebak, M. A novel native store-operated calcium channel encoded by Orai3: Selective requirement of Orai3 versus Orai1 in estrogen receptor-positive versus estrogen receptor-negative breast cancer cells. J. Biol. Chem. 2010, 285, 19173–19183. [Google Scholar] [CrossRef]
- Faouzi, M.; Hague, F.; Potier, M.; Ahidouch, A.; Sevestre, H.; Ouadid-Ahidouch, H. Down-regulation of Orai3 arrests cell-cycle progression and induces apoptosis in breast cancer cells but not in normal breast epithelial cells. J. Cell Physiol. 2011, 226, 542–551. [Google Scholar] [CrossRef]
- Motiani, R.K.; Zhang, X.; Harmon, K.E.; Keller, R.S.; Matrougui, K.; Bennett, J.A.; Trebak, M. Orai3 is an estrogen receptor alpha-regulated Ca(2)(+) channel that promotes tumorigenesis. FASEB J. 2013, 27, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Ay, A.S.; Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 constitutes a native store-operated calcium entry that regulates non small cell lung adenocarcinoma cell proliferation. PLoS ONE 2013, 8, e72889. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Vanden Abeele, F.; Lehen’kyi, V.; Gkika, D.; Guarmit, B.; Lepage, G.; Slomianny, C.; Borowiec, A.S.; Bidaux, G.; Benahmed, M.; et al. Remodeling of channel-forming ORAI proteins determines an oncogenic switch in prostate cancer. Cancer Cell 2014, 26, 19–32. [Google Scholar] [CrossRef] [PubMed]
- Benzerdjeb, N.; Sevestre, H.; Ahidouch, A.; Ouadid-Ahidouch, H. Orai3 is a predictive marker of metastasis and survival in resectable lung adenocarcinoma. Oncotarget 2016, 7, 81588–81597. [Google Scholar] [CrossRef]
- Tanwar, J.; Arora, S.; Motiani, R.K. Orai3: Oncochannel with therapeutic potential. Cell Calcium 2020, 90, 102247. [Google Scholar] [CrossRef]
- Arora, S.; Tanwar, J.; Sharma, N.; Saurav, S.; Motiani, R.K. Orai3 Regulates Pancreatic Cancer Metastasis by Encoding a Functional Store Operated Calcium Entry Channel. Cancers 2021, 13, 5937. [Google Scholar] [CrossRef] [PubMed]
- Dubois, C.; Kondratska, K.; Kondratskyi, A.; Morabito, A.; Mesilmany, L.; Farfariello, V.; Toillon, R.A.; Ziental Gelus, N.; Laurenge, E.; Vanden Abeele, F.; et al. ORAI3 silencing alters cell proliferation and promotes mitotic catastrophe and apoptosis in pancreatic adenocarcinoma. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 119023. [Google Scholar] [CrossRef]
- Zhu, H.; Zhang, H.; Jin, F.; Fang, M.; Huang, M.; Yang, C.S.; Chen, T.; Fu, L.; Pan, Z. Elevated Orai1 expression mediates tumor-promoting intracellular Ca2+ oscillations in human esophageal squamous cell carcinoma. Oncotarget 2014, 5, 3455–3471. [Google Scholar] [CrossRef]
- Srikanth, S.; Gwack, Y. Chapter Eight—Molecular Regulation of the Pore Component of CRAC Channels, Orai1. In Current Topics in Membranes; Murali, P., Ed.; Academic Press: Cambridge, MA, USA, 2013; Volume 71, pp. 181–207. [Google Scholar]
- Cantonero, C.; Sanchez-Collado, J.; Gonzalez-Nunez, M.A.; Salido, G.M.; Lopez, J.J.; Jardin, I.; Rosado, J.A. Store-independent Orai1-mediated Ca(2+) entry and cancer. Cell Calcium 2019, 80, 1–7. [Google Scholar] [CrossRef]
- Singh, A.K.; Roy, N.K.; Bordoloi, D.; Padmavathi, G.; Banik, K.; Khwairakpam, A.D.; Kunnumakkara, A.B.; Sukumar, P. Orai-1 and Orai-2 regulate oral cancer cell migration and colonisation by suppressing Akt/mTOR/NF-kappaB signalling. Life Sci. 2020, 261, 118372. [Google Scholar] [CrossRef]
- Shin, K.H.; Bae, S.D.; Hong, H.S.; Kim, R.H.; Kang, M.K.; Park, N.H. miR-181a shows tumor suppressive effect against oral squamous cell carcinoma cells by downregulating K-ras. Biochem. Biophys. Res. Commun. 2011, 404, 896–902. [Google Scholar] [CrossRef]
- Gwack, Y.; Srikanth, S.; Feske, S.; Cruz-Guilloty, F.; Oh-hora, M.; Neems, D.S.; Hogan, P.G.; Rao, A. Biochemical and functional characterization of Orai proteins. J. Biol. Chem. 2007, 282, 16232–16243. [Google Scholar] [CrossRef]
- Kim, K.D.; Srikanth, S.; Yee, M.K.; Mock, D.C.; Lawson, G.W.; Gwack, Y. ORAI1 deficiency impairs activated T cell death and enhances T cell survival. J. Immunol. 2011, 187, 3620–3630. [Google Scholar] [CrossRef]
- Chen, W.; Kang, K.L.; Alshaikh, A.; Varma, S.; Lin, Y.-L.; Shin, K.-H.; Kim, R.; Wang, C.-Y.; Park, N.-H.; Walentin, K.; et al. Grainyhead-like 2 (GRHL2) knockout abolishes oral cancer development through reciprocal regulation of the MAP kinase and TGF-β signaling pathways. Oncogenesis 2018, 7, 38. [Google Scholar] [CrossRef]
- Park, N.H.; Gujuluva, C.N.; Baek, J.H.; Cherrick, H.M.; Shin, K.H.; Min, B.M. Combined oral carcinogenicity of HPV-16 and benzo(a)pyrene: An in vitro multistep carcinogenesis model. Oncogene 1995, 10, 2145–2153. [Google Scholar] [PubMed]
- Najafi, M.; Farhood, B.; Mortezaee, K. Cancer stem cells (CSCs) in cancer progression and therapy. J. Cell Physiol. 2019, 234, 8381–8395. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.E.; Nguyen, A.; Kang, M.K.; Kim, R.H.; Park, N.H.; Shin, K.H. DYRK1A is required for maintenance of cancer stemness, contributing to tumorigenic potential in oral/oropharyngeal squamous cell carcinoma. Exp. Cell Res. 2021, 405, 112656. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Chang, J.T.; Andrechek, E.R.; Matsumura, N.; Baba, T.; Yao, G.; Kim, J.W.; Gatza, M.; Murphy, S.; Nevins, J.R. Anchorage-independent cell growth signature identifies tumors with metastatic potential. Oncogene 2009, 28, 2796–2805. [Google Scholar] [CrossRef]
- Lasorella, A.; Benezra, R.; Iavarone, A. The ID proteins: Master regulators of cancer stem cells and tumour aggressiveness. Nat. Rev. Cancer 2014, 14, 77–91. [Google Scholar] [CrossRef]
- Nayak, S.; Goel, M.M.; Makker, A.; Bhatia, V.; Chandra, S.; Kumar, S.; Agarwal, S.P. Fibroblast Growth Factor (FGF-2) and Its Receptors FGFR-2 and FGFR-3 May Be Putative Biomarkers of Malignant Transformation of Potentially Malignant Oral Lesions into Oral Squamous Cell Carcinoma. PLoS ONE 2015, 10, e0138801. [Google Scholar] [CrossRef]
- Ozretic, L.; Wagner, S.; Huebbers, C.U.; Gattenlohner, S.; Klussmann, J.P.; Beutner, D.; Zander, T.; Buettner, R.; Quaas, A. FGFR1 amplification and co-overexpression of c-MYC in oropharyngeal squamous cell carcinoma. Oral. Oncol. 2016, 54, e7–e9. [Google Scholar] [CrossRef]
- Dvorak, P.; Dvorakova, D.; Hampl, A. Fibroblast growth factor signaling in embryonic and cancer stem cells. FEBS Lett. 2006, 580, 2869–2874. [Google Scholar] [CrossRef]
- Gotoh, N. Control of stemness by fibroblast growth factor signaling in stem cells and cancer stem cells. Curr. Stem Cell Res. Ther. 2009, 4, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kieu, C.; Martin, C.E.; Han, J.; Chen, W.; Kim, J.S.; Kang, M.K.; Kim, R.H.; Park, N.H.; Kim, Y.; et al. NFATc3 plays an oncogenic role in oral/oropharyngeal squamous cell carcinomas by promoting cancer stemness via expression of OCT4. Oncotarget 2019, 10, 2306–2319. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.R.; Lee, S.H.; Rigas, N.K.; Kim, R.H.; Kang, M.K.; Park, N.H.; Shin, K.H. Elevated expression of JMJD6 is associated with oral carcinogenesis and maintains cancer stemness properties. Carcinogenesis 2016, 37, 119–128. [Google Scholar] [CrossRef]
- Patel, S.S.; Shah, K.A.; Shah, M.J.; Kothari, K.C.; Rawal, R.M. Cancer stem cells and stemness markers in oral squamous cell carcinomas. Asian Pac. J. Cancer Prev. 2014, 15, 8549–8556. [Google Scholar] [CrossRef]
- Meyer, M.J.; Fleming, J.M.; Lin, A.F.; Hussnain, S.A.; Ginsburg, E.; Vonderhaar, B.K. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. 2010, 70, 4624–4633. [Google Scholar] [CrossRef]
- Sanchez-Collado, J.; Jardin, I.; Lopez, J.J.; Ronco, V.; Salido, G.M.; Dubois, C.; Prevarskaya, N.; Rosado, J.A. Role of Orai3 in the Pathophysiology of Cancer. Int. J. Mol. Sci. 2021, 22, 11426. [Google Scholar] [CrossRef]
- Clay, M.R.; Tabor, M.; Owen, J.H.; Carey, T.E.; Bradford, C.R.; Wolf, G.T.; Wicha, M.S.; Prince, M.E. Single-Marker Identification of Head and Neck Squamous Cell Carcinoma Cancer Stem Cells with Aldehyde Dehydrogenase. Head Neck J. Sci. Spec. Head Neck 2010, 32, 1195–1201. [Google Scholar] [CrossRef]
- Ota, N.; Ohno, J.; Seno, K.; Taniguchi, K.; Ozeki, S. In vitro and in vivo expression of aldehyde dehydrogenase 1 in oral squamous cell carcinoma. Int. J. Oncol. 2014, 44, 435–442. [Google Scholar] [CrossRef]
- Richard, V.; Sebastian, P.; Nair, M.G.; Nair, S.N.; Malieckal, T.T.; Kumar, T.R.S.; Pillai, M.R. Multiple drug resistant, tumorigenic stem-like cells in oral cancer. Cancer Lett. 2013, 338, 300–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.F.; Miller, L.D.; Chan, X.B.; Black, M.A.; Pang, B.; Ong, C.W.; Salto-Tellez, M.; Liu, E.T.; Desai, K.V. JMJD6 is a driver of cellular proliferation and motility and a marker of poor prognosis in breast cancer. Breast Cancer Res. 2012, 14, 3001. [Google Scholar] [CrossRef]
- Davis, F.M.; Peters, A.A.; Grice, D.M.; Cabot, P.J.; Parat, M.O.; Roberts-Thomson, S.J.; Monteith, G.R. Non-stimulated, agonist-stimulated and store-operated Ca2+ influx in MDA-MB-468 breast cancer cells and the effect of EGF-induced EMT on calcium entry. PLoS ONE 2012, 7, e36923. [Google Scholar] [CrossRef] [PubMed]
- Perk, J.; Iavarone, A.; Benezra, R. Id family of helix-loop-helix proteins in cancer. Nat. Rev. Cancer 2005, 5, 603–614. [Google Scholar] [CrossRef]
- Zhao, Z.; Bo, Z.; Gong, W.; Guo, Y. Inhibitor of Differentiation 1 (Id1) in Cancer and Cancer Therapy. Int. J. Med. Sci. 2020, 17, 995–1005. [Google Scholar] [CrossRef]
- Lai, X.; Liao, J.; Lin, W.; Huang, C.; Li, J.; Lin, J.; Chen, Q.; Ye, Y. Inhibitor of DNA-binding protein 1 knockdown arrests the growth of colorectal cancer cells and suppresses hepatic metastasis in vivo. Oncol. Rep. 2014, 32, 79–88. [Google Scholar] [CrossRef]
- Cheng, Y.J.; Tsai, J.W.; Hsieh, K.C.; Yang, Y.C.; Chen, Y.J.; Huang, M.S.; Yuan, S.S. Id1 promotes lung cancer cell proliferation and tumor growth through Akt-related pathway. Cancer Lett. 2011, 307, 191–199. [Google Scholar] [CrossRef]
- Gumireddy, K.; Li, A.P.; Kossenkov, A.V.; Cai, K.Q.; Liu, Q.; Yan, J.C.; Xu, H.; Showe, L.; Zhang, L.; Huang, Q.H. ID1 Promotes Breast Cancer Metastasis by S100A9 Regulation. Mol. Cancer Res. 2014, 12, 1334–1343. [Google Scholar] [CrossRef]
- Sachdeva, R.; Wu, M.; Smiljanic, S.; Kaskun, O.; Ghannad-Zadeh, K.; Celebre, A.; Isaev, K.; Morrissy, A.S.; Guan, J.; Tong, J.F.; et al. ID1 Is Critical for Tumorigenesis and Regulates Chemoresistance in Glioblastoma. Cancer Res. 2019, 79, 4057–4071. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lai, X.; Yu, Y.; Li, J.; Cao, L.; Lin, W.; Huang, C.; Liao, J.; Chen, W.; Li, C.; et al. Inhibitor of DNA binding 1 (Id1) mediates stemness of colorectal cancer cells through the Id1-c-Myc-PLAC8 axis via the Wnt/beta-catenin and Shh signaling pathways. Cancer Manag. Res. 2019, 11, 6855–6869. [Google Scholar] [CrossRef]
- Shin, D.H.; Park, J.H.; Lee, J.Y.; Won, H.Y.; Jang, K.S.; Min, K.W.; Jang, S.H.; Woo, J.K.; Oh, S.H.; Kong, G. Overexpression of Id1 in transgenic mice promotes mammary basal stem cell activity and breast tumorigenesis. Oncotarget 2015, 6, 17276–17290. [Google Scholar] [CrossRef]
- Parekh, A.B.; Putney, J.W., Jr. Store-operated calcium channels. Physiol. Rev. 2005, 85, 757–810. [Google Scholar] [CrossRef]
- Mancini, M.; Toker, A. NFAT proteins: Emerging roles in cancer progression. Nat. Rev. Cancer 2009, 9, 810–820. [Google Scholar] [CrossRef] [PubMed]
- Daniel, C.; Gerlach, K.; Väth, M.; Neurath, M.F.; Weigmann, B. Nuclear factor of activated T cells—A transcription factor family as critical regulator in lung and colon cancer. Int. J. Cancer 2014, 134, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Chamlali, M.; Kouba, S.; Rodat-Despoix, L.; Todesca, L.M.; Petho, Z.; Schwab, A.; Ouadid-Ahidouch, H. Orai3 Calcium Channel Regulates Breast Cancer Cell Migration through Calcium-Dependent and -Independent Mechanisms. Cells 2021, 10, 3487. [Google Scholar] [CrossRef]
- Kim, K.D.; Srikanth, S.; Tan, Y.V.; Yee, M.K.; Jew, M.; Damoiseaux, R.; Jung, M.E.; Shimizu, S.; An, D.S.; Ribalet, B.; et al. Calcium signaling via Orai1 is essential for induction of the nuclear orphan receptor pathway to drive Th17 differentiation. J. Immunol. 2014, 192, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Mistry, H.; Hsieh, G.; Buhrlage, S.; Huang, M.; Park, E.; Cuny, G.; Galinsky, I.; Stone, R.M.; Gray, N.S.; Parmar, K.; et al. Small Molecule Inhibitors of USP1 Target ID1 Degradation in Leukemic Cells and Cause Cytotoxicity. Blood 2013, 122, 2906. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nguyen, A.; Sung, Y.; Lee, S.H.; Martin, C.E.; Srikanth, S.; Chen, W.; Kang, M.K.; Kim, R.H.; Park, N.-H.; Gwack, Y.; et al. Orai3 Calcium Channel Contributes to Oral/Oropharyngeal Cancer Stemness through the Elevation of ID1 Expression. Cells 2023, 12, 2225. https://doi.org/10.3390/cells12182225
Nguyen A, Sung Y, Lee SH, Martin CE, Srikanth S, Chen W, Kang MK, Kim RH, Park N-H, Gwack Y, et al. Orai3 Calcium Channel Contributes to Oral/Oropharyngeal Cancer Stemness through the Elevation of ID1 Expression. Cells. 2023; 12(18):2225. https://doi.org/10.3390/cells12182225
Chicago/Turabian StyleNguyen, Anthony, Youngjae Sung, Sung Hee Lee, Charlotte Ellen Martin, Sonal Srikanth, Wei Chen, Mo K. Kang, Reuben H. Kim, No-Hee Park, Yousang Gwack, and et al. 2023. "Orai3 Calcium Channel Contributes to Oral/Oropharyngeal Cancer Stemness through the Elevation of ID1 Expression" Cells 12, no. 18: 2225. https://doi.org/10.3390/cells12182225
APA StyleNguyen, A., Sung, Y., Lee, S. H., Martin, C. E., Srikanth, S., Chen, W., Kang, M. K., Kim, R. H., Park, N.-H., Gwack, Y., Kim, Y., & Shin, K.-H. (2023). Orai3 Calcium Channel Contributes to Oral/Oropharyngeal Cancer Stemness through the Elevation of ID1 Expression. Cells, 12(18), 2225. https://doi.org/10.3390/cells12182225