

Amyloid β1–42 Oligomers Induce Galectin–1S8 O–GlcNAcylation Leading to Microglia Migration

 and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Production and Purification of Amyloid-β1–42 (Aβ1–42) and Oligomers Preparation
2.3. Plasmid Construct and TAT-OBD-Gal-1 Protein Purification
2.4. Site-Directed Mutagenesis
2.5. Cell Culture
2.6. Microglia Transient Transfection
2.7. Wheat Germ Agglutinin-Affinity Precipitation and Protein Identification by LC-MS/MS
2.8. In Vitro O-GlcNAcTransferase (OGT) Assay
2.9. Scratch Wound Healing Assay
2.10. Transwell Migration Assay
2.11. Statistical Analysis
3. Results
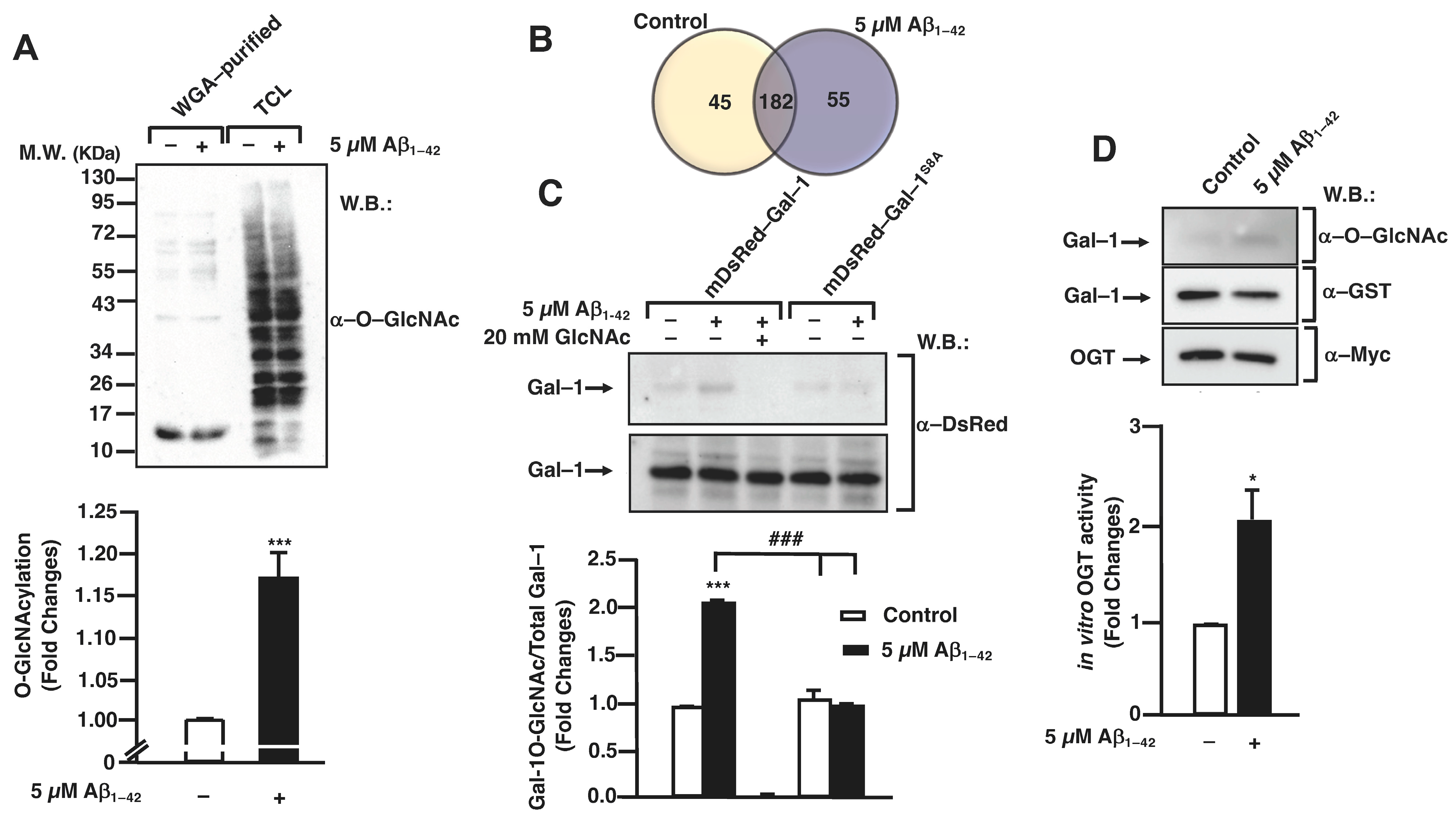
3.1. Aβ1–42 Oligomers Increase Protein O-GlcNAcylation in Human Microglia
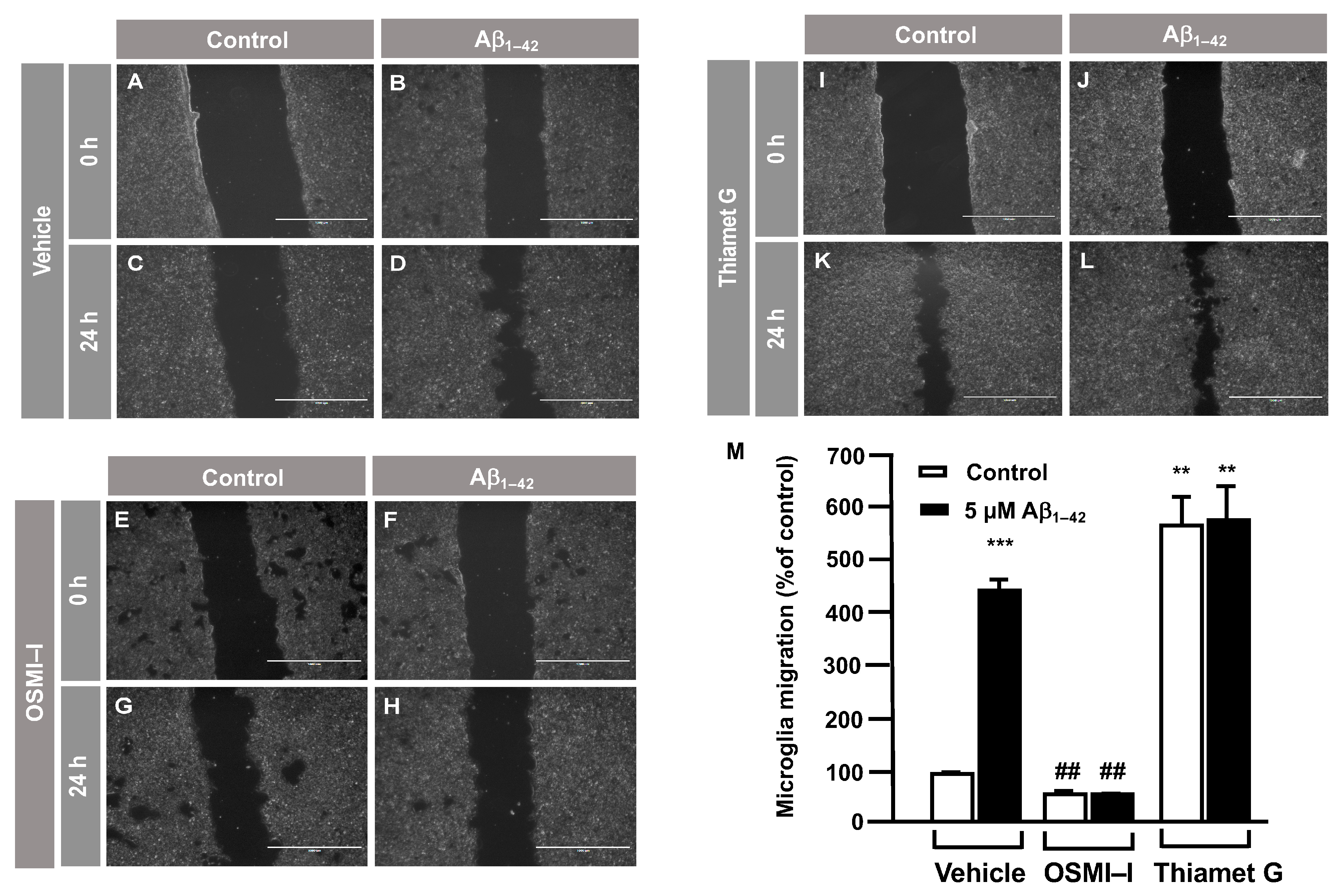
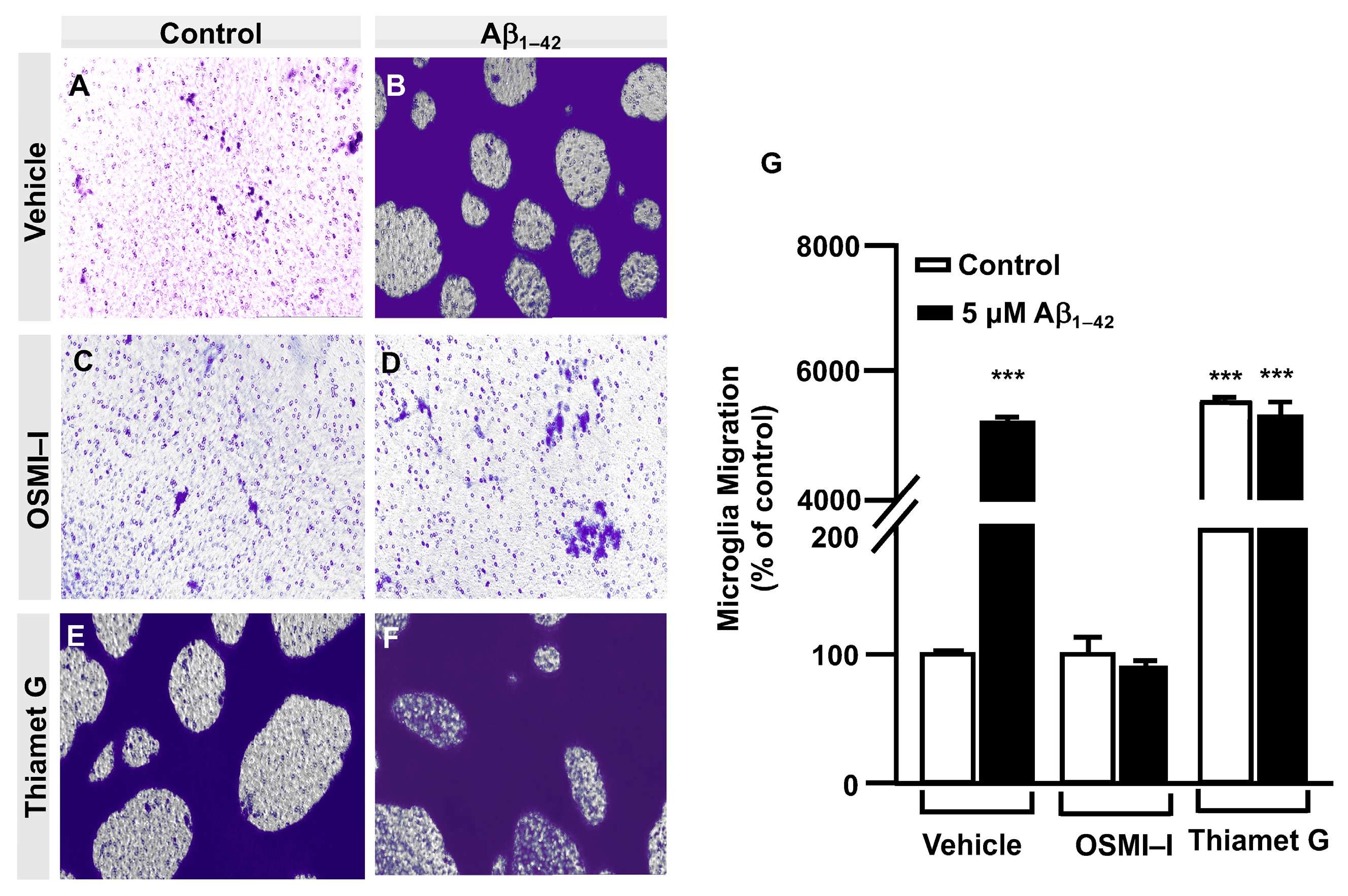
3.2. O-GlcNAc Homeostasis Controls Aβ1–42-Induced Microglial Migration
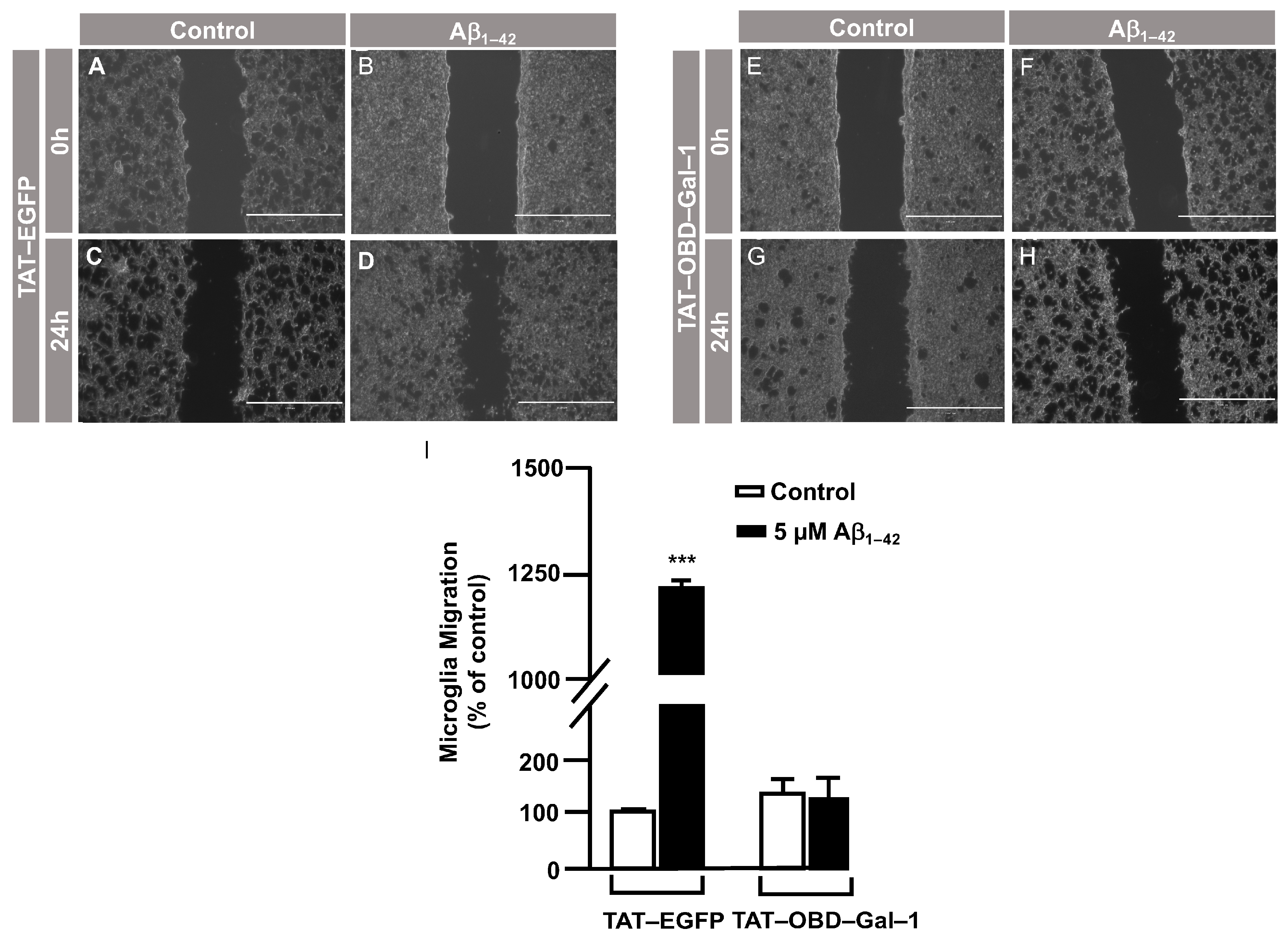
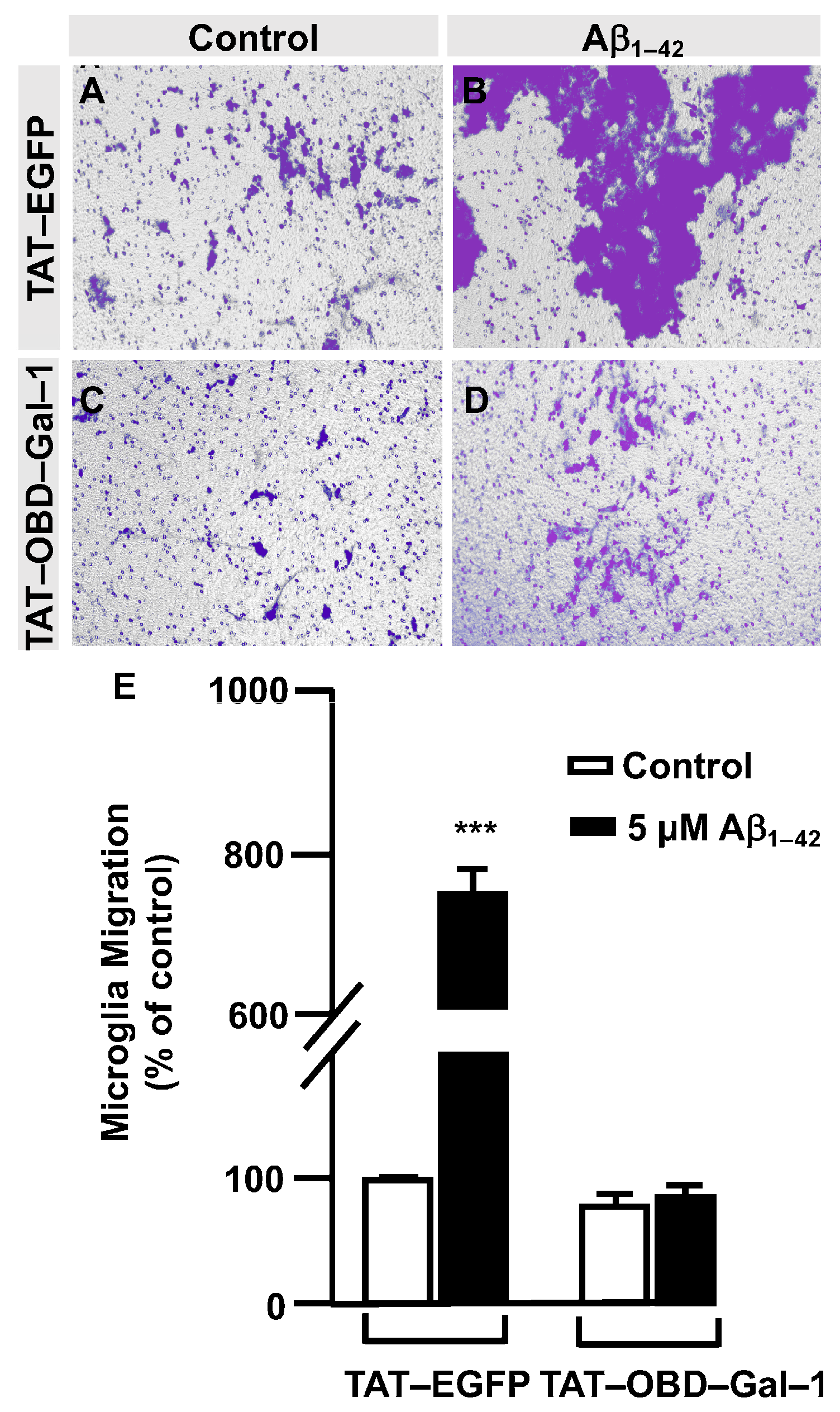
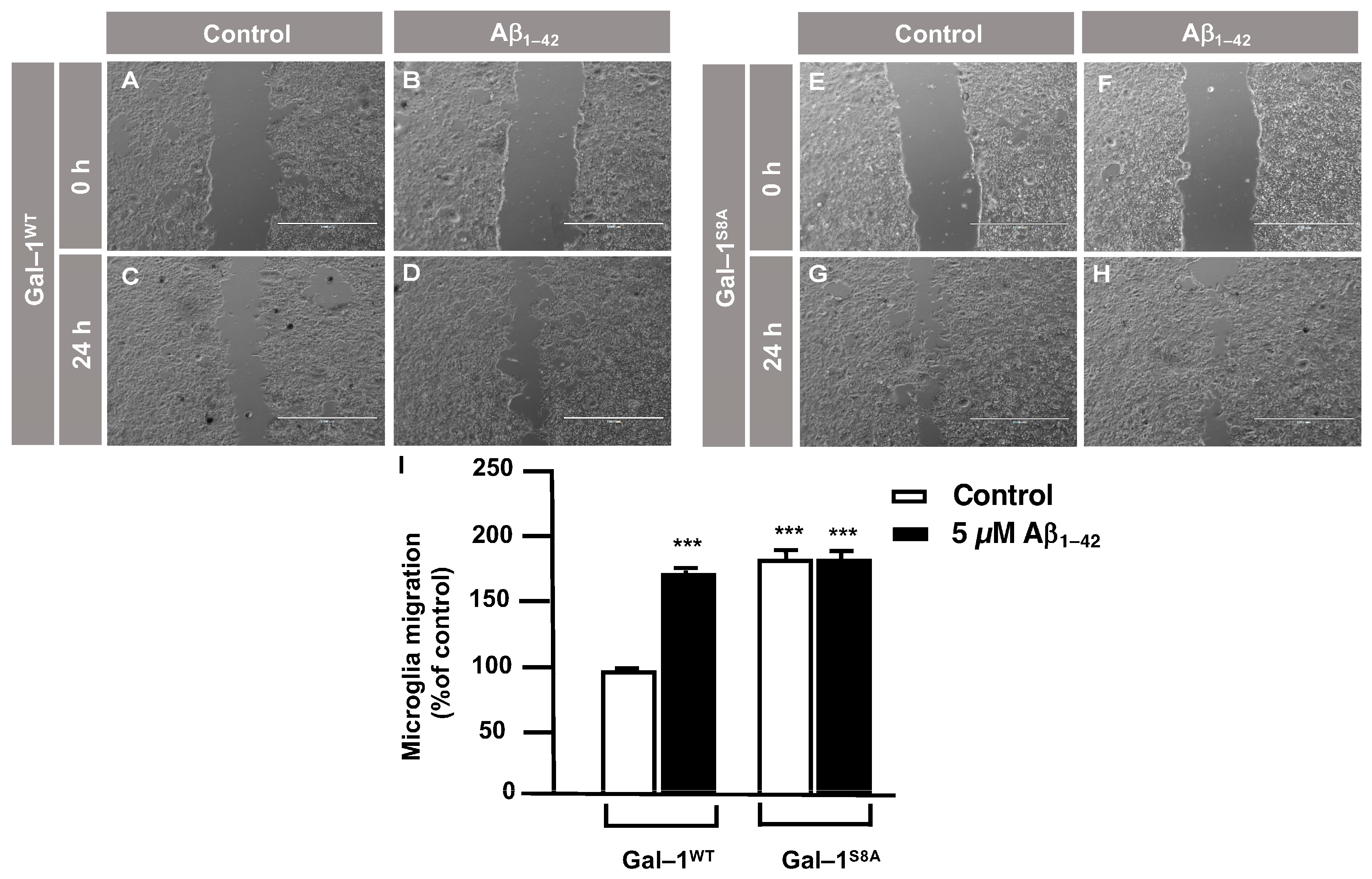
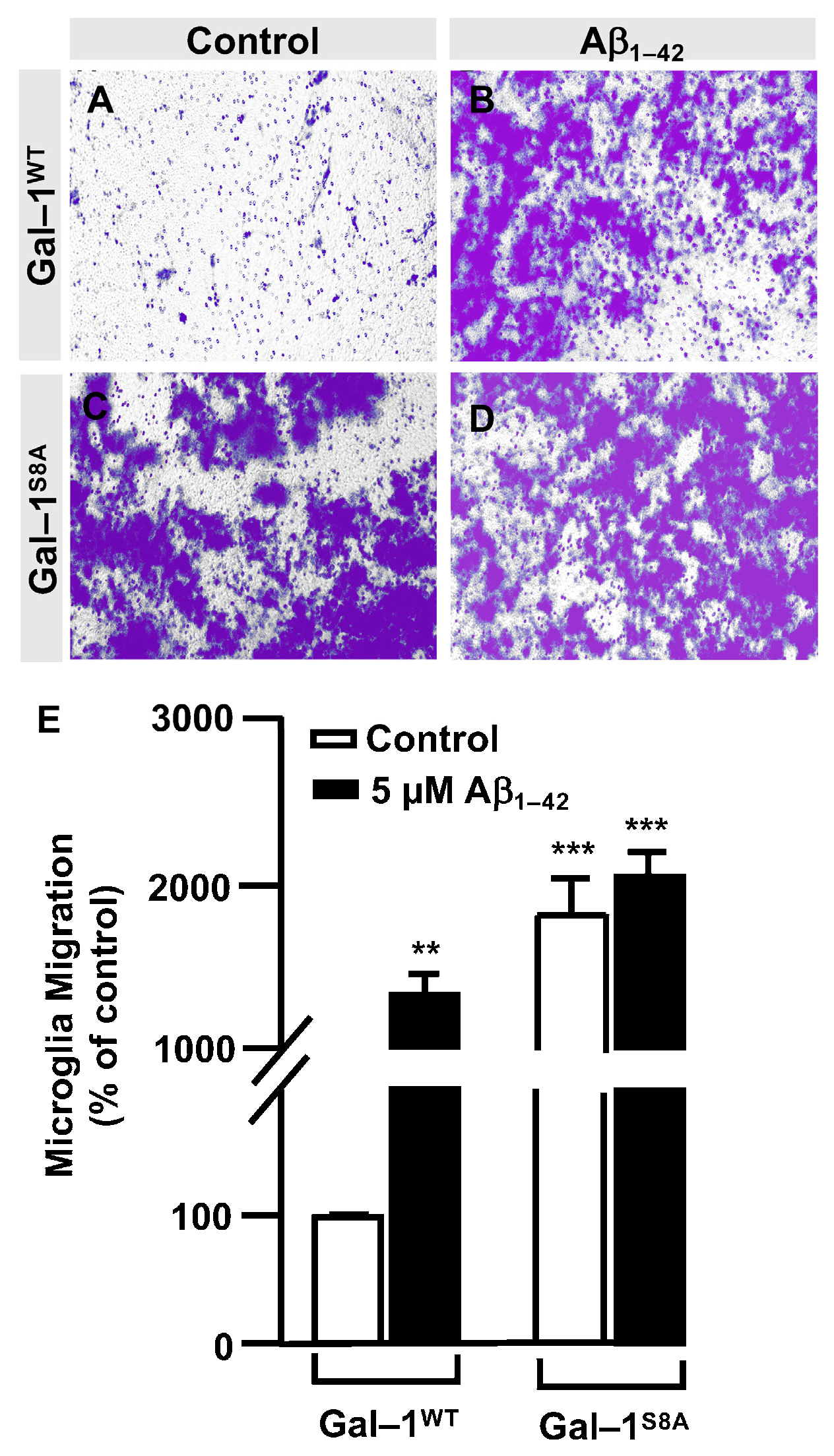
3.3. O-GlcNAc Homeostasis Controls Aβ1–42 Oligomers-Induced Microglial Migration through Galectin-1 Ser8 O-GlcNAcylation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, D.M.; Olivier-Van Stichelen, S. The O-GlcNAc cycling in neurodevelopment and associated diseases. Biochem. Soc. Trans. 2022, 50, 1693–1702. [Google Scholar]
- Bond, M.R.; Hanover, J.A. A little sugar goes a long way: The cell biology of O-GlcNAc. J. Cell Biol. 2015, 208, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Slawson, C.; Hart, G.W. O-GlcNAc signalling: Implications for cancer cell biology. Nat. Rev. Cancer 2011, 11, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Pravata, V.M.; Omelkova, M.; Stavridis, M.P.; Desbiens, C.M.; Stephen, H.M.; Lefeber, D.J.; Gecz, J.; Gundogdu, M.; Õunap, K.; Joss, S.; et al. An intellectual disability syndrome with single-nucleotide variants in O-GlcNAc transferase. Eur. J. Hum. Genet. 2020, 28, 706–714. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Vocadlo, D.J. O-GlcNAc and neurodegeneration: Biochemical mechanisms and potential roles in Alzheimer’s disease and beyond. Chem. Soc. Rev. 2014, 43, 6839–6858. [Google Scholar] [CrossRef]
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88. [Google Scholar] [CrossRef]
- Forster, S.; Welleford, A.S.; Triplett, J.C.; Sultana, R.; Schmitz, B.; Butterfield, D.A. Increased O-GlcNAc levels correlate with decreased O-GlcNAcase levels in Alzheimer disease brain. Biochim. Biophys. Acta 2014, 1842, 1333–1339. [Google Scholar] [CrossRef]
- Chun, Y.S.; Kwon, O.H.; Chung, S. O-GlcNAcylation of amyloid-beta precursor protein at threonine 576 residue regulates trafficking and processing. Biochem. Biophys. Res. Commun. 2017, 490, 486–491. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.X.; Iqbal, K. Hyperphosphorylation of microtubule-associated protein tau: A promising therapeutic target for Alzheimer disease. Curr. Med. Chem. 2008, 15, 2321–2328. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Ha, H.J.; Chung, E.S.; Baek, S.H.; Cho, Y.; Kim, H.K.; Han, J.; Sul, J.H.; Lee, J.; Kim, E.; et al. O-GlcNAcylation ameliorates the pathological manifestations of Alzheimer’s disease by inhibiting necroptosis. Sci. Adv. 2021, 7, eabd3207. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A.; Zorec, R.; Rodriguez, J.J.; Parpura, V. Astroglia dynamics in ageing and Alzheimer’s disease. Curr. Opin. Pharmacol. 2016, 26, 74–79. [Google Scholar] [CrossRef]
- Rodríguez-Gómez, J.A.; Kavanagh, E.; Engskog-Vlachos, P.; Engskog, M.K.R.; Herrera, A.J.; Espinosa-Oliva, A.M.; Joseph, B.; Hajji, N.; Venero, J.L.; Burguillos, M.A. Microglia: Agents of the CNS Pro-Inflammatory Response. Cells 2020, 9, 1717. [Google Scholar] [CrossRef]
- Lai, A.Y.; McLaurin, J. Clearance of amyloid-beta peptides by microglia and macrophages: The issue of what, when and where. Future Neurol. 2012, 7, 165–176. [Google Scholar] [CrossRef]
- Feng, W.; Zhang, Y.; Wang, Z.; Xu, H.; Wu, T.; Marshall, C.; Gao, J.; Xiao, M. Microglia prevent beta-amyloid plaque formation in the early stage of an Alzheimer’s disease mouse model with suppression of glymphatic clearance. Alzheimers Res. Ther. 2020, 12, 125. [Google Scholar] [CrossRef]
- Streit, W.J.; Sammons, N.W.; Kuhns, A.J.; Sparks, D.L. Dystrophic microglia in the aging human brain. Glia 2004, 45, 208–212. [Google Scholar] [CrossRef]
- Hickman, S.E.; Allison, E.K.; El Khoury, J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J. Neurosci. 2008, 28, 8354–8360. [Google Scholar] [CrossRef]
- Walsh, D.M.; Thulin, E.; Minogue, A.M.; Gustavsson, N.; Pang, E.; Teplow, D.B.; Linse, S. A facile method for expression and purification of the Alzheimer’s disease-associated amyloid beta-peptide. FEBS J. 2009, 276, 1266–1281. [Google Scholar] [CrossRef]
- Klein, W.L. Abeta toxicity in Alzheimer’s disease: Globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem. Int. 2002, 41, 345–352. [Google Scholar] [CrossRef]
- Haddad, E.; Zugaza, J.L.; Louache, F.; Debili, N.; Crouin, C.; Schwarz, K.; Fischer, A.; Vainchenker, W.; Bertoglio, J. The interaction between Cdc42 and WASP is required for SDF-1-induced T-lymphocyte chemotaxis. Blood 2001, 97, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Manterola, L.; Hernando-Rodríguez, M.; Ruiz, A.; Apraiz, A.; Arrizabalaga, O.; Vellón, L.; Alberdi, E.; Cavaliere, F.; Lacerda, H.M.; Jimenez, S.; et al. 1-42 β-amyloid peptide requires PDK1/nPKC/Rac 1 pathway to induce neuronal death. Transl. Psychiatry 2013, 3, e219. [Google Scholar] [CrossRef] [PubMed]
- Llavero, F.; Luque Montoro, M.; Arrazola Sastre, A.; Fernandez-Moreno, D.; Lacerda, H.M.; Parada, L.A.; Lucia, A.; Zugaza, J.L. Epidermal growth factor receptor controls glycogen phosphorylase in T cells through small GTPases of the RAS family. J. Biol. Chem. 2019, 294, 4345–4358. [Google Scholar] [CrossRef] [PubMed]
- Wyssenbach, A.; Quintela, T.; Llavero, F.; Zugaza, J.L.; Matute, C.; Alberdi, E. Amyloid β-induced astrogliosis is mediated by β1-integrin via NADPH oxidase 2 in Alzheimer’ disease. Aging Cell 2016, 15, 1140–1152. [Google Scholar] [CrossRef]
- Han, C.; Gu, Y.; Shan, H.; Mi, W.; Sun, J.; Shi, M.; Zhang, X.; Lu, X.; Han, F.; Gong, Q.; et al. O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nat. Commun. 2017, 8, 1491. [Google Scholar] [CrossRef]
- Xu, F.; Xu, Y.; Zhu, L.; Rao, P.; Wen, J.; Sang, Y.; Shang, F.; Liu, Y. Fasudil inhibits LPS- induced migration of retinal microglial cells via regulating p38-MAPK signaling pathway. Mol. Vis. 2016, 22, 836–846. [Google Scholar]
- Ortiz-Meoz, R.F.; Jiang, J.; Lazarus, M.B.; Orman, M.; Janetzko, J.; Fan, C.; Duveau, D.Y.; Tan, Z.W.; Thomas, C.J.; Walker, S. A small molecule that inhibits OGT activity in cells. ACS Chem. Biol. 2015, 10, 1392–1397. [Google Scholar] [CrossRef]
- Yuzwa, S.A.; Macauley, M.S.; Heinonen, J.E.; Shan, X.; Dennis, R.J.; He, Y.; Whitworth, G.E.; Stubbs, K.A.; McEachern, E.J.; Davies, G.J.; et al. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nat. Chem. Biol. 2008, 4, 483–490. [Google Scholar] [CrossRef]
- Camby, I.; Le Mercier, M.; Lefranc, F.; Kiss, R. Galectin-1: A small protein with major Functions. Glycobiology 2006, 16, 137R–157R. [Google Scholar] [CrossRef]
- Nio-Kobayashi, J.; Itabashi, T. Galectins and Their Ligand Glycoconjugates in the Central Nervous System Under Physiological and Pathological Conditions. Front. Neuroanat. 2021, 15, 767330. [Google Scholar] [CrossRef] [PubMed]
- Mathew, M.P.; Abramowitz, L.K.; Donaldson, J.G.; Hanover, J.A. Nutrient-responsive O-GlcNAcylation dynamically modulates the secretion of glycan-binding protein galectin 3. J. Biol. Chem. 2012, 298, 101743. [Google Scholar] [CrossRef] [PubMed]
- Cedeno-Laurent, F.; Dimitroff, C.J. Galectin-1 research in T cell immunity: Past, present and future. Clin. Immunol. 2021, 142, 107–116. [Google Scholar] [CrossRef]
- Mertins, P.; Yang, F.; Liu, T.; Mani, D.R.; Petyuk, V.A.; Gillette, M.A.; Clauser, K.R.; Qiao, J.W.; Gritsenko, M.A.; Moore, R.J.; et al. Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol. Cell. Proteom. 2014, 13, 1690–1704. [Google Scholar] [CrossRef]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Selnick, H.G.; Hess, J.F.; Tang, C.; Liu, K.; Schachter, J.B.; Ballard, J.E.; Marcus, J.; Klein, D.J.; Wang, X.; Pearson, M.; et al. Discovery of MK-8719, a Potent O-GlcNAcase Inhibitor as a Potential Treatment for Tauopathies. J. Med. Chem. 2019, 62, 10062–10097. [Google Scholar] [CrossRef]
- Robertson, L.A.; Moya, K.L.; Breen, K.C. The potential role of tau protein O glycosylation in Alzheimer’s disease. J. Alzheimers Dis. 2004, 6, 489–495. [Google Scholar] [CrossRef]
- Blaževitš, O.; Mideksa, Y.G.; Šolman, M.; Ligabue, A.; Ariotti, N.; Nakhaeizadeh, H.; Fansa, E.K.; Papageorgiou, A.C.; Wittinghofer, A.; Ahmadian, M.R.; et al. Galectin-1 dimers can scaffold Raf-effectors to increase H-ras nanoclustering. Sci. Rep. 2016, 6, 24165. [Google Scholar] [CrossRef]
- Starossom, S.C.; Mascanfroni, I.D.; Imitola, J.; Cao, L.; Raddassi, K.; Hernandez, S.F.; Bassil, R.; Croci, D.O.; Cerliani, J.P.; Delacour, D.; et al. Galectin-1 Deactivates Classically Activated Microglia and Protects from Inflammation-Induced Neurodegeneration. Immunity 2012, 37, 249–263. [Google Scholar] [CrossRef]
- Elola, M.T.; Ferragut, F.; Mendez-Huergo, S.P.; Croci, D.O.; Bracalente, C.; Rabinovich, G.A. Galectins: Multitask signaling molecules linking fibroblast, endothelial and immune cell programs in the tumor microenvironment. Cell. Immunol. 2018, 333, 34–45. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arrazola Sastre, A.; Luque Montoro, M.; Llavero, F.; Zugaza, J.L. Amyloid β1–42 Oligomers Induce Galectin–1S8 O–GlcNAcylation Leading to Microglia Migration. Cells 2023, 12, 1876. https://doi.org/10.3390/cells12141876
Arrazola Sastre A, Luque Montoro M, Llavero F, Zugaza JL. Amyloid β1–42 Oligomers Induce Galectin–1S8 O–GlcNAcylation Leading to Microglia Migration. Cells. 2023; 12(14):1876. https://doi.org/10.3390/cells12141876
Chicago/Turabian StyleArrazola Sastre, Alazne, Miriam Luque Montoro, Francisco Llavero, and José L. Zugaza. 2023. "Amyloid β1–42 Oligomers Induce Galectin–1S8 O–GlcNAcylation Leading to Microglia Migration" Cells 12, no. 14: 1876. https://doi.org/10.3390/cells12141876
APA StyleArrazola Sastre, A., Luque Montoro, M., Llavero, F., & Zugaza, J. L. (2023). Amyloid β1–42 Oligomers Induce Galectin–1S8 O–GlcNAcylation Leading to Microglia Migration. Cells, 12(14), 1876. https://doi.org/10.3390/cells12141876