
PFKFB3-Mediated Glycolysis Boosts Fibroblast Activation and Subsequent Kidney Fibrosis
 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Single-Cell Data Sequencing Analysis
2.2. Animals
2.3. Unilateral Ureteral Obstruction (UUO) Mouse Model
2.4. Unilateral Kidney Ischemia/Reperfusion Mouse Model
2.5. Cells
2.6. Reverse Transcription and Quantitative Real-Time PCR (RT-qPCR)
2.7. Western Blot Analysis
2.8. Immunofluorescence
2.9. Immunohistochemistry
2.10. Histological Staining
2.11. Metabolomics Assay
2.12. Statistical Analysis
3. Results
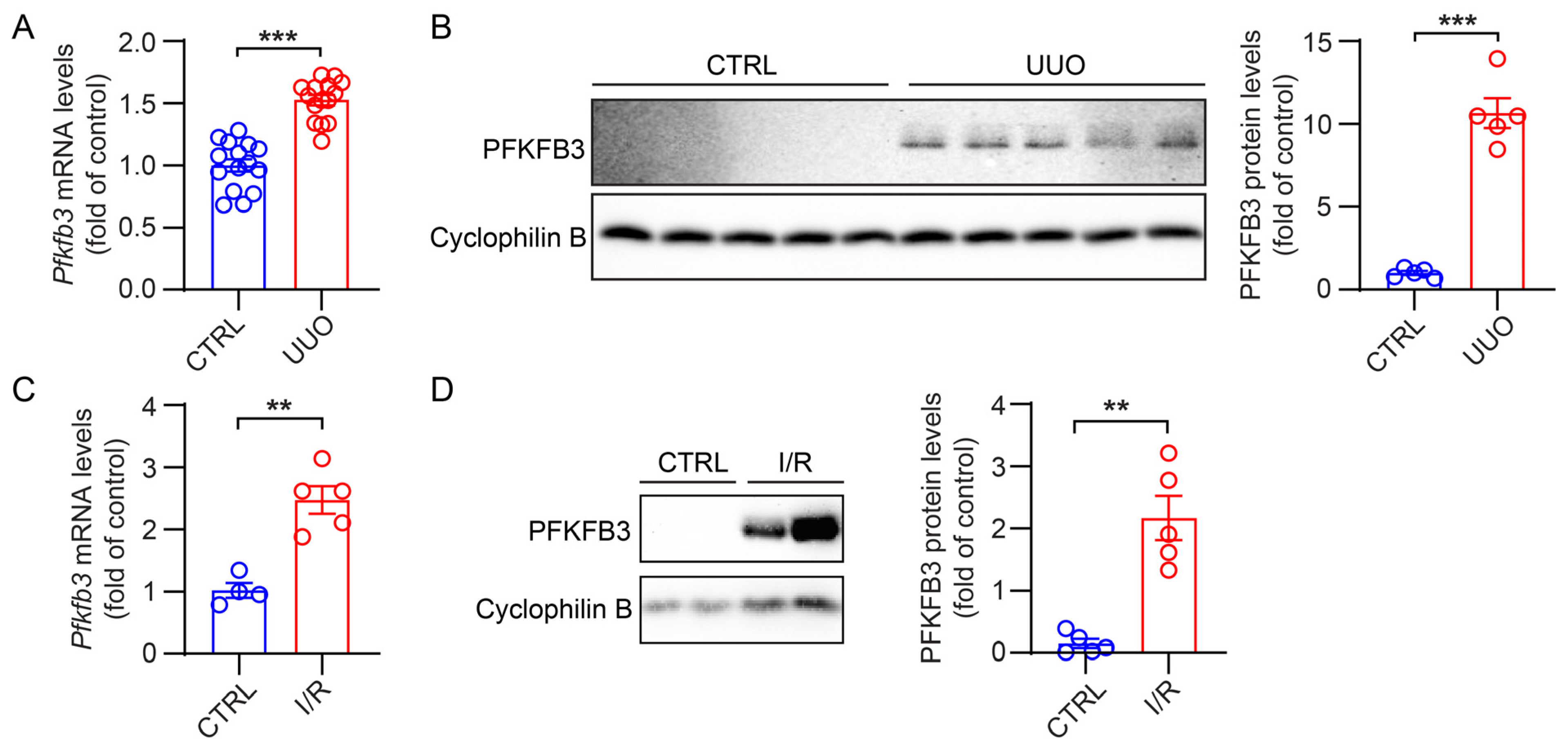
3.1. Induction of PFKFB3 Expression in Fibrotic Kidneys
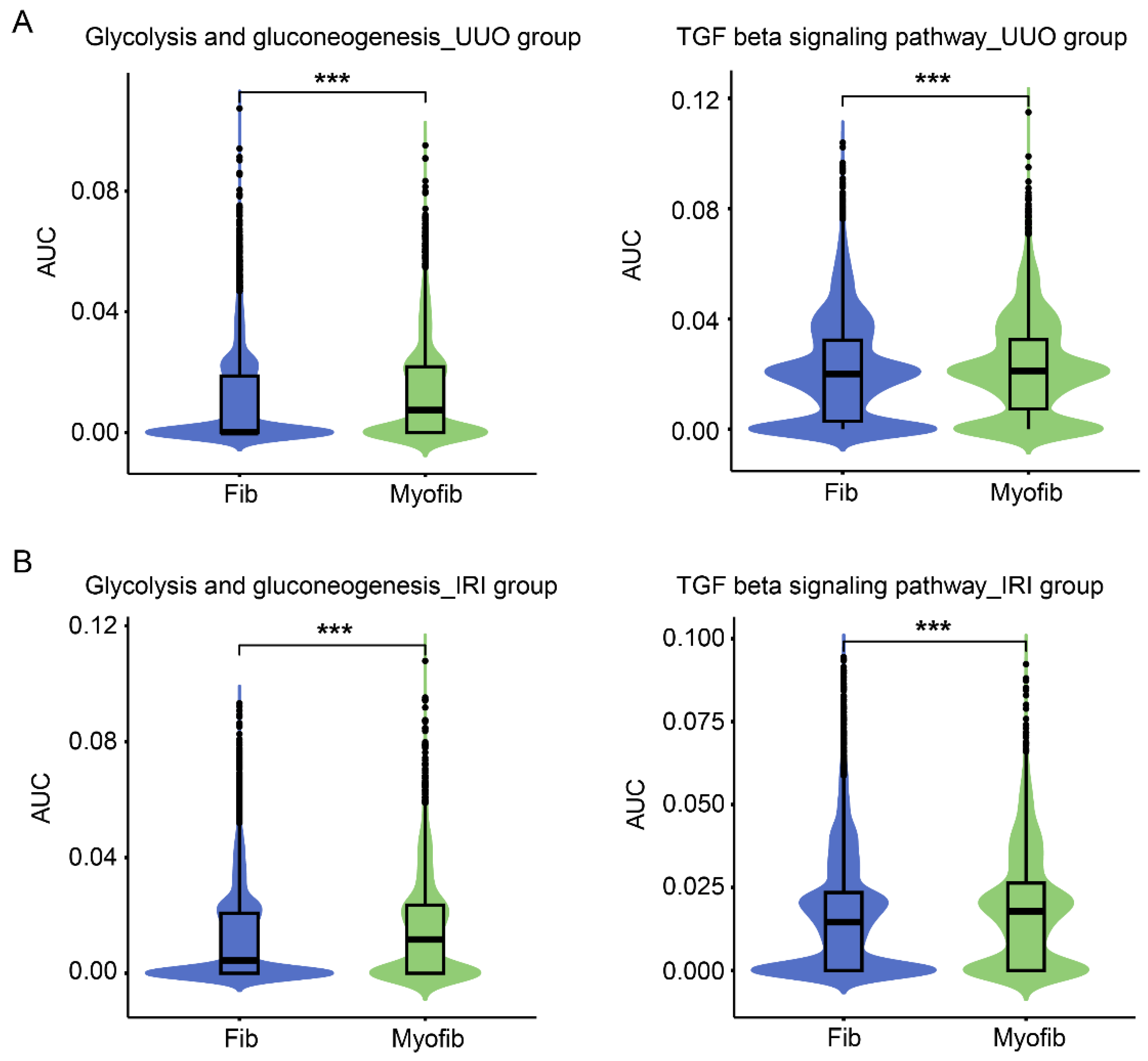
3.2. Increased Glycolysis and TGFβ Pathways in Renal Myofibroblasts
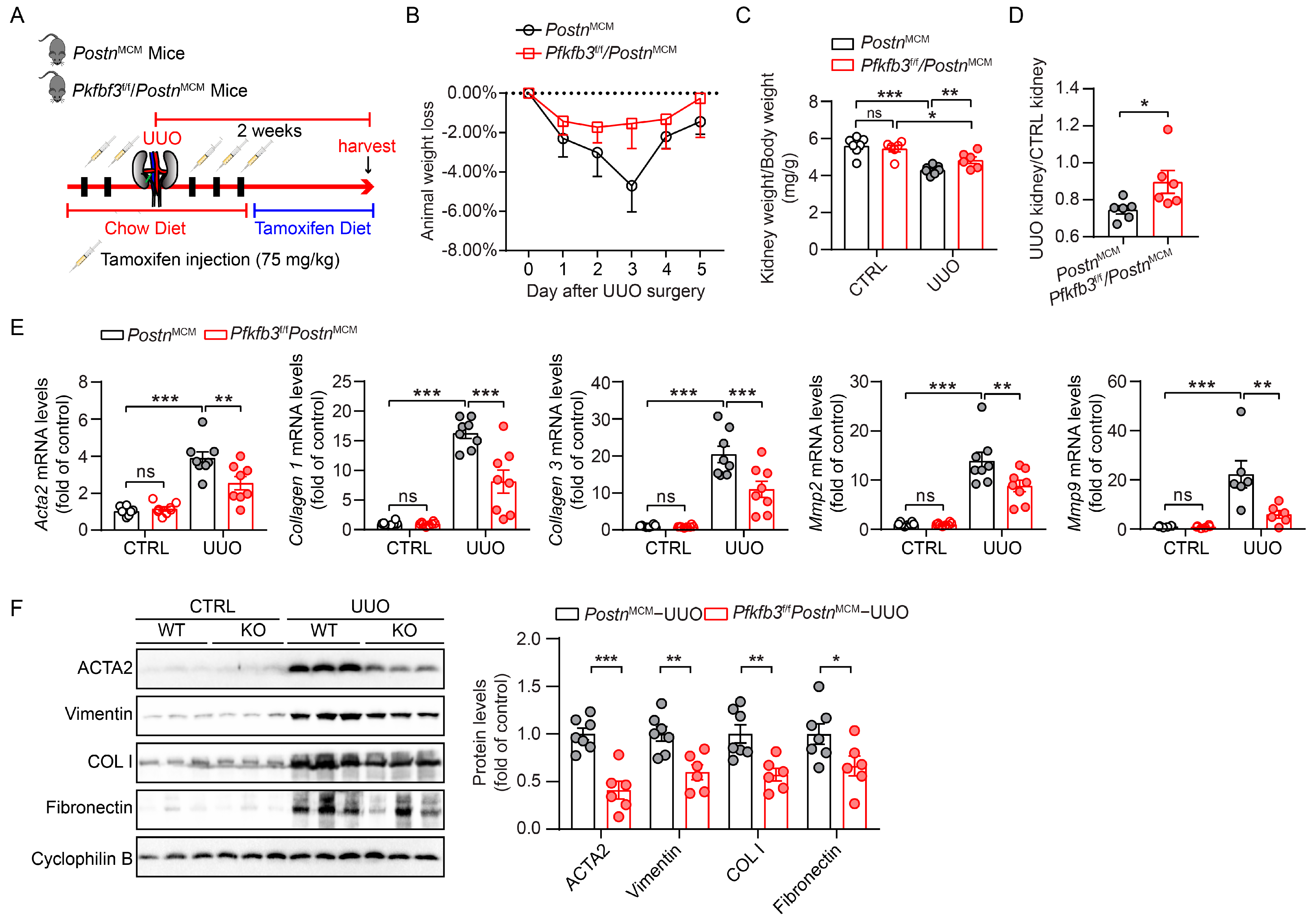
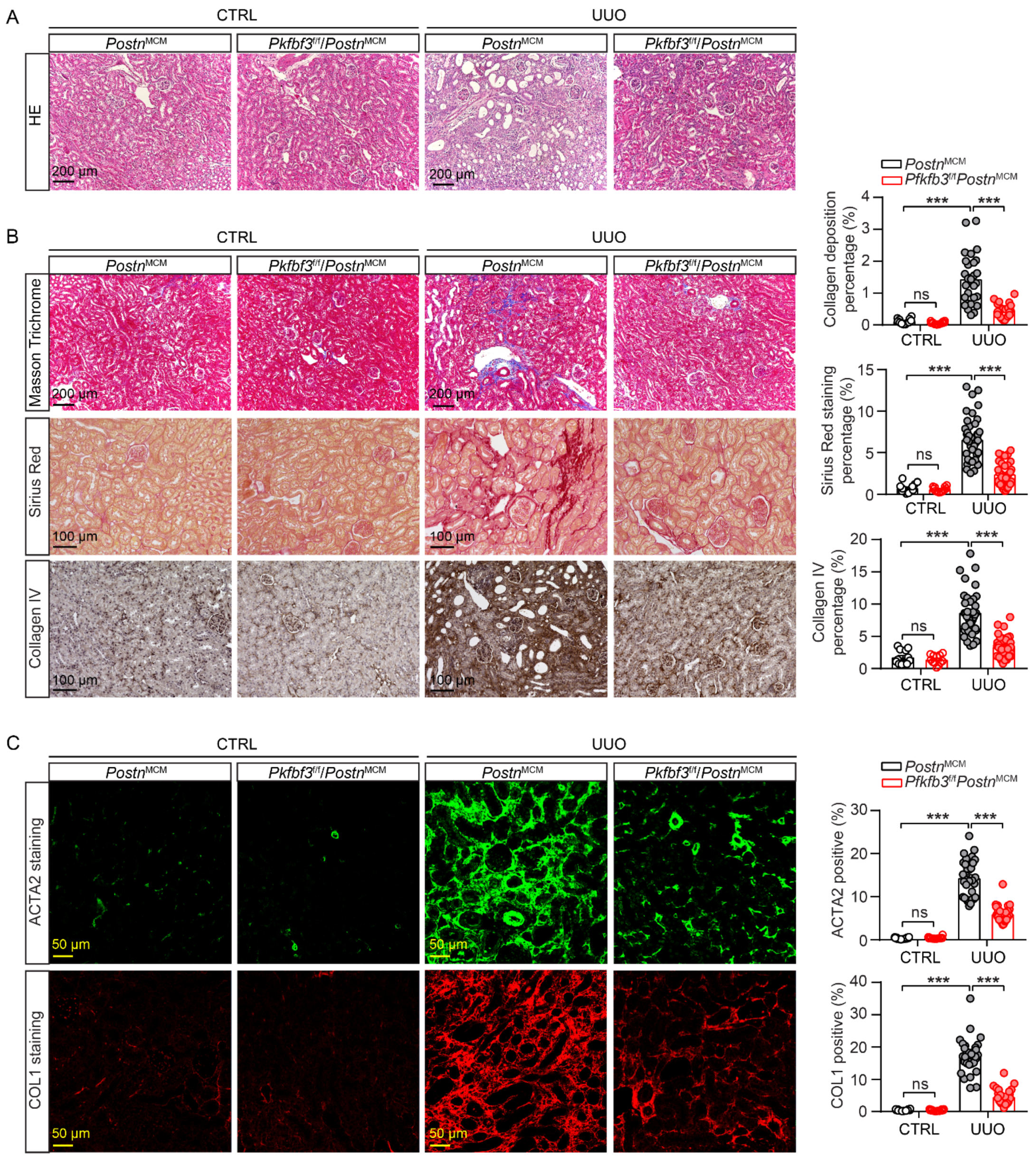
3.3. Myofibroblast-Specific Pfkfb3 Knockout Reduces Kidney Atrophy and Renal Fibrosis following UUO
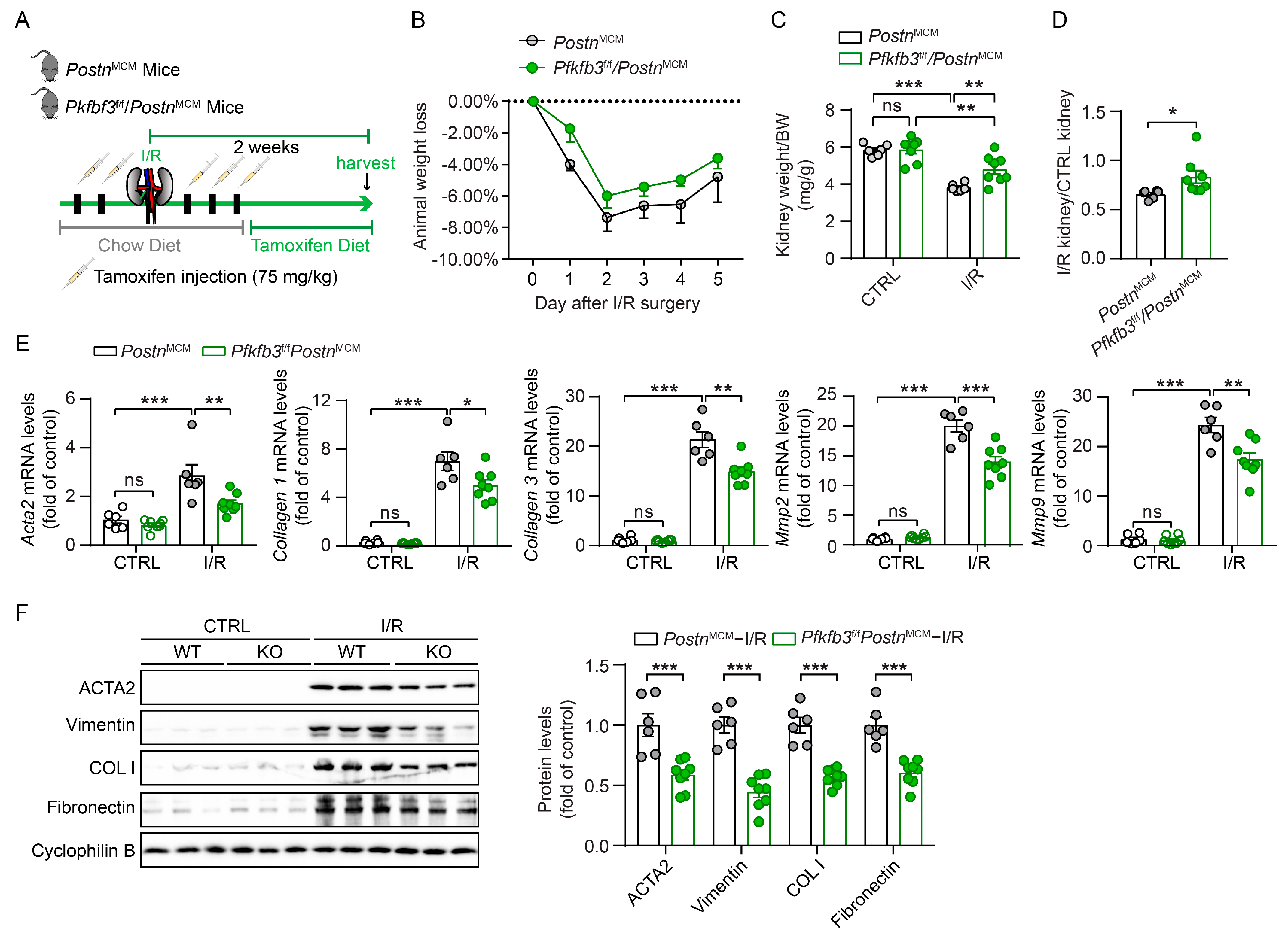
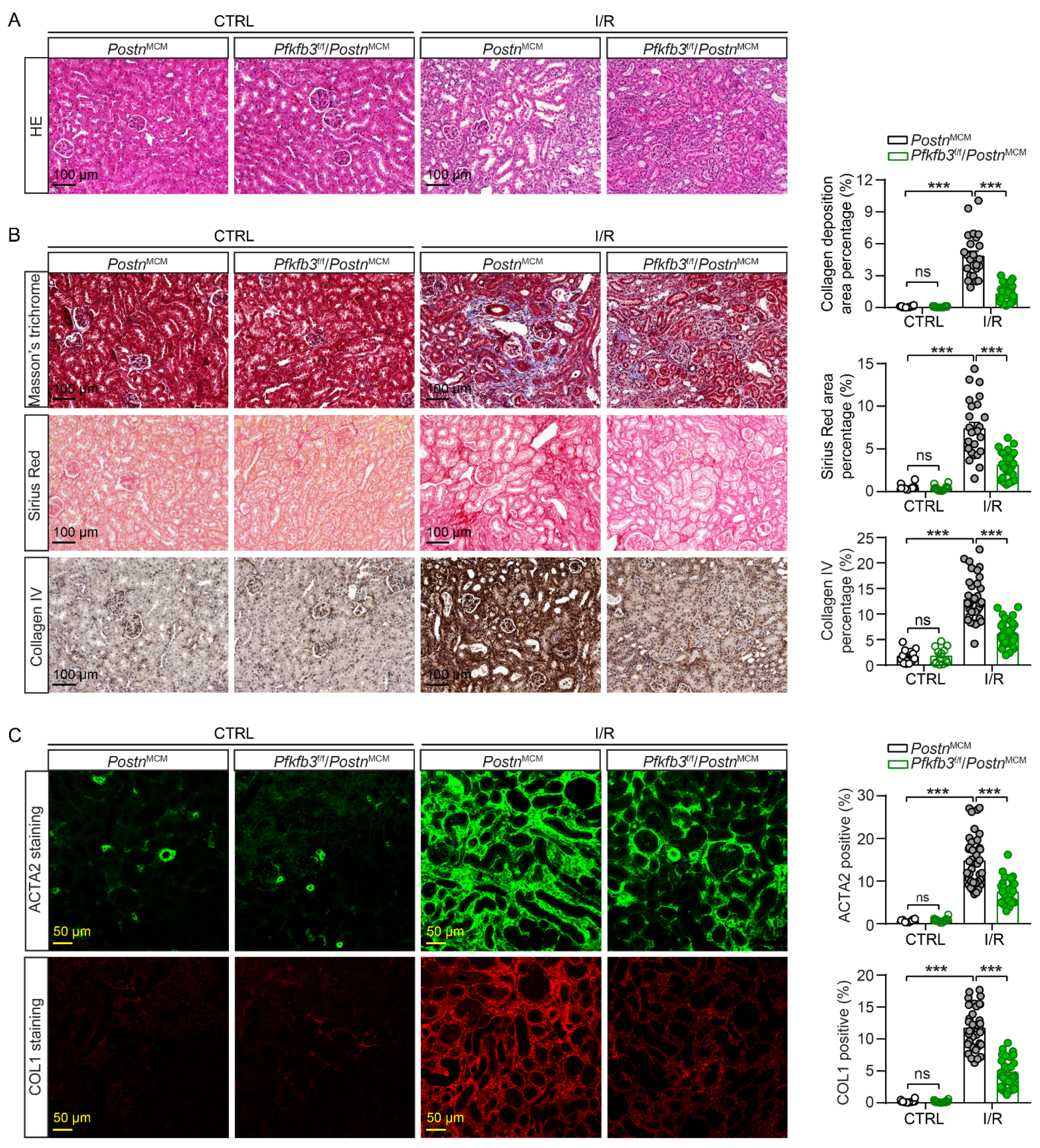
3.4. Myofibroblast-Specific Pfkfb3 Knockout Suppresses Kidney Atrophy and Fibrosis after Kidney Ischemia/Reperfusion
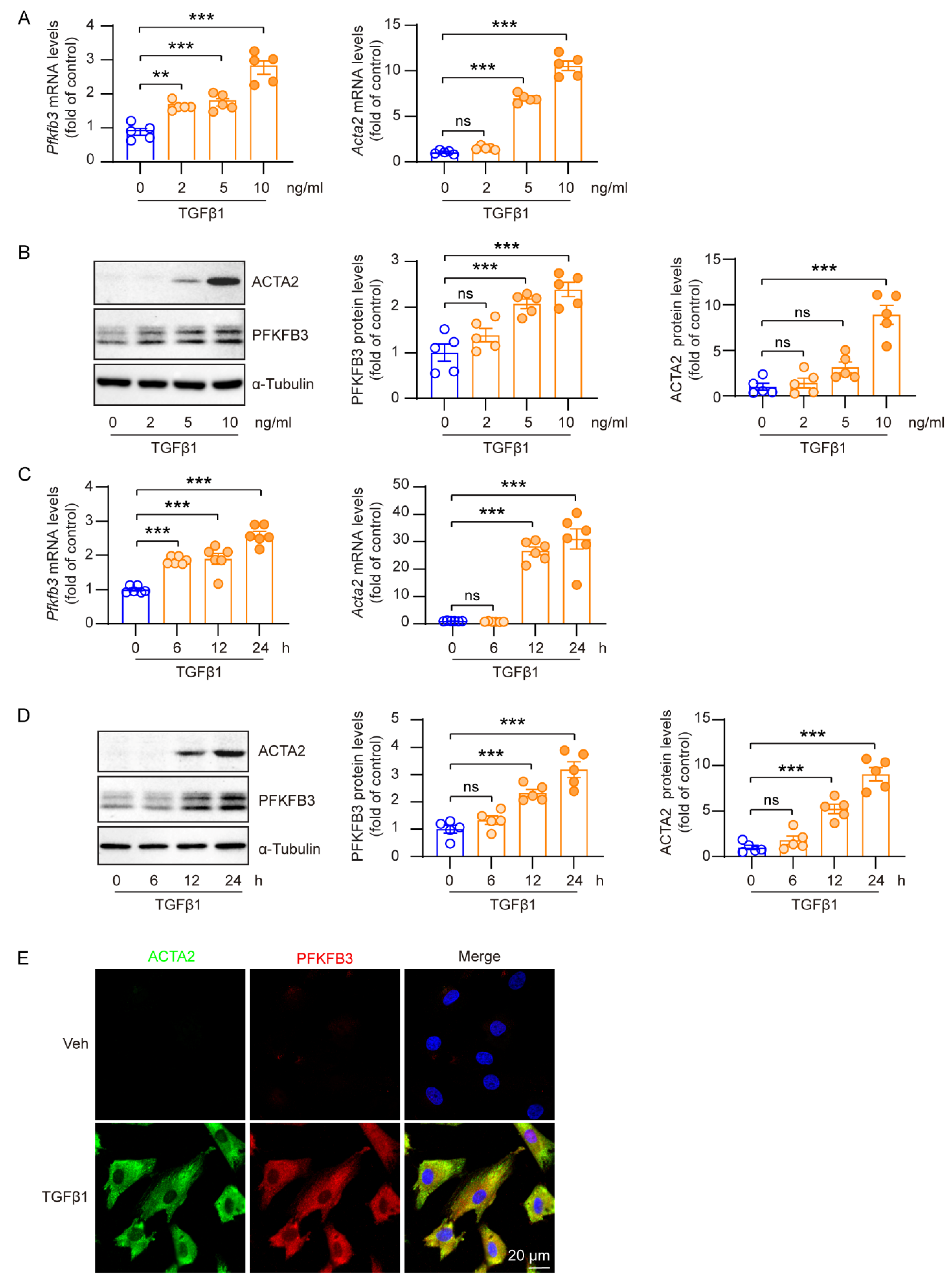
3.5. Increased PFKFB3 Expression in TGFβ1-Activated Renal Fibroblasts
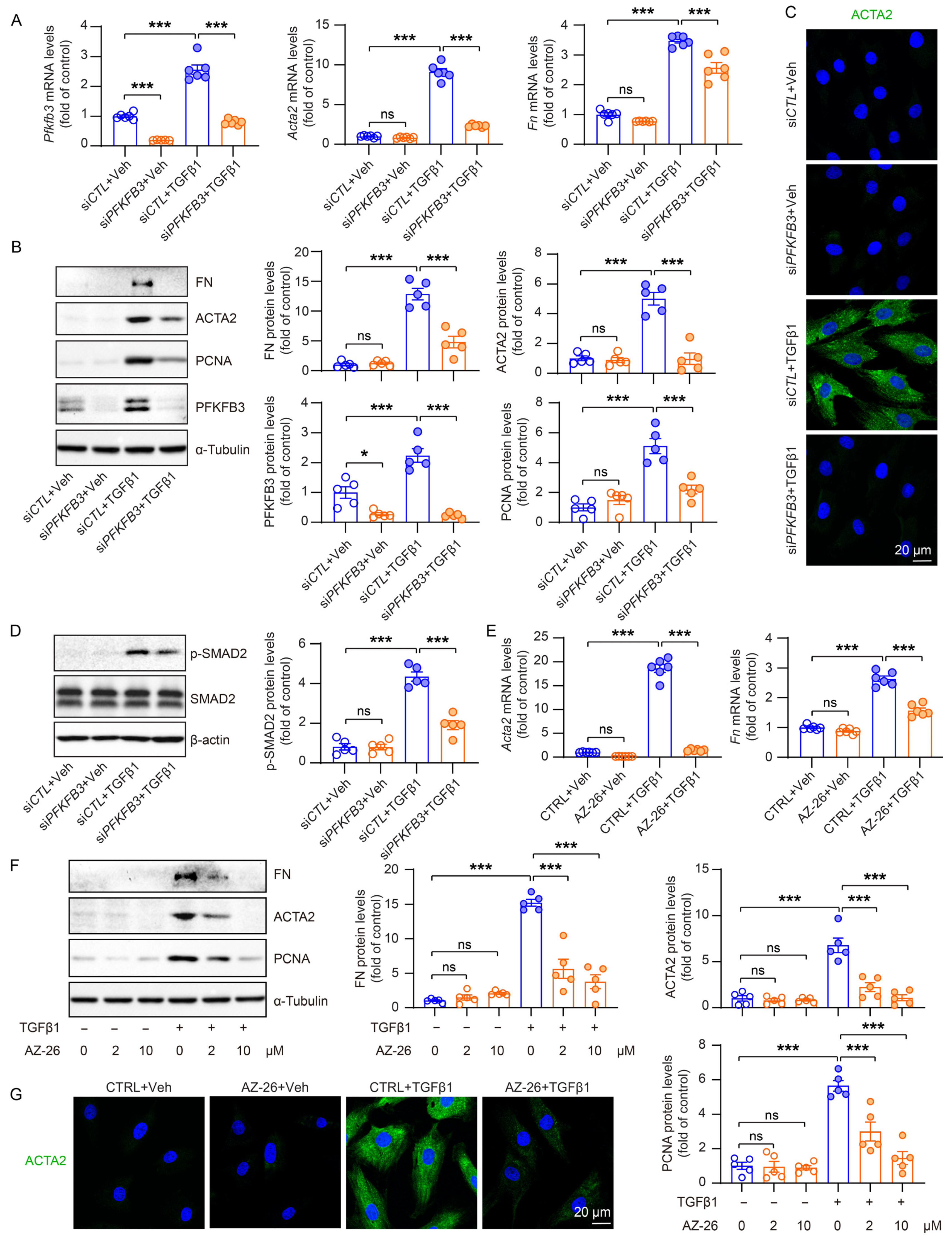
3.6. PFKFB3 Induction Promotes TGFβ1-Induced Renal Fibroblast Activation
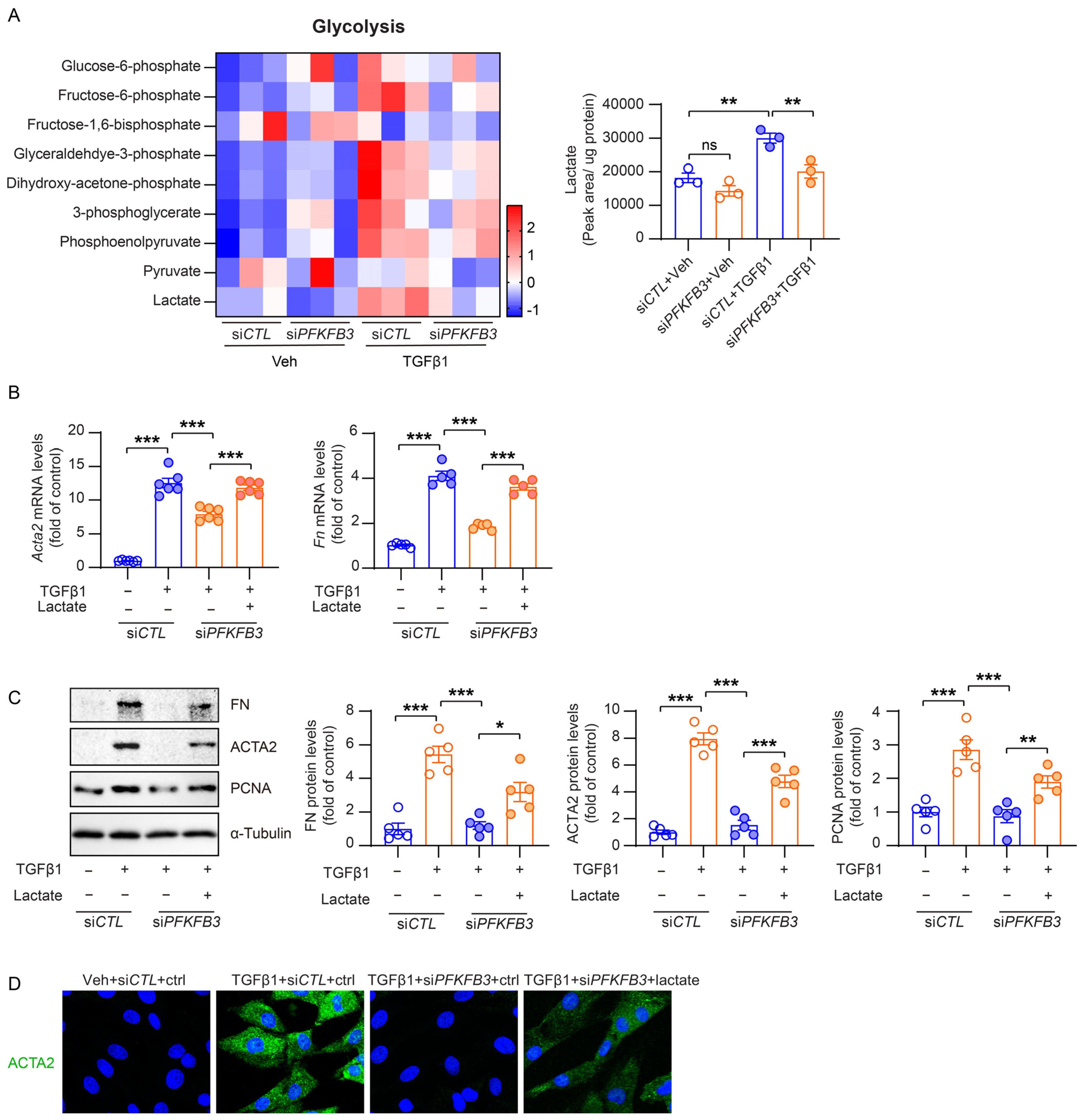
3.7. PFKFB3-Medicated Glycolysis Induction Is Responsible for Renal Fibroblast Activation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Rockey, D.C.; Bell, P.D.; Hill, J.A. Fibrosis—A common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [PubMed]
- Sato, Y.; Yanagita, M. Resident fibroblasts in the kidney: A major driver of fibrosis and inflammation. Inflamm. Regen. 2017, 37, 17. [Google Scholar]
- Cao, Q.; Wang, Y.; Harris, D.C. Macrophage heterogeneity, phenotypes, and roles in renal fibrosis. Kidney Int. Suppl. 2014, 4, 16–19. [Google Scholar] [CrossRef]
- Meng, X.M.; Wang, S.; Huang, X.R.; Yang, C.; Xiao, J.; Zhang, Y.; To, K.F.; Nikolic-Paterson, D.J.; Lan, H.Y. Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 2016, 7, e2495. [Google Scholar] [CrossRef] [PubMed]
- Kuppe, C.; Ibrahim, M.M.; Kranz, J.; Zhang, X.; Ziegler, S.; Perales-Paton, J.; Jansen, J.; Reimer, K.C.; Smith, J.R.; Dobie, R.; et al. Decoding myofibroblast origins in human kidney fibrosis. Nature 2021, 589, 281–286. [Google Scholar]
- Mack, M.; Yanagita, M. Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 2015, 87, 297–307. [Google Scholar]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar]
- LeBleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Zhu, X.; Jiang, L.; Long, M.; Wei, X.; Hou, Y.; Du, Y. Metabolic Reprogramming and Renal Fibrosis. Front. Med. 2021, 8, 746920. [Google Scholar]
- Liu, H.; Li, W.; He, Q.; Xue, J.; Wang, J.; Xiong, C.; Pu, X.; Nie, Z. Mass Spectrometry Imaging of Kidney Tissue Sections of Rat Subjected to Unilateral Ureteral Obstruction. Sci. Rep. 2017, 7, 41954. [Google Scholar] [PubMed]
- Lan, R.; Geng, H.; Singha, P.K.; Saikumar, P.; Bottinger, E.P.; Weinberg, J.M.; Venkatachalam, M.A. Mitochondrial Pathology and Glycolytic Shift during Proximal Tubule Atrophy after Ischemic AKI. J. Am. Soc. Nephrol. 2016, 27, 3356–3367. [Google Scholar] [PubMed]
- Qi, W.; Keenan, H.A.; Li, Q.; Ishikado, A.; Kannt, A.; Sadowski, T.; Yorek, M.A.; Wu, I.-H.; Lockhart, S.; Coppey, L.J.; et al. Pyruvate kinase M2 activation may protect against the progression of diabetic glomerular pathology and mitochondrial dysfunction. Nat. Med. 2017, 23, 753–762. [Google Scholar] [PubMed]
- Ding, H.; Jiang, L.; Xu, J.; Bai, F.; Zhou, Y.; Yuan, Q.; Luo, J.; Zen, K.; Yang, J. Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 2017, 313, F561–F575. [Google Scholar]
- Wei, Q.; Su, J.; Dong, G.; Zhang, M.; Huo, Y.; Dong, Z. Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. Physiol. Ren. Physiol. 2019, 316, F1162–F1172. [Google Scholar]
- Van Schaftingen, E.; Lederer, B.; Bartrons, R.; Hers, H.G. A kinetic study of pyrophosphate: Fructose-6-phosphate phosphotransferase from potato tubers. Application to a microassay of fructose 2,6-bisphosphate. Eur. J. Biochem. 1982, 129, 191–195. [Google Scholar] [CrossRef]
- Ghesquiere, B.; Wong, B.W.; Kuchnio, A.; Carmeliet, P. Metabolism of stromal and immune cells in health and disease. Nature 2014, 511, 167–176. [Google Scholar]
- Wang, L.; Zhang, X.; Cao, Y.; Ma, Q.; Mao, X.; Xu, J.; Yang, Q.; Zhou, Y.; Lucas, R.; Fulton, D.J.; et al. Mice with a specific deficiency of Pfkfb3 in myeloid cells are protected from hypoxia-induced pulmonary hypertension. Br. J. Pharmacol. 2021, 178, 1055–1072. [Google Scholar] [CrossRef]
- Liu, Z.; Mao, X.; Yang, Q.; Zhang, X.; Xu, J.; Ma, Q.; Zhou, Y.; Da, Q.; Cai, Y.; Sopeyin, A.; et al. Suppression of myeloid PFKFB3-driven glycolysis protects mice from choroidal neovascularization. Br. J. Pharmacol. 2022, 179, 5109–5131. [Google Scholar] [CrossRef]
- Liu, Z.; Xu, J.; Ma, Q.; Zhang, X.; Yang, Q.; Wang, L.; Cao, Y.; Xu, Z.; Tawfik, A.; Sun, Y.; et al. Glycolysis links reciprocal activation of myeloid cells and endothelial cells in the retinal angiogenic niche. Sci. Transl. Med. 2020, 12, eaay1371. [Google Scholar]
- Guo, S.; Li, A.; Fu, X.; Li, Z.; Cao, K.; Song, M.; Huang, S.; Li, Z.; Yan, J.; Wang, L.; et al. Gene-dosage effect of Pfkfb3 on monocyte/macrophage biology in atherosclerosis. Br. J. Pharmacol. 2022, 179, 4974–4991. [Google Scholar] [PubMed]
- Xie, N.; Tan, Z.; Banerjee, S.; Cui, H.; Ge, J.; Liu, R.M.; Bernard, K.; Thannickal, V.J.; Liu, G. Glycolytic Reprogramming in Myofibroblast Differentiation and Lung Fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1462–1474. [Google Scholar] [PubMed]
- Wen, L.; Wei, Q.; Livingston, M.J.; Dong, G.; Li, S.; Hu, X.; Li, Y.; Huo, Y.; Dong, Z. PFKFB3 mediates tubular cell death in cisplatin nephrotoxicity by activating CDK4. Transl. Res. 2023, 253, 31–40. [Google Scholar] [PubMed]
- Li, H.; Dixon, E.E.; Wu, H.; Humphreys, B.D. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022, 34, 1977–1998.e9. [Google Scholar]
- Aibar, S.; Gonzalez-Blas, C.B.; Moerman, T.; Huynh-Thu, V.A.; Imrichova, H.; Hulselmans, G.; Rambow, F.; Marine, J.-C.; Geurts, P.; Aerts, J.; et al. SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 2017, 14, 1083–1086. [Google Scholar]
- Xu, Y.; An, X.; Guo, X.; Habtetsion, T.G.; Wang, Y.; Xu, X.; Kandala, S.; Li, Q.; Li, H.; Zhang, C.; et al. Endothelial PFKFB3 plays a critical role in angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1231–1239. [Google Scholar] [CrossRef]
- Livingston, M.J.; Shu, S.; Fan, Y.; Li, Z.; Jiao, Q.; Yin, X.M.; Venkatachalam, M.A.; Dong, Z. Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis. Autophagy 2023, 19, 256–277. [Google Scholar] [CrossRef]
- Yang, Q.; Ma, Q.; Xu, J.; Liu, Z.; Mao, X.; Zhou, Y.; Cai, Y.; Da, Q.; Hong, M.; Weintraub, N.L.; et al. Endothelial AMPKα1/PRKAA1 exacerbates inflammation in HFD-fed mice. Br. J. Pharmacol. 2022, 179, 1661–1678. [Google Scholar]
- Yang, Q.; Xu, J.; Ma, Q.; Liu, Z.; Zhou, Y.; Cai, Y.; Mao, X.; Stepp, D.; Weintraub, N.; Fulton, D.J.; et al. Disruption of endothelial Pfkfb3 ameliorates diet-induced murine insulin resistance. J. Endocrinol. 2021, 250, 93–104. [Google Scholar]
- Yang, Q.; Xu, J.; Ma, Q.; Liu, Z.; Sudhahar, V.; Cao, Y.; Wang, L.; Zeng, X.; Zhou, Y.; Zhang, M.; et al. PRKAA1/AMPKα1-driven glycolysis in endothelial cells exposed to disturbed flow protects against atherosclerosis. Nat. Commun. 2018, 9, 4667. [Google Scholar]
- Ma, Q.; Yang, Q.; Xu, J.; Zhang, X.; Kim, D.; Liu, Z.; Da, Q.; Mao, X.; Zhou, Y.; Cai, Y.; et al. ATIC-Associated De Novo Purine Synthesis Is Critically Involved in Proliferative Arterial Disease. Circulation 2022, 146, 1444–1460. [Google Scholar] [CrossRef]
- Haug, K.; Cochrane, K.; Nainala, V.C.; Williams, M.; Chang, J.; Jayaseelan, K.V.; O’donovan, C. MetaboLights: A resource evolving in response to the needs of its scientific community. Nucleic Acids Res. 2020, 48, D440–D444. [Google Scholar] [CrossRef] [PubMed]
- Gewin, L. The many talents of transforming growth factor-beta in the kidney. Curr. Opin. Nephrol. Hypertens. 2019, 28, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef]
- Edeling, M.; Ragi, G.; Huang, S.; Pavenstadt, H.; Susztak, K. Developmental signalling pathways in renal fibrosis: The roles of Notch, Wnt and Hedgehog. Nat. Rev. Nephrol. 2016, 12, 426–439. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhou, L.; Wang, Y.; Miao, J.; Hong, X.; Hou, F.F.; Liu, Y. (Pro)renin Receptor Is an Amplifier of Wnt/beta-Catenin Signaling in Kidney Injury and Fibrosis. J. Am. Soc. Nephrol. 2017, 28, 2393–2408. [Google Scholar] [CrossRef]
- Tan, R.J.; Zhou, D.; Zhou, L.; Liu, Y. Wnt/beta-catenin signaling and kidney fibrosis. Kidney Int. Suppl. 2014, 4, 84–90. [Google Scholar] [CrossRef]
- Ding, H.; Zhou, D.; Hao, S.; Zhou, L.; He, W.; Nie, J.; Hou, F.F.; Liu, Y. Sonic hedgehog signaling mediates epithelial-mesenchymal communication and promotes renal fibrosis. J. Am. Soc. Nephrol. 2012, 23, 801–813. [Google Scholar] [CrossRef]
- Haase, V.H. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis Tissue Repair 2012, 5 (Suppl. S1), S16. [Google Scholar] [CrossRef]
- Higgins, D.F.; Kimura, K.; Bernhardt, W.M.; Shrimanker, N.; Akai, Y.; Hohenstein, B.; Saito, Y.; Johnson, R.S.; Kretzler, M.; Cohen, C.D.; et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J. Clin. Investig. 2007, 117, 3810–3820. [Google Scholar] [CrossRef]
- Minchenko, A.; Leshchinsky, I.; Opentanova, I.; Sang, N.; Srinivas, V.; Armstead, V.; Caro, J. Hypoxia-inducible factor-1-mediated expression of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase-3 (PFKFB3) gene. Its possible role in the Warburg effect. J. Biol. Chem. 2002, 277, 6183–6187. [Google Scholar]
- Leung, R.W.H.; Lee, T.K.W. Wnt/beta-Catenin Signaling as a Driver of Stemness and Metabolic Reprogramming in Hepatocellular Carcinoma. Cancers 2022, 14, 5468. [Google Scholar] [PubMed]
- Esen, E.; Chen, J.; Karner, C.M.; Okunade, A.L.; Patterson, B.W.; Long, F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013, 17, 745–755. [Google Scholar] [PubMed]
- De Bock, K.; Georgiadou, M.; Schoors, S.; Kuchnio, A.; Wong, B.W.; Cantelmo, A.R.; Quaegebeur, A.; Ghesquière, B.; Cauwenberghs, S.; Eelen, G.; et al. Role of PFKFB3-driven glycolysis in vessel sprouting. Cell 2013, 154, 651–663. [Google Scholar] [PubMed]
- Delle, H.; Rocha, J.R.; Cavaglieri, R.C.; Vieira, J.M., Jr.; Malheiros, D.M.; Noronha, I.L. Antifibrotic effect of tamoxifen in a model of progressive renal disease. J. Am. Soc. Nephrol. 2012, 23, 37–48. [Google Scholar]
- Kim, C.S.; Kim, I.J.; Choi, J.S.; Bae, E.H.; Ma, S.K.; Kim, S.W. Tamoxifen ameliorates obstructive nephropathy through Src and the PI3K/Akt/mTOR pathway. Biol. Cell 2019, 111, 18–27. [Google Scholar]
- Zhou, D.; Fu, H.; Liu, S.; Zhang, L.; Xiao, L.; Bastacky, S.I.; Liu, Y. Early activation of fibroblasts is required for kidney repair and regeneration after injury. FASEB J. 2019, 33, 12576–12587. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Huo, E.; Cai, Y.; Zhang, Z.; Dong, C.; Asara, J.M.; Wei, Q. PFKFB3-Mediated Glycolysis Boosts Fibroblast Activation and Subsequent Kidney Fibrosis. Cells 2023, 12, 2081. https://doi.org/10.3390/cells12162081
Yang Q, Huo E, Cai Y, Zhang Z, Dong C, Asara JM, Wei Q. PFKFB3-Mediated Glycolysis Boosts Fibroblast Activation and Subsequent Kidney Fibrosis. Cells. 2023; 12(16):2081. https://doi.org/10.3390/cells12162081
Chicago/Turabian StyleYang, Qiuhua, Emily Huo, Yongfeng Cai, Zhidan Zhang, Charles Dong, John M. Asara, and Qingqing Wei. 2023. "PFKFB3-Mediated Glycolysis Boosts Fibroblast Activation and Subsequent Kidney Fibrosis" Cells 12, no. 16: 2081. https://doi.org/10.3390/cells12162081
APA StyleYang, Q., Huo, E., Cai, Y., Zhang, Z., Dong, C., Asara, J. M., & Wei, Q. (2023). PFKFB3-Mediated Glycolysis Boosts Fibroblast Activation and Subsequent Kidney Fibrosis. Cells, 12(16), 2081. https://doi.org/10.3390/cells12162081