
Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Lifespan Assays
2.3. Climbing Assay
2.4. Microscopy
2.5. Western Blotting
2.6. Immunohistochemistry
2.7. PCR and Quantitative PCR
2.8. Statistical Analysis
3. Results
3.1. Results
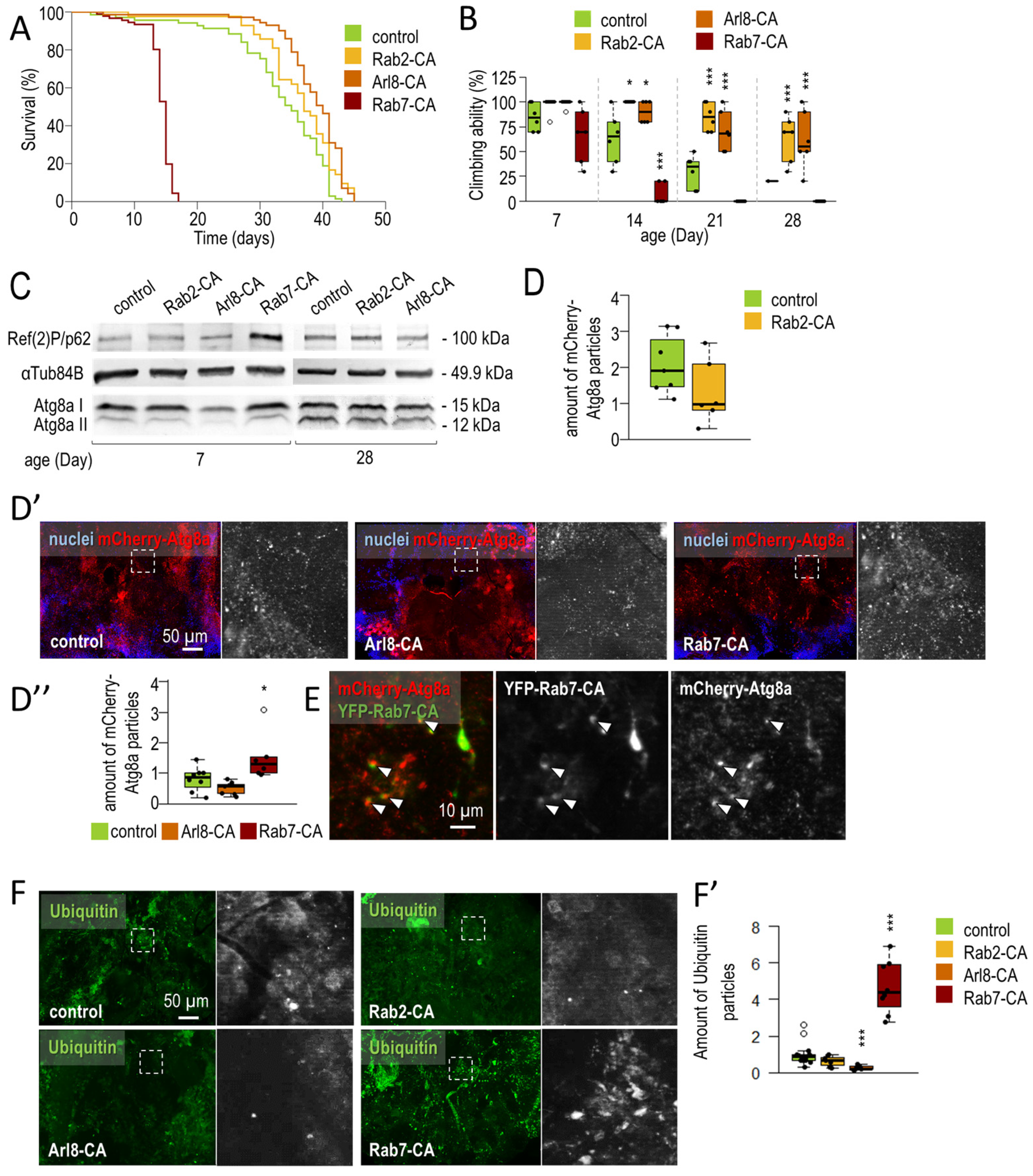
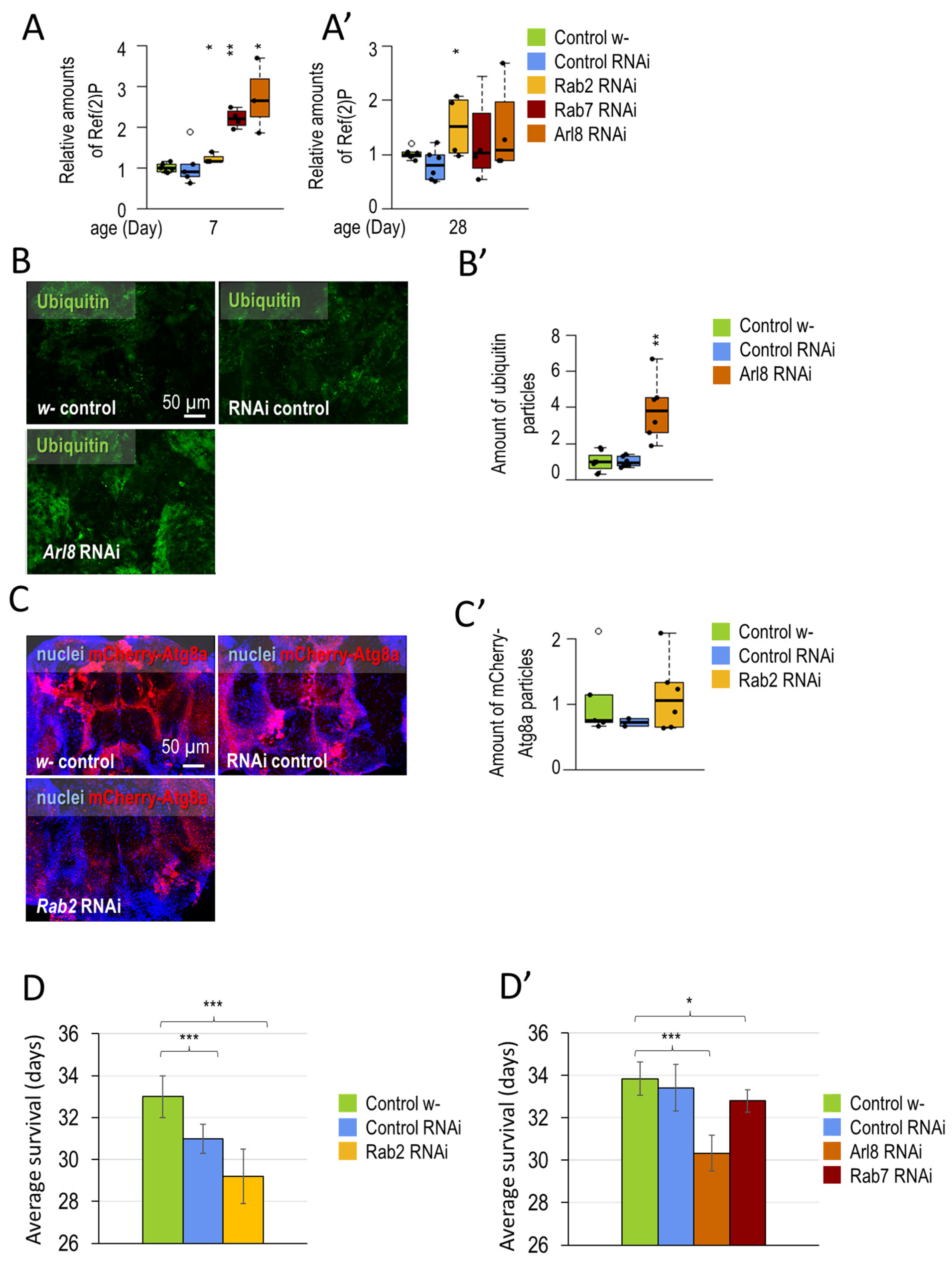
3.1.1. Small-GTPase Proteins Involved in the Lysosomal Degradation Pathway Are Required for Normal Longevity and Viability of Flies
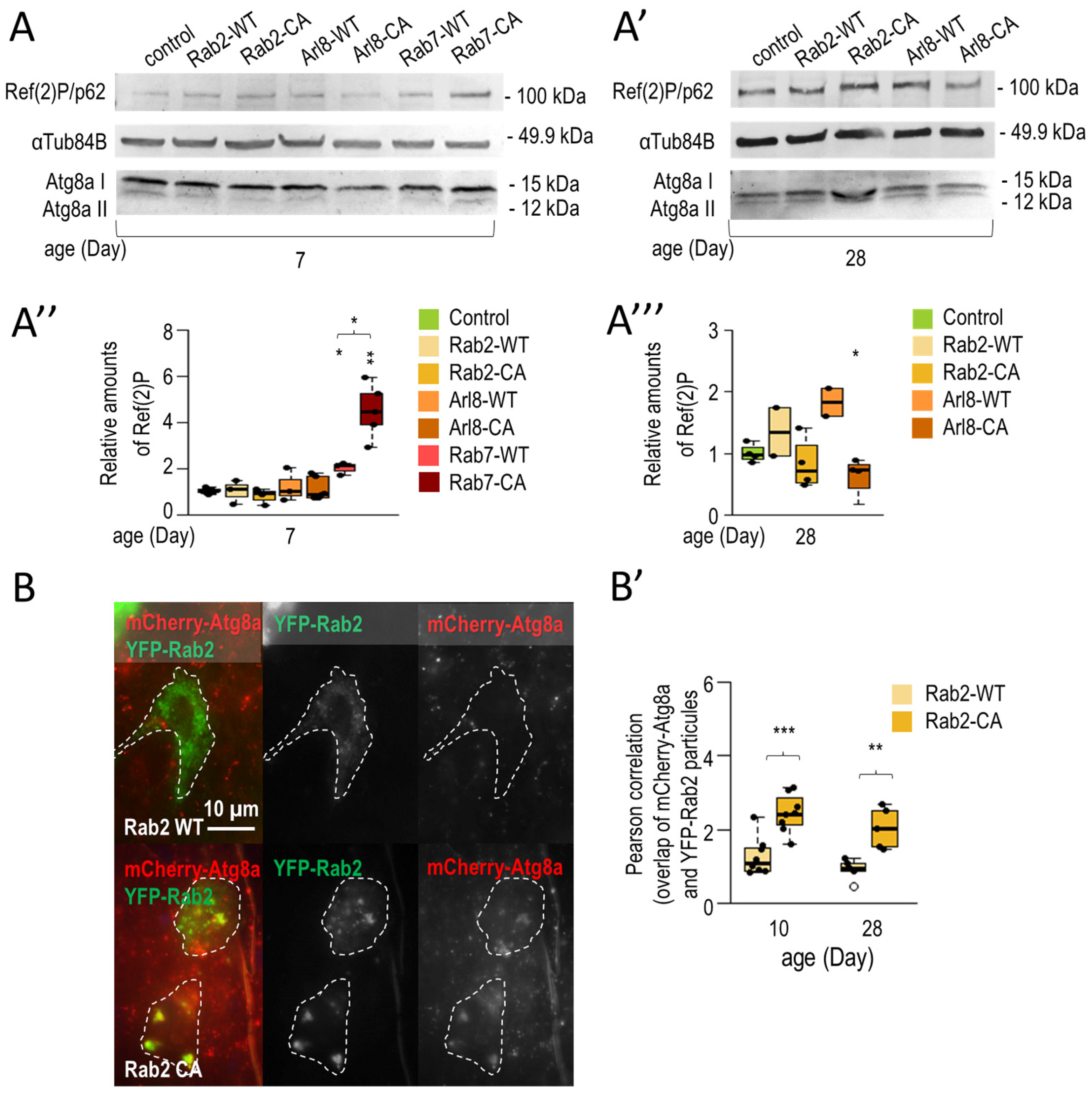
3.1.2. Rab2 and Arl8 Activation Extends Lifespan and Improves Climbing Ability
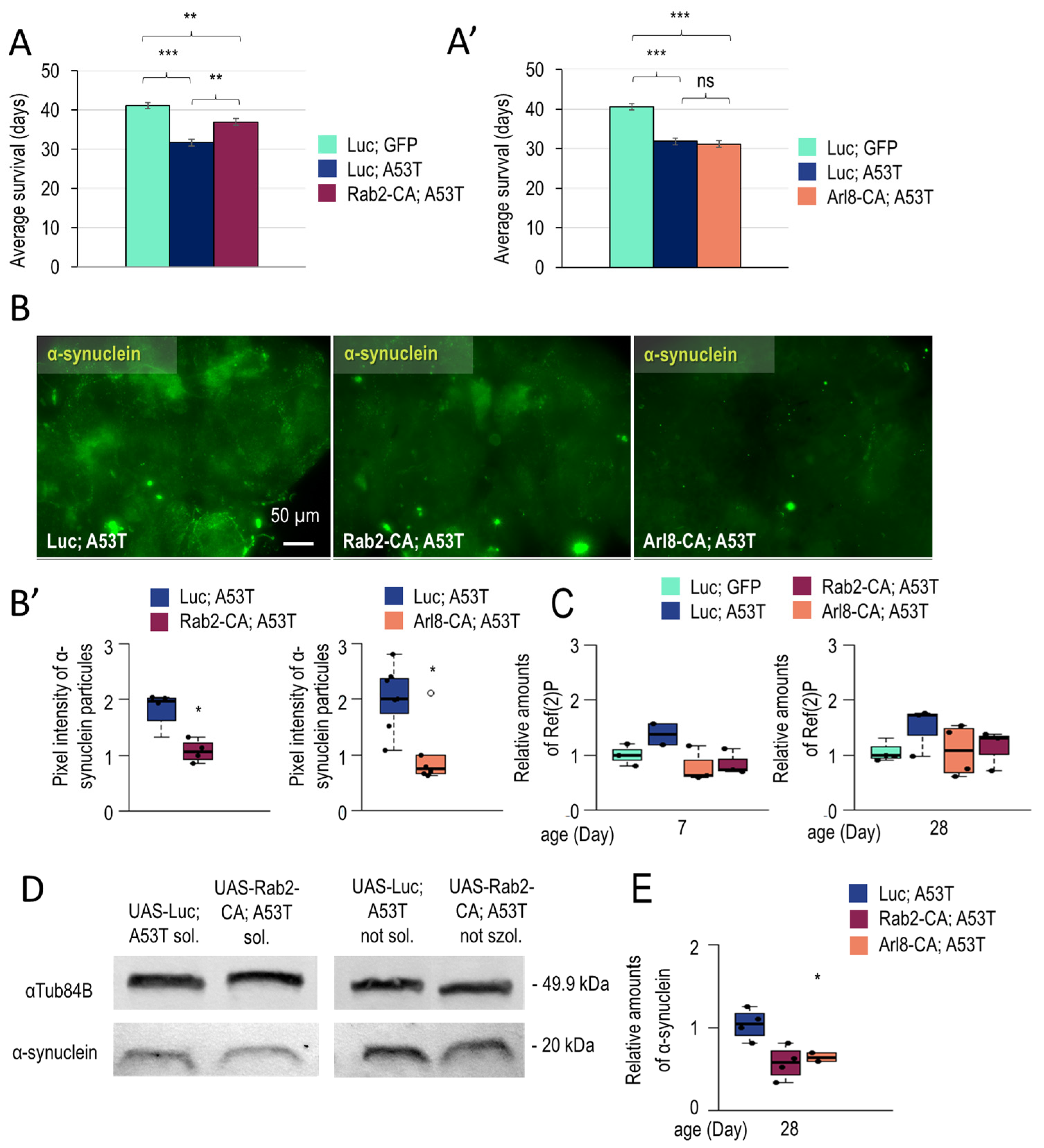
3.1.3. Rab2 Activation Promotes Longevity in a Parkinson’s Disease Model
4. Discussion
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A





References
- Adams, J. The Proteasome: Structure, Function, and Role in the Cell. Cancer Treat. Rev. 2003, 29, 3–9. [Google Scholar] [CrossRef]
- Parzych, K.R.; Klionsky, D.J. An Overview of Autophagy: Morphology, Mechanism, and Regulation. Antioxid. Redox Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Pedro, J.M.B.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in Major Human Diseases. EMBO J. 2021, 40, 108863. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Codogno, P. The Mechanism and Physiological Function of Macroautophagy. J. Innate Immun. 2013, 5, 427–433. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [PubMed]
- Rae, M.J.; Butler, R.N.; Campisi, J.; De Grey, A.D.N.J.; Finch, C.E.; Gough, M.; Martin, G.M.; Vijg, J.; Perrott, K.M.; Logan, B.J. The Demographic and Biomedical Case for Late-Life Interventions in Aging. Sci. Transl. Med. 2010, 2, 40cm21. [Google Scholar] [CrossRef] [PubMed]
- Meléndez, A.; Tallóczy, Z.; Seaman, M.; Eskelinen, E.L.; Hall, D.H.; Levine, B. Autophagy Genes Are Essential for Dauer Development and Life-Span Extension in C. Elegans. Science 2003, 301, 1387–1391. [Google Scholar] [CrossRef]
- Donati, A.; Cavallini, G.; Paradiso, C.; Vittorini, S.; Pollera, M.; Gori, Z.; Bergamini, E. Age-Related Changes in the Autophagic Proteolysis of Rat Isolated Liver Cells: Effects of Antiaging Dietary Restrictions. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2001, 56, B375–B383. [Google Scholar] [CrossRef]
- Tóth, M.L.; Sigmond, T.; Borsos, É.; Barna, J.; Erdélyi, P.; Takács-Vellai, K.; Orosz, L.; Kovács, A.L.; Csikós, G.; Sass, M.; et al. Longevity Pathways Converge on Autophagy Genes to Regulate Life Span in Caenorhabditis Elegans. Autophagy 2008, 4, 330–338. [Google Scholar] [CrossRef]
- Vellai, T.; Takács-Vellai, K. Regulation of Protein Turnover by Longevity Pathways. In Advances in Experimental Medicine and Biology; Springer: Berlin/Heidelberg, Germany, 2010; Volume 694, pp. 69–80. [Google Scholar]
- Vellai, T. Autophagy Genes and Ageing. Cell Death Differ. 2009, 16, 94–102. [Google Scholar] [CrossRef]
- Sigmond, T.; Barna, J.; Tóth, M.L.; Takács-Vellai, K.; Pásti, G.; Kovács, A.L.; Vellai, T. Chapter 30 Autophagy in Caenorhabditis Elegans. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2008; Volume 451, pp. 521–540. [Google Scholar]
- Simonsen, A.; Cumming, R.C.; Brech, A.; Isakson, P.; Schubert, D.R.; Finley, K.D. Promoting Basal Levels of Autophagy in the Nervous System Enhances Longevity and Oxidant Resistance in Adult Drosophila. Autophagy 2008, 4, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Pyo, J.O.; Yoo, S.M.; Ahn, H.H.; Nah, J.; Hong, S.H.; Kam, T.I.; Jung, S.; Jung, Y.K. Overexpression of Atg5 in Mice Activates Autophagy and Extends Lifespan. Nat. Commun. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J.; Warner, H.R. Aging in Mitotic and Post-Mitotic Cells. In Advances in Cell Aging and Gerontology; Gilchrest, B.A., Bohr, V.A., Eds.; Elsevier Science B.V.: Amsterdam, The Netherlands, 2001; pp. 1–16. [Google Scholar]
- Rubinsztein, D.C. The Roles of Intracellular Protein-Degradation Pathways in Neurodegeneration. Nature 2006, 443, 780–786. [Google Scholar] [CrossRef]
- Vellai, T.; Tóth, M.L.; Kovács, A.L. Janus-Faced Autophagy: A Dual Role of Cellular Self-Eating in Neurodegeneration? Autophagy 2007, 3, 461–463. [Google Scholar] [CrossRef]
- Tominaga, K.; Suzuki, H.I. TGF-β Signaling in Cellular Senescence and Aging-Related Pathology. Int. J. Mol. Sci. 2019, 20, 5002. [Google Scholar] [CrossRef]
- Shaw, W.M.; Luo, S.; Landis, J.; Ashraf, J.; Murphy, C.T. The C. Elegans TGF-β Dauer Pathway Regulates Longevity via Insulin Signaling. Curr. Biol. 2007, 17, 1635. [Google Scholar] [CrossRef] [PubMed]
- Oh, S.W.; Mukhopadhyay, A.; Svrzikapa, N.; Jiang, F.; Davis, R.J.; Tissenbaum, H.A. JNK Regulates Lifespan in Caenorhabditis Elegans by Modulating Nuclear Translocation of Forkhead Transcription Factor/DAF-16. Proc. Natl. Acad. Sci. USA 2005, 102, 4494. [Google Scholar] [CrossRef]
- Liu, J.K. Antiaging Agents: Safe Interventions to Slow Aging and Healthy Life Span Extension. Nat. Prod. Bioprospect. 2022, 12, 18. [Google Scholar] [CrossRef]
- Kiechl, S.; Pechlaner, R.; Willeit, P.; Notdurfter, M.; Paulweber, B.; Willeit, K.; Werner, P.; Ruckenstuhl, C.; Iglseder, B.; Weger, S.; et al. Higher Spermidine Intake Is Linked to Lower Mortality: A Prospective Population-Based Study. Am. J. Clin. Nutr. 2018, 108, 371–380. [Google Scholar] [CrossRef]
- Madeo, F.; Eisenberg, T.; Pietrocola, F.; Kroemer, G. Spermidine in Health and Disease. Science 2018, 359, eaan2788. [Google Scholar] [CrossRef]
- Gupta, V.K.; Scheunemann, L.; Eisenberg, T.; Mertel, S.; Bhukel, A.; Koemans, T.S.; Kramer, J.M.; Liu, K.S.Y.; Schroeder, S.; Stunnenberg, H.G.; et al. Restoring Polyamines Protects from Age-Induced Memory Impairment in an Autophagy-Dependent Manner. Nat. Neurosci. 2013, 16, 1453–1460. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, A.; Zheng, Y.R.; Wu, X.L.; Tang, W.D.; Liu, M.R.; Ma, S.J.; Jiang, L.; Hu, W.W.; Zhang, X.N.; Chen, Z. Urolithin A-activated Autophagy but Not Mitophagy Protects against Ischemic Neuronal Injury by Inhibiting ER Stress in Vitro and in Vivo. CNS Neurosci. Ther. 2019, 25, 976. [Google Scholar] [CrossRef] [PubMed]
- Bakhtiari, N.; Hosseinkhani, S.; Tashakor, A.; Hemmati, R. Ursolic Acid Ameliorates Aging-Metabolic Phenotype through Promoting of Skeletal Muscle Rejuvenation. Med. Hypotheses 2015, 85, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Chen, S.Y.; Lu, C.C.; Lin, J.A.; Yen, G.C. Ursolic Acid Promotes Apoptosis, Autophagy, and Chemosensitivity in Gemcitabine-Resistant Human Pancreatic Cancer Cells. Phytother. Res. 2020, 34, 2053–2066. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Alimova, I.N.; Baturin, D.A.; Popovich, I.G.; Zabezhinski, M.A.; Rosenfeld, S.V.; Manton, K.G.; Semenchenko, A.V.; Yashin, A.I. Dose-Dependent Effect of Melatonin on Life Span and Spontaneous Tumor Incidence in Female SHR Mice. Exp. Gerontol. 2003, 38, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Izmaylov, D.M.; Obukhova, L.K. Geroprotector Effectiveness of Melatonin: Investigation of Lifespan of Drosophila melanogaster. Mech. Ageing Dev. 1999, 106, 233–240. [Google Scholar] [CrossRef]
- Luo, F.; Sandhu, A.F.; Rungratanawanich, W.; Williams, G.E.; Akbar, M.; Zhou, S.; Song, B.J.; Wang, X. Melatonin and Autophagy in Aging-Related Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 7174. [Google Scholar] [CrossRef]
- Maday, S. Mechanisms of Neuronal Homeostasis: Autophagy in the Axon. Brain Res. 2016, 1649, 143–150. [Google Scholar] [CrossRef]
- Maday, S.; Holzbaur, E.L.F. Autophagosome Assembly and Cargo Capture in the Distal Axon. Autophagy 2012, 8, 858–860. [Google Scholar] [CrossRef]
- Cheng, X.T.; Zhou, B.; Lin, M.Y.; Cai, Q.; Sheng, Z.H. Axonal Autophagosomes Recruit Dynein for Retrograde Transport through Fusion with Late Endosomes. J. Cell Biol. 2015, 209, 377–386. [Google Scholar] [CrossRef]
- Maday, S.; Wallace, K.E.; Holzbaur, E.L.F. Autophagosomes Initiate Distally and Mature during Transport toward the Cell Soma in Primary Neurons. J. Cell Biol. 2012, 196, 407–417. [Google Scholar] [CrossRef]
- Hara, T.; Nakamura, K.; Matsui, M.; Yamamoto, A.; Nakahara, Y.; Suzuki-Migishima, R.; Yokoyama, M.; Mishima, K.; Saito, I.; Okano, H.; et al. Suppression of Basal Autophagy in Neural Cells Causes Neurodegenerative Disease in Mice. Nature 2006, 441, 885–889. [Google Scholar] [CrossRef]
- Maria Fimia, G.; Stoykova, A.; Romagnoli, A.; Giunta, L.; Di Bartolomeo, S.; Nardacci, R.; Corazzari, M.; Fuoco, C.; Ucar, A.; Schwartz, P.; et al. Ambra1 Regulates Autophagy and Development of the Nervous System. Nature 2007, 447, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Reimer, R.J.; Schneider, K.; Leal-Ortiz, S.; Montenegro-Venegas, C.; Kim, S.A.; Garner, L.C.; Gundelfinger, E.D.; Reimer, R.J.; Garner, C.C. Bassoon Controls Presynaptic Autophagy through Atg5. Neuron 2017, 93, 897–913.e7. [Google Scholar] [CrossRef]
- Konstantinidis, G.; Tavernarakis, N. Molecular Basis of Neuronal Autophagy in Ageing: Insights from Caenorhabditis Elegans. Cells 2021, 10, 694. [Google Scholar] [CrossRef]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef]
- Komatsu, M.; Waguri, S.; Chiba, T.; Murata, S.; Iwata, J.; Tanida, I.; Ueno, T.; Koike, M.; Uchiyama, Y.; Kominami, E.; et al. Loss of Autophagy in the Central Nervous System Causes Neurodegeneration in Mice. Nature 2006, 441, 880–884. [Google Scholar] [CrossRef] [PubMed]
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired Degradation of Mutant Alpha-Synuclein by Chaperone-Mediated Autophagy. Science 2004, 305, 1292–1295. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.M.; Youle, R.J. PINK1- and Parkin-Mediated Mitophagy at a Glance. J. Cell Sci. 2012, 125, 795–799. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. MTOR Signaling at a Glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef]
- Vellai, T. How the Amino Acid Leucine Activates the Key Cell-Growth Regulator MTOR. Nature 2021, 596, 192–194. [Google Scholar] [CrossRef]
- Boutouja, F.; Stiehm, C.M.; Platta, H.W. MTOR: A Cellular Regulator Interface in Health and Disease. Cells 2019, 8, 18. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and Molecular Mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef]
- Papp, D.; Kovács, T.; Billes, V.; Varga, M.; Tarnóci, A.; Hackler, L.; Puskás, L.G.; Liliom, H.; Tárnok, K.; Schlett, K.; et al. AUTEN-67, an Autophagy-Enhancing Drug Candidate with Potent Antiaging and Neuroprotective Effects. Autophagy 2016, 12, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Kovács, T.; Billes, V.; Komlós, M.; Hotzi, B.; Manzéger, A.; Tarnóci, A.; Papp, D.; Szikszai, F.; Szinyákovics, J.; Rácz, Á.; et al. The Small Molecule AUTEN-99 (Autophagy Enhancer-99) Prevents the Progression of Neurodegenerative Symptoms. Sci. Rep. 2017, 7, 42014. [Google Scholar] [CrossRef] [PubMed]
- Komlós, M.; Szinyákovics, J.; Falcsik, G.; Sigmond, T.; Jezsó, B.; Vellai, T.; Kovács, T. The Small-Molecule Enhancers of Autophagy AUTEN-67 and -99 Delay Ageing in Drosophila Striated Muscle Cells. Int. J. Mol. Sci. 2023, 24, 8100. [Google Scholar] [CrossRef]
- Billes, V.; Kovács, T.; Hotzi, B.; Manzéger, A.; Tagscherer, K.; Komlós, M.; Tarnóci, A.; Pádár, Z.; Erdős, A.; Bjelik, A.; et al. AUTEN-67 (Autophagy Enhancer-67) Hampers the Progression of Neurodegenerative Symptoms in a Drosophila Model of Huntington’s Disease. J. Huntingt. Dis. 2016, 5, 133–147. [Google Scholar] [CrossRef] [PubMed]
- Button, R.W.; Roberts, S.L.; Willis, T.L.; Oliver Hanemann, C.; Luo, S. Accumulation of Autophagosomes Confers Cytotoxicity. J. Biol. Chem. 2017, 292, 13599–13614. [Google Scholar] [CrossRef]
- Kovács, T.; Szinyákovics, J.; Billes, V.; Murányi, G.; Varga, V.B.; Bjelik, A.; Légrádi, Á.; Szabó, M.; Sándor, S.; Kubinyi, E.; et al. A Conserved MTMR Lipid Phosphatase Increasingly Suppresses Autophagy in Brain Neurons during Aging. Sci. Rep. 2022, 12, 21817. [Google Scholar] [CrossRef]
- Stenmark, H. Rab GTPases as Coordinators of Vesicle Traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef]
- Stroupe, C. This Is the End: Regulation of Rab7 Nucleotide Binding in Endolysosomal Trafficking and Autophagy. Front. Cell Dev. Biol. 2018, 6, 129. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Jiang, X.; Tian, R.; Zhao, P.; Li, L.; Wang, X.; Chen, S.; Zhu, Y.; Mei, M.; Bao, S.; et al. RAB2 Regulates the Formation of Autophagosome and Autolysosome in Mammalian Cells. Autophagy 2019, 15, 1774–1786. [Google Scholar] [CrossRef] [PubMed]
- Boda, A.; Lőrincz, P.; Takáts, S.; Csizmadia, T.; Tóth, S.; Kovács, A.L.; Juhász, G. Drosophila Arl8 Is a General Positive Regulator of Lysosomal Fusion Events. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, M.G.; Munafó, D.B.; Berón, W.; Colombo, M.I. Rab7 Is Required for the Normal Progression of the Autophagic Pathway in Mammalian Cells. J. Cell Sci. 2004, 117, 2687–2697. [Google Scholar] [CrossRef]
- Kirisako, T.; Baba, M.; Ishihara, N.; Miyazawa, K.; Ohsumi, M.; Yoshimori, T.; Noda, T.; Ohsumi, Y. Formation Process of Autophagosome Is Traced with Apg8/Aut7p in Yeast. J. Cell Biol. 1999, 147, 435–446. [Google Scholar] [CrossRef]
- Lund, V.K.; Madsen, K.L.; Kjaerulff, O. Drosophila Rab2 Controls Endosome-Lysosome Fusion and LAMP Delivery to Late Endosomes. Autophagy 2018, 14, 1520–1542. [Google Scholar] [CrossRef]
- Fujita, N.; Huang, W.; Lin, T.H.; Groulx, J.F.; Jean, S.; Nguyen, J.; Kuchitsu, Y.; Koyama-Honda, I.; Mizushima, N.; Fukuda, M.; et al. Genetic Screen in Drosophila Muscle Identifies Autophagy-Mediated T-Tubule Remodeling and a Rab2 Role in Autophagy. eLife 2017, 6, e23367. [Google Scholar] [CrossRef]
- Zhao, X.; Liberti, R.; Jian, J.; Fu, W.; Hettinghouse, A.; Sun, Y.; Liu, C.J. Progranulin Associates with Rab2 and Is Involved in Autophagosome-Lysosome Fusion in Gaucher Disease. J. Mol. Med. 2021, 99, 1639–1654. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, A.K.; Sinka, R.; Torres, I.L.; Lilley, K.S.; Munro, S. Toward a Comprehensive Map of the Effectors of Rab GTPases. Dev. Cell 2014, 31, 358. [Google Scholar] [CrossRef] [PubMed]
- Kajiho, H.; Kajiho, Y.; Frittoli, E.; Confalonieri, S.; Bertalot, G.; Viale, G.; Di Fiore, P.P.; Oldani, A.; Garre, M.; Beznoussenko, G.V.; et al. RAB2A Controls MT1-MMP Endocytic and E-cadherin Polarized Golgi Trafficking to Promote Invasive Breast Cancer Programs. EMBO Rep. 2016, 17, 1061. [Google Scholar] [CrossRef]
- Lőrincz, P.; Mauvezin, C.; Juhász, G. Exploring Autophagy in Drosophila. Cells 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed]
- Jäger, S.; Bucci, C.; Tanida, I.; Ueno, T.; Kominami, E.; Saftig, P.; Eskelinen, E.L. Role for Rab7 in Maturation of Late Autophagic Vacuoles. J. Cell Sci. 2004, 117, 4837–4848. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Glatz, G.; Szenci, G.; Boda, A.; Horváth, G.V.; Hegedűs, K.; Kovács, A.L.; Juhász, G. Non-Canonical Role of the SNARE Protein Ykt6 in Autophagosome-Lysosome Fusion. PLoS Genet. 2018, 14, e1007359. [Google Scholar] [CrossRef] [PubMed]
- Lund, V.K.; Lycas, M.D.; Schack, A.; Andersen, R.C.; Gether, U.; Kjaerulff, O. Rab2 Drives Axonal Transport of Dense Core Vesicles and Lysosomal Organelles. Cell Rep. 2021, 35, 108973. [Google Scholar] [CrossRef]
- Kuchitsu, Y.; Fukuda, M. Revisiting Rab7 Functions in Mammalian Autophagy: Rab7 Knockout Studies. Cells 2018, 7, 215. [Google Scholar] [CrossRef]
- Bhargava, H.K.; Tabata, K.; Byck, J.M.; Hamasaki, M.; Farrell, D.P.; Anishchenko, I.; DiMaio, F.; Im, Y.J.; Yoshimori, T.; Hurley, J.H. Structural Basis for Autophagy Inhibition by the Human Rubicon-Rab7 Complex. Proc. Natl. Acad. Sci. USA 2020, 117, 17003–17010. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Multiple Roles of the Small GTPase Rab7. Cells 2016, 5, 34. [Google Scholar] [CrossRef]
- Oka, M.; Hashimoto, K.; Yamaguchi, Y.; Saitoh, S.I.; Sugiura, Y.; Motoi, Y.; Honda, K.; Kikko, Y.; Ohata, S.; Suematsu, M.; et al. Arl8b Is Required for Lysosomal Degradation of Maternal Proteins in the Visceral Yolk Sac Endoderm of Mouse Embryos. J. Cell Sci. 2017, 130, 3568–3577. [Google Scholar] [CrossRef] [PubMed]
- Rosa-Ferreira, C.; Sweeney, S.T.; Munro, S. The Small G Protein Arl8 Contributes to Lysosomal Function and Long-Range Axonal Transport in Drosophila. Biol. Open 2018, 7, bio035964. [Google Scholar] [CrossRef]
- Mrakovic, A.; Kay, J.G.; Furuya, W.; Brumell, J.H.; Botelho, R.J. Rab7 and Arl8 GTPases Are Necessary for Lysosome Tubulation in Macrophages. Traffic 2012, 13, 1667–1679. [Google Scholar] [CrossRef]
- Cherry, S.; Jin, E.J.; Özel, M.N.; Lu, Z.; Agi, E.; Wang, D.; Jung, W.-H.; Epstein, D.; Meinertzhagen, I.A.; Chan, C.-C.; et al. Charcot-Marie-Tooth 2B Mutations in Rab7 Cause Dosage-Dependent Neurodegeneration Due to Partial Loss of Function. eLife 2013, 2, e01064. [Google Scholar] [CrossRef]
- Szegö, E.M.; Van den Haute, C.; Höfs, L.; Baekelandt, V.; Van der Perren, A.; Falkenburger, B.H. Rab7 Reduces α-Synuclein Toxicity in Rats and Primary Neurons. Exp. Neurol. 2022, 347, 113900. [Google Scholar] [CrossRef]
- Griffin, E.F.; Caldwell, K.A.; Caldwell, G.A. Vacuolar Protein Sorting Protein 41 (VPS41) at an Intersection of Endosomal Traffic in Neurodegenerative Disease. Neural Regen. Res. 2019, 14, 1210–1212. [Google Scholar] [CrossRef]
- Griffin, E.F.; Yan, X.; Caldwell, K.A.; Caldwell, G.A. Distinct Functional Roles of Vps41-Mediated Neuroprotection in Alzheimer’s and Parkinson’s Disease Models of Neurodegeneration. Hum. Mol. Genet. 2018, 27, 4176–4193. [Google Scholar] [CrossRef]
- Inoshita, T.; Liu, J.Y.; Taniguchi, D.; Ishii, R.; Shiba-Fukushima, K.; Hattori, N.; Imai, Y. Parkinson Disease-Associated Leucine-Rich Repeat Kinase Regulates UNC-104-Dependent Axonal Transport of Arl8-Positive Vesicles in Drosophila. iScience 2022, 25, 105476. [Google Scholar] [CrossRef]
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Schink, M.K.O.; Theodossiou, T.A.; Johansen, T.; Juhász, G.; Bilder, D.; et al. Microenvironmental Autophagy Promotes Tumour Growth. Nature 2017, 541, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Pircs, K.; Nagy, P.; Varga, A.; Venkei, Z.; Erdi, B.; Hegedus, K.; Juhasz, G. Advantages and Limitations of Different P62-Based Assays for Estimating Autophagic Activity in Drosophila. PLoS ONE 2012, 7, e44214. [Google Scholar] [CrossRef] [PubMed]
- Takáts, S.; Nagy, P.; Varga, Á.; Pircs, K.; Kárpáti, M.; Varga, K.; Kovács, A.L.; Hegedűs, K.; Juhász, G. Autophagosomal Syntaxin17-Dependent Lysosomal Degradation Maintains Neuronal Function in Drosophila. J. Cell Biol. 2013, 201, 531–539. [Google Scholar] [CrossRef]
- Lőrincz, P.; Tóth, S.; Benkő, P.; Lakatos, Z.; Boda, A.; Glatz, G.; Zobel, M.; Bisi, S.; Hegedűs, K.; Takáts, S.; et al. Rab2 Promotes Autophagic and Endocytic Lysosomal Degradation. J. Cell Biol. 2017, 216, 1937–1947. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A New Mathematical Model for Relative Quantification in Real-Time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.L.D.; Kaun, K.R.; Edgar, B.A. The Small GTPase Rheb Affects Central Brain Neuronal Morphology and Memory Formation in Drosophila. PLoS ONE 2012, 7, e44888. [Google Scholar] [CrossRef]
- Shi, G.X.; Cai, W.; Andres, D.A. Rit Subfamily Small GTPases: Regulators in Neuronal Differentiation and Survival. Cell Signal. 2013, 25, 2060. [Google Scholar] [CrossRef] [PubMed]
- Mavromatakis, Y.E.; Tomlinson, A. The Role of the Small GTPase Rap in Drosophila R7 Photoreceptor Specification. Proc. Natl. Acad. Sci. USA 2012, 109, 3844–3849. [Google Scholar] [CrossRef] [PubMed]
- van der Voet, M.; Nijhof, B.; Oortveld, M.A.W.; Schenck, A. Drosophila Models of Early Onset Cognitive Disorders and Their Clinical Applications. Neurosci. Biobehav. Rev. 2014, 46 Pt 2, 326–342. [Google Scholar] [CrossRef] [PubMed]
- Riemensperger, T.; Issa, A.R.; Pech, U.; Coulom, H.; Nguyễn, M.V.; Cassar, M.; Jacquet, M.; Fiala, A.; Birman, S. A Single Dopamine Pathway Underlies Progressive Locomotor Deficits in a Drosophila Model of Parkinson Disease. Cell Rep. 2013, 5, 952–960. [Google Scholar] [CrossRef] [PubMed]
- Nagy, P.; Kárpáti, M.; Varga, Á.; Pircs, K.; Venkei, Z.; Takáts, S.; Varga, K.; Érdi, B.; Hegedus, K.; Juhász, G. Atg17/FIP200 Localizes to Perilysosomal Ref(2)P Aggregates and Promotes Autophagy by Activation of Atg1 in Drosophila. Autophagy 2014, 10, 453–467. [Google Scholar] [CrossRef] [PubMed]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Øvervatn, A.; Bjørkøy, G.; Johansen, T. P62/SQSTM1 Binds Directly to Atg8/LC3 to Facilitate Degradation of Ubiquitinated Protein Aggregates by Autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Zhang, J.; Schulze, K.L.; Robin Hiesinger, P.; Suyama, K.; Wang, S.; Fish, M.; Acar, M.; Hoskins, R.A.; Bellen, H.J.; Scott, M.P. Thirty-One Flavors of Drosophila Rab Proteins. Genetics 2007, 176, 1307–1322. [Google Scholar] [CrossRef]
- Kurz, A.; Double, K.L.; Lastres-Becker, I.; Tozzi, A.; Tantucci, M.; Bockhart, V.; Bonin, M.; García-Arencibia, M.; Nuber, S.; Schlaudraff, F.; et al. A53T-Alpha-Synuclein Overexpression Impairs Dopamine Signaling and Striatal Synaptic Plasticity in Old Mice. PLoS ONE 2010, 5, e11464. [Google Scholar] [CrossRef]
- Kotzbauer, P.T.; Giasson, B.I.; Kravitz, A.V.; Golbe, L.I.; Mark, M.H.; Trojanowski, J.Q.; Lee, V.M.Y. Fibrillization of α-Synuclein and Tau in Familial Parkinson’s Disease Caused by the A53T α-Synuclein Mutation. Exp. Neurol. 2004, 187, 279–288. [Google Scholar] [CrossRef]
- Billes, V.; Kovács, T.; Manzéger, A.; Lőrincz, P.; Szincsák, S.; Regős, Á.; Kulcsár, P.I.; Korcsmáros, T.; Lukácsovich, T.; Hoffmann, G.; et al. Developmentally Regulated Autophagy Is Required for Eye Formation in Drosophila. Autophagy 2018, 14, 1499–1519. [Google Scholar] [CrossRef]
- Lo Piccolo, L.; Bonaccorso, R.; Onorati, M.C. Nuclear and Cytoplasmic Soluble Proteins Extraction from a Small Quantity of Drosophila’s Whole Larvae and Tissues. Int. J. Mol. Sci. 2015, 16, 12360. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, R.; Arya, S.B.; Jagga, D.; Kaur, H.; Tuli, A.; Sharma, M. The Rab7 Effector PLEKHM1 Binds Arl8b to Promote Cargo Traffic to Lysosomes. J. Cell Biol. 2017, 216, 1051–1070. [Google Scholar] [CrossRef]
- Jongsma, M.L.; Bakker, J.; Cabukusta, B.; Liv, N.; van Elsland, D.; Fermie, J.; Akkermans, J.L.; Kuijl, C.; van der Zanden, S.Y.; Janssen, L.; et al. SKIP-HOPS Recruits TBC1D15 for a Rab7-to-Arl8b Identity Switch to Control Late Endosome Transport. EMBO J. 2020, 39, e102301. [Google Scholar] [CrossRef]
- Sharon, R.; Bar-Joseph, I.; Frosch, M.P.; Walsh, D.M.; Hamilton, J.A.; Selkoe, D.J. The Formation of Highly Soluble Oligomers of α-Synuclein Is Regulated by Fatty Acids and Enhanced in Parkinson’s Disease. Neuron 2003, 37, 583–595. [Google Scholar] [CrossRef] [PubMed]
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In Vivo Demonstration That α-Synuclein Oligomers Are Toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199. [Google Scholar] [CrossRef]
- Dinter, E.; Saridaki, T.; Nippold, M.; Plum, S.; Diederichs, L.; Komnig, D.; Fensky, L.; May, C.; Marcus, K.; Voigt, A.; et al. Rab7 Induces Clearance of α-Synuclein Aggregates. J. Neurochem. 2016, 138, 758–774. [Google Scholar] [CrossRef] [PubMed]
- Khatter, D.; Sindhwani, A.; Sharma, M. Arf-like GTPase Arl8: Moving from the Periphery to the Center of Lysosomal Biology. Cell. Logist. 2015, 5, e1086501. [Google Scholar] [CrossRef]
- Donaldson, J.G.; Jackson, C.L. ARF Family G Proteins and Their Regulators: Roles in Membrane Transport, Development and Disease. Nat. Rev. Mol. Cell Biol. 2011, 12, 362. [Google Scholar] [CrossRef]
- Götz, T.W.B.; Puchkov, D.; Lysiuk, V.; Lützkendorf, J.; Nikonenko, A.G.; Quentin, C.; Lehmann, M.; Sigrist, S.J.; Petzoldt, A.G. Rab2 Regulates Presynaptic Precursor Vesicle Biogenesis at the Trans-Golgi. J. Cell Biol. 2021, 220, e202006040. [Google Scholar] [CrossRef]
- Guerra, F.; Bucci, C. Role of the RAB7 Protein in Tumor Progression and Cisplatin Chemoresistance. Cancers 2019, 11, 1096. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szinyákovics, J.; Keresztes, F.; Kiss, E.A.; Falcsik, G.; Vellai, T.; Kovács, T. Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing. Cells 2023, 12, 1753. https://doi.org/10.3390/cells12131753
Szinyákovics J, Keresztes F, Kiss EA, Falcsik G, Vellai T, Kovács T. Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing. Cells. 2023; 12(13):1753. https://doi.org/10.3390/cells12131753
Chicago/Turabian StyleSzinyákovics, Janka, Fanni Keresztes, Eszter Anna Kiss, Gergő Falcsik, Tibor Vellai, and Tibor Kovács. 2023. "Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing" Cells 12, no. 13: 1753. https://doi.org/10.3390/cells12131753
APA StyleSzinyákovics, J., Keresztes, F., Kiss, E. A., Falcsik, G., Vellai, T., & Kovács, T. (2023). Potent New Targets for Autophagy Enhancement to Delay Neuronal Ageing. Cells, 12(13), 1753. https://doi.org/10.3390/cells12131753