
Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes


 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Mouse Spinal Cord Astrocyte Primary Cell Cultures
2.3. Western Blot
2.4. Immunofluorescence Experiments
2.5. RT-qPCR
2.6. Cytosolic Calcium Concentration
2.7. In Vitro Treatments with a Specific mGluR5 Antisense Oligonucleotide and a Selective mGluR5 Negative Allosteric Modulator
2.8. Evaluation of the Energetic Profile
2.9. Enzyme-Linked Immunosorbent Assay
2.10. Glutamate Quantification
2.11. Spinal Cord Motor Neuron Preparation and Co-Cultures with Astrocytes
2.12. Statistics
3. Results
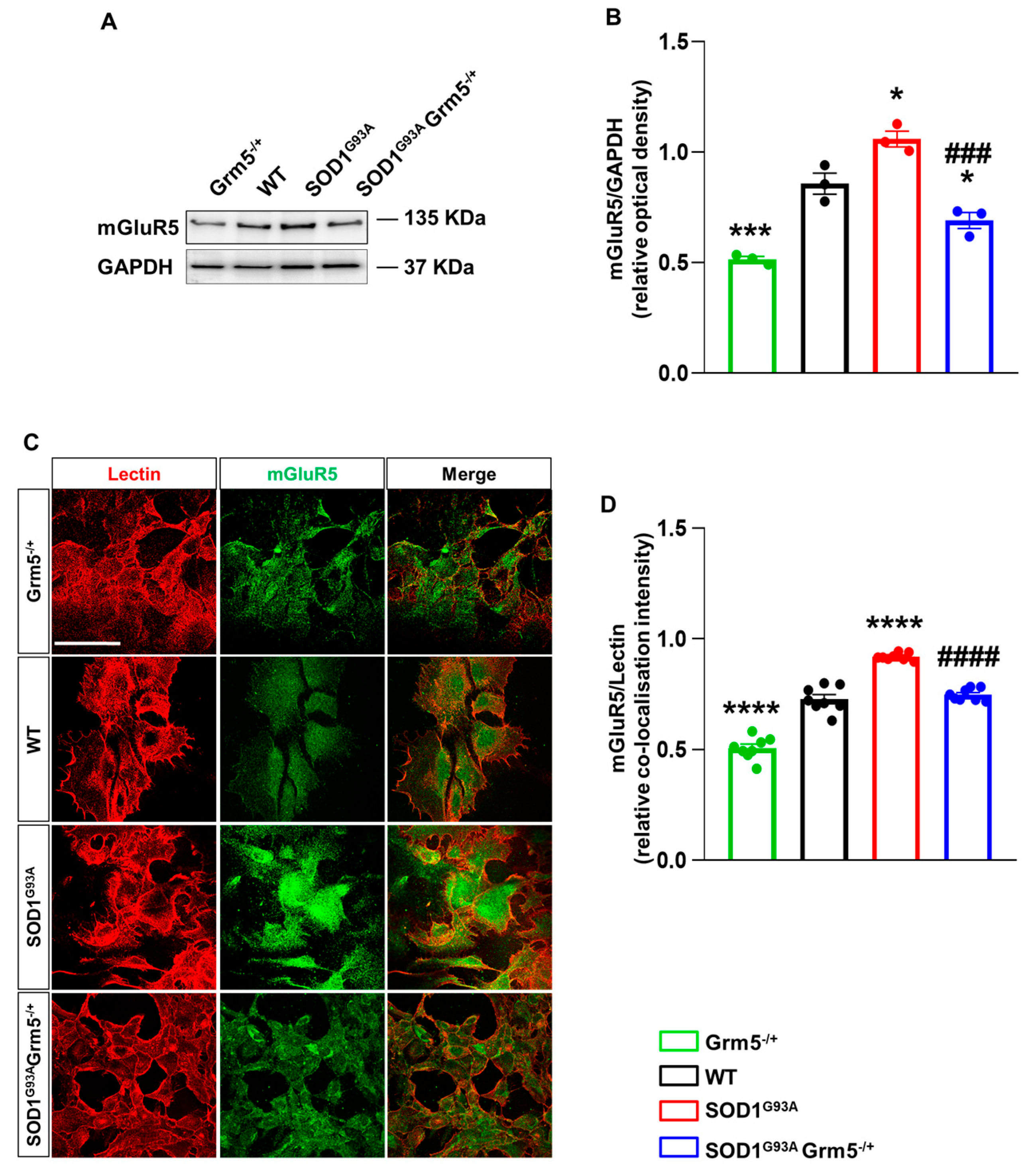
3.1. Genetic mGluR5 Ablation Reduces the Receptor Expression in SOD1G93A Spinal Cord Astrocytes
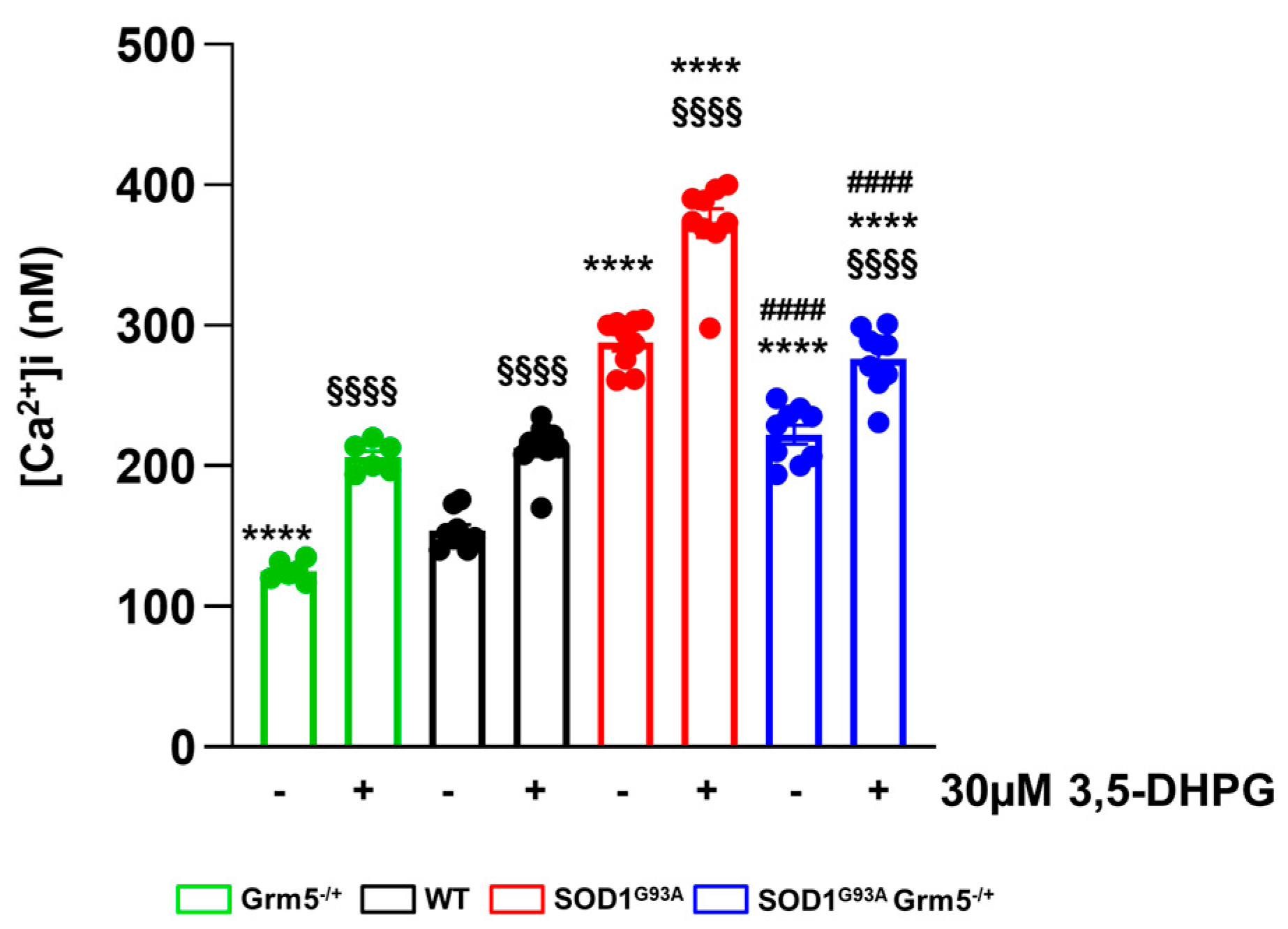
3.2. Downregulation of mGluR5 Reduces the Elevated Cytoplasmic Ca2+ Levels in SOD1G93A Spinal Cord Astrocytes
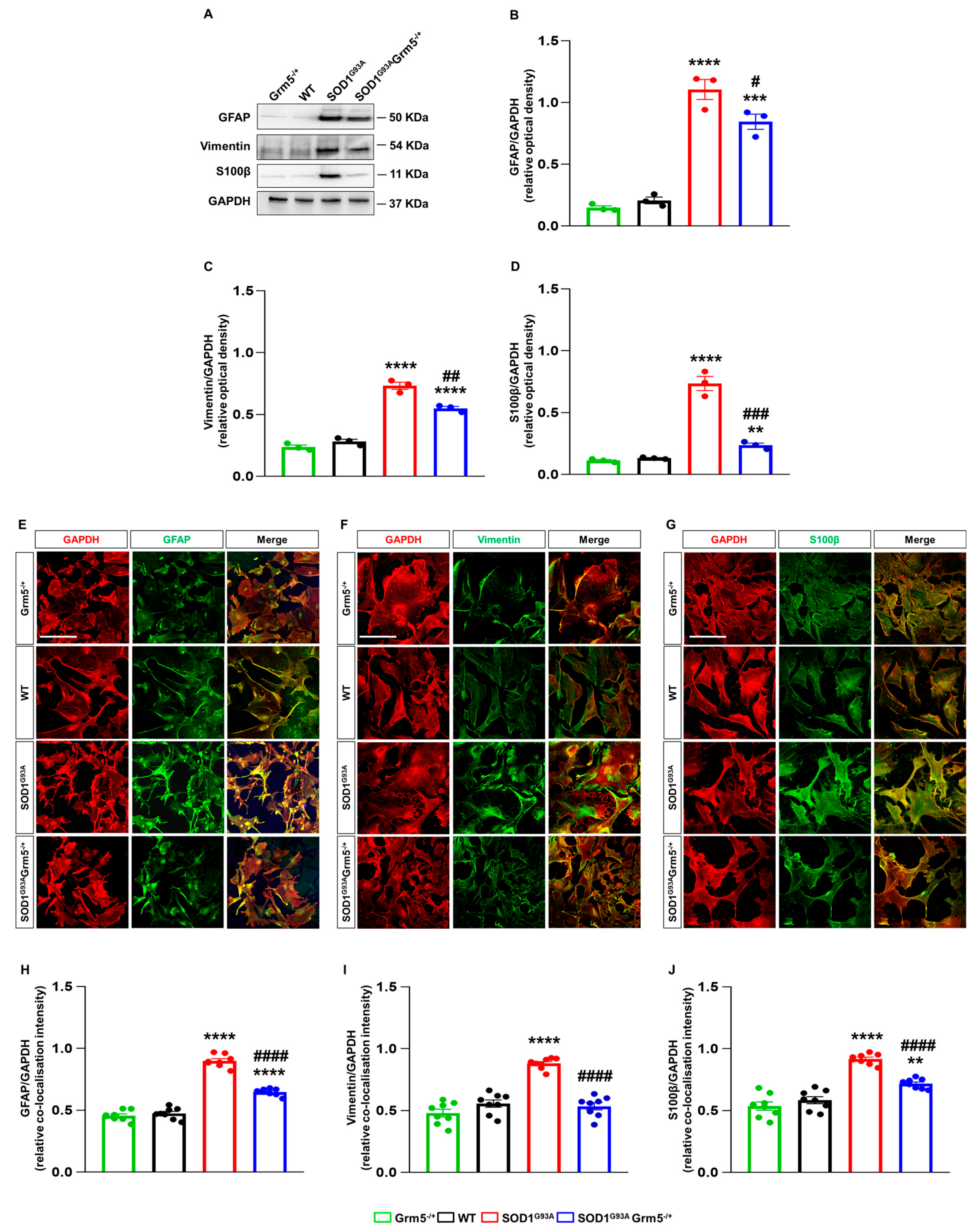
3.3. Downregulation of mGluR5 Reduces the Reactive Phenotype of SOD1G93A Spinal Cord Astrocytes
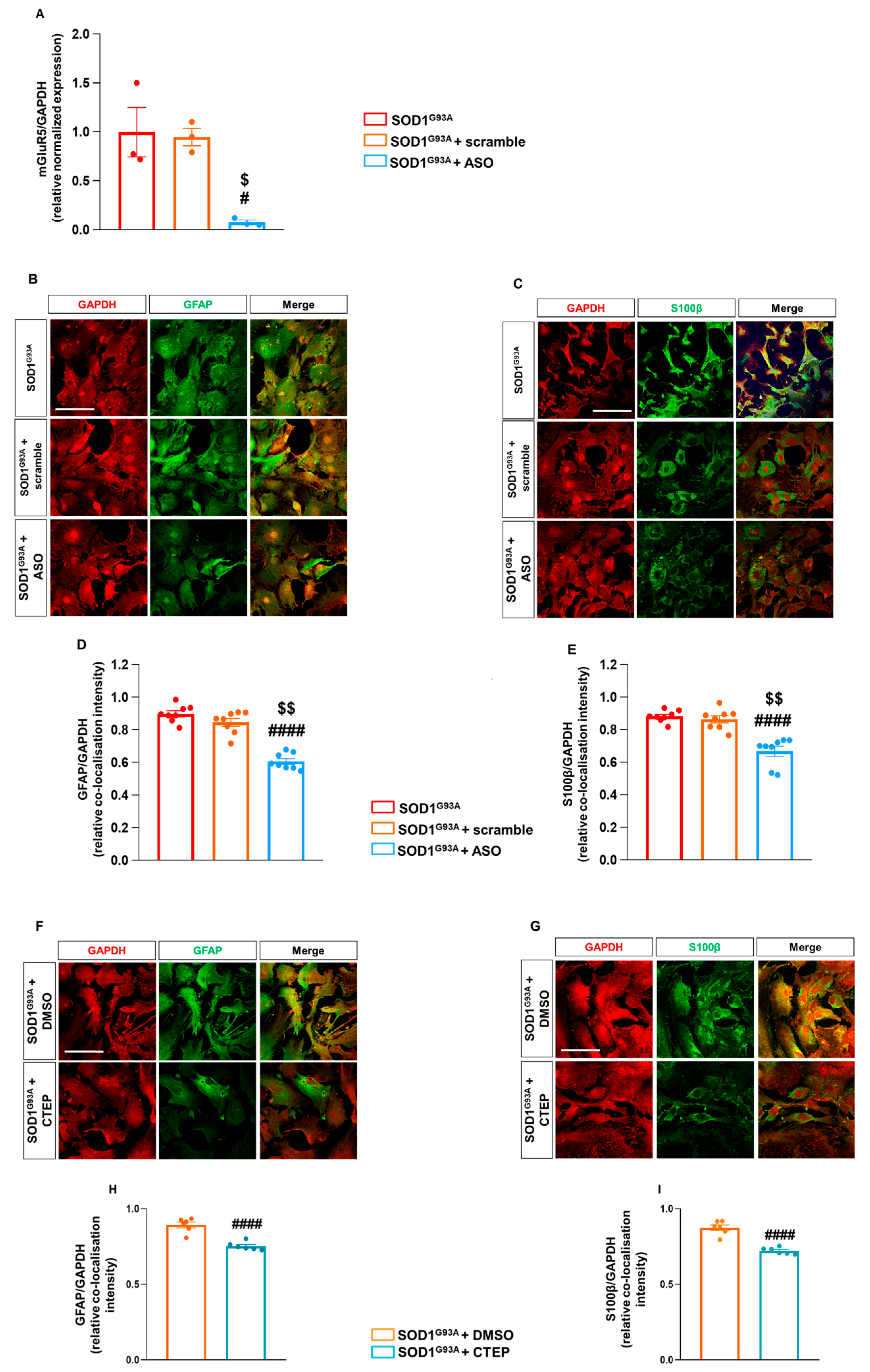
3.4. Reducing mGluR5 In Vitro by Antisense Oligonucleotide and Negative Allosteric Modulation Mimics the In Vivo Genetic Downregulation
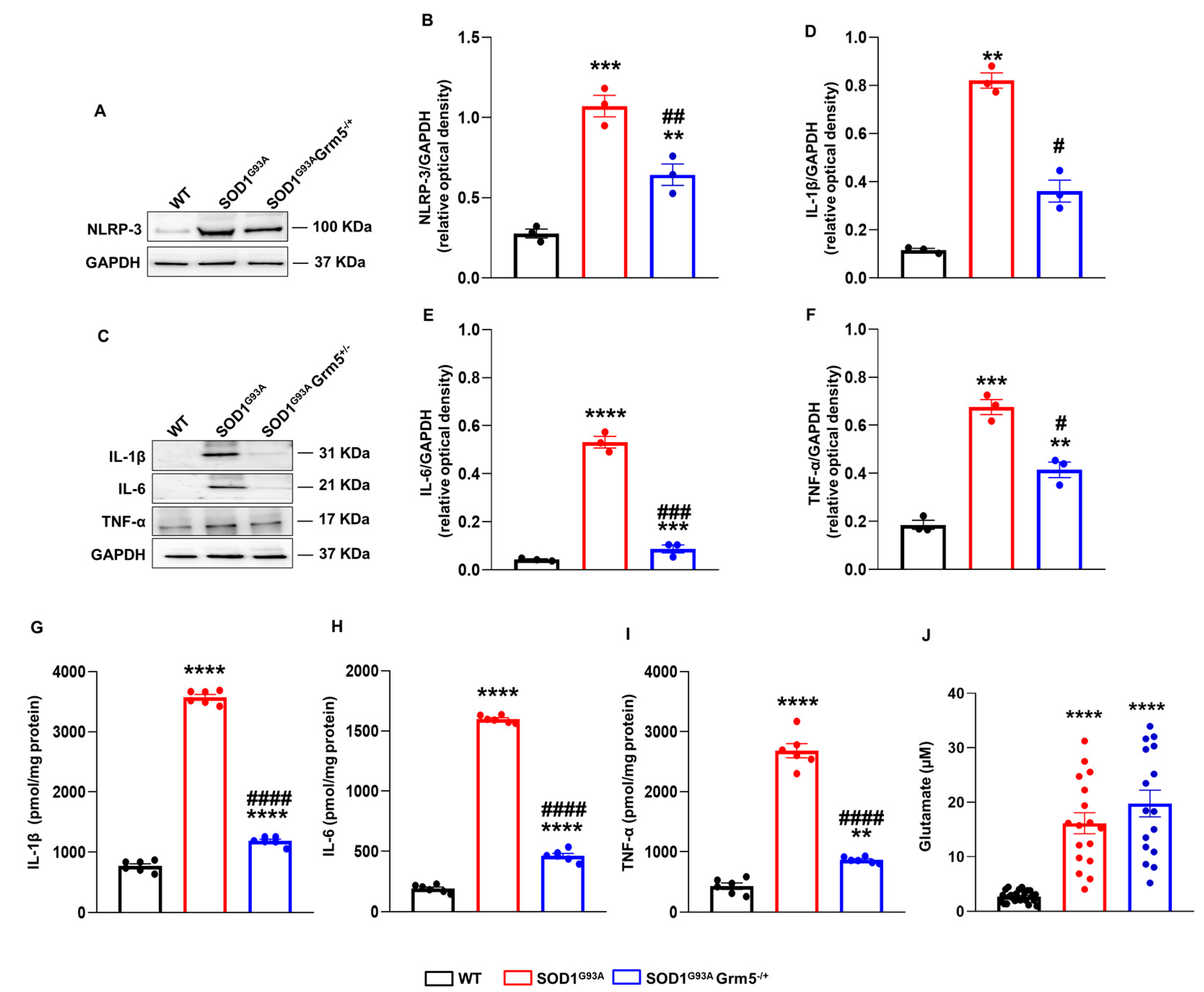
3.5. Genetic Downregulation of mGluR5 Dampens NLRP-3 Inflammasome and Pro-Inflammatory Cytokines
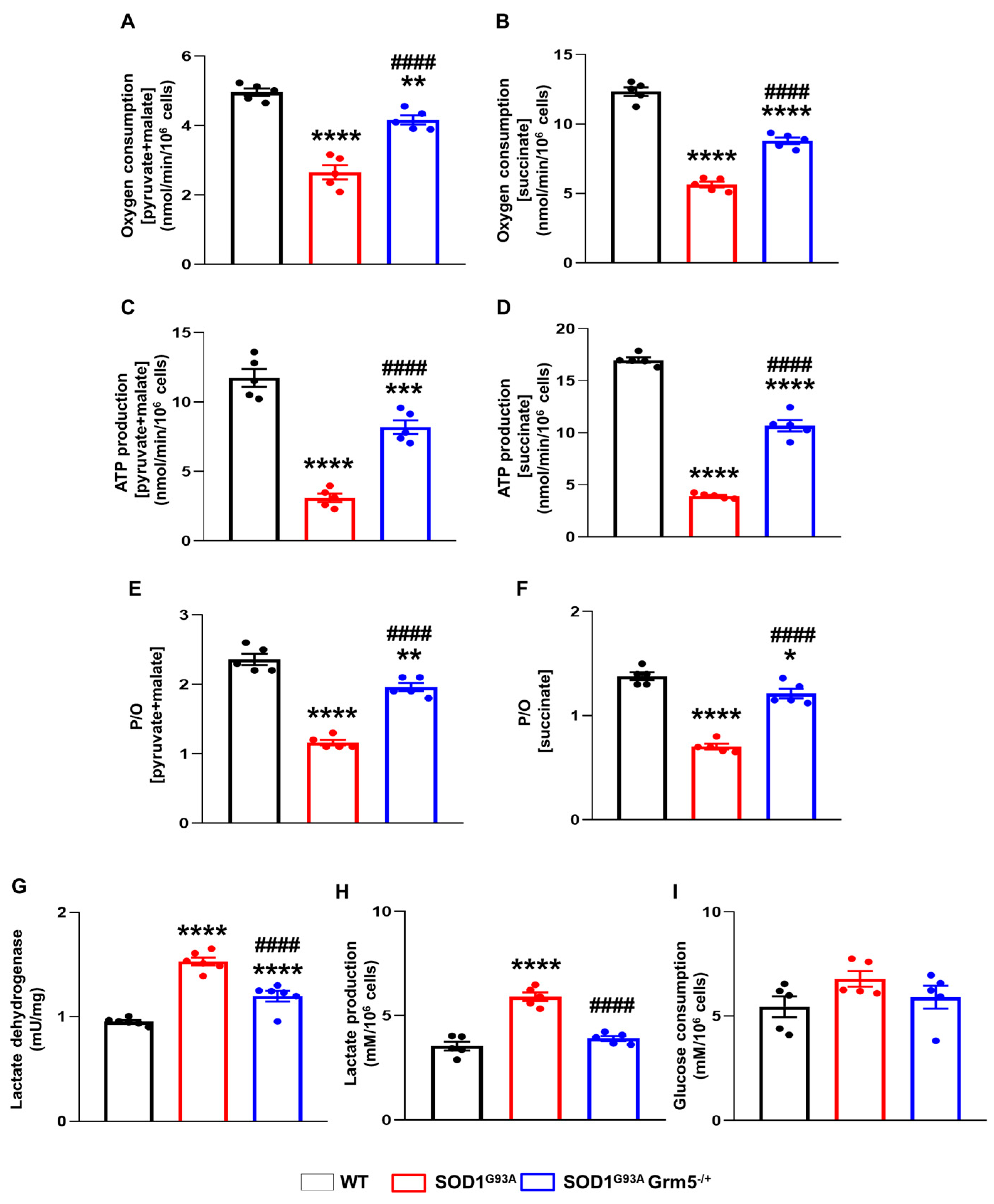
3.6. Genetic Downregulation of mGluR5 Positively Affects the Bioenergetic Metabolism in SOD1G93A Spinal Cord Astrocytes
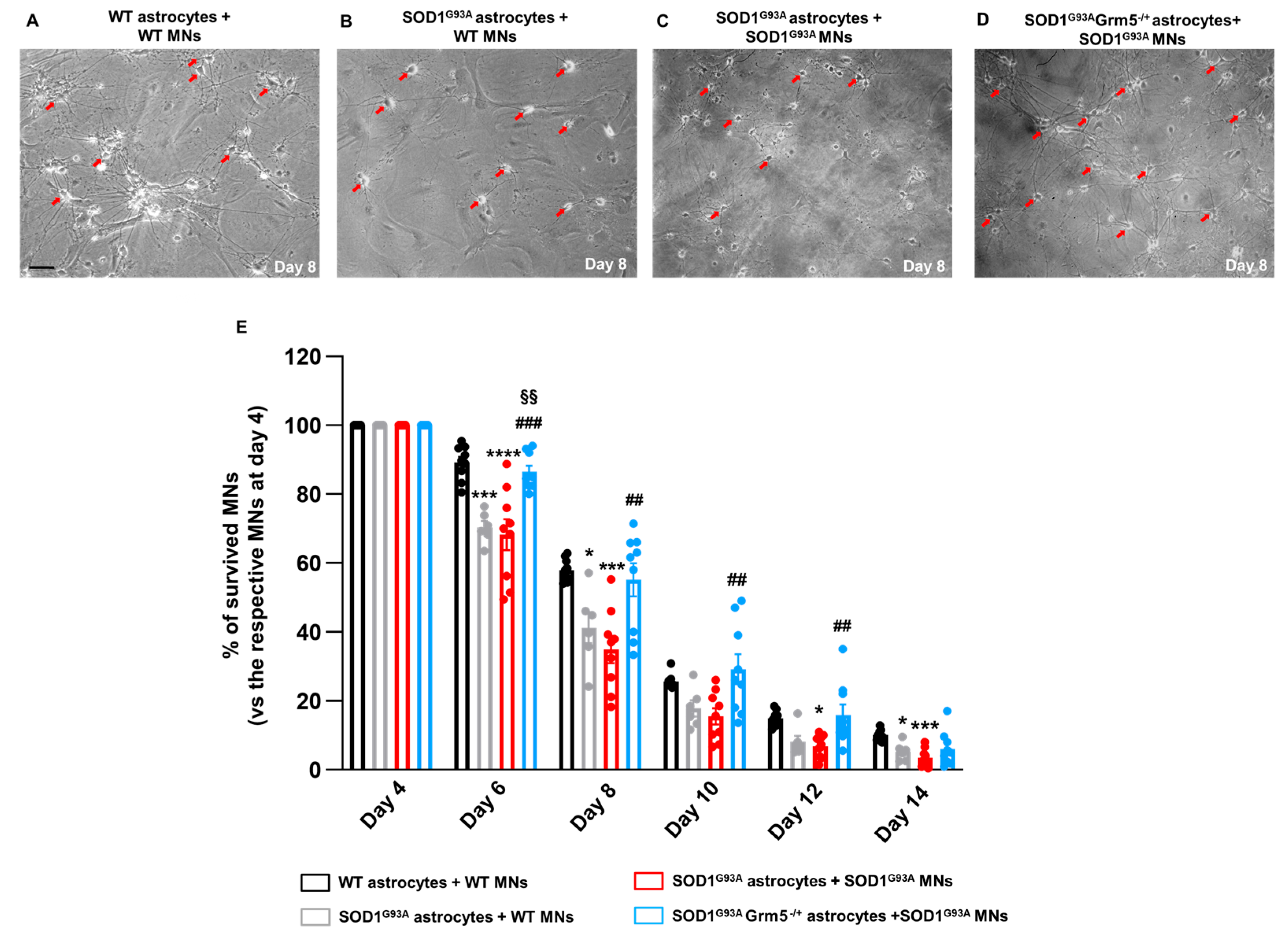
3.7. The Genetic Downregulation of mGluR5 Reduces the Neurotoxicity of SOD1G93A Astrocytes towards Spinal MNs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brown, R.H., Jr. Amyotrophic Lateral Sclerosis: Recent Insights from Genetics and Transgenic Mice. Cell 1995, 80, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Eisen, A. Amyotrophic Lateral Sclerosis-Evolutionary and Other Perspectives. Muscle Nerve 2009, 40, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Rowland, L.P.; Shneider, N.A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2001, 344, 1688–1700. [Google Scholar] [CrossRef]
- Swinnen, B.; Robberecht, W. The Phenotypic Variability of Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2014, 10, 661–670. [Google Scholar] [CrossRef] [PubMed]
- Huynh, W.; Ahmed, R.; Mahoney, C.J.; Nguyen, C.; Tu, S.; Caga, J.; Loh, P.; Lin, C.S.-Y.; Kiernan, M.C. The Impact of Cognitive and Behavioral Impairment in Amyotrophic Lateral Sclerosis. Expert Rev. Neurother. 2020, 20, 281–293. [Google Scholar] [CrossRef]
- Grad, L.I.; Rouleau, G.A.; Ravits, J.; Cashman, N.R. Clinical Spectrum of Amyotrophic Lateral Sclerosis (ALS). Cold Spring Harb. Perspect. Med. 2017, 7, a024117. [Google Scholar] [CrossRef]
- Pecoraro, V.; Mandrioli, J.; Carone, C.; Chiò, A.; Traynor, B.J.; Trenti, T. The NGS Technology for the Identification of Genes Associated with the ALS. A Systematic Review. Eur. J. Clin. Investig. 2020, 50, e13228. [Google Scholar] [CrossRef]
- Chio, A.; Battistini, S.; Calvo, A.; Caponnetto, C.; Conforti, F.L.; Corbo, M.; Giannini, F.; Mandrioli, J.; Mora, G.; Sabatelli, M.; et al. Genetic Counselling in ALS: Facts, Uncertainties and Clinical Suggestions. J. Neurol. Neurosurg. Psychiatry 2014, 85, 478–485. [Google Scholar] [CrossRef]
- Bonifacino, T.; Zerbo, R.A.; Balbi, M.; Torazza, C.; Frumento, G.; Fedele, E.; Bonanno, G.; Milanese, M. Nearly 30 Years of Animal Models to Study Amyotrophic Lateral Sclerosis: A Historical Overview and Future Perspectives. Int. J. Mol. Sci. 2021, 22, 12236. [Google Scholar] [CrossRef]
- Lu, H.; Dong Le, W.; Xie, Y.-Y.; Wang, X.-P. Current Therapy of Drugs in Amyotrophic Lateral Sclerosis. Curr. Neuropharmacol. 2016, 14, 314–321. [Google Scholar] [CrossRef]
- Abe, K.; Aoki, M.; Tsuji, S.; Itoyama, Y.; Sobue, G.; Togo, M.; Hamada, C.; Tanaka, M.; Akimoto, M.; Nakamura, K.; et al. Safety and Efficacy of Edaravone in Well Defined Patients with Amyotrophic Lateral Sclerosis: A Randomised, Double-Blind, Placebo-Controlled Trial. Lancet Neurol. 2017, 16, 505–512. [Google Scholar] [CrossRef]
- Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Miller, T.M.; Cudkowicz, M.E.; Genge, A.; Shaw, P.J.; Sobue, G.; Bucelli, R.C.; Chiò, A.; et al. Trial of Antisense Oligonucleotide Tofersen for SOD1 ALS. N. Engl. J. Med. 2022, 387, 1099–1110. [Google Scholar] [CrossRef]
- Benatar, M.; Wuu, J.; Andersen, P.M.; Bucelli, R.C.; Andrews, J.A.; Otto, M.; Farahany, N.A.; Harrington, E.A.; Chen, W.; Mitchell, A.A.; et al. Correction to: Design of a Randomized, Placebo-Controlled, Phase 3 Trial of Tofersen Initiated in Clinically Presymptomatic SOD1 Variant Carriers: The ATLAS Study. Neurotherapeutics 2022, 19, 1686. [Google Scholar] [CrossRef]
- Morrison, B.M.; Morrison, J.H. Amyotrophic Lateral Sclerosis Associated with Mutations in Superoxide Dismutase: A Putative Mechanism of Degeneration. Brain Res. Rev. 1999, 29, 121–135. [Google Scholar] [CrossRef]
- Milanese, M.; Zappettini, S.; Onofri, F.; Musazzi, L.; Tardito, D.; Bonifacino, T.; Messa, M.; Racagni, G.; Usai, C.; Benfenati, F.; et al. Abnormal Exocytotic Release of Glutamate in a Mouse Model of Amyotrophic Lateral Sclerosis. J. Neurochem. 2011, 116, 1028–1042. [Google Scholar] [CrossRef]
- Ferraiuolo, L.; Kirby, J.; Grierson, A.J.; Sendtner, M.; Shaw, P.J. Molecular Pathways of Motor Neuron Injury in Amyotrophic Lateral Sclerosis. Nat. Rev. Neurol. 2011, 7, 616–630. [Google Scholar] [CrossRef]
- Nijssen, J.; Comley, L.H.; Hedlund, E. Motor Neuron Vulnerability and Resistance in Amyotrophic Lateral Sclerosis. Acta Neuropathol. 2017, 133, 863–885. [Google Scholar] [CrossRef]
- Le Gall, L.; Anakor, E.; Connolly, O.; Vijayakumar, U.G.; Duddy, W.J.; Duguez, S. Molecular and Cellular Mechanisms Affected in ALS. J. Pers. Med. 2020, 10, 101. [Google Scholar] [CrossRef]
- Lee, W.; Parpura, V. Spatio-Temporal Characteristics of Metabotropic Glutamate Receptor 5 Traffic at or near the Plasma Membrane in Astrocytes. Glia 2016, 64, 1050–1065. [Google Scholar] [CrossRef]
- Gong, Z.; Ba, L.; Zhang, M. Dysfunction of the Oligodendrocytes in Amyotrophic Lateral Sclerosis. J. Biomed. Res. 2022, 36, 336–342. [Google Scholar] [CrossRef]
- Boillée, S.; Velde, C.V.; Cleveland, D.W. ALS: A Disease of Motor Neurons and Their Nonneuronal Neighbors. Neuron 2006, 52, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Ilieva, H.; Polymenidou, M.; Cleveland, D.W. Non–Cell Autonomous Toxicity in Neurodegenerative Disorders: ALS and Beyond. J. Cell Biol. 2009, 187, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Schweingruber, C.; Hedlund, E. The Cell Autonomous and Non-Cell Autonomous Aspects of Neuronal Vulnerability and Resilience in Amyotrophic Lateral Sclerosis. Biology 2022, 11, 1191. [Google Scholar] [CrossRef] [PubMed]
- Van Harten, A.C.M.M.; Phatnani, H.; Przedborski, S. Non-Cell-Autonomous Pathogenic Mechanisms in Amyotrophic Lateral Sclerosis. Trends Neurosci. 2021, 44, 658–668. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, F.P.; Carrasco, M.A.; Siao, M.C.; Maniatis, T.; Eggan, K. Non-Cell Autonomous Effect of Glia on Motor Neurons in an Embryonic Stem Cell-Based ALS Model. Nat. Neurosci. 2007, 10, 608–614. [Google Scholar] [CrossRef]
- Beers, D.R.; Appel, S.H. Immune Dysregulation in Amyotrophic Lateral Sclerosis: Mechanisms and Emerging Therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Yamanaka, K.; Chun, S.J.; Boillee, S.; Fujimori-Tonou, N.; Yamashita, H.; Gutmann, D.H.; Takahashi, R.; Misawa, H.; Cleveland, D.W. Astrocytes as Determinants of Disease Progression in Inherited Amyotrophic Lateral Sclerosis. Nat. Neurosci. 2008, 11, 251–253. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Van Kammen, M.; Levey, A.I.; Martin, L.J.; Kuncl, R.W. Selective Loss of Glial Glutamate Transporter GLT-1 in Amyotrophic Lateral Sclerosis. Ann. Neurol. 1995, 38, 73–84. [Google Scholar] [CrossRef]
- Bonifacino, T.; Musazzi, L.; Milanese, M.; Seguini, M.; Marte, A.; Gallia, E.; Cattaneo, L.; Onofri, F.; Popoli, M.; Bonanno, G. Altered Mechanisms Underlying the Abnormal Glutamate Release in Amyotrophic Lateral Sclerosis at a Pre-Symptomatic Stage of the Disease. Neurobiol. Dis. 2016, 95, 122–133. [Google Scholar] [CrossRef]
- Bonifacino, T.; Rebosio, C.; Provenzano, F.; Torazza, C.; Balbi, M.; Milanese, M.; Raiteri, L.; Usai, C.; Fedele, E.; Bonanno, G. Enhanced Function and Overexpression of Metabotropic Glutamate Receptors 1 and 5 in the Spinal Cord of the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis during Disease Progression. Int. J. Mol. Sci. 2019, 20, 4552. [Google Scholar] [CrossRef]
- Shaw, P.J.; Ince, P.G. Glutamate, Excitotoxicity and Amyotrophic Lateral Sclerosis; Springer: Berlin/Heidelberg, Germany, 1997. [Google Scholar]
- Spreux-Varoquaux, O.; Bensimon, G.; Lacomblez, L.; Salachas, F.; Pradat, P.F.; Le Forestier, N.; Marouan, A.; Dib, M.; Meininger, V. Glutamate Levels in Cerebrospinal Fluid in Amyotrophic Lateral Sclerosis: A Reappraisal Using a New HPLC Method with Coulometric Detection in a Large Cohort of Patients. J. Neurol. Sci. 2002, 193, 73–78. [Google Scholar] [CrossRef]
- Wuolikainen, A.; Andersen, P.M.; Moritz, T.; Marklund, S.L.; Antti, H. ALS Patients with Mutations in the SOD1 Gene Have an Unique Metabolomic Profile in the Cerebrospinal Fluid Compared with ALS Patients without Mutations. Mol. Genet. Metab. 2012, 105, 472–478. [Google Scholar] [CrossRef]
- Danbolt, N.C. Glutamate Uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Dykes-Hoberg, M.; Rothstein, J.D. Altered Expression of the Glutamate Transporter EAAT2b in Neurological Disease. Ann. Neurol. 2004, 55, 469–477. [Google Scholar] [CrossRef]
- Raiteri, L.; Paolucci, E.; Prisco, S.; Raiteri, M.; Bonanno, G. Activation of a Glycine Transporter on Spinal Cord Neurons Causes Enhanced Glutamate Release in a Mouse Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2003, 138, 1021–1025. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Fedele, E.; Rebosio, C.; Cattaneo, L.; Benfenati, F.; Usai, C.; Bonanno, G. Exocytosis Regulates Trafficking of GABA and Glycine Heterotransporters in Spinal Cord Glutamatergic Synapses: A Mechanism for the Excessive Heterotransporter-Induced Release of Glutamate in Experimental Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2015, 74, 314–324. [Google Scholar] [CrossRef]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Kazama, M.; Kato, Y.; Kakita, A.; Noguchi, N.; Urano, Y.; Masui, K.; Niida-Kawaguchi, M.; Yamamoto, T.; Watabe, K.; Kitagawa, K.; et al. Astrocytes Release Glutamate via Cystine/Glutamate Antiporter Upregulated in Response to Increased Oxidative Stress Related to Sporadic Amyotrophic Lateral Sclerosis. Neuropathology 2020, 40, 587–598. [Google Scholar] [CrossRef]
- Van Den Bosch, L.; Van Damme, P.; Bogaert, E.; Robberecht, W. The Role of Excitotoxicity in the Pathogenesis of Amyotrophic Lateral Sclerosis. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 1068–1082. [Google Scholar] [CrossRef]
- Pieri, M.; Carunchio, I.; Curcio, L.; Mercuri, N.B.; Zona, C. Increased Persistent Sodium Current Determines Cortical Hyperexcitability in a Genetic Model of Amyotrophic Lateral Sclerosis. Exp. Neurol. 2009, 215, 368–379. [Google Scholar] [CrossRef]
- King, A.E.; Woodhouse, A.; Kirkcaldie, M.T.K.; Vickers, J.C. Excitotoxicity in ALS: Overstimulation, or Overreaction? Exp. Neurol. 2016, 275 Pt 1, 162–171. [Google Scholar] [CrossRef]
- Bruno, V.; Caraci, F.; Copani, A.; Matrisciano, F.; Nicoletti, F.; Battaglia, G. The Impact of Metabotropic Glutamate Receptors into Active Neurodegenerative Processes: A “Dark Side” in the Development of New Symptomatic Treatments for Neurologic and Psychiatric Disorders. Neuropharmacology 2017, 115, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Van Vliet, E.A.; Mayboroda, O.A.; Troost, D.; Lopes Da Silva, F.H.; Gorter, J.A. Upregulation of Metabotropic Glutamate Receptor Subtype MGluR3 and MGluR5 in Reactive Astrocytes in a Rat Model of Mesial Temporal Lobe Epilepsy. Eur. J. Neurosci. 2000, 12, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Gorter, J.A.; Ijlst-Keizers, H.; Rozemuller, A.J.; Yankaya, B.; Leenstra, S.; Troost, D. Expression and Functional Role of MGluR3 and MGluR5 in Human Astrocytes and Glioma Cells: Opposite Regulation of Glutamate Transporter Proteins. Eur. J. Neurosci. 2003, 17, 2106–2118. [Google Scholar] [CrossRef] [PubMed]
- Aronica, E.; Catania, M.V.; Geurts, J.; Yankaya, B.; Troost, D. Immunohistochemical Localization of Group I and II Metabotropic Glutamate Receptors in Control and Amyotrophic Lateral Sclerosis Human Spinal Cord: Upregulation in Reactive Astrocytes. Neuroscience 2001, 105, 509–520. [Google Scholar] [CrossRef]
- Brownell, A.L.; Kuruppu, D.; Kil, K.E.; Jokivarsi, K.; Poutiainen, P.; Zhu, A.; Maxwell, M. PET Imaging Studies Show Enhanced Expression of MGluR5 and Inflammatory Response during Progressive Degeneration in ALS Mouse Model Expressing SOD1-G93A Gene. J. Neuroinflamm. 2015, 12, 217. [Google Scholar] [CrossRef]
- Rossi, D.; Brambilla, L.; Valori, C.F.; Roncoroni, C.; Crugnola, A.; Yokota, T.; Bredesen, D.E.; Volterra, A. Focal Degeneration of Astrocytes in Amyotrophic Lateral Sclerosis. Cell Death Differ. 2008, 15, 1691–1700. [Google Scholar] [CrossRef]
- Martorana, F.; Brambilla, L.; Valori, C.F.; Bergamaschi, C.; Roncoroni, C.; Aronica, E.; Volterra, A.; Bezzi, P.; Rossi, D. The BH4 Domain of Bcl-X(L) Rescues Astrocyte Degeneration in Amyotrophic Lateral Sclerosis by Modulating Intracellular Calcium Signals. Hum. Mol. Genet. 2012, 21, 826–840. [Google Scholar] [CrossRef]
- Bonifacino, T.; Cattaneo, L.; Gallia, E.; Puliti, A.; Melone, M.; Provenzano, F.; Bossi, S.; Musante, I.; Usai, C.; Conti, F.; et al. In-Vivo Effects of Knocking-down Metabotropic Glutamate Receptor 5 in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neuropharmacology 2017, 123, 433–445. [Google Scholar] [CrossRef]
- Milanese, M.; Bonifacino, T.; Torazza, C.; Provenzano, F.; Kumar, M.; Ravera, S.; Zerbo, A.R.; Frumento, G.; Balbi, M.; Nguyen, T.P.N.; et al. Blocking Glutamate MGlu(5) Receptors with the Negative Allosteric Modulator CTEP Improves Disease Course in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Br. J. Pharmacol. 2021, 178, 3747–3764. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.-X.; et al. Motor Neuron Degeneration in Mice That Express a Human Cu,Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Lu, Y.-M.; Jia, Z.; Janus, C.; Henderson, J.T.; Gerlai, R.; Wojtowicz, J.M.; Roder, J.C. Mice Lacking Metabotropic Glutamate Receptor 5 Show Impaired Learning and Reduced CA1 Long-Term Potentiation (LTP) But Normal CA3 LTP. J. Neurosci. 1997, 17, 5196–5205. [Google Scholar] [CrossRef]
- Bonifacino, T.; Provenzano, F.; Gallia, E.; Ravera, S.; Torazza, C.; Bossi, S.; Ferrando, S.; Puliti, A.; Van Den Bosch, L.; Bonanno, G.; et al. In-Vivo Genetic Ablation of Metabotropic Glutamate Receptor Type 5 Slows down Disease Progression in the SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Neurobiol. Dis. 2019, 129, 79–92. [Google Scholar] [CrossRef]
- Uccelli, A.; Milanese, M.; Principato, M.C.; Morando, S.; Bonifacino, T.; Vergani, L.; Giunti, D.; Voci, A.; Carminati, E.; Giribaldi, F.; et al. Intravenous Mesenchymal Stem Cells Improve Survival and Motor Function in Experimental Amyotrophic Lateral Sclerosis. Mol. Med. 2012, 18, 794–804. [Google Scholar] [CrossRef]
- Provenzano, F.; Nyberg, S.; Giunti, D.; Torazza, C.; Parodi, B.; Bonifacino, T.; Usai, C.; Kerlero de Rosbo, N.; Milanese, M.; Uccelli, A.; et al. Micro-RNAs Shuttled by Extracellular Vesicles Secreted from Mesenchymal Stem Cells Dampen Astrocyte Pathological Activation and Support Neuroprotection in In-Vitro Models of ALS. Cells 2022, 11, 3923. [Google Scholar] [CrossRef]
- Marini, C.; Cossu, V.; Kumar, M.; Milanese, M.; Cortese, K.; Bruno, S.; Bellese, G.; Carta, S.; Zerbo, R.A.; Torazza, C.; et al. The Role of Endoplasmic Reticulum in the Differential Endurance against Redox Stress in Cortical and Spinal Astrocytes from the Newborn SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Antioxidants 2021, 10, 1392. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of Co-Localization of Objects in Dual-Colour Confocal Images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and Quantitative Measurement of Protein-Protein Colocalization in Live Cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef]
- McCloy, R.A.; Rogers, S.; Caldon, C.E.; Lorca, T.; Castro, A.; Burgess, A. Partial Inhibition of Cdk1 in G 2 Phase Overrides the SAC and Decouples Mitotic Events. Cell Cycle 2014, 13, 1400–1412. [Google Scholar] [CrossRef]
- Jakic, B.; Buszko, M.; Cappellano, G.; Wick, G. Elevated Sodium Leads to the Increased Expression of HSP60 and Induces Apoptosis in HUVECs. PLoS ONE 2017, 12, e0179383. [Google Scholar] [CrossRef] [PubMed]
- Rossi, P.I.A.; Musante, I.; Summa, M.; Pittaluga, A.; Emionite, L.; Ikehata, M.; Rastaldi, M.P.; Ravazzolo, R.; Puliti, A. Compensatory Molecular and Functional Mechanisms in Nervous System of the Grm1crv4 Mouse Lacking the MGlu1 Receptor: A Model for Motor Coordination Deficits. Cereb. Cortex 2013, 23, 2179–2189. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A New Generation of Ca2+ Indicators with Greatly Improved Fluorescence Properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Swayze, E.E.; Siwkowski, A.M.; Wancewicz, E.V.; Migawa, M.T.; Wyrzykiewicz, T.K.; Hung, G.; Monia, B.P.; Bennett, C.F. Antisense Oligonucleotides Containing Locked Nucleic Acid Improve Potency but Cause Significant Hepatotoxicity in Animals. Nucleic Acids Res. 2007, 35, 687–700. [Google Scholar] [CrossRef]
- Lindemann, L.; Jaeschke, G.; Michalon, A.; Vieira, E.; Honer, M.; Spooren, W.; Porter, R.; Hartung, T.; Kolczewski, S.; Büttelmann, B.; et al. CTEP: A Novel, Potent, Long-Acting, and Orally Bioavailable Metabotropic Glutamate Receptor 5 Inhibitor. J. Pharmacol. Exp. Ther. 2011, 339, 474–486. [Google Scholar] [CrossRef]
- Alfei, S.; Baig, I. An Optimized and Very Detailed, Grams Scale Synthesis of CTEP, through a Complete Characterization of All the Isolated and Purified Intermediates. Org. Commun. 2017, 10, 114–121. [Google Scholar] [CrossRef]
- Ravera, S.; Cossu, V.; Tappino, B.; Nicchia, E.; Dufour, C.; Cavani, S.; Sciutto, A.; Bolognesi, C.; Columbaro, M.; Degan, P.; et al. Concentration-dependent Metabolic Effects of Metformin in Healthy and Fanconi Anemia Lymphoblast Cells. J. Cell. Physiol. 2018, 233, 1736–1751. [Google Scholar] [CrossRef]
- Hinkle, P.C. P/O Ratios of Mitochondrial Oxidative Phosphorylation. Biochim. Biophys. Acta Bioenerg. 2005, 1706, 1–11. [Google Scholar] [CrossRef]
- Bianchi, G.; Martella, R.; Ravera, S.; Marini, C.; Capitanio, S.; Orengo, A.; Emionite, L.; Lavarello, C.; Amaro, A.; Petretto, A.; et al. Fasting Induces Anti-Warburg Effect That Increases Respiration but Reduces ATP-Synthesis to Promote Apoptosis in Colon. Cancer Models 2015, 6, 11806–11819. [Google Scholar] [CrossRef]
- Ravera, S.; Bonifacino, T.; Bartolucci, M.; Milanese, M.; Gallia, E.; Provenzano, F.; Cortese, K.; Panfoli, I.; Bonanno, G. Characterization of the Mitochondrial Aerobic Metabolism in the Pre- and Perisynaptic Districts of the SOD1 G93A Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 9220–9233. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in Mitochondrial Energetic Function Compels Fanconi Anaemia Cells to Glycolytic Metabolism. Biochim. Biophys. Acta—Mol. Basis Dis. 2017, 1863, 1214–1221. [Google Scholar] [CrossRef]
- Milanese, M.; Tardito, D.; Musazzi, L.; Treccani, G.; Mallei, A.; Bonifacino, T.; Gabriel, C.; Mocaer, E.; Racagni, G.; Popoli, M.; et al. Chronic Treatment with Agomelatine or Venlafaxine Reduces Depolarization-Evoked Glutamate Release from Hippocampal Synaptosomes. BMC Neurosci. 2013, 14, 75. [Google Scholar] [CrossRef]
- Bonalume, V.; Caffino, L.; Castelnovo, L.F.; Faroni, A.; Liu, S.; Hu, J.; Milanese, M.; Bonanno, G.; Sohns, K.; Hoffmann, T.; et al. Axonal GABAA Stabilizes Excitability in Unmyelinated Sensory Axons Secondary to NKCC1 Activity. J. Physiol. 2021, 599, 4065–4084. [Google Scholar] [CrossRef]
- Vandenberghe, W.; Van Den Bosch, L.; Robberecht, W. Glial Cells Potentiate Kainate-Induced Neuronal Death in a Motoneuron-Enriched Spinal Coculture System. Brain Res. 1998, 807, 1–10. [Google Scholar] [CrossRef]
- Wiese, S.; Herrmann, T.; Drepper, C.; Jablonka, S.; Funk, N.; Klausmeyer, A.; Rogers, M.-L.L.; Rush, R.; Sendtner, M. Isolation and Enrichment of Embryonic Mouse Motoneurons from the Lumbar Spinal Cord of Individual Mouse Embryos. Nat. Protoc. 2010, 5, 31–38. [Google Scholar] [CrossRef]
- D’Antoni, S.; Berretta, A.; Bonaccorso, C.M.; Bruno, V.; Aronica, E.; Nicoletti, F.; Catania, M.V. Metabotropic Glutamate Receptors in Glial Cells. Neurochem. Res. 2008, 33, 2436–2443. [Google Scholar] [CrossRef]
- Kawamata, H.; Ng, S.K.; Diaz, N.; Burstein, S.; Morel, L.; Osgood, A.; Sider, B.; Higashimori, H.; Haydon, P.G.; Manfredi, G.; et al. Abnormal Intracellular Calcium Signaling and SNARE Dependent Exocytosis Contributes to SOD1G93A Astrocyte- Mediated Toxicity in Amyotrophic Lateral Sclerosis. J. Neurosci. 2014, 34, 2331–2348. [Google Scholar] [CrossRef]
- Tripathi, P.; Rodriguez-Muela, N.; Klim, J.R.; de Boer, A.S.; Agrawal, S.; Sandoe, J.; Lopes, C.S.; Ogliari, K.S.; Williams, L.A.; Shear, M.; et al. Reactive Astrocytes Promote ALS-like Degeneration and Intracellular Protein Aggregation in Human Motor Neurons by Disrupting Autophagy through TGF-β1. Stem Cell Reports 2017, 9, 667–680. [Google Scholar] [CrossRef]
- Kia, A.; McAvoy, K.; Krishnamurthy, K.; Trotti, D.; Pasinelli, P. Astrocytes Expressing ALS-Linked Mutant FUS Induce Motor Neuron Death through Release of Tumor Necrosis Factor-Alpha. Glia 2018, 66, 1016–1033. [Google Scholar] [CrossRef]
- Schiffer, D.; Cordera, S.; Cavalla, P.; Migheli, A. Reactive Astrogliosis of the Spinal Cord in Amyotrophic Lateral Sclerosis. J. Neurol. Sci. 1996, 139, 27–33. [Google Scholar] [CrossRef] [PubMed]
- Goldman, R.D.; Khuon, S.; Chou, Y.H.; Opal, P.; Steinert, P.M. The Function of Intermediate Filaments in Cell Shape and Cytoskeletal Integrity. J. Cell Biol. 1996, 134, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Benninger, F.; Glat, M.J.; Offen, D.; Steiner, I. Glial Fibrillary Acidic Protein as a Marker of Astrocytic Activation in the Cerebrospinal Fluid of Patients with Amyotrophic Lateral Sclerosis. J. Clin. Neurosci. 2016, 26, 75–78. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; Donno, C.; Giannetti, S.; Perić, M.; Andjus, P.; D’Ambrosi, N.; Michetti, F. The Astrocytic S100B Protein with Its Receptor RAGE Is Aberrantly Expressed in SOD1G93A Models, and Its Inhibition Decreases the Expression of Proinflammatory Genes. Mediat. Inflamm. 2017, 2017, 1626204. [Google Scholar] [CrossRef]
- Abd-Elrahman, K.S.; Hamilton, A.; Hutchinson, S.R.; Liu, F.; Russell, R.C.; Ferguson, S.S.G. MGluR5 Antagonism Increases Autophagy and Prevents Disease Progression in the ZQ175 Mouse Model of Huntington’s Disease. Sci. Signal 2017, 10, eaan6387. [Google Scholar] [CrossRef]
- Farmer, K.; Abd-Elrahman, K.S.; Derksen, A.; Rowe, E.M.; Thompson, A.M.; Rudyk, C.A.; Prowse, N.A.; Dwyer, Z.; Bureau, S.C.; Fortin, T.; et al. MGluR5 Allosteric Modulation Promotes Neurorecovery in a 6-OHDA-Toxicant Model of Parkinson’s Disease. Mol. Neurobiol. 2020, 57, 1418–1431. [Google Scholar] [CrossRef]
- Meissner, F.; Molawi, K.; Zychlinsky, A. Mutant Superoxide Dismutase 1-Induced IL-1β Accelerates ALS Pathogenesis. Proc. Natl. Acad. Sci. USA 2010, 107, 13046–13050. [Google Scholar] [CrossRef]
- Bellezza, I.; Grottelli, S.; Costanzi, E.; Scarpelli, P.; Pigna, E.; Morozzi, G.; Mezzasoma, L.; Peirce, M.J.; Moresi, V.; Adamo, S.; et al. Peroxynitrite Activates the NLRP3 Inflammasome Cascade in SOD1(G93A) Mouse Model of Amyotrophic Lateral Sclerosis. Mol. Neurobiol. 2018, 55, 2350–2361. [Google Scholar] [CrossRef]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes Are Active Players in Cerebral Innate Immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef]
- Holden, C. Astrocytes Secrete Substance That Kills Motor Neurons in ALS. Science 2007, 316, 353. [Google Scholar] [CrossRef]
- Tefera, T.W.; Borges, K. Metabolic Dysfunctions in Amyotrophic Lateral Sclerosis Pathogenesis and Potential Metabolic Treatments. Front. Neurosci. 2017, 10, 611. [Google Scholar] [CrossRef]
- Vandoorne, T.; De Bock, K.; Van Den Bosch, L. Energy Metabolism in ALS: An Underappreciated Opportunity? Acta Neuropathol. 2018, 135, 489–509. [Google Scholar] [CrossRef]
- Stoklund Dittlau, K.; Van Den Bosch, L. Why Should We Care about Astrocytes in a Motor Neuron Disease? Front. Mol. Med. 2023, 3, 1047540. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Supplie, L.M.; Düking, T.; Campbell, G.; Diaz, F.; Moraes, C.T.; Götz, M.; Hamprecht, B.; Boretius, S.; Mahad, D.; Nave, K.-A. Respiration-Deficient Astrocytes Survive as Glycolytic Cells In Vivo. J. Neurosci. 2017, 37, 4231–4242. [Google Scholar] [CrossRef]
- Yin, S.; Niswender, C.M. Progress toward Advanced Understanding of Metabotropic Glutamate Receptors: Structure, Signaling and Therapeutic Indications. Cell Signal 2014, 26, 2284–2297. [Google Scholar] [CrossRef]
- Liu, H.-N.; Sanelli, T.; Horne, P.; Pioro, E.P.; Strong, M.J.; Rogaeva, E.; Bilbao, J.; Zinman, L.; Robertson, J. Lack of Evidence of Monomer/Misfolded Superoxide Dismutase-1 in Sporadic Amyotrophic Lateral Sclerosis. Ann. Neurol. 2009, 66, 75–80. [Google Scholar] [CrossRef]
- Luyt, K.; Váradi, A.; Durant, C.F.; Molnár, E. Oligodendroglial Metabotropic Glutamate Receptors Are Developmentally Regulated and Involved in the Prevention of Apoptosis. J. Neurochem. 2006, 99, 641–656. [Google Scholar] [CrossRef]
- Nicoletti, F.; Bockaert, J.; Collingridge, G.L.; Conn, P.J.; Ferraguti, F.; Schoepp, D.D.; Wroblewski, J.T.; Pin, J.P. Metabotropic Glutamate Receptors: From the Workbench to the Bedside. Neuropharmacology 2011, 60, 1017–1041. [Google Scholar] [CrossRef]
- Anneser, J.M.H.; Ince, P.G.; Shaw, P.J.; Borasio, G.D. Differential Expression of MGluR5 in Human Lumbosacral Motoneurons. Neuroreport 2004, 15, 271–273. [Google Scholar] [CrossRef]
- Anneser, J.M.H.; Chahli, C.; Ince, P.G.; Borasio, G.D.; Shaw, P.J. Glial Proliferation and Metabotropic Glutamate Receptor Expression in Amyotrophic Lateral Sclerosis. J. Neuropathol. Exp. Neurol. 2004, 63, 831–840. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Vergouts, M.; Doyen, P.J.; Peeters, M.; Opsomer, R.; Hermans, E. Constitutive Downregulation Protein Kinase C Epsilon in HSOD1(G93A) Astrocytes Influences MGluR5 Signaling and the Regulation of Glutamate Uptake. Glia 2018, 66, 749–761. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Kirchhoff, F. Glutamate-Mediated Neuronal-Glial Transmission. J. Anat. 2007, 210, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Viwatpinyo, K.; Chongthammakun, S. Activation of Group I Metabotropic Glutamate Receptors Leads to Brain-Derived Neurotrophic Factor Expression in Rat C6 Cells. Neurosci. Lett. 2009, 467, 127–130. [Google Scholar] [CrossRef]
- Kanumilli, S.; Roberts, P.J. Mechanisms of Glutamate Receptor Induced Proliferation of Astrocytes. Neuroreport 2006, 17, 1877–1881. [Google Scholar] [CrossRef] [PubMed]
- Jean, Y.Y.; Lercher, L.D.; Dreyfus, C.F. Glutamate Elicits Release of BDNF from Basal Forebrain Astrocytes in a Process Dependent on Metabotropic Receptors and the PLC Pathway. Neuron Glia Biol. 2008, 4, 35–42. [Google Scholar] [CrossRef]
- Bezzi, P.; Volterra, A. Imaging Exocytosis and Recycling of Synaptic-Like Microvesicles in Astrocytes. Cold Spring Harb. Protoc. 2014, 2014. [Google Scholar] [CrossRef]
- Panatier, A.; Vallée, J.; Haber, M.; Murai, K.K.; Lacaille, J.-C.; Robitaille, R. Astrocytes Are Endogenous Regulators of Basal Transmission at Central Synapses. Cell 2011, 146, 785–798. [Google Scholar] [CrossRef]
- Shah, A.; Silverstein, P.S.; Singh, D.P.; Kumar, A. Involvement of Metabotropic Glutamate Receptor 5, AKT/PI3K Signaling and NF-ΚB Pathway in Methamphetamine-Mediated Increase in IL-6 and IL-8 Expression in Astrocytes. J. Neuroinflamm. 2012, 9, 52. [Google Scholar] [CrossRef]
- Vermeiren, C.; Najimi, M.; Vanhoutte, N.; Tilleux, S.; De Hemptinne, I.; Maloteaux, J.M.; Hermans, E. Acute Up-Regulation of Glutamate Uptake Mediated by MGluR5a in Reactive Astrocytes. J. Neurochem. 2005, 94, 405–416. [Google Scholar] [CrossRef]
- Ribeiro, F.M.; DeVries, R.A.; Hamilton, A.; Guimaraes, I.M.; Cregan, S.P.; Pires, R.G.W.; Ferguson, S.S.G. Metabotropic Glutamate Receptor 5 Knockout Promotes Motor and Biochemical Alterations in a Mouse Model of Huntington’s Disease. Hum. Mol. Genet. 2014, 23, 2030–2042. [Google Scholar] [CrossRef]
- Hamilton, A.; Vasefi, M.; Vander Tuin, C.; McQuaid, R.J.; Anisman, H.; Ferguson, S.S.G. Chronic Pharmacological MGluR5 Inhibition Prevents Cognitive Impairment and Reduces Pathogenesis in an Alzheimer Disease Mouse Model. Cell Rep. 2016, 15, 1859–1865. [Google Scholar] [CrossRef]
- Ure, J.; Baudry, M.; Perassolo, M. Metabotropic Glutamate Receptors and Epilepsy. J. Neurol. Sci. 2006, 247, 1–9. [Google Scholar] [CrossRef]
- Dölen, G.; Bear, M.F. Role for Metabotropic Glutamate Receptor 5 (MGluR5) in the Pathogenesis of Fragile X Syndrome. J. Physiol. 2008, 586, 1503–1508. [Google Scholar] [CrossRef]
- Paquet, M.; Ribeiro, F.M.; Guadagno, J.; Esseltine, J.L.; Ferguson, S.S.G.; Cregan, S.P. Role of Metabotropic Glutamate Receptor 5 Signaling and Homer in Oxygen Glucose Deprivation-Mediated Astrocyte Apoptosis. Mol. Brain 2013, 6, 9. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Zorec, R. Astroglial Signalling in Health and Disease. Neurosci. Lett. 2019, 689, 1–4. [Google Scholar] [CrossRef]
- Ziff, O.J.; Clarke, B.E.; Taha, D.M.; Crerar, H.; Luscombe, N.M.; Patani, R. Meta-Analysis of Human and Mouse ALS Astrocytes Reveals Multi-Omic Signatures of Inflammatory Reactive States. Genome Res. 2022, 32, 71–84. [Google Scholar] [CrossRef]
- Patani, R.; Hardingham, G.E.; Liddelow, S.A. Functional Roles of Reactive Astrocytes in Neuroinflammation and Neurodegeneration. Nat. Rev. Neurol. 2023, 19, 395–409. [Google Scholar] [CrossRef]
- Migheli, A.; Cordera, S.; Bendotti, C.; Atzori, C.; Piva, R.; Schiffer, D. S-100b Protein Is Upregulated in Astrocytes and Motor Neurons in the Spinal Cord of Patients with Amyotrophic Lateral Sclerosis. Neurosci. Lett. 1999, 261, 25–28. [Google Scholar] [CrossRef]
- Díaz-Amarilla, P.; Olivera-Bravo, S.; Trias, E.; Cragnolini, A.; Martínez-Palma, L.; Cassina, P.; Beckman, J.; Barbeito, L. Phenotypically Aberrant Astrocytes That Promote Motoneuron Damage in a Model of Inherited Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. USA 2011, 108, 18126–18131. [Google Scholar] [CrossRef]
- Korobeynikov, V.A.; Lyashchenko, A.K.; Blanco-Redondo, B.; Jafar-Nejad, P.; Shneider, N.A. Antisense Oligonucleotide Silencing of FUS Expression as a Therapeutic Approach in Amyotrophic Lateral Sclerosis. Nat. Med. 2022, 28, 104–116. [Google Scholar] [CrossRef]
- Corey, D.R. Nusinersen, an Antisense Oligonucleotide Drug for Spinal Muscular Atrophy. Nat. Neurosci. 2017, 20, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Krieg, A.M. FDA Approves Eteplirsen for Duchenne Muscular Dystrophy: The Next Chapter in the Eteplirsen Saga. Nucleic Acid. Ther. 2017, 27, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Reilmann, R.; Rouzade-Dominguez, M.; Saft, C.; Süssmuth, S.D.; Priller, J.; Rosser, A.; Rickards, H.; Schöls, L.; Pezous, N.; Gasparini, F.; et al. A Randomized, Placebo-controlled Trial of AFQ056 for the Treatment of Chorea in Huntington’s Disease. Mov. Disord. 2015, 30, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Tison, F.; Keywood, C.; Wakefield, M.; Durif, F.; Corvol, J.-C.; Eggert, K.; Lew, M.; Isaacson, S.; Bezard, E.; Poli, S.-M.; et al. A Phase 2A Trial of the Novel MGluR5-Negative Allosteric Modulator Dipraglurant for Levodopa-Induced Dyskinesia in Parkinson’s Disease. Mov. Disord. 2016, 31, 1373–1380. [Google Scholar] [CrossRef] [PubMed]
- Quiroz, J.A.; Tamburri, P.; Deptula, D.; Banken, L.; Beyer, U.; Rabbia, M.; Parkar, N.; Fontoura, P.; Santarelli, L. Efficacy and Safety of Basimglurant as Adjunctive Therapy for Major Depression. JAMA Psychiatry 2016, 73, 675–684. [Google Scholar] [CrossRef]
- Youssef, E.A.; Berry-Kravis, E.; Czech, C.; Hagerman, R.J.; Hessl, D.; Wong, C.Y.; Rabbia, M.; Deptula, D.; John, A.; Kinch, R.; et al. Effect of the MGluR5-NAM Basimglurant on Behavior in Adolescents and Adults with Fragile X Syndrome in a Randomized, Double-Blind, Placebo-Controlled Trial: FragXis Phase 2 Results. Neuropsychopharmacology 2018, 43, 503–512. [Google Scholar] [CrossRef]
- Trenkwalder, C.; Stocchi, F.; Poewe, W.; Dronamraju, N.; Kenney, C.; Shah, A.; von Raison, F.; Graf, A. Mavoglurant in Parkinson’s Patients with l-Dopa-Induced Dyskinesias: Two Randomized Phase 2 Studies. Mov. Disord. 2016, 31, 1054–1058. [Google Scholar] [CrossRef]
- Jaeschke, G.; Kolczewski, S.; Spooren, W.; Vieira, E.; Bitter-Stoll, N.; Boissin, P.; Borroni, E.; Büttelmann, B.; Ceccarelli, S.; Clemann, N.; et al. Metabotropic Glutamate Receptor 5 Negative Allosteric Modulators: Discovery of 2-Chloro-4-[1-(4-Fluorophenyl)-2,5-Dimethyl-1 H-Imidazol-4-Ylethynyl]Pyridine (Basimglurant, RO4917523), a Promising Novel Medicine for Psychiatric Diseases. J. Med. Chem. 2015, 58, 1358–1371. [Google Scholar] [CrossRef]
- Lindemann, L.; Porter, R.H.; Scharf, S.H.; Kuennecke, B.; Bruns, A.; von Kienlin, M.; Harrison, A.C.; Paehler, A.; Funk, C.; Gloge, A.; et al. Pharmacology of Basimglurant (RO4917523, RG7090), a Unique Metabotropic Glutamate Receptor 5 Negative Allosteric Modulator in Clinical Development for Depression. J. Pharmacol. Exp. Ther. 2015, 353, 213–233. [Google Scholar] [CrossRef]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Beccaria, F.; Salamino, F.; Milanese, M.; Bonanno, G.; Pontremoli, S.; Melloni, E. Adaptive Modifications in the Calpain/Calpastatin System in Brain Cells after Persistent Alteration in Ca2+ Homeostasis. J. Biol. Chem. 2010, 285, 631–643. [Google Scholar] [CrossRef]
- Stifanese, R.; Averna, M.; De Tullio, R.; Pedrazzi, M.; Milanese, M.; Bonifacino, T.; Bonanno, G.; Salamino, F.; Pontremoli, S.; Melloni, E. Role of Calpain-1 in the Early Phase of Experimental ALS. Arch. Biochem. Biophys. 2014, 562, 1–8. [Google Scholar] [CrossRef]
- Rivest, S. Regulation of Innate Immune Responses in the Brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Glial Cells in Amyotrophic Lateral Sclerosis. Exp. Neurol. 2014, 262, 111–120. [Google Scholar] [CrossRef]
- Bendotti, C.; Carrì, M.T. Lessons from Models of SOD1-Linked Familial ALS. Trends Mol. Med. 2004, 10, 393–400. [Google Scholar] [CrossRef]
- Zuo, T.; Gong, B.; Gao, Y.; Yuan, L. An in Vitro Study on the Stimulatory Effects of Extracellular Glutamate on Astrocytes. Mol. Biol. Rep. 2023. ahead of print. [Google Scholar] [CrossRef]
- Mahmoud, S.; Gharagozloo, M.; Simard, C.; Gris, D. Astrocytes Maintain Glutamate Homeostasis in the CNS by Controlling the Balance between Glutamate Uptake and Release. Cells 2019, 8, 184. [Google Scholar] [CrossRef]
- Satarker, S.; Bojja, S.L.; Gurram, P.C.; Mudgal, J.; Arora, D.; Nampoothiri, M. Astrocytic Glutamatergic Transmission and Its Implications in Neurodegenerative Disorders. Cells 2022, 11, 1139. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Aubert, A.; Costalat, R.; Magistretti, P.J.; Pellerin, L. Brain Lactate Kinetics: Modeling Evidence for Neuronal Lactate Uptake upon Activation. Proc. Natl. Acad. Sci. USA 2005, 102, 16448–16453. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial Free Radical Generation, Oxidative Stress, and Aging11This Article Is Dedicated to the Memory of Our Dear Friend, Colleague, and Mentor Lars Ernster (1920–1998), in Gratitude for All He Gave to Us. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Percie du Sert, N.; Hurst, V.; Ahluwalia, A.; Alam, S.; Avey, M.T.; Baker, M.; Browne, W.J.; Clark, A.; Cuthill, I.C.; Dirnagl, U.; et al. The ARRIVE Guidelines 2.0: Updated Guidelines for Reporting Animal Research. Br. J. Pharmacol. 2020, 177, 3617–3624. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Torazza, C.; Provenzano, F.; Gallia, E.; Cerminara, M.; Balbi, M.; Bonifacino, T.; Tessitore, S.; Ravera, S.; Usai, C.; Musante, I.; et al. Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes. Cells 2023, 12, 1952. https://doi.org/10.3390/cells12151952
Torazza C, Provenzano F, Gallia E, Cerminara M, Balbi M, Bonifacino T, Tessitore S, Ravera S, Usai C, Musante I, et al. Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes. Cells. 2023; 12(15):1952. https://doi.org/10.3390/cells12151952
Chicago/Turabian StyleTorazza, Carola, Francesca Provenzano, Elena Gallia, Maria Cerminara, Matilde Balbi, Tiziana Bonifacino, Sara Tessitore, Silvia Ravera, Cesare Usai, Ilaria Musante, and et al. 2023. "Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes" Cells 12, no. 15: 1952. https://doi.org/10.3390/cells12151952
APA StyleTorazza, C., Provenzano, F., Gallia, E., Cerminara, M., Balbi, M., Bonifacino, T., Tessitore, S., Ravera, S., Usai, C., Musante, I., Puliti, A., Van Den Bosch, L., Jafar-nejad, P., Rigo, F., Milanese, M., & Bonanno, G. (2023). Genetic Downregulation of the Metabotropic Glutamate Receptor Type 5 Dampens the Reactive and Neurotoxic Phenotype of Adult ALS Astrocytes. Cells, 12(15), 1952. https://doi.org/10.3390/cells12151952