

Heroin Self-Administration and Extinction Increase Prelimbic Cortical Astrocyte–Synapse Proximity and Alter Dendritic Spine Morphometrics That Are Reversed by N-Acetylcysteine

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Subjects and Surgery
2.2. Drugs
2.3. Heroin Self-Administration, Extinction, and Cued-Reinstatement Procedures
2.4. Whole-Cell Recordings from Astrocytes
2.5. Transcardial Perfusions and Immunohistochemistry
2.6. Confocal Microscopy
2.7. Image Analysis
2.8. Statistical Analyses
3. Results
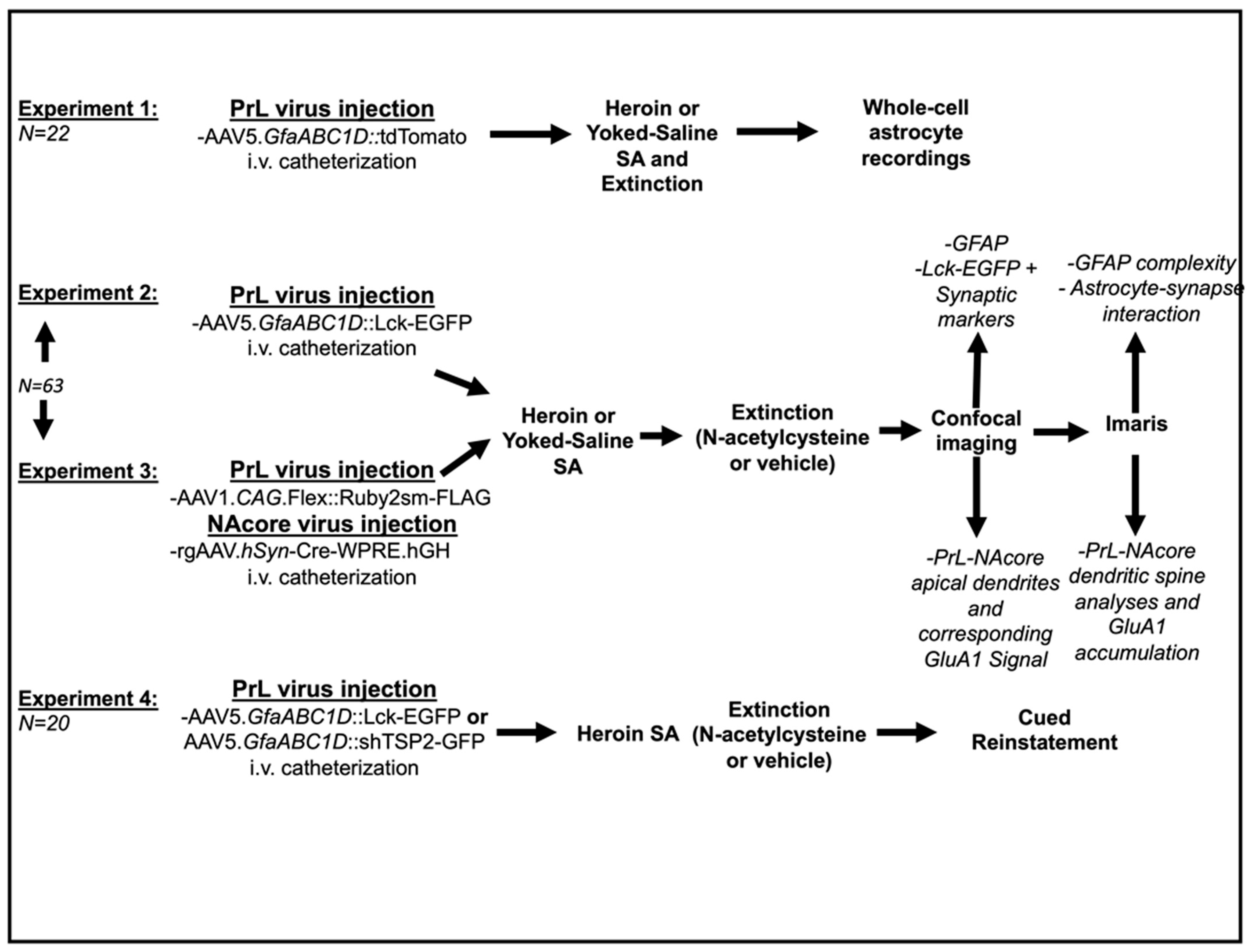
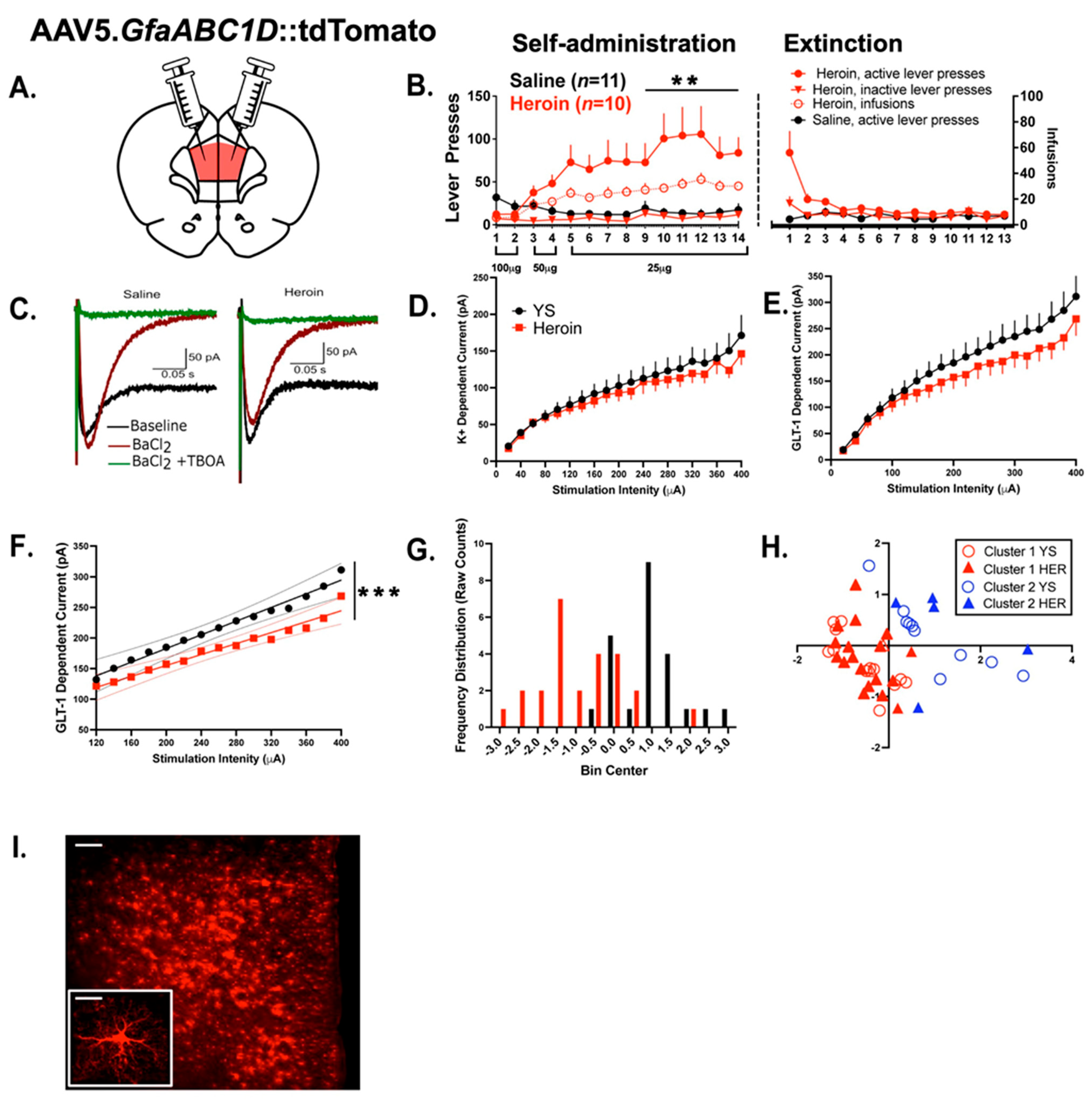
3.1. Experiment 1—Heroin SA and Extinction
3.2. Experiment 1—Heroin SA and Extinction Produced Alterations in Evoked Astrocytic GLT—1 Current
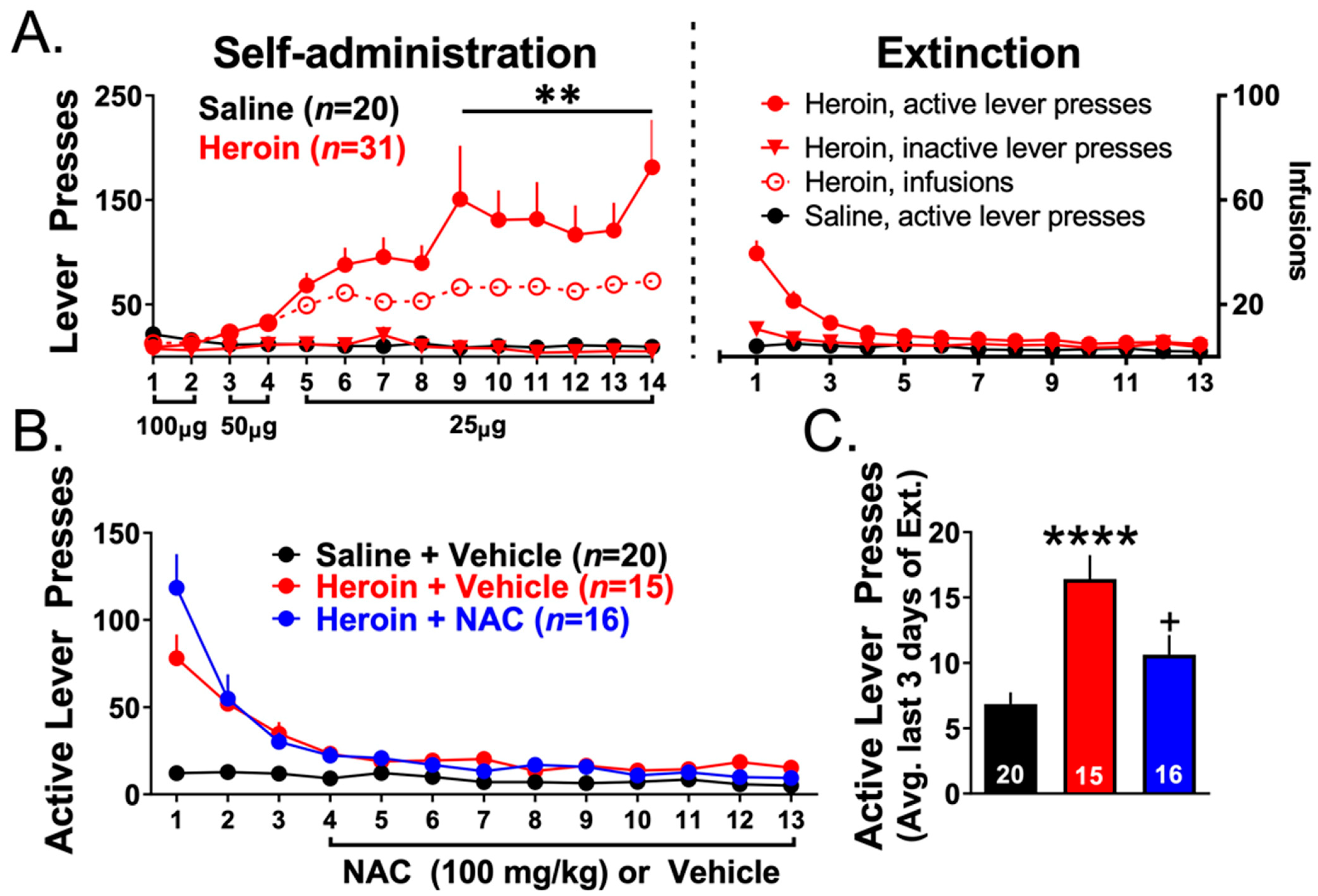
3.3. Experiments 2–3— Heroin Self-Administration and Extinction
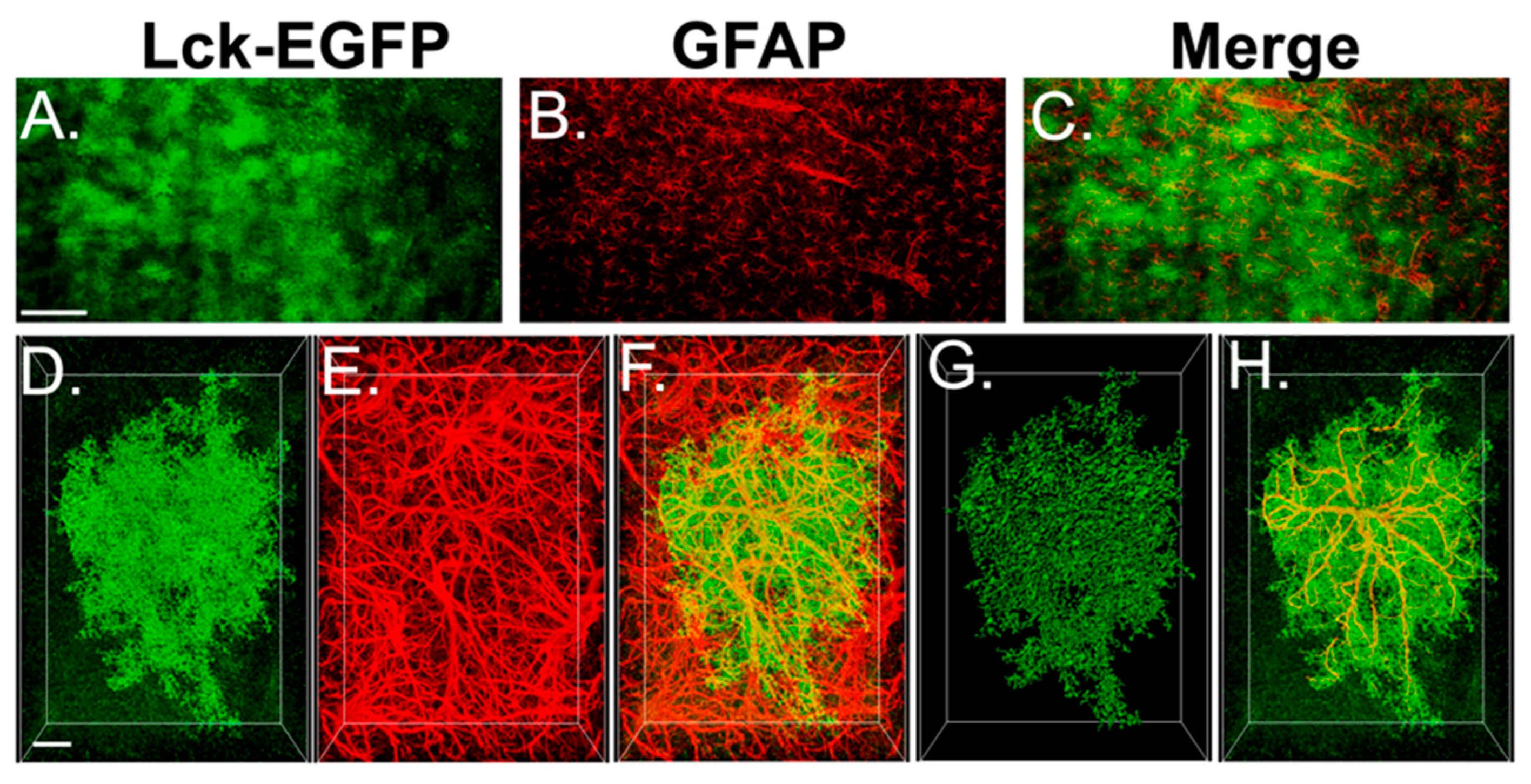
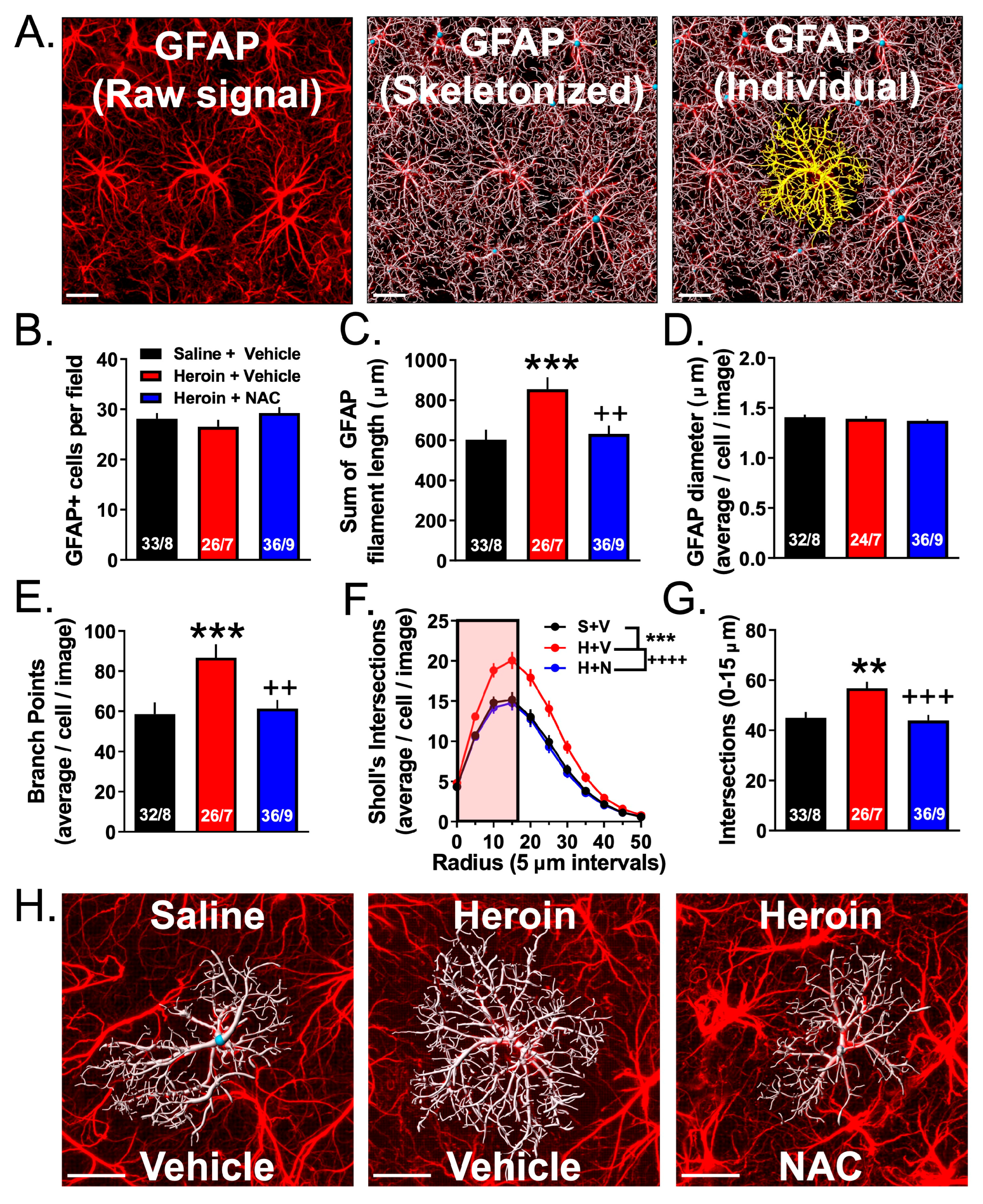
3.4. Experiment 2A—Heroin SA and Extinction Increased Astrocyte GFAP Arbor Complexity, an Effect That Was Prevented by Repeated NAC Treatment during Extinction
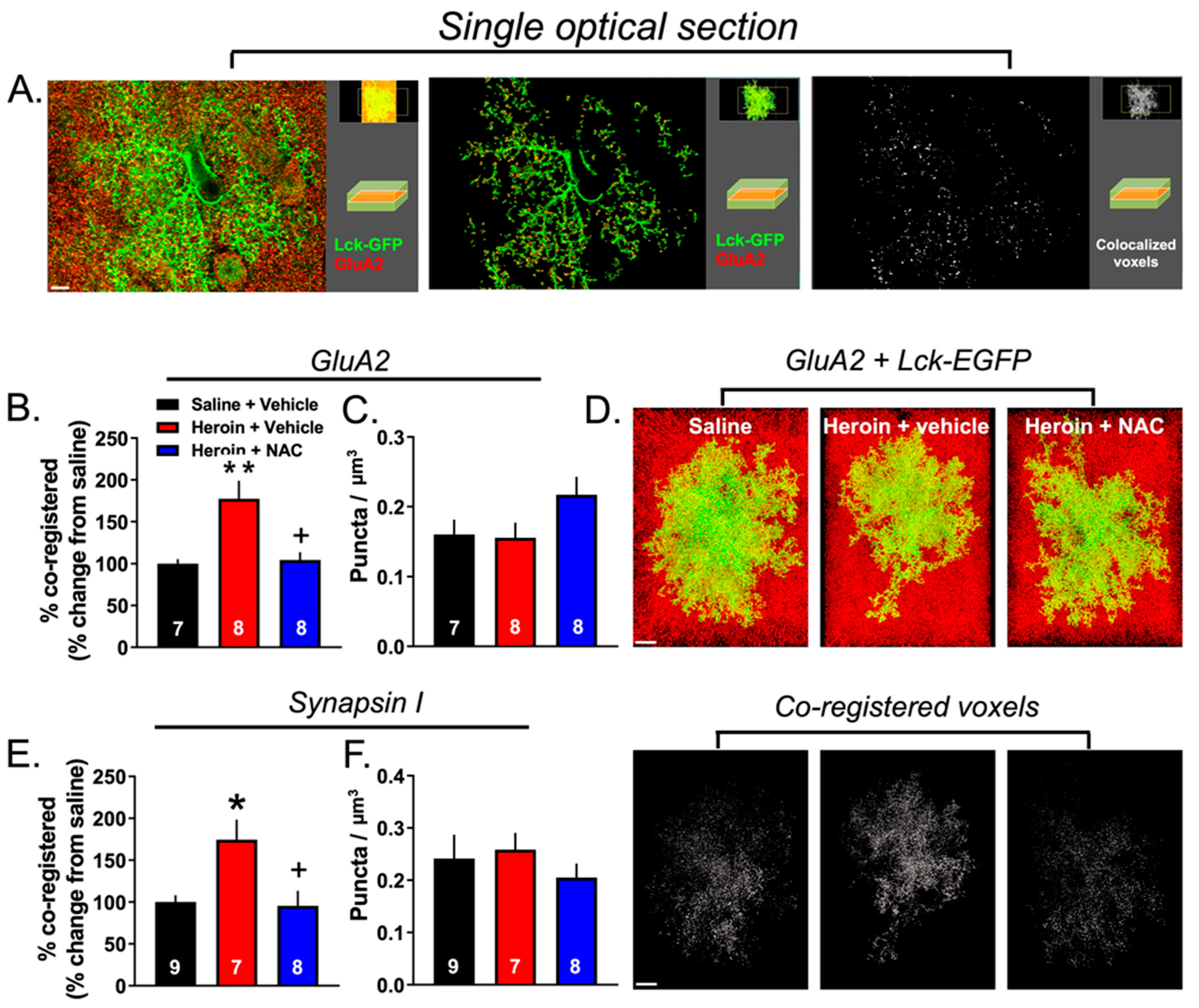
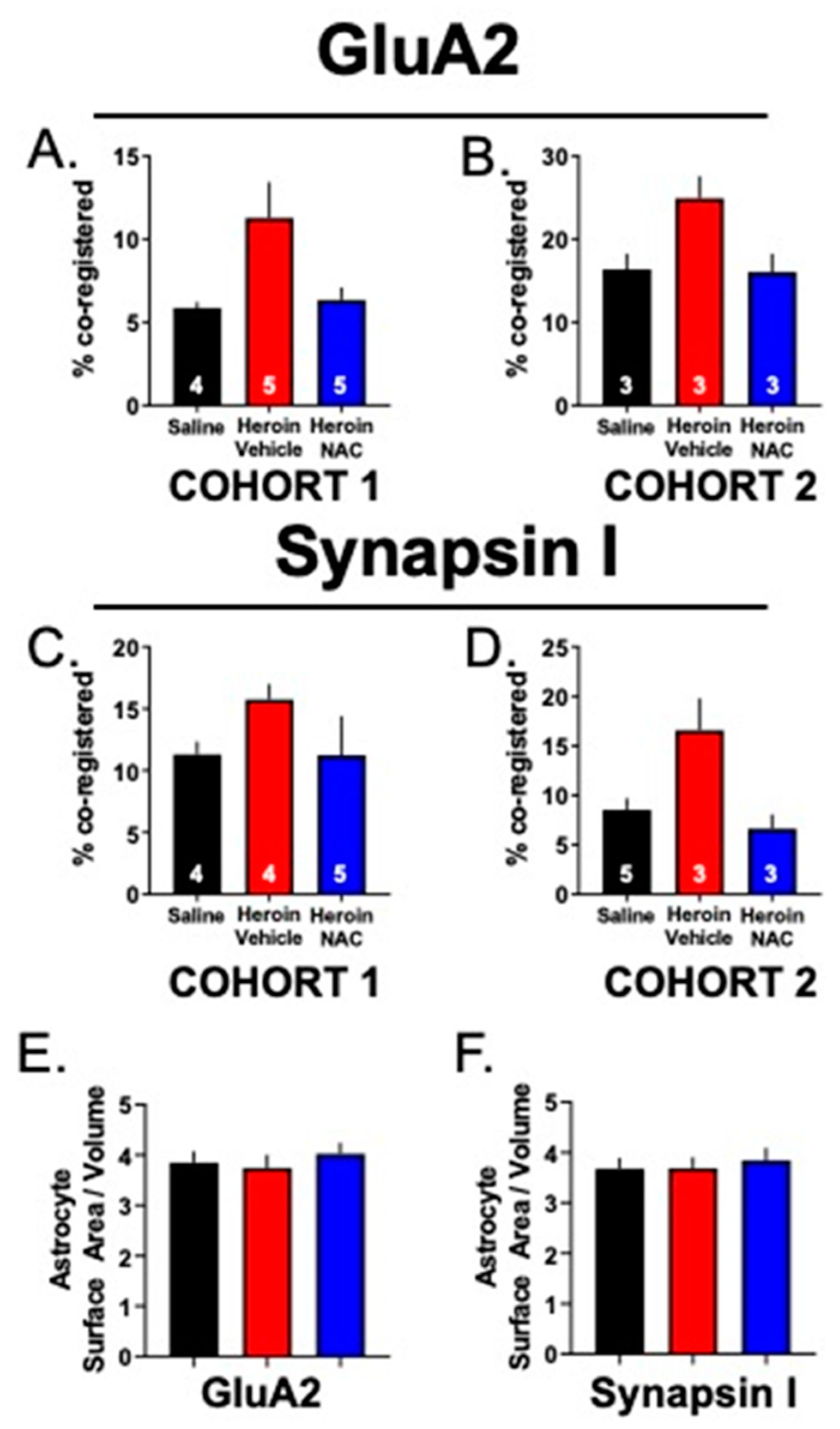
3.5. Experiment 2B—Heroin SA and Extinction Increased Astrocyte Association with Multiple Synaptic Markers, an Effect That Was Prevented by Repeated NAC Treatment during Extinction
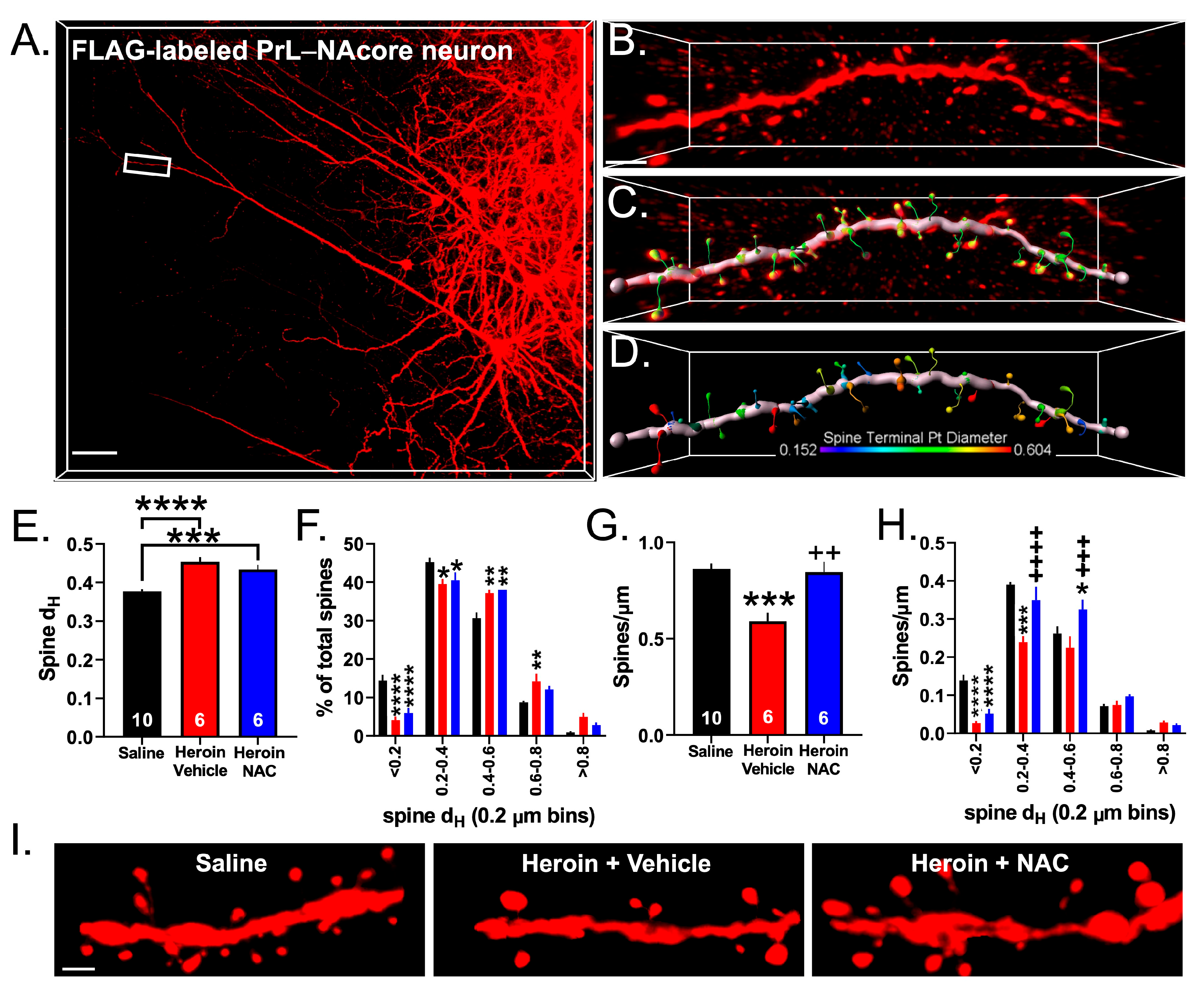
3.6. Experiment 3A—Heroin SA and Extinction Impacts Spine Density and Spine Head Diameter on PrL-NAcore Neurons, an Effect That Was Partially Reversed by Repeated NAC Treatment during Extinction
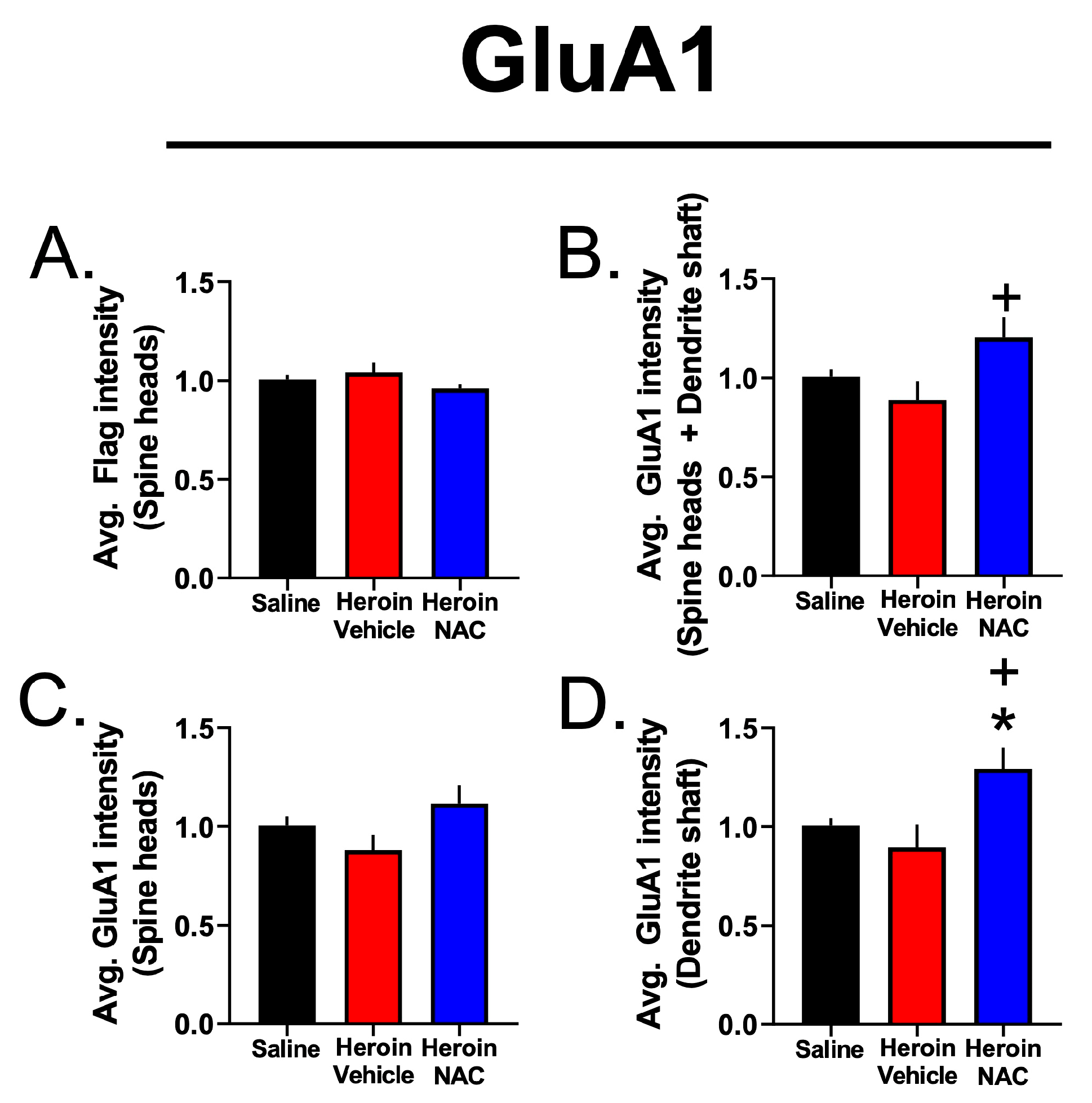
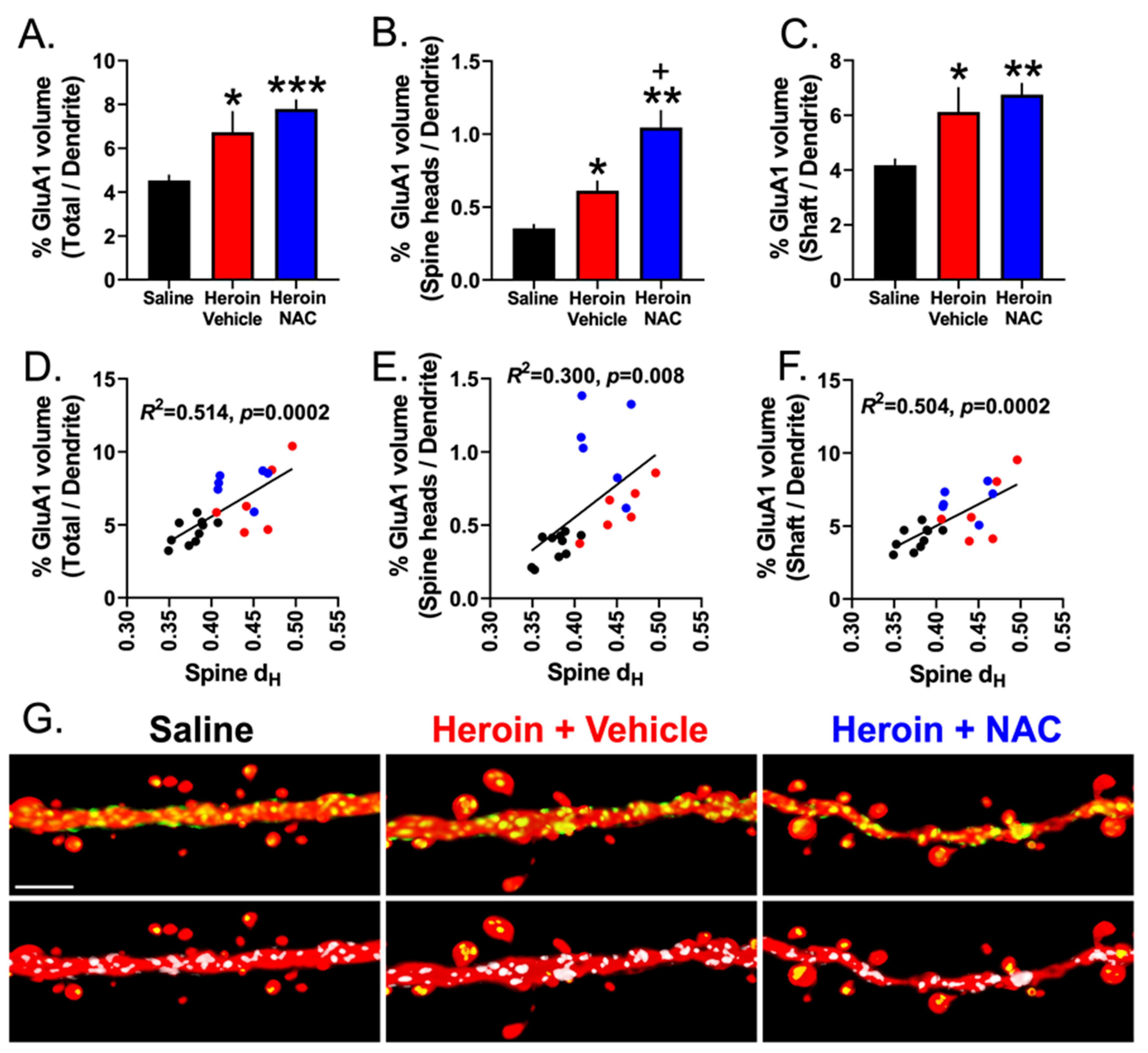
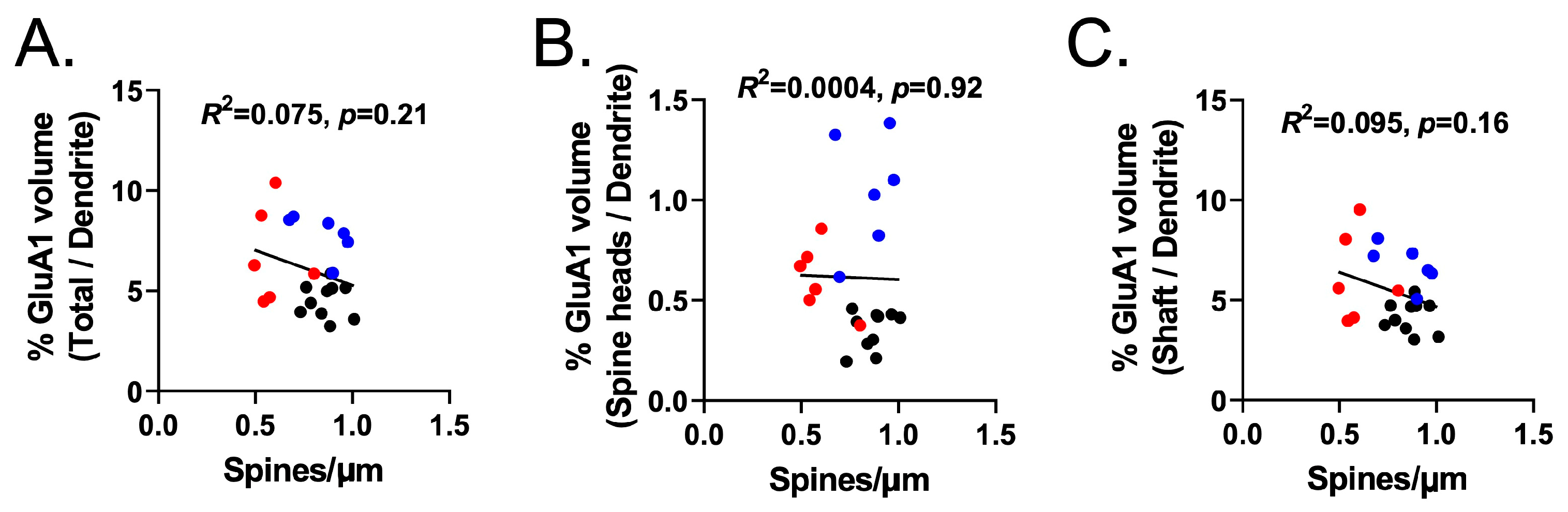
3.7. Experiment 3B—Heroin SA and Extinction Increased GluA1 Immunoreactivity in Dendritic Shafts and Spine Heads of PrL-NAcore Neurons, and NAC Potentiated Spine-Head-Specific GluA1
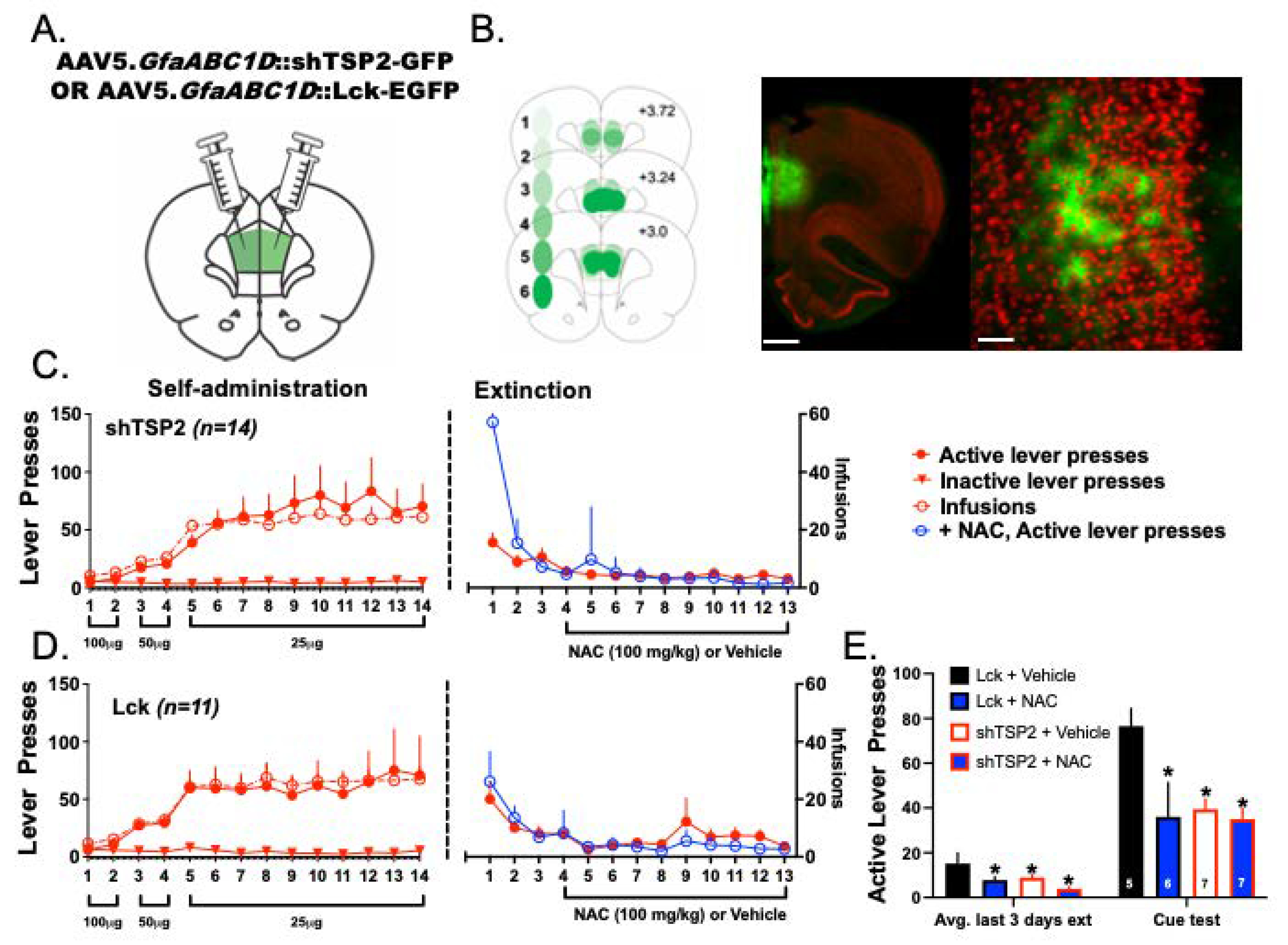
3.8. Experiment 4—Both NAC Treatment and Delivery of shTSP-2 to PrL Astrocytes Blunt Cue-Induced Relapse to Heroin Seeking
4. Discussion
4.1. Heroin SA and Extinction Evoke Functional Alterations in PrL Astrocytes
4.2. Evoked GLT-1-Dependent Current Reveals a Functional Clustering of Cortical Astrocytes, Independent of Heroin Treatment
4.3. Repeated NAC Treatment Facilitated Extinction Training
4.4. Repeated NAC Treatment during Extinction Reversed Increases in Astrocyte Complexity and Association of PAPs with Synapses in PrL Cortex Induced by Heroin SA and Extinction
4.5. Extinction from Heroin SA Altered Dendritic Spine Morphological Profiles and GluA1 Immunoreactivity in Discrete Dendritic Subcompartments of Prelimbic Cortical Neurons Projecting to the Nucleus Accumbens Core
4.6. NAC Treatment and Downregulation of Astrocyte TSP-2 Decreased Cued-Relapse to Heroin Seeking
4.7. Limitations and Methodological Considerations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, L.; Li, N.; Ge, S.; Lozano, A.M.; Lee, D.J.; Yang, C.; Li, L.; Bai, Q.; Lu, H.; Wang, J.; et al. Long-term results after deep brain stimulation of nucleus accumbens and the anterior limb of the internal capsule for preventing heroin relapse: An open-label pilot study. Brain Stimul. 2019, 12, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, Y.; Zhang, Y.; Li, W.; Yang, W.; Zhu, J.; Wu, N.; Chang, H.; Zheng, Y.; Qin, W.; et al. Craving correlates with mesolimbic responses to heroin-related cues in short-term abstinence from heroin: An event-related fMRI study. Brain Res. 2012, 1469, 63–72. [Google Scholar] [CrossRef]
- Lamontagne, S.J.; Olmstead, M.C. Animal models in addiction research: A dimensional approach. Neurosci. Biobehav. Rev. 2019, 106, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Hearing, M. Prefrontal-accumbens opioid plasticity: Implications for relapse and dependence. Pharmacol. Res. 2019, 139, 158–165. [Google Scholar] [CrossRef]
- Hamel, L.; Thangarasa, T.; Samadi, O.; Ito, R. Caudal Nucleus Accumbens Core Is Critical in the Regulation of Cue-Elicited Approach-Avoidance Decisions. eNeuro 2017, 4, ENEURO.0330-16.2017. [Google Scholar] [CrossRef] [PubMed]
- Bossert, J.M.; Ghitza, U.E.; Lu, L.; Epstein, D.H.; Shaham, Y. Neurobiology of relapse to heroin and cocaine seeking: An update and clinical implications. Eur. J. Pharmacol. 2005, 526, 36–50. [Google Scholar] [CrossRef]
- Russo, S.J.; Dietz, D.M.; Dumitriu, D.; Morrison, J.H.; Malenka, R.C.; Nestler, E.J. The addicted synapse: Mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010, 33, 267–276. [Google Scholar] [CrossRef]
- Shen, H.; Kalivas, P.W. Reduced LTP and LTD in prefrontal cortex synapses in the nucleus accumbens after heroin self-administration. Int. J. Neuropsychopharmacol. 2013, 16, 1165–1167. [Google Scholar] [CrossRef]
- Moussawi, K.; Pacchioni, A.; Moran, M.; Olive, M.F.; Gass, J.T.; Lavin, A.; Kalivas, P.W. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat. Neurosci. 2009, 12, 182–189. [Google Scholar] [CrossRef]
- Knackstedt, L.A.; Melendez, R.I.; Kalivas, P.W. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol. Psychiatry 2010, 67, 81–84. [Google Scholar] [CrossRef]
- Shen, H.W.; Scofield, M.D.; Boger, H.; Hensley, M.; Kalivas, P.W. Synaptic glutamate spillover due to impaired glutamate uptake mediates heroin relapse. J. Neurosci. 2014, 34, 5649–5657. [Google Scholar] [CrossRef]
- Kruyer, A.; Scofield, M.D.; Wood, D.; Reissner, K.J.; Kalivas, P.W. Heroin Cue-Evoked Astrocytic Structural Plasticity at Nucleus Accumbens Synapses Inhibits Heroin Seeking. Biol. Psychiatry 2019, 86, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Scofield, M.D.; Li, H.; Siemsen, B.M.; Healey, K.L.; Tran, P.K.; Woronoff, N.; Boger, H.A.; Kalivas, P.W.; Reissner, K.J. Cocaine Self-Administration and Extinction Leads to Reduced Glial Fibrillary Acidic Protein Expression and Morphometric Features of Astrocytes in the Nucleus Accumbens Core. Biol. Psychiatry 2016, 80, 207–215. [Google Scholar] [CrossRef]
- Siemsen, B.M.; Reichel, C.M.; Leong, K.C.; Garcia-Keller, C.; Gipson, C.D.; Spencer, S.; McFaddin, J.A.; Hooker, K.N.; Kalivas, P.W.; Scofield, M.D. Effects of Methamphetamine Self-Administration and Extinction on Astrocyte Structure and Function in the Nucleus Accumbens Core. Neuroscience 2019, 406, 528–541. [Google Scholar] [CrossRef]
- Scofield, M.D.; Kalivas, P.W. Astrocytic dysfunction and addiction: Consequences of impaired glutamate homeostasis. Neuroscientist 2014, 20, 610–622. [Google Scholar] [CrossRef] [PubMed]
- LaLumiere, R.T.; Kalivas, P.W. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J. Neurosci. 2008, 28, 3170–3177. [Google Scholar] [CrossRef]
- Kalivas, P.W.; Volkow, N.D. New medications for drug addiction hiding in glutamatergic neuroplasticity. Mol. Psychiatry 2011, 16, 974–986. [Google Scholar] [CrossRef]
- Roberts-Wolfe, D.J.; Kalivas, P.W. Glutamate Transporter GLT-1 as a Therapeutic Target for Substance Use Disorders. CNS Neurol. Disord. Drug Targets 2015, 14, 745–756. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Kalivas, P.W. N-acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biol. Psychiatry 2008, 63, 338–340. [Google Scholar] [CrossRef]
- Hodebourg, R.; Murray, J.E.; Fouyssac, M.; Puaud, M.; Everitt, B.J.; Belin, D. Heroin seeking becomes dependent on dorsal striatal dopaminergic mechanisms and can be decreased by N-acetylcysteine. Eur. J. Neurosci. 2019, 50, 2036–2044. [Google Scholar] [CrossRef]
- LaRowe, S.D.; Kalivas, P.W. The Role of N-Acetylcysteine in Inhibiting Responding During Extinction in Rats Trained to Self-Administer Cocaine. Open. Addict. J. 2010, 3, 88–91. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kupchik, Y.M.; Moussawi, K.; Tang, X.C.; Wang, X.; Kalivas, B.C.; Kolokithas, R.; Ogburn, K.B.; Kalivas, P.W. The effect of N-acetylcysteine in the nucleus accumbens on neurotransmission and relapse to cocaine. Biol. Psychiatry 2012, 71, 978–986. [Google Scholar] [CrossRef] [PubMed]
- Powell, G.L.; Leyrer-Jackson, J.M.; Goenaga, J.; Namba, M.D.; Pina, J.; Spencer, S.; Stankeviciute, N.; Schwartz, D.; Allen, N.P.; Del Franco, A.P.; et al. Chronic treatment with N-acetylcysteine decreases extinction responding and reduces cue-induced nicotine-seeking. Physiol. Rep. 2019, 7, e13958. [Google Scholar] [CrossRef] [PubMed]
- Reissner, K.J.; Gipson, C.D.; Tran, P.K.; Knackstedt, L.A.; Scofield, M.D.; Kalivas, P.W. Glutamate transporter GLT-1 mediates N-acetylcysteine inhibition of cocaine reinstatement. Addict. Biol. 2015, 20, 316–323. [Google Scholar] [CrossRef]
- Gomes, F.C.; Paulin, D.; Moura Neto, V. Glial fibrillary acidic protein (GFAP): Modulation by growth factors and its implication in astrocyte differentiation. Braz. J. Med. Biol. Res. 1999, 32, 619–631. [Google Scholar] [CrossRef]
- Garrido, E.; Perez-Garcia, C.; Alguacil, L.F.; Diez-Fernandez, C. The alpha2-adrenoceptor antagonist yohimbine reduces glial fibrillary acidic protein upregulation induced by chronic morphine administration. Neurosci. Lett. 2005, 383, 141–144. [Google Scholar] [CrossRef]
- Stiene-Martin, A.; Gurwell, J.A.; Hauser, K.F. Morphine alters astrocyte growth in primary cultures of mouse glial cells: Evidence for a direct effect of opiates on neural maturation. Brain Res. Dev. Brain Res. 1991, 60, 1–7. [Google Scholar] [CrossRef]
- Shigetomi, E.; Kracun, S.; Sofroniew, M.V.; Khakh, B.S. A genetically targeted optical sensor to monitor calcium signals in astrocyte processes. Nat. Neurosci. 2010, 13, 759–766. [Google Scholar] [CrossRef]
- Viswanathan, S.; Williams, M.E.; Bloss, E.B.; Stasevich, T.J.; Speer, C.M.; Nern, A.; Pfeiffer, B.D.; Hooks, B.M.; Li, W.P.; English, B.P.; et al. High-performance probes for light and electron microscopy. Nat. Methods 2015, 12, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, K.L.; Shukla, A.; Beroun, A.; Ishikawa, M.; Huang, X.; Wang, Y.; Wang, Y.Q.; Yang, Y.; Bastola, N.D.; et al. Cocaine Triggers Astrocyte-Mediated Synaptogenesis. Biol. Psychiatry 2021, 89, 386–397. [Google Scholar] [CrossRef]
- Siemsen, B.M.; Giannotti, G.; McFaddin, J.A.; Scofield, M.D.; McGinty, J.F. Biphasic effect of abstinence duration following cocaine self-administration on spine morphology and plasticity-related proteins in prelimbic cortical neurons projecting to the nucleus accumbens core. Brain Struct. Funct. 2019, 224, 741–758. [Google Scholar] [CrossRef] [PubMed]
- Costes, S.V.; Daelemans, D.; Cho, E.H.; Dobbin, Z.; Pavlakis, G.; Lockett, S. Automatic and quantitative measurement of protein-protein colocalization in live cells. Biophys. J. 2004, 86, 3993–4003. [Google Scholar] [CrossRef] [PubMed]
- Testen, A.; Ali, M.; Sexton, H.G.; Hodges, S.; Dubester, K.; Reissner, K.J.; Swartzwelder, H.S.; Risher, M.L. Region-Specific Differences in Morphometric Features and Synaptic Colocalization of Astrocytes During Development. Neuroscience 2019, 400, 98–109. [Google Scholar] [CrossRef]
- Testen, A.; Sepulveda-Orengo, M.T.; Gaines, C.H.; Reissner, K.J. Region-Specific Reductions in Morphometric Properties and Synaptic Colocalization of Astrocytes Following Cocaine Self-Administration and Extinction. Front. Cell. Neurosci. 2018, 12, 246. [Google Scholar] [CrossRef]
- Robinson, T.E.; Gorny, G.; Savage, V.R.; Kolb, B. Widespread but regionally specific effects of experimenter- versus self-administered morphine on dendritic spines in the nucleus accumbens, hippocampus, and neocortex of adult rats. Synapse 2002, 46, 271–279. [Google Scholar] [CrossRef]
- John Lin, C.C.; Yu, K.; Hatcher, A.; Huang, T.W.; Lee, H.K.; Carlson, J.; Weston, M.C.; Chen, F.; Zhang, Y.; Zhu, W.; et al. Identification of diverse astrocyte populations and their malignant analogs. Nat. Neurosci. 2017, 20, 396–405. [Google Scholar] [CrossRef]
- Hoft, S.; Griemsmann, S.; Seifert, G.; Steinhauser, C. Heterogeneity in expression of functional ionotropic glutamate and GABA receptors in astrocytes across brain regions: Insights from the thalamus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130602. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- LaRowe, S.D.; Kalivas, P.W.; Nicholas, J.S.; Randall, P.K.; Mardikian, P.N.; Malcolm, R.J. A double-blind placebo-controlled trial of N-acetylcysteine in the treatment of cocaine dependence. Am. J. Addict. 2013, 22, 443–452. [Google Scholar] [CrossRef]
- Nocito Echevarria, M.A.; Andrade Reis, T.; Ruffo Capatti, G.; Siciliano Soares, V.; da Silveira, D.X.; Fidalgo, T.M. N-acetylcysteine for treating cocaine addiction—A systematic review. Psychiatry Res. 2017, 251, 197–203. [Google Scholar] [CrossRef]
- Mardikian, P.N.; LaRowe, S.D.; Hedden, S.; Kalivas, P.W.; Malcolm, R.J. An open-label trial of N-acetylcysteine for the treatment of cocaine dependence: A pilot study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2007, 31, 389–394. [Google Scholar] [CrossRef]
- Schmidt-Kastner, R.; Wietasch, K.; Weigel, H.; Eysel, U.T. Immunohistochemical staining for glial fibrillary acidic protein (GFAP) after deafferentation or ischemic infarction in rat visual system: Features of reactive and damaged astrocytes. Int. J. Dev. Neurosci. 1993, 11, 157–174. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, B.B.; Fox, C.A.; Thompson, R.C.; Meng, F.; Watson, S.J.; Akil, H. Primary astroglial cultures derived from several rat brain regions differentially express mu, delta and kappa opioid receptor mRNA. Brain Res. Mol. Brain Res. 1995, 34, 209–220. [Google Scholar] [CrossRef]
- Kelly, E. Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol. 2013, 169, 1430–1446. [Google Scholar] [CrossRef]
- Hauser, K.F.; Stiene-Martin, A.; Mattson, M.P.; Elde, R.P.; Ryan, S.E.; Godleske, C.C. mu-Opioid receptor-induced Ca2+ mobilization and astroglial development: Morphine inhibits DNA synthesis and stimulates cellular hypertrophy through a Ca(2+)-dependent mechanism. Brain Res. 1996, 720, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Stiene-Martin, A.; Mattson, M.P.; Hauser, K.F. Opiates selectively increase intracellular calcium in developing type-1 astrocytes: Role of calcium in morphine-induced morphologic differentiation. Brain Res. Dev. Brain Res. 1993, 76, 189–196. [Google Scholar] [CrossRef]
- Nam, M.H.; Han, K.S.; Lee, J.; Won, W.; Koh, W.; Bae, J.Y.; Woo, J.; Kim, J.; Kwong, E.; Choi, T.Y.; et al. Activation of Astrocytic mu-Opioid Receptor Causes Conditioned Place Preference. Cell Rep. 2019, 28, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Madayag, A.; Lobner, D.; Kau, K.S.; Mantsch, J.R.; Abdulhameed, O.; Hearing, M.; Grier, M.D.; Baker, D.A. Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J. Neurosci. 2007, 27, 13968–13976. [Google Scholar] [CrossRef]
- Kau, K.S.; Madayag, A.; Mantsch, J.R.; Grier, M.D.; Abdulhameed, O.; Baker, D.A. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience 2008, 155, 530–537. [Google Scholar] [CrossRef]
- Clavreul, S.; Abdeladim, L.; Hernandez-Garzon, E.; Niculescu, D.; Durand, J.; Ieng, S.H.; Barry, R.; Bonvento, G.; Beaurepaire, E.; Livet, J.; et al. Cortical astrocytes develop in a plastic manner at both clonal and cellular levels. Nat. Commun. 2019, 10, 4884. [Google Scholar] [CrossRef]
- Stogsdill, J.A.; Ramirez, J.; Liu, D.; Kim, Y.H.; Baldwin, K.T.; Enustun, E.; Ejikeme, T.; Ji, R.R.; Eroglu, C. Astrocytic neuroligins control astrocyte morphogenesis and synaptogenesis. Nature 2017, 551, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, R.; Rauen, T.; Fischer, F.; Wiessner, M.; Grewer, C.; Bicho, A.; Pow, D.V. Cloning, transport properties, and differential localization of two splice variants of GLT-1 in the rat CNS: Implications for CNS glutamate homeostasis. Glia 2004, 45, 155–169. [Google Scholar] [CrossRef]
- Testen, A.; Kim, R.; Reissner, K.J. High-Resolution Three-Dimensional Imaging of Individual Astrocytes Using Confocal Microscopy. Curr. Protoc. Neurosci. 2020, 91, e92. [Google Scholar] [CrossRef] [PubMed]
- Scofield, M.D. Exploring the Role of Astroglial Glutamate Release and Association With Synapses in Neuronal Function and Behavior. Biol. Psychiatry 2018, 84, 778–786. [Google Scholar] [CrossRef]
- Scofield, M.D.; Heinsbroek, J.A.; Gipson, C.D.; Kupchik, Y.M.; Spencer, S.; Smith, A.C.; Roberts-Wolfe, D.; Kalivas, P.W. The Nucleus Accumbens: Mechanisms of Addiction across Drug Classes Reflect the Importance of Glutamate Homeostasis. Pharmacol. Rev. 2016, 68, 816–871. [Google Scholar] [CrossRef]
- Kim, R.; Sepulveda-Orengo, M.T.; Healey, K.L.; Williams, E.A.; Reissner, K.J. Regulation of glutamate transporter 1 (GLT-1) gene expression by cocaine self-administration and withdrawal. Neuropharmacology 2018, 128, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fattore, L.; Puddu, M.C.; Picciau, S.; Cappai, A.; Fratta, W.; Serra, G.P.; Spiga, S. Astroglial in vivo response to cocaine in mouse dentate gyrus: A quantitative and qualitative analysis by confocal microscopy. Neuroscience 2002, 110, 1–6. [Google Scholar] [CrossRef]
- SheikhBahaei, S.; Morris, B.; Collina, J.; Anjum, S.; Znati, S.; Gamarra, J.; Zhang, R.; Gourine, A.V.; Smith, J.C. Morphometric analysis of astrocytes in brainstem respiratory regions. J. Comp. Neurol. 2018, 526, 2032–2047. [Google Scholar] [CrossRef]
- Zhang, S.; Wu, M.; Peng, C.; Zhao, G.; Gu, R. GFAP expression in injured astrocytes in rats. Exp. Ther. Med. 2017, 14, 1905–1908. [Google Scholar] [CrossRef]
- Haseleu, J.; Anlauf, E.; Blaess, S.; Endl, E.; Derouiche, A. Studying subcellular detail in fixed astrocytes: Dissociation of morphologically intact glial cells (DIMIGs). Front. Cell. Neurosci. 2013, 7, 54. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Jones, Y.Z.; Ellisman, M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002, 22, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [Google Scholar] [CrossRef]
- Bernardinelli, Y.; Randall, J.; Janett, E.; Nikonenko, I.; Konig, S.; Jones, E.V.; Flores, C.E.; Murai, K.K.; Bochet, C.G.; Holtmaat, A.; et al. Activity-dependent structural plasticity of perisynaptic astrocytic domains promotes excitatory synapse stability. Curr. Biol. 2014, 24, 1679–1688. [Google Scholar] [CrossRef]
- Genoud, C.; Quairiaux, C.; Steiner, P.; Hirling, H.; Welker, E.; Knott, G.W. Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol. 2006, 4, e343. [Google Scholar] [CrossRef] [PubMed]
- Pannasch, U.; Freche, D.; Dallerac, G.; Ghezali, G.; Escartin, C.; Ezan, P.; Cohen-Salmon, M.; Benchenane, K.; Abudara, V.; Dufour, A.; et al. Connexin 30 sets synaptic strength by controlling astroglial synapse invasion. Nat. Neurosci. 2014, 17, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Tso, M.C.; Herzog, E.D. Was Cajal right about sleep? BMC Biol. 2015, 13, 67. [Google Scholar] [CrossRef] [PubMed]
- Liao, D.; Lin, H.; Law, P.Y.; Loh, H.H. Mu-opioid receptors modulate the stability of dendritic spines. Proc. Natl. Acad. Sci. USA 2005, 102, 1725–1730. [Google Scholar] [CrossRef]
- Gipson, C.D.; Kupchik, Y.M.; Shen, H.; Reissner, K.J.; Thomas, C.A.; Kalivas, P.W. Relapse induced by cues predicting cocaine depends on rapid, transient synaptic potentiation. Neuron 2013, 77, 867–872. [Google Scholar] [CrossRef] [PubMed]
- Shi, S.H.; Hayashi, Y.; Petralia, R.S.; Zaman, S.H.; Wenthold, R.J.; Svoboda, K.; Malinow, R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 1999, 284, 1811–1816. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Ellis-Davies, G.C.; Nemoto, T.; Miyashita, Y.; Iino, M.; Kasai, H. Dendritic spine geometry is critical for AMPA receptor expression in hippocampal CA1 pyramidal neurons. Nat. Neurosci. 2001, 4, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, M.; Honkura, N.; Ellis-Davies, G.C.; Kasai, H. Structural basis of long-term potentiation in single dendritic spines. Nature 2004, 429, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Lambe, E.K.; Aghajanian, G.K. Hypocretin (orexin) induces calcium transients in single spines postsynaptic to identified thalamocortical boutons in prefrontal slice. Neuron 2003, 40, 139–150. [Google Scholar] [CrossRef] [PubMed]
- Paspalas, C.D.; Wang, M.; Arnsten, A.F. Constellation of HCN channels and cAMP regulating proteins in dendritic spines of the primate prefrontal cortex: Potential substrate for working memory deficits in schizophrenia. Cereb. Cortex 2013, 23, 1643–1654. [Google Scholar] [CrossRef]
- Kuroda, M.; Murakami, K.; Igarashi, H.; Okada, A. The convergence of axon terminals from the mediodorsal thalamic nucleus and ventral tegmental area on pyramidal cells in layer V of the rat prelimbic cortex. Eur. J. Neurosci. 1996, 8, 1340–1349. [Google Scholar] [CrossRef] [PubMed]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef]
- Santello, M.; Cali, C.; Bezzi, P. Gliotransmission and the tripartite synapse. Adv. Exp. Med. Biol. 2012, 970, 307–331. [Google Scholar] [CrossRef]
- Risher, W.C.; Kim, N.; Koh, S.; Choi, J.E.; Mitev, P.; Spence, E.F.; Pilaz, L.J.; Wang, D.; Feng, G.; Silver, D.L.; et al. Thrombospondin receptor alpha2delta-1 promotes synaptogenesis and spinogenesis via postsynaptic Rac1. J. Cell Biol. 2018, 217, 3747–3765. [Google Scholar] [CrossRef]
- Garcia, O.; Torres, M.; Helguera, P.; Coskun, P.; Busciglio, J. A role for thrombospondin-1 deficits in astrocyte-mediated spine and synaptic pathology in Down’s syndrome. PLoS ONE 2010, 5, e14200. [Google Scholar] [CrossRef]
- Ikeda, H.; Miyatake, M.; Koshikawa, N.; Ochiai, K.; Yamada, K.; Kiss, A.; Donlin, M.J.; Panneton, W.M.; Churchill, J.D.; Green, M.; et al. Morphine modulation of thrombospondin levels in astrocytes and its implications for neurite outgrowth and synapse formation. J. Biol. Chem. 2010, 285, 38415–38427. [Google Scholar] [CrossRef]
- Pereska, Z.; Dejanova, B.; Bozinovska, C.; Petkovska, L. Prooxidative/antioxidative homeostasis in heroin addiction and detoxification. Bratisl. Lek. Listy 2007, 108, 393–398. [Google Scholar]
- Cunha-Oliveira, T.; Rego, A.C.; Garrido, J.; Borges, F.; Macedo, T.; Oliveira, C.R. Street heroin induces mitochondrial dysfunction and apoptosis in rat cortical neurons. J. Neurochem. 2007, 101, 543–554. [Google Scholar] [CrossRef]
- Cunha-Oliveira, T.; Rego, A.C.; Garrido, J.; Borges, F.; Macedo, T.; Oliveira, C.R. Neurotoxicity of heroin-cocaine combinations in rat cortical neurons. Toxicology 2010, 276, 11–17. [Google Scholar] [CrossRef]
- Chen, J.K.; Zhan, Y.J.; Yang, C.S.; Tzeng, S.F. Oxidative stress-induced attenuation of thrombospondin-1 expression in primary rat astrocytes. J. Cell Biochem. 2011, 112, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, R.J.; Meredith, T.J. Use of N-acetylcysteine in clinical toxicology. Am. J. Med. 1991, 30, 5131–5139. [Google Scholar] [CrossRef] [PubMed]
- Schmaal, L.; Veltman, D.J.; Nederveen, A.; van den Brink, W.; Goudriaan, A.E. N-acetylcysteine normalizes glutamate levels in cocaine-dependent patients: A randomized crossover magnetic resonance spectroscopy study. Neuropsychopharmacology 2012, 37, 2143–2152. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- Haber, M.; Zhou, L.; Murai, K.K. Cooperative astrocyte and dendritic spine dynamics at hippocampal excitatory synapses. J. Neurosci. 2006, 26, 8881–8891. [Google Scholar] [CrossRef] [PubMed]
- Zuber, B.; Nikonenko, I.; Klauser, P.; Muller, D.; Dubochet, J. The mammalian central nervous synaptic cleft contains a high density of periodically organized complexes. Proc. Natl. Acad. Sci. USA 2005, 102, 19192–19197. [Google Scholar] [CrossRef]












Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siemsen, B.M.; Denton, A.R.; Parrila-Carrero, J.; Hooker, K.N.; Carpenter, E.A.; Prescot, M.E.; Brock, A.G.; Westphal, A.M.; Leath, M.-N.; McFaddin, J.A.; et al. Heroin Self-Administration and Extinction Increase Prelimbic Cortical Astrocyte–Synapse Proximity and Alter Dendritic Spine Morphometrics That Are Reversed by N-Acetylcysteine. Cells 2023, 12, 1812. https://doi.org/10.3390/cells12141812
Siemsen BM, Denton AR, Parrila-Carrero J, Hooker KN, Carpenter EA, Prescot ME, Brock AG, Westphal AM, Leath M-N, McFaddin JA, et al. Heroin Self-Administration and Extinction Increase Prelimbic Cortical Astrocyte–Synapse Proximity and Alter Dendritic Spine Morphometrics That Are Reversed by N-Acetylcysteine. Cells. 2023; 12(14):1812. https://doi.org/10.3390/cells12141812
Chicago/Turabian StyleSiemsen, Benjamin M., Adam R. Denton, Jeffrey Parrila-Carrero, Kaylee N. Hooker, Eilish A. Carpenter, Meagan E. Prescot, Ashley G. Brock, Annaka M. Westphal, Mary-Nan Leath, John A. McFaddin, and et al. 2023. "Heroin Self-Administration and Extinction Increase Prelimbic Cortical Astrocyte–Synapse Proximity and Alter Dendritic Spine Morphometrics That Are Reversed by N-Acetylcysteine" Cells 12, no. 14: 1812. https://doi.org/10.3390/cells12141812
APA StyleSiemsen, B. M., Denton, A. R., Parrila-Carrero, J., Hooker, K. N., Carpenter, E. A., Prescot, M. E., Brock, A. G., Westphal, A. M., Leath, M.-N., McFaddin, J. A., Jhou, T. C., McGinty, J. F., & Scofield, M. D. (2023). Heroin Self-Administration and Extinction Increase Prelimbic Cortical Astrocyte–Synapse Proximity and Alter Dendritic Spine Morphometrics That Are Reversed by N-Acetylcysteine. Cells, 12(14), 1812. https://doi.org/10.3390/cells12141812