
Migration of Myogenic Cells Is Highly Influenced by Cytoskeletal Septin7

 , , , ,
, , , ,  , , , and
, , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Silencing
2.3. Immunofluorescent Staining of Cultured Cells
2.4. Determination of Cellular Proliferation
2.5. Fusion Protein Design and Expression
2.6. Examination of the Septin7-N-mCherry Fusion Protein Expression in Living Cells
2.7. Migration Assay and Analysis
2.8. Measurement of Intracellular Ca2+ Concentration ([Ca2+]i) in Migrating Cells
2.9. Quantification and Statistical Analysis
3. Results
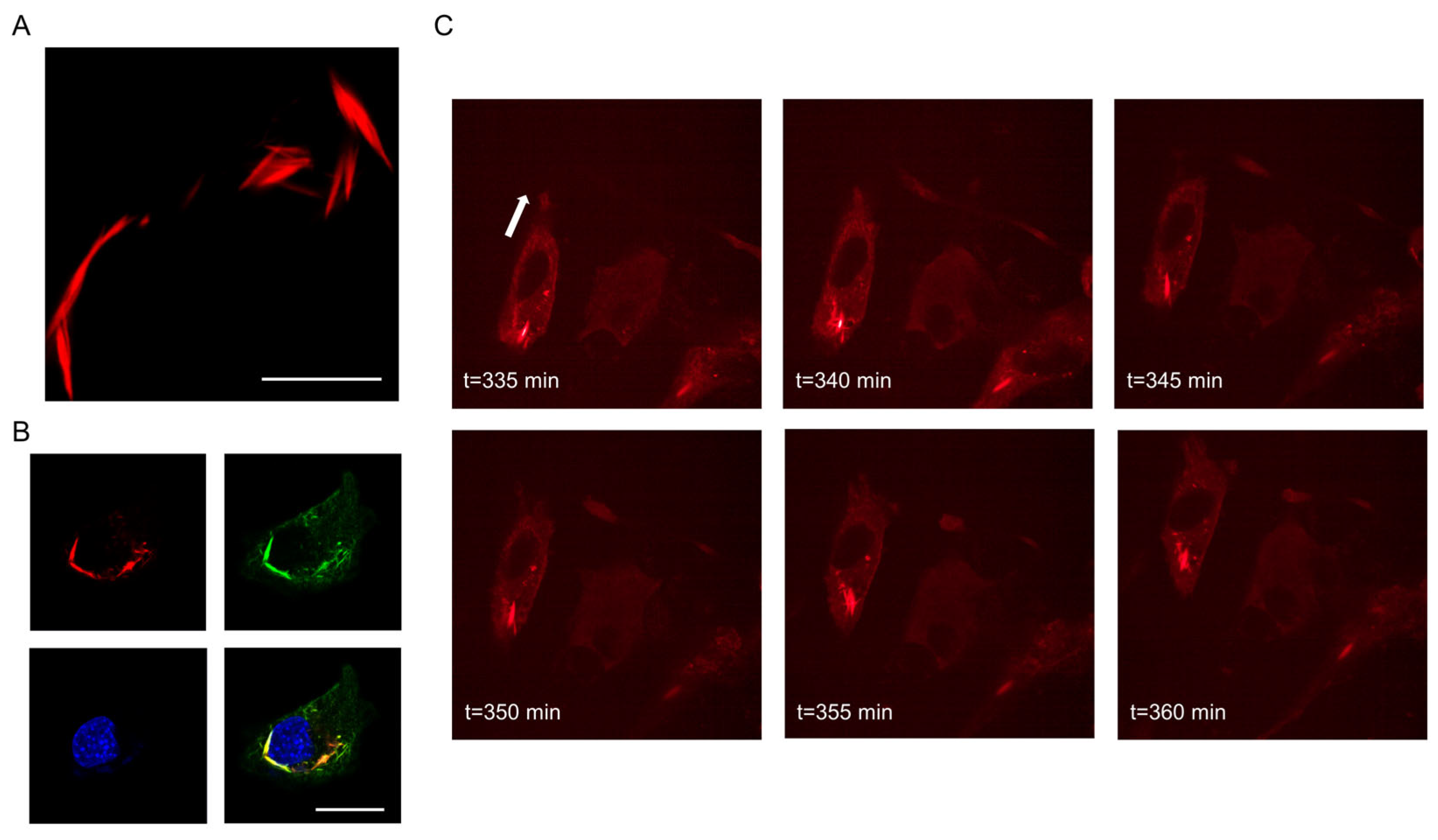
3.1. Intracellular Appearance and Structure of Septin7 Are Different in Migrating and Non-Migrating Myoblasts
3.2. Migration of Myogenic Cells Is Accompanied by Changes in Intracellular [Ca2+]
3.3. Downregulation of Septin7 Protein Expression Altered the Migration of C2C12 Myoblasts
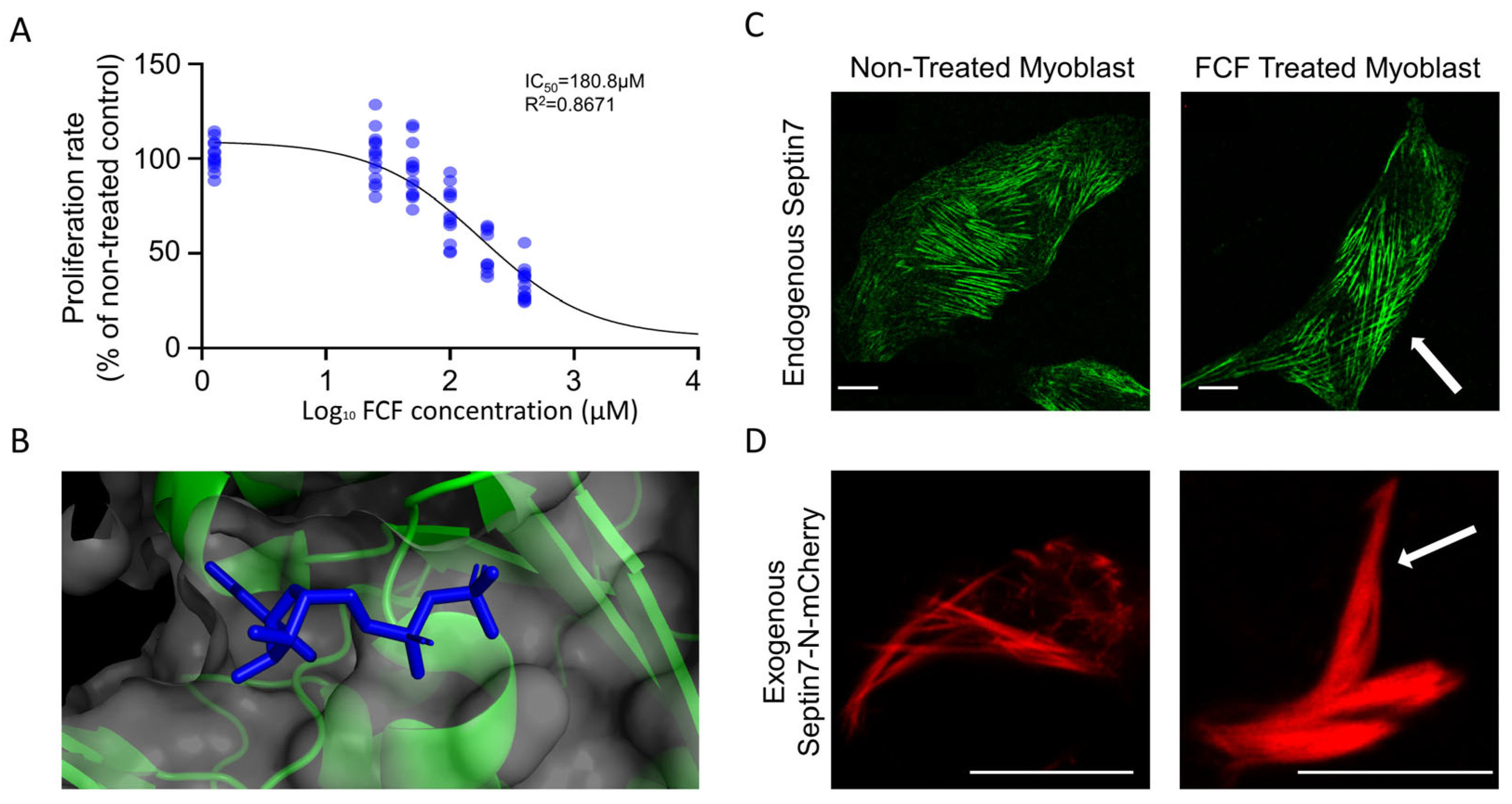
3.4. FCF Stabilizes Septin7 Filaments in C2C12 Myoblasts
3.5. FCF Treatment Inhibits the Migration of C2C12 Myoblasts; S7-KD Cultures Are More Resistant to FCF
4. Discussion
4.1. Septin7 Filaments Are Important Cytoskeletal Parts of Myoblasts and Rearranged during Migration
4.2. Modified Septin7 Expression Alters [Ca2+]i Homeostasis in Migrating Myoblasts
4.3. Amount of Septin7 Regulate Migration Parameters in Myoblasts
4.4. Dynamic Rearrangement of Septin Assembly Is Required for Myoblast Migration
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choi, S.; Ferrari, G.; Tedesco, F.S. Cellular dynamics of myogenic cell migration: Molecular mechanisms and implications for skeletal muscle cell therapies. EMBO Mol. Med. 2020, 12, e12357. [Google Scholar] [CrossRef] [PubMed]
- Baghdadi, M.B.; Tajbakhsh, S. Regulation and phylogeny of skeletal muscle regeneration. Dev. Biol. 2018, 433, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Le Moal, E.; Pialoux, V.; Juban, G.; Groussard, C.; Zouhal, H.; Chazaud, B.; Mounier, R. Redox Control of Skeletal Muscle Regeneration. Antioxidants Redox Signal. 2017, 27, 276–310. [Google Scholar] [CrossRef]
- Wang, Y.X.; Rudnicki, M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2012, 13, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.J.; Badylak, S.F. Regeneration of skeletal muscle. Cell Tissue Res. 2012, 347, 759–774. [Google Scholar] [CrossRef] [PubMed]
- Mazzotti, A.L.; Coletti, D. The need for a consensus on the locution “central nuclei” in striated muscle myopathies. Front. Physiol. 2016, 7, 577. [Google Scholar] [CrossRef]
- Mostowy, S.; Cossart, P. Septins: The fourth component of the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2012, 13, 183–194. [Google Scholar] [CrossRef]
- Cleary, J.M.; Hancock, W.O. Molecular mechanisms underlying microtubule growth dynamics. Curr. Biol. 2021, 31, R560–R573. [Google Scholar] [CrossRef]
- Lehka, L.; Rędowicz, M.J. Mechanisms regulating myoblast fusion: A multilevel interplay. Semin. Cell Dev. Biol. 2020, 104, 81–92. [Google Scholar] [CrossRef]
- Farrugia, A.J.; Rodríguez, J.; Orgaz, J.L.; Lucas, M.; Sanz-Moreno, V.; Calvo, F. CDC42EP5/BORG3 modulates SEPT9 to promote actomyosin function, migration, and invasion. J. Cell Biol. 2020, 219, e201912159. [Google Scholar] [CrossRef]
- Liu, Z.; Vong, Q.P.; Liu, C.; Zheng, Y. Borg5 is required for angiogenesis by regulating persistent directional migration of the cardiac microvascular endothelial cells. Mol. Biol. Cell 2014, 25, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Connolly, D.; Abdesselam, I.; Verdier-Pinard, P.; Montagna, C. Septin roles in tumorigenesis. Biol. Chem. 2011, 392, 725–738. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fei, F.; Qu, J.; Li, C.; Li, Y.; Zhang, S. The role of septin 7 in physiology and pathological disease: A systematic review of current status. J. Cell Mol. Med. 2018, 22, 3298–3307. [Google Scholar] [CrossRef] [PubMed]
- Angelis, D.; Karasmanis, E.P.; Bai, X.; Spiliotis, E.T. In silico docking of forchlorfenuron (FCF) to septins suggests that FCF interferes with GTP binding. PLoS ONE 2014, 9, e96390. [Google Scholar] [CrossRef] [PubMed]
- Woods, B.L.; Gladfelter, A.S. The state of the septin cytoskeleton from assembly to function. Curr. Opin. Cell Biol. 2021, 68, 105–112. [Google Scholar] [CrossRef]
- Spiliotis, E.T.; Nakos, K. Cellular functions of actin- and microtubule-associated septins. Curr. Biol. 2021, 31, R651–R666. [Google Scholar] [CrossRef]
- Tooley, A.J.; Gilden, J.; Jacobelli, J.; Beemiller, P.; Trimble, W.S.; Kinoshita, M.; Krummel, M.F. Amoeboid T lymphocytes require the septin cytoskeleton for cortical integrity and persistent motility. Nat. Cell Biol. 2009, 11, 17–26. [Google Scholar] [CrossRef]
- Füchtbauer, A.; Lassen, L.B.; Jensen, A.B.; Howard, J.; De Salas Quiroga, A.; Warming, S.; Sørensen, A.B.; Pedersen, F.S.; Füchtbauer, E.M. Septin9 is involved in septin filament formation and cellular stability. Biol. Chem. 2011, 392, 769–777. [Google Scholar] [CrossRef]
- Zeng, Y.; Cao, Y.; Liu, L.; Zhao, J.; Zhang, T.; Xiao, L.; Jia, M.; Tian, Q.; Yu, H.; Chen, S.; et al. SEPT9_i1 regulates human breast cancer cell motility through cytoskeletal and RhoA/FAK signaling pathway regulation. Cell Death Dis. 2019, 10, 720. [Google Scholar] [CrossRef]
- Zhang, N.; Liu, L.; Fan, N.; Zhang, Q.; Wang, W.; Zheng, M.; Ma, L.; Li, Y.; Shi, L. The requirement of SEPT2 and SEPT7 for migration and invasion in human breast cancer via MEK/ERK activation. Oncotarget 2016, 7, 61587–61600. [Google Scholar] [CrossRef]
- Verdier-Pinard, P.; Salaun, D.; Bouguenina, H.; Shimada, S.; Pophillat, M.; Audebert, S.; Agavnian, E.; Coslet, S.; Charafe-Jauffret, E.; Tachibana, T.; et al. Septin 9-i2 is downregulated in tumors, impairs cancer cell migration and alters subnuclear actin filaments. Sci. Rep. 2017, 7, 44976. [Google Scholar] [CrossRef]
- Fan, Y.; Du, Z.; Ding, Q.; Zhang, J.; Op Den Winkel, M.; Gerbes, A.L.; Liu, M.; Steib, C.J. SEPT6 drives hepatocellular carcinoma cell proliferation, migration and invasion via the Hippo/YAP signaling pathway. Int. J. Oncol. 2021, 58, 25. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.; Li, X.; Zhu, J. LSD1-mediated stabilization of SEPT6 protein activates the TGF-β1 pathway and regulates non-small-cell lung cancer metastasis. Cancer Gene Ther. 2022, 29, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Hou, M.; Liu, X.; Cao, J.; Chen, B. SEPT7 overexpression inhibits glioma cell migration by targeting the actin cytoskeleton pathway. Oncol. Rep. 2016, 35, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Hua, D.; Zhang, J.; Lan, Q.; Huang, Q.; Yoon, J.G.; Han, X.; Li, L.; Foltz, G.; Zheng, S.; et al. MicroRNA-127-3p promotes glioblastoma cell migration and invasion by targeting the tumor-suppressor gene SEPT7. Oncol. Rep. 2014, 31, 2261–2269. [Google Scholar] [CrossRef]
- Chen, T.Y.; Lin, T.C.; Kuo, P.L.; Chen, Z.R.; Cheng, H.L.; Chao, Y.Y.; Syu, J.S.; Lu, F.I.; Wang, C.Y. Septin 7 is a centrosomal protein that ensures S phase entry and microtubule nucleation by maintaining the abundance of p150glued. J. Cell Physiol. 2021, 236, 2706–2724. [Google Scholar] [CrossRef]
- Iwase, M.; Okada, S.; Oguchi, T.; Tohe, A. Forchlorfenuron, a phenylurea cytokinin, disturbs septin organization in Saccharomyces cerevisiae. Genes Genet. Syst. 2004, 79, 199–206. [Google Scholar] [CrossRef]
- DeMay, B.S.; Meseroll, R.A.; Occhipinti, P.; Gladfelter, A.S. Cellular requirements for the small molecule forchlorfenuron to stabilize the septin cytoskeleton. Cytoskeleton 2010, 67, 383–399. [Google Scholar] [CrossRef]
- Heasley, L.R.; Garcia, G.; Mcmurray, M.A. Off-target effects of the septin drug forchlorfenuron on nonplant eukaryotes. Eukaryot. Cell 2014, 13, 1411–1420. [Google Scholar] [CrossRef]
- Hu, Q.; Nelson, W.J.; Spiliotis, E.T. Forchlorfenuron alters mammalian septin assembly, organization, and dynamics. J. Biol. Chem. 2008, 283, 29563–29571. [Google Scholar] [CrossRef]
- Kim, K.K.; Singh, R.K.; Khazan, N.; Kodza, A.; Singh, N.A.; Jones, A.; Sivagnanalingam, U.; Towner, M.; Itamochi, H.; Turner, R.; et al. Development of Potent Forchlorfenuron Analogs and Their Cytotoxic Effect in Cancer Cell Lines. Sci. Rep. 2020, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Pomorski, P.; Watson, J.M.; Haskill, S.; Jacobson, K.A. How adhesion, migration, and cytoplasmic calcium transients influence interleukin-1β mRNA stabilization in human monocytes. Cell Motil. 2004, 57, 143–157. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Ishihara, A.; Oxford, G.; Johnson, B.; Jacobson, K. Regulation of cell movement is mediated by stretch-activated calcium channels. Nature 1999, 400, 382–386. [Google Scholar] [CrossRef]
- Wei, C.; Wang, X.; Chen, M.; Ouyang, K.; Song, L.-S.; Cheng, H. Calcium flickers steer cell migration. Nature 2009, 457, 901–905. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, P.; Giovannelli, A.; Ragozzino, D.; Eusebi, F.; Ruzzier, F. Spontaneous and Repetitive Calcium Transients in C2C12 Mouse Myotubes during In Vitro Myogenesis. Eur. J. Neurosci. 1997, 9, 800–808. [Google Scholar] [CrossRef] [PubMed]
- Deb, B.K.; Hasan, G. Regulation of store-operated Ca2+ entry by septins. Front. Cell Dev. Biol. 2016, 4, 142. [Google Scholar] [CrossRef]
- Deb, B.K.; Chakraborty, P.; Gopurappilly, R.; Hasan, G. SEPT7 regulates Ca2+ entry through Orai channels in human neural progenitor cells and neurons. Cell Calcium 2020, 90, 102252. [Google Scholar] [CrossRef]
- Porter, G.A.; Makuck, R.F.; Rivkees, S.A. Reduction in intracellular calcium levels inhibits myoblast differentiation. J. Biol. Chem. 2002, 277, 28942–28947. [Google Scholar] [CrossRef]
- Formigli, L.; Francini, F.; Meacci, E.; Vassalli, M.; Nosi, D.; Quercioli, F.; Tiribilli, B.; Bencini, C.; Piperio, C.; Bruni, P.; et al. Sphingosine 1-phosphate induces Ca2+ transients and cytoskeletal rearrangement in C2C12 myoblastic cells. Am. J. Physiol. Physiol. 2002, 282, C1361–C1373. [Google Scholar] [CrossRef]
- Louis, M.; Zanou, N.; Van Schoor, M.; Gailly, P. TRPC1 regulates skeletal myoblast migration and differentiation. J. Cell Sci. 2008, 121, 3951–3959. [Google Scholar] [CrossRef]
- Gönczi, M.; Ráduly, Z.; Szabó, L.; Fodor, J.; Telek, A.; Dobrosi, N.; Balogh, N.; Szentesi, P.; Kis, G.; Antal, M.; et al. Septin7 is indispensable for proper skeletal muscle architecture and function. eLife 2022, 11, e75863. [Google Scholar] [CrossRef] [PubMed]
- Piccinini, F.; Kiss, A.; Horvath, P. CellTracker (not only) for dummies. Bioinformatics 2016, 32, 955–957. [Google Scholar] [CrossRef] [PubMed]
- Gönczi, M.; Dienes, B.; Dobrosi, N.; Fodor, J.; Balogh, N.; Oláh, T.; Csernoch, L. Septins, a cytoskeletal protein family, with emerging role in striated muscle. J. Muscle Res. Cell Motil. 2021, 42, 251–265. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.N.; Narumanchi, S.; Paavola, J.; Perttunen, S.; Wang, H.; Lakkisto, P.; Tikkanen, I.; Lehtonen, S. Sept7b is required for the subcellular organization of cardiomyocytes and cardiac function in zebrafish. Am. J. Physiol.-Hear. Circ. Physiol. 2017, 312, H1085–H1095. [Google Scholar] [CrossRef]
- Jia, Z.F.; Huang, Q.; Kang, C.S.; Yang, W.D.; Wang, G.X.; Yu, S.Z.; Jiang, H.; Pu, P.Y. Overexpression of septin 7 suppresses glioma cell growth. J. Neurooncol. 2010, 98, 329–340. [Google Scholar] [CrossRef]
- Becsky, D.; Szabo, K.; Gyulai-Nagy, S.; Gajdos, T.; Bartos, Z.; Balind, A.; Dux, L.; Horvath, P.; Erdelyi, M.; Homolya, L.; et al. Syndecan-4 Modulates Cell Polarity and Migration by Influencing Centrosome Positioning and Intracellular Calcium Distribution. Front. Cell Dev. Biol. 2020, 8, 575227. [Google Scholar] [CrossRef]
- KOYANO, T.; KUME, K.; KONISHI, M.; TODA, T.; HIRATA, D. Search for Kinases Related to Transition of Growth Polarity in Fission Yeast. Biosci. Biotechnol. Biochem. 2010, 74, 1129–1133. [Google Scholar] [CrossRef]
- Tsai, F.C.; Kuo, G.H.; Chang, S.W.; Tsai, P.J. Ca2+ signaling in cytoskeletal reorganization, cell migration, and cancer metastasis. Biomed Res. Int. 2015, 2015, 409245. [Google Scholar] [CrossRef]
- Kim, J.M.; Lee, M.; Kim, N.; Heo, W. Do Optogenetic toolkit reveals the role of Ca2+ sparklets in coordinated cell migration. Proc. Natl. Acad. Sci. USA 2016, 113, 5952–5957. [Google Scholar] [CrossRef]
- Sztretye, M.; Singlár, Z.; Ganbat, N.; Al-Gaadi, D.; Szabó, K.; Köhler, Z.M.; Dux, L.; Keller-Pintér, A.; Csernoch, L.; Szentesi, P. Unravelling the Effects of Syndecan-4 Knockdown on Skeletal Muscle Functions. Int. J. Mol. Sci. 2023, 24, 6933. [Google Scholar] [CrossRef]
- Deb, B.K.; Pathak, T.; Hasan, G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat. Commun. 2016, 7, 11751. [Google Scholar] [CrossRef] [PubMed]
- Kandi, R.; Senger, K.; Grigoryan, A.; Soller, K.; Sakk, V.; Schuster, T.; Eiwen, K.; Menon, M.B.; Gaestel, M.; Zheng, Y.; et al. Cdc42-Borg4-Septin7 axis regulates HSC polarity and function. EMBO Rep. 2021, 22, e52931. [Google Scholar] [CrossRef] [PubMed]
- Kramer, N.; Walzl, A.; Unger, C.; Rosner, M.; Krupitza, G.; Hengstschläger, M.; Dolznig, H. In vitro cell migration and invasion assays. Mutat. Res.-Rev. Mutat. Res. 2013, 752, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Gönczi, M.; Teixeira, J.M.C.; Barrera-Vilarmau, S.; Mediani, L.; Antoniani, F.; Nagy, T.M.; Fehér, K.; Ráduly, Z.; Ambrus, V.; Tőzsér, J.; et al. Alternatively spliced exon regulates context-dependent MEF2D higher-order assembly during myogenesis. Nat. Commun. 2023, 14, 1329. [Google Scholar] [CrossRef]
- Kang, J.-S.; Bae, G.-U.; Yi, M.-J.; Yang, Y.-J.; Oh, J.-E.; Takaesu, G.; Zhou, Y.T.; Low, B.C.; Krauss, R.S. A Cdo–Bnip-2–Cdc42 signaling pathway regulates p38α/β MAPK activity and myogenic differentiation. J. Cell Biol. 2008, 182, 497–507. [Google Scholar] [CrossRef]
- Hall, P.A.; Russell, S.H. Mammalian septins: Dynamic heteromers with roles in cellular morphogenesis and compartmentalization. J. Pathol. 2012, 226, 287–299. [Google Scholar] [CrossRef]
- Sirajuddin, M.; Farkasovsky, M.; Hauer, F.; Kühlmann, D.; Macara, I.G.; Weyand, M.; Stark, H.; Wittinghofer, A. Structural insight into filament formation by mammalian septins. Nature 2007, 449, 311–315. [Google Scholar] [CrossRef]
- Mendonça, D.C.; Macedo, J.N.; Guimarães, S.L.; Barroso da Silva, F.L.; Cassago, A.; Garratt, R.C.; Portugal, R.V.; Araujo, A.P.U. A revised order of subunits in mammalian septin complexes. Cytoskeleton 2019, 76, 457–466. [Google Scholar] [CrossRef]
- Sun, L.; Cao, X.; Lechuga, S.; Feygin, A.; Naydenov, N.G.; Ivanov, A.I. A Septin Cytoskeleton-Targeting Small Molecule, Forchlorfenuron, Inhibits Epithelial Migration via Septin-Independent Perturbation of Cellular Signaling. Cells 2020, 9, 84. [Google Scholar] [CrossRef]
- Henzi, T.; Diep, K.-L.; Oberson, A.; Salicio, V.; Bochet, C.G.; Schwaller, B. Forchlorfenuron and Novel Analogs Cause Cytotoxic Effects in Untreated and Cisplatin-Resistant Malignant Mesothelioma-Derived Cells. Int. J. Mol. Sci. 2022, 23, 3963. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ráduly, Z.; Szabó, L.; Dienes, B.; Szentesi, P.; Bana, Á.V.; Hajdú, T.; Kókai, E.; Hegedűs, C.; Csernoch, L.; Gönczi, M. Migration of Myogenic Cells Is Highly Influenced by Cytoskeletal Septin7. Cells 2023, 12, 1825. https://doi.org/10.3390/cells12141825
Ráduly Z, Szabó L, Dienes B, Szentesi P, Bana ÁV, Hajdú T, Kókai E, Hegedűs C, Csernoch L, Gönczi M. Migration of Myogenic Cells Is Highly Influenced by Cytoskeletal Septin7. Cells. 2023; 12(14):1825. https://doi.org/10.3390/cells12141825
Chicago/Turabian StyleRáduly, Zsolt, László Szabó, Beatrix Dienes, Péter Szentesi, Ágnes Viktória Bana, Tibor Hajdú, Endre Kókai, Csaba Hegedűs, László Csernoch, and Mónika Gönczi. 2023. "Migration of Myogenic Cells Is Highly Influenced by Cytoskeletal Septin7" Cells 12, no. 14: 1825. https://doi.org/10.3390/cells12141825
APA StyleRáduly, Z., Szabó, L., Dienes, B., Szentesi, P., Bana, Á. V., Hajdú, T., Kókai, E., Hegedűs, C., Csernoch, L., & Gönczi, M. (2023). Migration of Myogenic Cells Is Highly Influenced by Cytoskeletal Septin7. Cells, 12(14), 1825. https://doi.org/10.3390/cells12141825