

Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain

 and
and
Abstract
1. Introduction
2. Materials and Methods
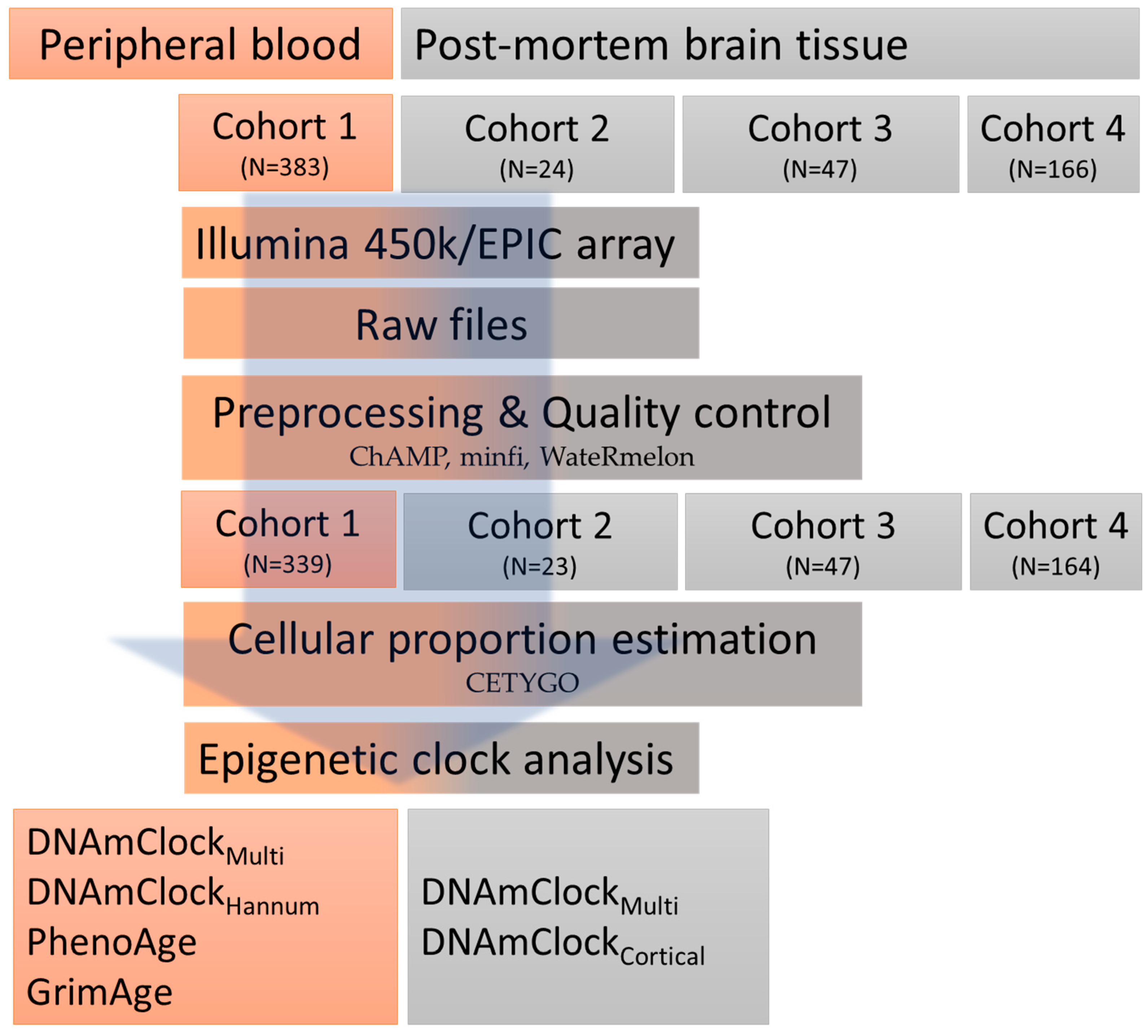
2.1. Study Overview/Design
2.1.1. Peripheral Blood Samples
2.1.2. Post-Mortem Brain Tissue Samples
2.2. DNA Methylation Data Pre-Processing
2.3. Epigenetic Clocks and Estimations of DNAm Age Acceleration
2.4. Statistical Analysis
3. Results
3.1. Correlation between DNAm Age and Chronological Age in the Peripheral Blood and Post-Mortem Brain Tissue Cohorts
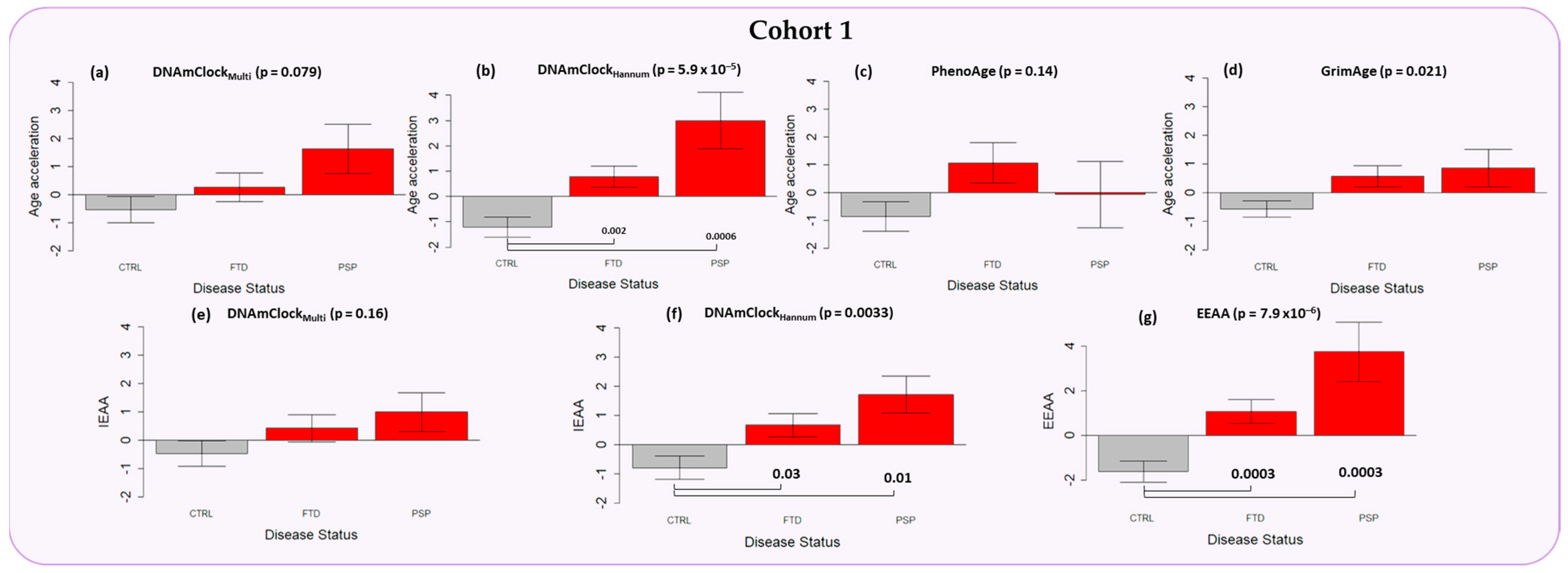
3.2. Epigenetic Age Acceleration in the Peripheral Blood of Individuals with a Clinical Diagnosis of FTD and PSP
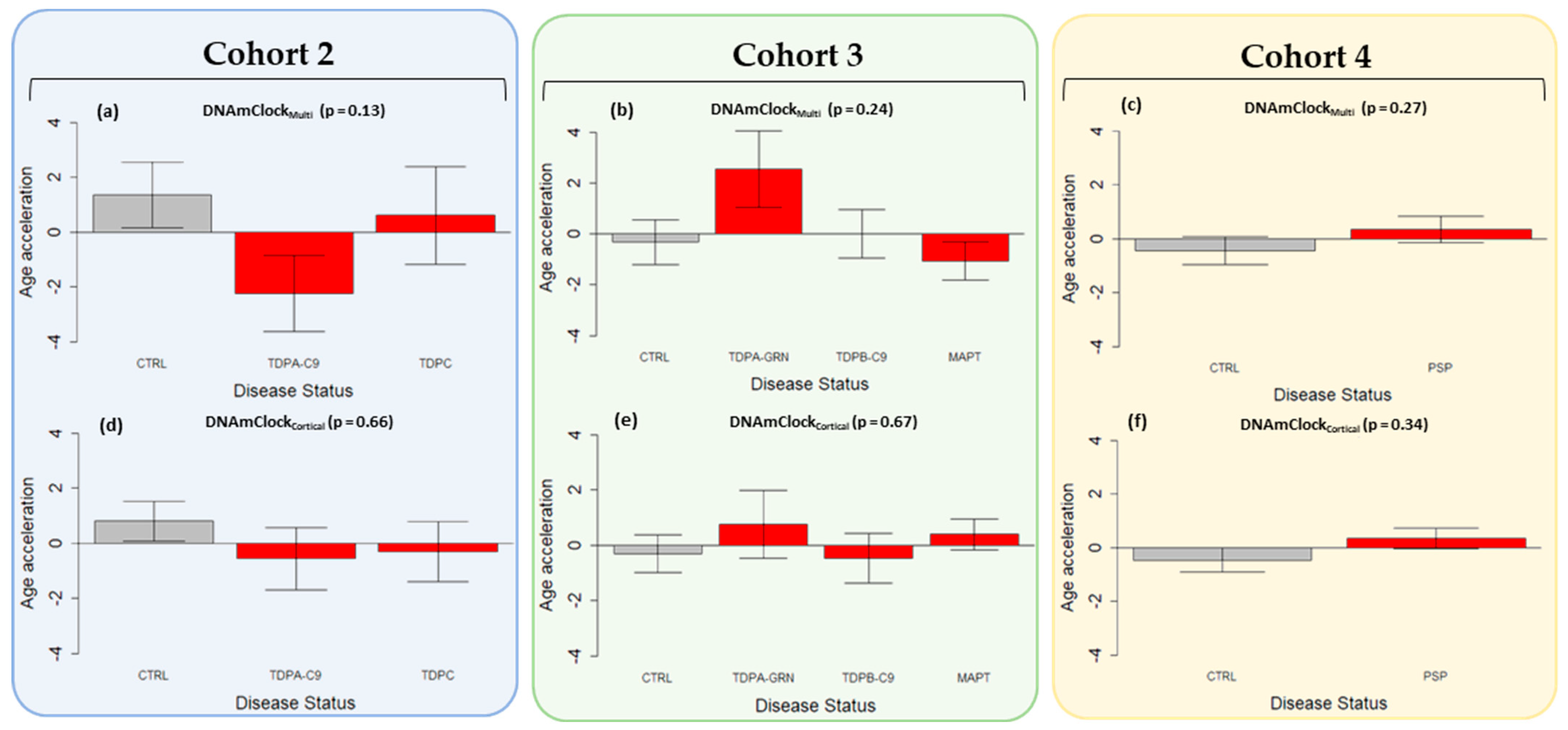
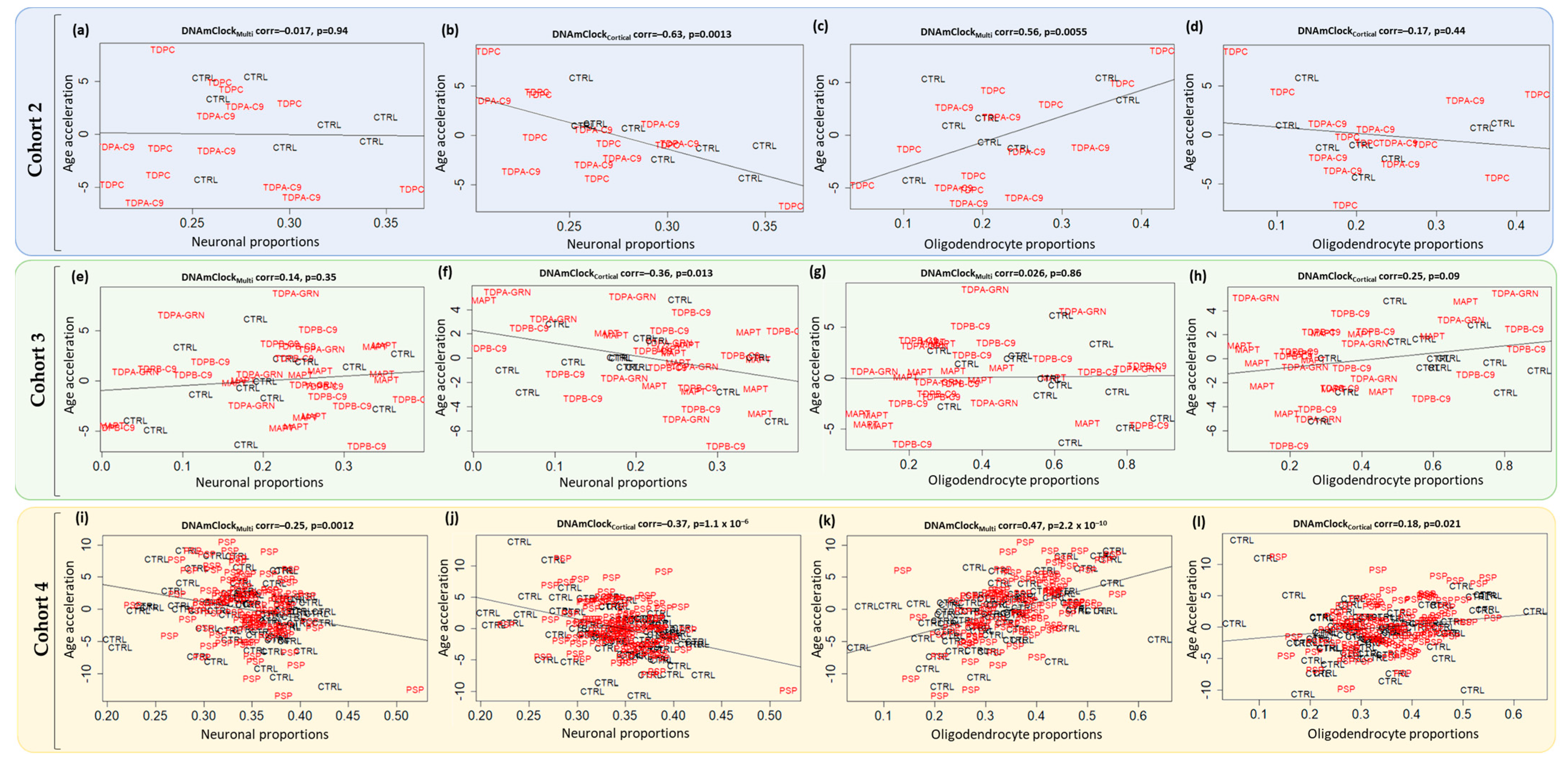
3.3. Epigenetic Age Acceleration in Post-Mortem Brain Tissue of Pathologically Confirmed FTLD Subtypes
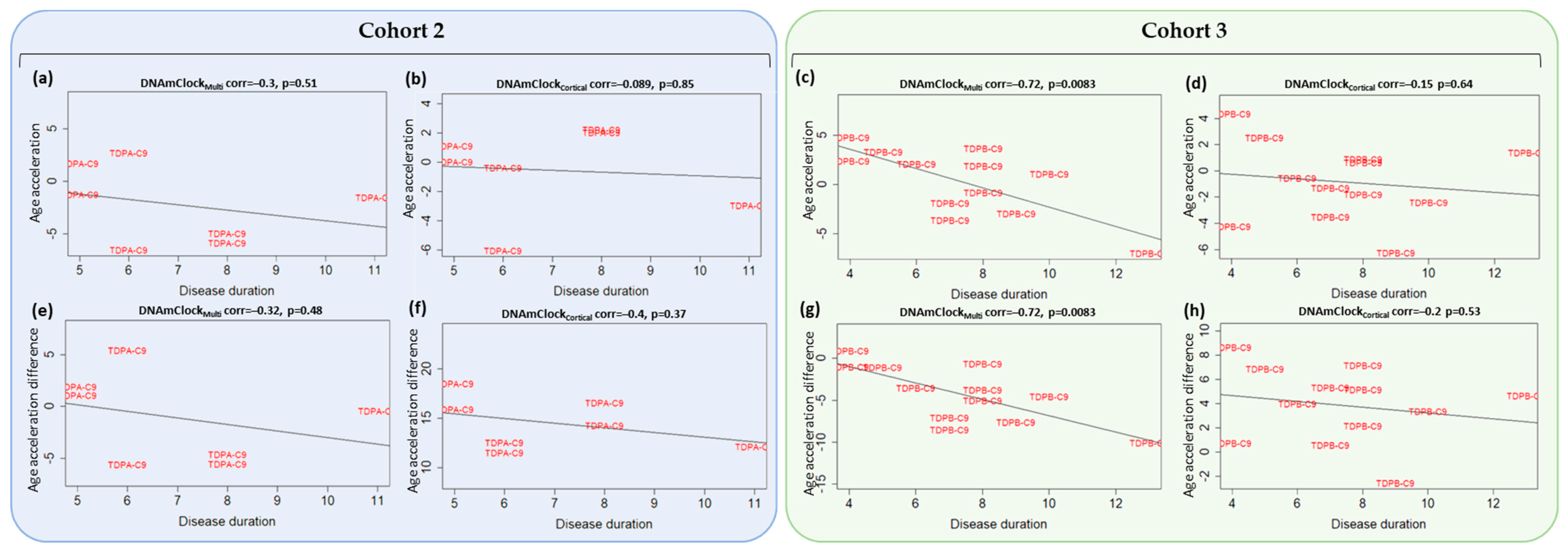
3.4. Association of Epigenetic Age Acceleration with Disease Onset and Duration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lashley, T.; Rohrer, J.D.; Mead, S.; Revesz, T. Review: An update on clinical, genetic and pathological aspects of frontotemporal lobar degenerations. Neuropathol. Appl. Neurobiol. 2015, 41, 858–881. [Google Scholar] [CrossRef]
- Rabinovici, G.D.; Miller, B.L. Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010, 24, 375–398. [Google Scholar] [CrossRef]
- Cairns, N.J.; Bigio, E.H.; Mackenzie, I.R.; Neumann, M.; Lee, V.M.; Hatanpaa, K.J.; White, C.L., 3rd; Schneider, J.A.; Grinberg, L.T.; Halliday, G.; et al. Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: Consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol. 2007, 114, 5–22. [Google Scholar] [CrossRef]
- Ferrari, R.; Manzoni, C.; Hardy, J. Genetics and molecular mechanisms of frontotemporal lobar degeneration: An update and future avenues. Neurobiol. Aging 2019, 78, 98–110. [Google Scholar] [CrossRef]
- Aswathy, P.M.; Jairani, P.S.; Mathuranath, P.S. Genetics of frontotemporal lobar degeneration. Ann. Indian Acad. Neurol. 2010, 13, S55–S62. [Google Scholar] [CrossRef]
- Li, Y.; Chen, J.A.; Sears, R.L.; Gao, F.; Klein, E.D.; Karydas, A.; Geschwind, M.D.; Rosen, H.J.; Boxer, A.L.; Guo, W.; et al. An epigenetic signature in peripheral blood associated with the haplotype on 17q21.31, a risk factor for neurodegenerative tauopathy. PLoS Genet. 2014, 10, e1004211. [Google Scholar] [CrossRef]
- Weber, A.; Schwarz, S.C.; Tost, J.; Trumbach, D.; Winter, P.; Busato, F.; Tacik, P.; Windhorst, A.C.; Fagny, M.; Arzberger, T.; et al. Epigenome-wide DNA methylation profiling in Progressive Supranuclear Palsy reveals major changes at DLX1. Nat. Commun. 2018, 9, 2929. [Google Scholar] [CrossRef]
- Fodder, K.; Murthy, M.; Rizzu, P.; Toomey, C.E.; Hasan, R.; Humphrey, J.; Raj, T.; Lunnon, K.; Mill, J.; Heutink, P.; et al. Brain DNA methylomic analysis of frontotemporal lobar degeneration reveals OTUD4 in shared dysregulated signatures across pathological subtypes. Acta Neuropathol. 2023, 146, 77–95. [Google Scholar] [CrossRef]
- Ratnavalli, E.; Brayne, C.; Dawson, K.; Hodges, J.R. The prevalence of frontotemporal dementia. Neurology 2002, 58, 1615–1621. [Google Scholar] [CrossRef]
- Lu, A.T.; Hannon, E.; Levine, M.E.; Crimmins, E.M.; Lunnon, K.; Mill, J.; Geschwind, D.H.; Horvath, S. Genetic architecture of epigenetic and neuronal ageing rates in human brain regions. Nat. Commun. 2017, 8, 15353. [Google Scholar] [CrossRef]
- Hannum, G.; Guinney, J.; Zhao, L.; Zhang, L.; Hughes, G.; Sadda, S.; Klotzle, B.; Bibikova, M.; Fan, J.B.; Gao, Y.; et al. Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 2013, 49, 359–367. [Google Scholar] [CrossRef]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef]
- Shireby, G.L.; Davies, J.P.; Francis, P.T.; Burrage, J.; Walker, E.M.; Neilson, G.W.A.; Dahir, A.; Thomas, A.J.; Love, S.; Smith, R.G.; et al. Recalibrating the epigenetic clock: Implications for assessing biological age in the human cortex. Brain 2020, 143, 3763–3775. [Google Scholar] [CrossRef]
- Levine, M.E.; Lu, A.T.; Quach, A.; Chen, B.H.; Assimes, T.L.; Bandinelli, S.; Hou, L.; Baccarelli, A.A.; Stewart, J.D.; Li, Y.; et al. An epigenetic biomarker of aging for lifespan and healthspan. Aging 2018, 10, 573–591. [Google Scholar] [CrossRef]
- Lu, A.T.; Quach, A.; Wilson, J.G.; Reiner, A.P.; Aviv, A.; Raj, K.; Hou, L.; Baccarelli, A.A.; Li, Y.; Stewart, J.D.; et al. DNA methylation GrimAge strongly predicts lifespan and healthspan. Aging 2019, 11, 303–327. [Google Scholar] [CrossRef]
- Horvath, S.; Raj, K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 2018, 19, 371–384. [Google Scholar] [CrossRef]
- Zhou, A.; Wu, Z.; Zaw Phyo, A.Z.; Torres, D.; Vishwanath, S.; Ryan, J. Epigenetic aging as a biomarker of dementia and related outcomes: A systematic review. Epigenomics 2022, 14, 1125–1138. [Google Scholar] [CrossRef]
- Zhang, M.; Tartaglia, M.C.; Moreno, D.; Sato, C.; McKeever, P.; Weichert, A.; Keith, J.; Robertson, J.; Zinman, L.; Rogaeva, E. DNA methylation age-acceleration is associated with disease duration and age at onset in C9orf72 patients. Acta Neuropathol. 2017, 134, 271–279. [Google Scholar] [CrossRef]
- Coppola, G.; Miller, B.L.; Chui, H.; Varpetian, A.; Levey, A.; Cotman, C.W.; DeCarli, C.; Mendez, M.F.; Bartzokis, G.; Kukull, W.A.; et al. Genetic investigation in frontotemporal dementia and Alzheimer’s disease: The GIFT study. Ann. Neurol. 2007, 62, S52. [Google Scholar]
- Menden, K.; Francescatto, M.; Nyima, T.; Blauwendraat, C.; Dhingra, A.; Lizardo, M.C.; Fernandes, N.; Kaurani, L.; Kronenberg-Versteeg, D.; Atarsu, B.; et al. Integrated multi-omics analysis reveals common and distinct dysregulated pathways for genetic subtypes of Frontotemporal Dementia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Pidsley, R.; CC, Y.W.; Volta, M.; Lunnon, K.; Mill, J.; Schalkwyk, L.C. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genom. 2013, 14, 293. [Google Scholar] [CrossRef]
- Aryee, M.J.; Jaffe, A.E.; Corrada-Bravo, H.; Ladd-Acosta, C.; Feinberg, A.P.; Hansen, K.D.; Irizarry, R.A. Minfi: A flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics 2014, 30, 1363–1369. [Google Scholar] [CrossRef]
- Tian, Y.; Morris, T.J.; Webster, A.P.; Yang, Z.; Beck, S.; Feber, A.; Teschendorff, A.E. ChAMP: Updated methylation analysis pipeline for Illumina BeadChips. Bioinformatics 2017, 33, 3982–3984. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2022; Available online: https://www.R-project.org/ (accessed on 16 July 2023).
- Houseman, E.A.; Accomando, W.P.; Koestler, D.C.; Christensen, B.C.; Marsit, C.J.; Nelson, H.H.; Wiencke, J.K.; Kelsey, K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinform. 2012, 13, 86. [Google Scholar] [CrossRef]
- Murthy, M.; Shireby, G.; Miki, Y.; Vire, E.; Lashley, T.; Warner, T.T.; Mill, J.; Bettencourt, C. Epigenetic age acceleration is associated with oligodendrocyte proportions in MSA and control brain tissue. Neuropathol. Appl. Neurobiol. 2023, 49, e12872. [Google Scholar] [CrossRef]
- Vellame, D.S.; Shireby, G.; MacCalman, A.; Dempster, E.L.; Burrage, J.; Gorrie-Stone, T.; Schalkwyk, L.S.; Mill, J.; Hannon, E. Uncertainty quantification of reference-based cellular deconvolution algorithms. Epigenetics 2023, 18, 2137659. [Google Scholar] [CrossRef]
- Chen, B.H.; Marioni, R.E.; Colicino, E.; Peters, M.J.; Ward-Caviness, C.K.; Tsai, P.C.; Roetker, N.S.; Just, A.C.; Demerath, E.W.; Guan, W.; et al. DNA methylation-based measures of biological age: Meta-analysis predicting time to death. Aging 2016, 8, 1844–1865. [Google Scholar] [CrossRef]
- Smith, J.A.; Raisky, J.; Ratliff, S.M.; Liu, J.; Kardia, S.L.R.; Turner, S.T.; Mosley, T.H.; Zhao, W. Intrinsic and extrinsic epigenetic age acceleration are associated with hypertensive target organ damage in older African Americans. BMC Med. Genom. 2019, 12, 141. [Google Scholar] [CrossRef]
- Quach, A.; Levine, M.E.; Tanaka, T.; Lu, A.T.; Chen, B.H.; Ferrucci, L.; Ritz, B.; Bandinelli, S.; Neuhouser, M.L.; Beasley, J.M.; et al. Epigenetic clock analysis of diet, exercise, education, and lifestyle factors. Aging 2017, 9, 419–446. [Google Scholar] [CrossRef]
- Dugue, P.A.; Bassett, J.K.; Joo, J.E.; Jung, C.H.; Ming Wong, E.; Moreno-Betancur, M.; Schmidt, D.; Makalic, E.; Li, S.; Severi, G.; et al. DNA methylation-based biological aging and cancer risk and survival: Pooled analysis of seven prospective studies. Int. J. Cancer 2018, 142, 1611–1619. [Google Scholar] [CrossRef]
- Grant, C.D.; Jafari, N.; Hou, L.F.; Li, Y.; Stewart, J.D.; Zhang, G.S.; Lamichhane, A.; Manson, J.E.; Baccarelli, A.A.; Whitsel, E.A.; et al. A longitudinal study of DNA methylation as a potential mediator of age-related diabetes risk. Geroscience 2017, 39, 475–489. [Google Scholar] [CrossRef]
- Thrush, K.L.; Bennett, D.A.; Gaiteri, C.; Horvath, S.; Dyck, C.H.V.; Higgins-Chen, A.T.; Levine, M.E. Aging the brain: Multi-region methylation principal component based clock in the context of Alzheimer’s disease. Aging 2022, 14, 5641–5668. [Google Scholar] [CrossRef]
- Shireby, G.; Dempster, E.L.; Policicchio, S.; Smith, R.G.; Pishva, E.; Chioza, B.; Davies, J.P.; Burrage, J.; Lunnon, K.; Seiler Vellame, D.; et al. DNA methylation signatures of Alzheimer’s disease neuropathology in the cortex are primarily driven by variation in non-neuronal cell-types. Nat. Commun. 2022, 13, 5620. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef]
- Sawyer, R.P.; Hill, E.J.; Yokoyama, J.; Medvedovic, M.; Ren, Y.; Zhang, X.; Choubey, D.; Shatz, R.S.; Miller, B.; Woo, D. Differences in peripheral immune system gene expression in frontotemporal degeneration. Medicine 2022, 101, e28645. [Google Scholar] [CrossRef]
- Horvath, S.; Ritz, B.R. Increased epigenetic age and granulocyte counts in the blood of Parkinson’s disease patients. Aging 2015, 7, 1130–1142. [Google Scholar] [CrossRef]
- Degerman, S.; Josefsson, M.; Adolfsson, A.N.; Wennstedt, S.; Landfors, M.; Haider, Z.; Pudas, S.; Hultdin, M.; Nyberg, L.; Adolfsson, R. Maintained memory in aging is associated with young epigenetic age. Neurobiol. Aging 2017, 55, 167–171. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peripheral Blood | |||
| Sample Group | No. of Individuals | Females (%) [% Unknowns] | Average Chronological Age (SD) |
| Cohort 1 | |||
| Controls | 178 | 53.4 [13.5] | 68.9 (10.4) |
| FTD | 117 | 26.5 [43.6] | 65.2 (9.0) |
| PSP | 44 | 15.9 [40.9] | 69.9 (7.3) |
| Total | 339 | 39.2 [27.4] | 67.7 (9.8) |
| Post-mortem brain tissue | |||
| Sample Group | No. of individuals | Females (%) [% unknowns] | Average Chronological age (SD) |
| Cohort 2 | |||
| Controls | 8 | 62.5 | 75.8 (5.6) |
| FTLD-TDPA (C9orf72) | 7 | 57.1 | 66.9 (4.8) |
| FTLD-TDPC (Sporadic) | 8 | 50.0 | 72.9 (4.8) |
| Total | 23 | 56.5 | 72.0 (6.1) |
| Cohort 3 | |||
| Controls | 14 | 64.3 | 78.4 (11.8) |
| FTLD-TDPA (GRN) | 7 | 71.4 | 64.6 (7.6) |
| FTLD-TDPB (C9orf72) | 13 | 61.5 | 63.8 (8.2) |
| FTLD-Tau (MAPT) | 13 | 46.2 | 60.9 (7.6) |
| Total | 47 | 59.6 | 67.5 (11.5) |
| Cohort 4 | |||
| Controls | 71 | 35.2 | 76.0 (8.0) |
| PSP | 93 | 41.9 | 71.6 (5.3) |
| Total | 164 | 39.0 | 73.5 (6.9) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murthy, M.; Rizzu, P.; Heutink, P.; Mill, J.; Lashley, T.; Bettencourt, C. Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain. Cells 2023, 12, 1922. https://doi.org/10.3390/cells12141922
Murthy M, Rizzu P, Heutink P, Mill J, Lashley T, Bettencourt C. Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain. Cells. 2023; 12(14):1922. https://doi.org/10.3390/cells12141922
Chicago/Turabian StyleMurthy, Megha, Patrizia Rizzu, Peter Heutink, Jonathan Mill, Tammaryn Lashley, and Conceição Bettencourt. 2023. "Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain" Cells 12, no. 14: 1922. https://doi.org/10.3390/cells12141922
APA StyleMurthy, M., Rizzu, P., Heutink, P., Mill, J., Lashley, T., & Bettencourt, C. (2023). Epigenetic Age Acceleration in Frontotemporal Lobar Degeneration: A Comprehensive Analysis in the Blood and Brain. Cells, 12(14), 1922. https://doi.org/10.3390/cells12141922