
Rewired Metabolism Caused by the Oncogenic Deregulation of MYC as an Attractive Therapeutic Target in Cancers


Abstract
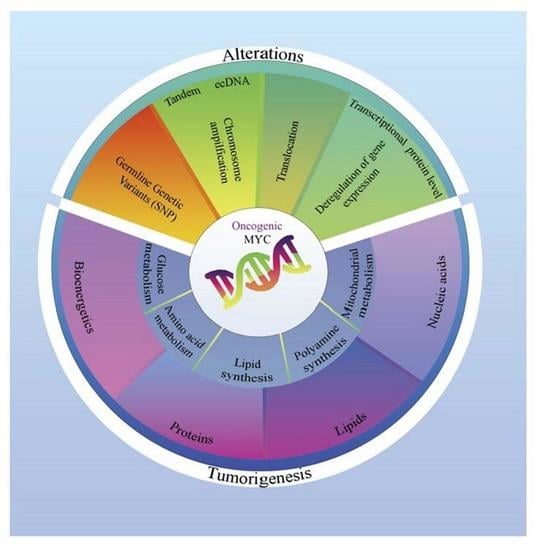
1. Introduction
2. Overview of MYC Regulation in the Physiological State
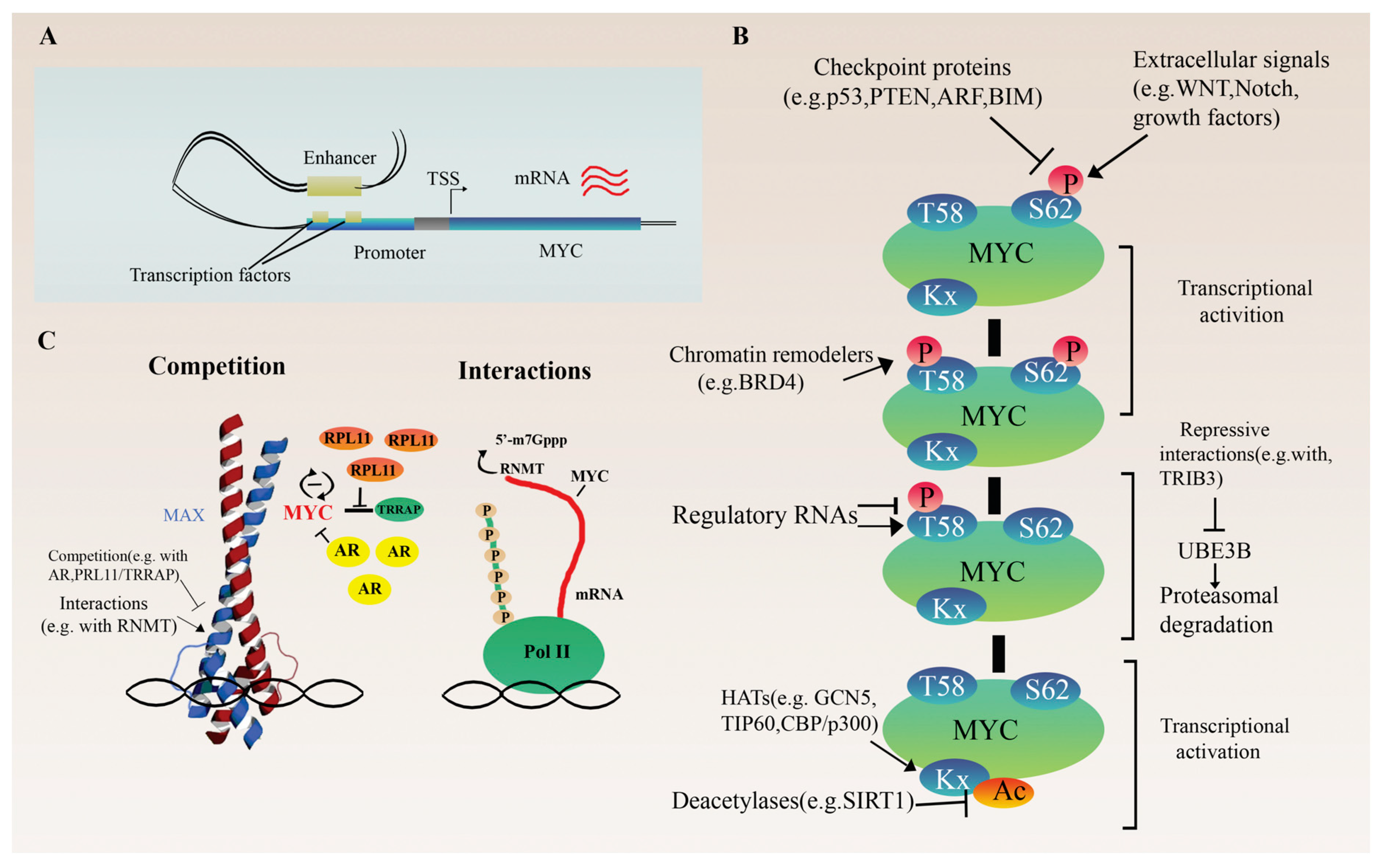
2.1. Regulation at the Transcriptional Level
2.2. Indirect and Direct Translation Regulation
3. Oncogenic Deregulation of MYC and Its Effect on Cancer Metabolism
3.1. General Aspects of the Metabolic Regulation by MYC
3.1.1. Glucose Metabolism
3.1.2. Amino Acid Metabolism
3.1.3. Lipid Metabolism
3.1.4. Polyamine Metabolism
3.1.5. Mitochondrial Metabolism
3.2. Distinct Processes of MYC Oncogenic Deregulation
3.2.1. Germline Genetic Variants: Single-Nucleotide Polymorphisms (SNPs)
3.2.2. Amplification
3.2.3. Translocation
3.2.4. Deregulation of Gene Expression
Transcriptional Regulation
Translational Deregulation and Protein Stability
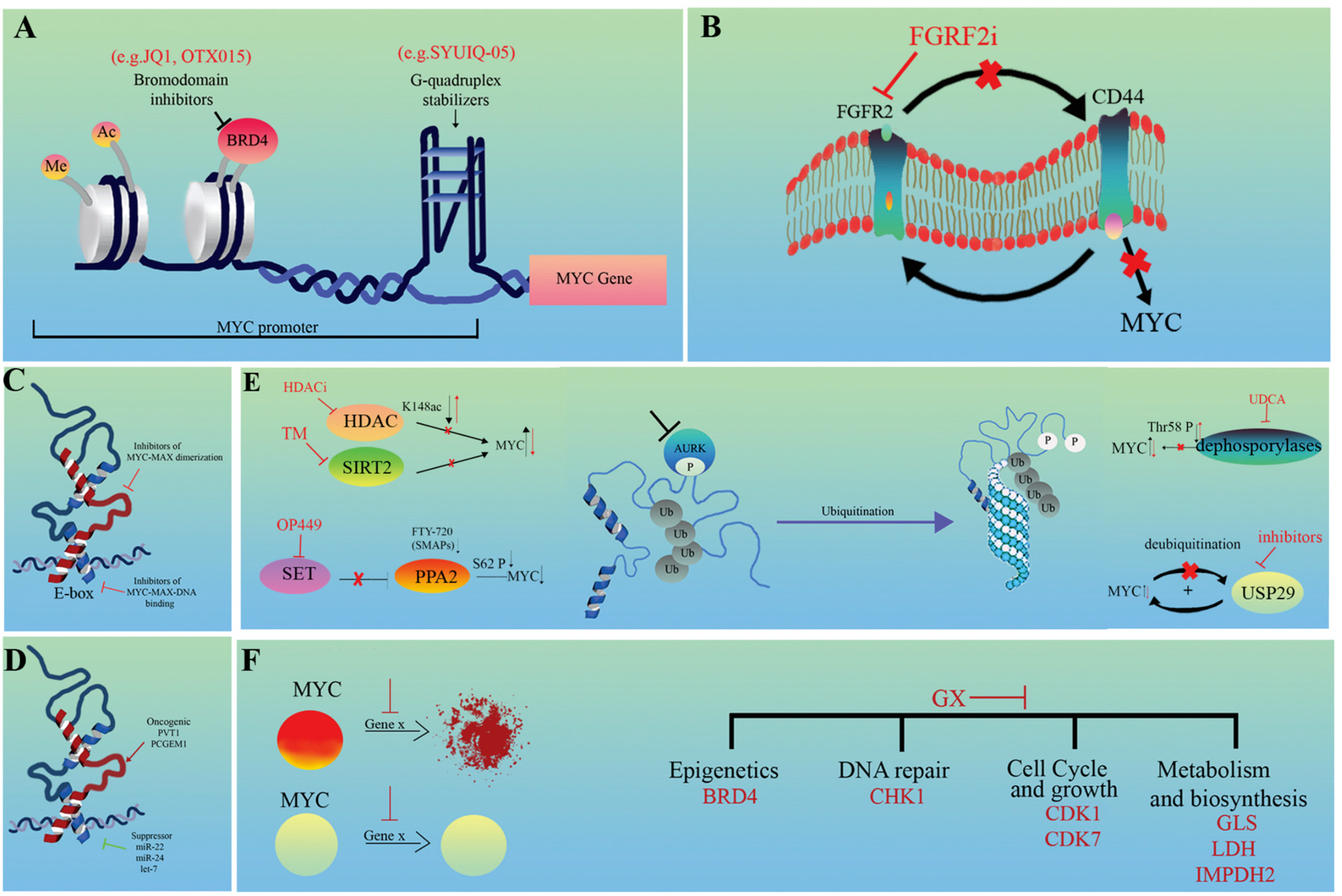
4. Conclusions and Potential Therapeutic Aspects
4.1. Targeting Upstream Features Influencing MYC Expression
4.1.1. Modulating Chromatin Interactions
4.1.2. Indirect Inhibition of MYC Expression
4.2. Targeting MYC Effector (Downstream) Features
4.3. Regulatory RNAs as Putative Therapeutic Targets
4.4. Protein Degradation
4.5. Synthetic Lethal Interactions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C.V. MYC, Metabolism, and Cancer. Cancer Discov. 2015, 5, 1024–1039. [Google Scholar] [CrossRef]
- Wolfer, A.; Wittner, B.S.; Irimia, D.; Flavin, R.J.; Lupien, M.; Gunawardane, R.N.; Meyer, C.A.; Lightcap, E.S.; Tamayo, P.; Mesirov, J.P.; et al. MYC regulation of a “poor-prognosis” metastatic cancer cell state. Proc. Natl. Acad. Sci. USA 2010, 107, 3698–3703. [Google Scholar] [CrossRef] [PubMed]
- Cory, S.; Gerondakis, S.; Adams, J.M. Interchromosomal recombination of the cellular oncogene c-myc with the immunoglobulin heavy chain locus in murine plasmacytomas is a reciprocal exchange. EMBO J. 1983, 2, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Gearhart, J.; Pashos, E.E.; Prasad, M.K. Pluripotency redux--advances in stem-cell research. N. Engl. J. Med. 2007, 357, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Conacci-Sorrell, M.; McFerrin, L.; Eisenman, R.N. An overview of MYC and its interactome. Cold Spring Harb. Perspect. Med. 2014, 4, a014357. [Google Scholar] [CrossRef] [PubMed]
- Eisenman, R.N. Deconstructing myc. Genes Dev. 2001, 15, 2023–2030. [Google Scholar] [CrossRef]
- Caforio, M.; Sorino, C.; Iacovelli, S.; Fanciulli, M.; Locatelli, F.; Folgiero, V. Recent advances in searching c-Myc transcriptional cofactors during tumorigenesis. J. Exp. Clin. Cancer Res. 2018, 37, 239. [Google Scholar] [CrossRef]
- Dang, C.V. Gene regulation: Fine-tuned amplification in cells. Nature 2014, 511, 417–418. [Google Scholar] [CrossRef]
- Li, Q.; Seo, J.H.; Stranger, B.; McKenna, A.; Pe’er, I.; Laframboise, T.; Brown, M.; Tyekucheva, S.; Freedman, M.L. Integrative eQTL-based analyses reveal the biology of breast cancer risk loci. Cell 2013, 152, 633–641. [Google Scholar] [CrossRef]
- Sabo, A.; Kress, T.R.; Pelizzola, M.; de Pretis, S.; Gorski, M.M.; Tesi, A.; Morelli, M.J.; Bora, P.; Doni, M.; Verrecchia, A.; et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature 2014, 511, 488–492. [Google Scholar] [CrossRef]
- Thomas, L.R.; Wang, Q.; Grieb, B.C.; Phan, J.; Foshage, A.M.; Sun, Q.; Olejniczak, E.T.; Clark, T.; Dey, S.; Lorey, S.; et al. Interaction with WDR5 promotes target gene recognition and tumorigenesis by MYC. Mol. Cell 2015, 58, 440–452. [Google Scholar] [CrossRef] [PubMed]
- Walz, S.; Lorenzin, F.; Morton, J.; Wiese, K.E.; von Eyss, B.; Herold, S.; Rycak, L.; Dumay-Odelot, H.; Karim, S.; Bartkuhn, M.; et al. Activation and repression by oncogenic MYC shape tumour-specific gene expression profiles. Nature 2014, 511, 483–487. [Google Scholar] [CrossRef] [PubMed]
- Soufi, A.; Garcia, M.F.; Jaroszewicz, A.; Osman, N.; Pellegrini, M.; Zaret, K.S. Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 2015, 161, 555–568. [Google Scholar] [CrossRef] [PubMed]
- Baluapuri, A.; Wolf, E.; Eilers, M. Target gene-independent functions of MYC oncoproteins. Nat. Rev. Mol. Cell Biol. 2020, 21, 255–267. [Google Scholar] [CrossRef]
- Bywater, M.J.; Burkhart, D.L.; Straube, J.; Sabo, A.; Pendino, V.; Hudson, J.E.; Quaife-Ryan, G.A.; Porrello, E.R.; Rae, J.; Parton, R.G.; et al. Reactivation of Myc transcription in the mouse heart unlocks its proliferative capacity. Nat. Commun. 2020, 11, 1827. [Google Scholar] [CrossRef] [PubMed]
- Kress, T.R.; Pellanda, P.; Pellegrinet, L.; Bianchi, V.; Nicoli, P.; Doni, M.; Recordati, C.; Bianchi, S.; Rotta, L.; Capra, T.; et al. Identification of MYC-Dependent Transcriptional Programs in Oncogene-Addicted Liver Tumors. Cancer Res. 2016, 76, 3463–3472. [Google Scholar] [CrossRef] [PubMed]
- Pellanda, P.; Dalsass, M.; Filipuzzi, M.; Loffreda, A.; Verrecchia, A.; Castillo Cano, V.; Thabussot, H.; Doni, M.; Morelli, M.J.; Soucek, L.; et al. Integrated requirement of non-specific and sequence-specific DNA binding in Myc-driven transcription. EMBO J. 2021, 40, e105464. [Google Scholar] [CrossRef]
- See, Y.X.; Chen, K.; Fullwood, M.J. MYC overexpression leads to increased chromatin interactions at super-enhancers and MYC binding sites. Genome Res. 2022, 32, 629–642. [Google Scholar] [CrossRef]
- Xin, B.; Rohs, R. Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. 2018, 28, 321–333. [Google Scholar] [CrossRef]
- Sauve, S.; Naud, J.-F.; Lavigne, P. The mechanism of discrimination between cognate and non-specific DNA by dimeric b/HLH/LZ transcription factors. J. Mol. Biol. 2007, 365, 1163–1175. [Google Scholar] [CrossRef]
- Allevato, M.; Bolotin, E.; Grossman, M.; Mane-Padros, D.; Sladek, F.M.; Martinez, E. Sequence-specific DNA binding by MYC/MAX to low-affinity non-E-box motifs. PLoS ONE 2017, 12, e0180147. [Google Scholar] [CrossRef] [PubMed]
- Bahr, C.; von Paleske, L.; Uslu, V.V.; Remeseiro, S.; Takayama, N.; Ng, S.W.; Murison, A.; Langenfeld, K.; Petretich, M.; Scognamiglio, R.; et al. A Myc enhancer cluster regulates normal and leukaemic haematopoietic stem cell hierarchies. Nature 2018, 553, 515–520. [Google Scholar] [CrossRef] [PubMed]
- van Riggelen, J.; Yetil, A.; Felsher, D.W. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat. Rev. Cancer 2010, 10, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Dai, M.-S.; Arnold, H.; Sun, X.-X.; Sears, R.; Lu, H. Inhibition of c-Myc activity by ribosomal protein L11. EMBO J. 2007, 26, 3332–3345. [Google Scholar] [CrossRef] [PubMed]
- Morcelle, C.; Menoyo, S.; Moron-Duran, F.D.; Tauler, A.; Kozma, S.C.; Thomas, G.; Gentilella, A. Oncogenic MYC Induces the Impaired Ribosome Biogenesis Checkpoint and Stabilizes p53 Independent of Increased Ribosome Content. Cancer Res. 2019, 79, 4348–4359. [Google Scholar] [CrossRef]
- Wang, R.; Peng, C.; Song, J.; Hua, Y.; Wu, Q.; Deng, L.; Cao, Y.; Zhang, J.; Zhang, L.; Wu, L.; et al. Downregulated RRS1 inhibits invasion and metastasis of BT549 through RPL11-c-Myc-SNAIL axis. Int. J. Oncol. 2022, 60, 33. [Google Scholar] [CrossRef]
- Van Bortle, K.; Marciano, D.P.; Liu, Q.; Chou, T.; Lipchik, A.M.; Gollapudi, S.; Geller, B.S.; Monte, E.; Kamakaka, R.T.; Snyder, M.P. A cancer-associated RNA polymerase III identity drives robust transcription and expression of snaR-A noncoding RNA. Nat. Commun. 2022, 13, 3007. [Google Scholar] [CrossRef]
- Devaiah, B.N.; Mu, J.; Akman, B.; Uppal, S.; Weissman, J.D.; Cheng, D.; Baranello, L.; Nie, Z.; Levens, D.; Singer, D.S. MYC protein stability is negatively regulated by BRD4. Proc. Natl. Acad. Sci. USA 2020, 117, 13457–13467. [Google Scholar] [CrossRef]
- Li, K.; Wang, F.; Yang, Z.-N.; Zhang, T.-T.; Yuan, Y.-F.; Zhao, C.-X.; Yeerjiang, Z.; Cui, B.; Hua, F.; Lv, X.-X.; et al. TRIB3 promotes MYC-associated lymphoma development through suppression of UBE3B-mediated MYC degradation. Nat. Commun. 2020, 11, 6316. [Google Scholar] [CrossRef]
- Smolarz, B.; Zadrozna-Nowak, A.; Romanowicz, H. The Role of lncRNA in the Development of Tumors, including Breast Cancer. Int. J. Mol. Sci. 2021, 22, 8427. [Google Scholar] [CrossRef]
- Tseng, Y.Y.; Moriarity, B.S.; Gong, W.; Akiyama, R.; Tiwari, A.; Kawakami, H.; Ronning, P.; Reuland, B.; Guenther, K.; Beadnell, T.C.; et al. PVT1 dependence in cancer with MYC copy-number increase. Nature 2014, 512, 82–86. [Google Scholar] [CrossRef]
- Cole, M.D.; Cowling, V.H. Specific regulation of mRNA cap methylation by the c-Myc and E2F1 transcription factors. Oncogene 2009, 28, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Posternak, V.; Ung, M.H.; Cheng, C.; Cole, M.D. MYC Mediates mRNA Cap Methylation of Canonical Wnt/β-Catenin Signaling Transcripts By Recruiting CDK7 and RNA Methyltransferase. Mol. Cancer Res. 2017, 15, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Sanchez, M.E.; Gonatopoulos-Pournatzis, T.; Preston, G.; Lawlor, M.A.; Cowling, V.H. S-Adenosyl homocysteine hydrolase is required for Myc-induced mRNA cap methylation, protein synthesis, and cell proliferation. Mol. Cell. Biol. 2009, 29, 6182–6191. [Google Scholar] [CrossRef] [PubMed]
- Roohinejad, Z.; Bahramian, S.; Shamsabadi, F.T.; Sahebi, R.; Amini, A.; Sabour, D.; Shafiee, M. Upregulation of the c-MYC oncogene and adjacent long noncoding RNAs PVT1 and CCAT1 in esophageal squamous cell carcinoma. BMC Cancer 2023, 23, 34. [Google Scholar] [CrossRef]
- Lal, A.; Navarro, F.; Maher, C.A.; Maliszewski, L.E.; Yan, N.; O’Day, E.; Chowdhury, D.; Dykxhoorn, D.M.; Tsai, P.; Hofmann, O.; et al. miR-24 Inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3’UTR microRNA recognition elements. Mol. Cell 2009, 35, 610–625. [Google Scholar] [CrossRef]
- Kim, H.H.; Kuwano, Y.; Srikantan, S.; Lee, E.K.; Martindale, J.L.; Gorospe, M. HuR recruits let-7/RISC to repress c-Myc expression. Genes Dev. 2009, 23, 1743–1748. [Google Scholar] [CrossRef]
- Paulmann, C.; Spallek, R.; Karpiuk, O.; Heider, M.; Schaffer, I.; Zecha, J.; Klaeger, S.; Walzik, M.; Ollinger, R.; Engleitner, T.; et al. The OTUD6B-LIN28B-MYC axis determines the proliferative state in multiple myeloma. EMBO J. 2022, 41, e110871. [Google Scholar] [CrossRef]
- Desi, N.; Teh, V.; Tong, Q.Y.; Lim, C.Y.; Tabatabaeian, H.; Chew, X.H.; Sanchez-Mejias, A.; Chan, J.J.; Zhang, B.; Pitcheshwar, P.; et al. MiR-138 is a potent regulator of the heterogenous MYC transcript population in cancers. Oncogene 2022, 41, 1178–1189. [Google Scholar] [CrossRef]
- Zhu, Q.; Zhang, C.; Qu, T.; Lu, X.; He, X.; Li, W.; Yin, D.; Han, L.; Guo, R.; Zhang, E. MNX1-AS1 Promotes Phase Separation of IGF2BP1 to Drive c-Myc-Mediated Cell-Cycle Progression and Proliferation in Lung Cancer. Cancer Res. 2022, 82, 4340–4358. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, Z.; Wen, W.; Liu, Z.; Zhang, C.; Li, M.; Hu, F.; Wei, S.; Bae, S.; Zhou, J.; et al. The microRNA-3622 family at the 8p21 locus exerts oncogenic effects by regulating the p53-downstream gene network in prostate cancer progression. Oncogene 2022, 41, 3186–3196. [Google Scholar] [CrossRef]
- Bowman, T.; Broome, M.A.; Sinibaldi, D.; Wharton, W.; Pledger, W.J.; Sedivy, J.M.; Irby, R.; Yeatman, T.; Courtneidge, S.A.; Jove, R. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc. Natl. Acad. Sci. USA 2001, 98, 7319–7324. [Google Scholar] [CrossRef] [PubMed]
- Chou, Y.-T.; Lin, H.-H.; Lien, Y.-C.; Wang, Y.-H.; Hong, C.-F.; Kao, Y.-R.; Lin, S.-C.; Chang, Y.-C.; Lin, S.-Y.; Chen, S.-J.; et al. EGFR promotes lung tumorigenesis by activating miR-7 through a Ras/ERK/Myc pathway that targets the Ets2 transcriptional repressor ERF. Cancer Res. 2010, 70, 8822–8831. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O’Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef]
- Tang, W.; Pei, M.; Li, J.; Xu, N.; Xiao, W.; Yu, Z.; Zhang, J.; Hong, L.; Guo, Z.; Lin, J.; et al. The miR-3648/FRAT1-FRAT2/c-Myc negative feedback loop modulates the metastasis and invasion of gastric cancer cells. Oncogene 2022, 41, 4823–4838. [Google Scholar] [CrossRef]
- Feng, Y.C.; Liu, X.Y.; Teng, L.; Ji, Q.; Wu, Y.; Li, J.M.; Gao, W.; Zhang, Y.Y.; La, T.; Tabatabaee, H.; et al. c-Myc inactivation of p53 through the pan-cancer lncRNA MILIP drives cancer pathogenesis. Nat. Commun. 2020, 11, 4980. [Google Scholar] [CrossRef]
- Lee, Y.-R.; Chen, M.; Lee, J.D.; Zhang, J.; Lin, S.-Y.; Fu, T.-M.; Chen, H.; Ishikawa, T.; Chiang, S.-Y.; Katon, J.; et al. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science 2019, 364, eaau0159. [Google Scholar] [CrossRef] [PubMed]
- Muthalagu, N.; Junttila, M.R.; Wiese, K.E.; Wolf, E.; Morton, J.; Bauer, B.; Evan, G.I.; Eilers, M.; Murphy, D.J. BIM is the primary mediator of MYC-induced apoptosis in multiple solid tissues. Cell Rep. 2014, 8, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wu, Y.; Nouri, M.; Spisak, S.; Russo, J.W.; Sowalsky, A.G.; Pomerantz, M.M.; Wei, Z.; Korthauer, K.; Seo, J.H.; et al. Androgen receptor and MYC equilibration centralizes on developmental super-enhancer. Nat. Commun. 2021, 12, 7308. [Google Scholar] [CrossRef]
- Luengo, A.; Gui, D.Y.; Vander Heiden, M.G. Targeting Metabolism for Cancer Therapy. Cell Chem. Biol. 2017, 24, 1161–1180. [Google Scholar] [CrossRef]
- Tu, R.; Kang, W.; Yang, M.; Wang, L.; Bao, Q.; Chen, Z.; Dong, Y.; Wang, J.; Jiang, J.; Liu, H.; et al. USP29 coordinates MYC and HIF1α stabilization to promote tumor metabolism and progression. Oncogene 2021, 40, 6417–6429. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.L.; Inguva, A.; Pei, S.; Jones, C.; Krug, A.; Ye, H.; Minhajuddin, M.; Winters, A.; Furtek, S.L.; Gamboni, F.; et al. The STAT3-MYC axis promotes survival of leukemia stem cells by regulating SLC1A5 and oxidative phosphorylation. Blood 2022, 139, 584–596. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Target c-Myc to treat pancreatic cancer. Cancer Biol. Ther. 2022, 23, 34–50. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Pyun, W.Y.; Park, H.W. Cancer Metabolism: Phenotype, Signaling and Therapeutic Targets. Cells 2020, 9, 2308. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Yan, T.; Bao, Y.; Shen, C.; Yu, C.; Zhu, X.; Tian, X.; Guo, F.; Liang, Q.; Liu, Q.; et al. LncRNA GLCC1 promotes colorectal carcinogenesis and glucose metabolism by stabilizing c-Myc. Nat. Commun. 2019, 10, 3499. [Google Scholar] [CrossRef]
- Valvona, C.J.; Fillmore, H.L.; Nunn, P.B.; Pilkington, G.J. The Regulation and Function of Lactate Dehydrogenase A: Therapeutic Potential in Brain Tumor. Brain Pathol. 2016, 26, 3–17. [Google Scholar] [CrossRef]
- Gan, L.; Xiu, R.; Ren, P.; Yue, M.; Su, H.; Guo, G.; Xiao, D.; Yu, J.; Jiang, H.; Liu, H.; et al. Metabolic targeting of oncogene MYC by selective activation of the proton-coupled monocarboxylate family of transporters. Oncogene 2016, 35, 3037–3048. [Google Scholar] [CrossRef]
- De Santis, M.C.; Porporato, P.E.; Martini, M.; Morandi, A. Signaling Pathways Regulating Redox Balance in Cancer Metabolism. Front. Oncol. 2018, 8, 126. [Google Scholar] [CrossRef]
- Santana-Codina, N.; Roeth, A.A.; Zhang, Y.; Yang, A.; Mashadova, O.; Asara, J.M.; Wang, X.; Bronson, R.T.; Lyssiotis, C.A.; Ying, H.; et al. Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nat. Commun. 2018, 9, 4945. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.C.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.-M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Morrish, F.; Isern, N.; Sadilek, M.; Jeffrey, M.; Hockenbery, D.M. c-Myc activates multiple metabolic networks to generate substrates for cell-cycle entry. Oncogene 2009, 28, 2485–2491. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Song, L.; Wan, Q.; Wu, G.; Li, X.; Wang, Y.; Wang, J.; Liu, Z.; Zhong, X.; He, X.; et al. cMyc-mediated activation of serine biosynthesis pathway is critical for cancer progression under nutrient deprivation conditions. Cell Res. 2015, 25, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Yue, M.; Jiang, J.; Gao, P.; Liu, H.; Qing, G. Oncogenic MYC Activates a Feedforward Regulatory Loop Promoting Essential Amino Acid Metabolism and Tumorigenesis. Cell Rep. 2017, 21, 3819–3832. [Google Scholar] [CrossRef]
- Venkateswaran, N.; Lafita-Navarro, M.C.; Hao, Y.-H.; Kilgore, J.A.; Perez-Castro, L.; Braverman, J.; Borenstein-Auerbach, N.; Kim, M.; Lesner, N.P.; Mishra, P.; et al. MYC promotes tryptophan uptake and metabolism by the kynurenine pathway in colon cancer. Genes Dev. 2019, 33, 1236–1251. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Tambay, V.; Raymond, V.-A.; Bilodeau, M. MYC Rules: Leading Glutamine Metabolism toward a Distinct Cancer Cell Phenotype. Cancers 2021, 13, 4484. [Google Scholar] [CrossRef]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y.; et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021, 502, 154–165. [Google Scholar] [CrossRef]
- Bensaad, K.; Favaro, E.; Lewis, C.A.; Peck, B.; Lord, S.; Collins, J.M.; Pinnick, K.E.; Wigfield, S.; Buffa, F.M.; Li, J.-L.; et al. Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 2014, 9, 349–365. [Google Scholar] [CrossRef]
- Bachmann, A.S.; Geerts, D. Polyamine synthesis as a target of MYC oncogenes. J. Biol. Chem. 2018, 293, 18757–18769. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Leon-Letelier, R.A.; Abdel Sater, A.H.; Vykoukal, J.; Dennison, J.B.; Hanash, S.; Fahrmann, J.F. c-MYC-Driven Polyamine Metabolism in Ovarian Cancer: From Pathogenesis to Early Detection and Therapy. Cancers 2023, 15, 623. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Ge, M.; Hu, J.; Li, X.; Che, L.; Sun, K.; Cheng, L.; Huang, Y.; Pilo, M.G.; Cigliano, A.; et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology 2017, 66, 167–181. [Google Scholar] [CrossRef] [PubMed]
- Lin, A.-P.; Qiu, Z.; Ethiraj, P.; Sasi, B.; Jaafar, C.; Rakheja, D.; Aguiar, R.C.T. MYC, mitochondrial metabolism and O-GlcNAcylation converge to modulate the activity and subcellular localization of DNA and RNA demethylases. Leukemia 2022, 36, 1150–1159. [Google Scholar] [CrossRef]
- Dang, C.V. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb. Perspect. Med. 2013, 3, a014217. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-L.; Wang, L.-Y.; Yu, Y.-L.; Chen, H.-W.; Srivastava, S.; Petrovics, G.; Kung, H.-J. A long noncoding RNA connects c-Myc to tumor metabolism. Proc. Natl. Acad. Sci. USA 2014, 111, 18697–18702. [Google Scholar] [CrossRef]
- Huarte, M. The emerging role of lncRNAs in cancer. Nat. Med. 2015, 21, 1253–1261. [Google Scholar] [CrossRef]
- Lancho, O.; Herranz, D. The MYC Enhancer-ome: Long-Range Transcriptional Regulation of MYC in Cancer. Trends Cancer 2018, 4, 810–822. [Google Scholar] [CrossRef]
- Haiman, C.A.; Le Marchand, L.; Yamamato, J.; Stram, D.O.; Sheng, X.; Kolonel, L.N.; Wu, A.H.; Reich, D.; Henderson, B.E. A common genetic risk factor for colorectal and prostate cancer. Nat. Genet. 2007, 39, 954–956. [Google Scholar] [CrossRef]
- Pirlog, R.; Drula, R.; Nutu, A.; Calin, G.A.; Berindan-Neagoe, I. The Roles of the Colon Cancer Associated Transcript 2 (CCAT2) Long Non-Coding RNA in Cancer: A Comprehensive Characterization of the Tumorigenic and Molecular Functions. Int. J. Mol. Sci. 2021, 22, 12491. [Google Scholar] [CrossRef]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 cancer risk variant rs6983267 shows long-range interaction with MYC in colorectal cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef]
- Freedman, M.L.; Haiman, C.A.; Patterson, N.; McDonald, G.J.; Tandon, A.; Waliszewska, A.; Penney, K.; Steen, R.G.; Ardlie, K.; John, E.M.; et al. Admixture mapping identifies 8q24 as a prostate cancer risk locus in African-American men. Proc. Natl. Acad. Sci. USA 2006, 103, 14068–14073. [Google Scholar] [CrossRef]
- Haiman, C.A.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple regions within 8q24 independently affect risk for prostate cancer. Nat. Genet. 2007, 39, 638–644. [Google Scholar] [CrossRef]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 prostate, breast, and colon cancer risk loci show tissue-specific long-range interaction with MYC. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 gene desert variant associated with prostate cancer risk confers differential in vivo activity to a MYC enhancer. Genome Res. 2010, 20, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Tian, J.; Yang, Y.; Zhong, R.; Li, J.; Zhai, K.; Ke, J.; Lou, J.; Chen, W.; Zhu, B.; et al. A Rare Missense Variant in TCF7L2 Associates with Colorectal Cancer Risk by Interacting with a GWAS-Identified Regulatory Variant in the MYC Enhancer. Cancer Res. 2018, 78, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Easton, D.F.; Pooley, K.A.; Dunning, A.M.; Pharoah, P.D.; Thompson, D.; Ballinger, D.G.; Struewing, J.P.; Morrison, J.; Field, H.; Luben, R.; et al. Genome-wide association study identifies novel breast cancer susceptibility loci. Nature 2007, 447, 1087–1093. [Google Scholar] [CrossRef]
- Jenkins, R.B.; Xiao, Y.; Sicotte, H.; Decker, P.A.; Kollmeyer, T.M.; Hansen, H.M.; Kosel, M.L.; Zheng, S.; Walsh, K.M.; Rice, T.; et al. A low-frequency variant at 8q24.21 is strongly associated with risk of oligodendroglial tumors and astrocytomas with IDH1 or IDH2 mutation. Nat. Genet. 2012, 44, 1122–1125. [Google Scholar] [CrossRef]
- Oktay, Y.; Ülgen, E.; Can, O.; Akyerli, C.B.; Yüksel, Ş.; Erdemgil, Y.; Durası, M.; Henegariu, O.I.; Nanni, E.P.; Selevsek, N.; et al. IDH-mutant glioma specific association of rs55705857 located at 8q24.21 involves MYC deregulation. Sci. Rep. 2016, 6, 27569. [Google Scholar] [CrossRef]
- Barrett, M.T.; Deiotte, R.; Lenkiewicz, E.; Malasi, S.; Holley, T.; Evers, L.; Posner, R.G.; Jones, T.; Han, H.; Sausen, M.; et al. Clinical study of genomic drivers in pancreatic ductal adenocarcinoma. Br. J. Cancer 2017, 117, 572–582. [Google Scholar] [CrossRef]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef]
- Zhang, X.; Choi, P.S.; Francis, J.M.; Imielinski, M.; Watanabe, H.; Cherniack, A.D.; Meyerson, M. Identification of focally amplified lineage-specific super-enhancers in human epithelial cancers. Nat. Genet. 2016, 48, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Herranz, D.; Ambesi-Impiombato, A.; Palomero, T.; Schnell, S.A.; Belver, L.; Wendorff, A.A.; Xu, L.; Castillo-Martin, M.; Llobet-Navás, D.; Cordon-Cardo, C.; et al. A NOTCH1-driven MYC enhancer promotes T cell development, transformation and acute lymphoblastic leukemia. Nat. Med. 2014, 20, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, J.; Zhou, Y.; Fasolino, M.; Goldman, N.; Schwartz, G.W.; Mumbach, M.R.; Nguyen, S.C.; Rome, K.S.; Sela, Y.; Zapataro, Z.; et al. Oncogenic Notch Promotes Long-Range Regulatory Interactions within Hyperconnected 3D Cliques. Mol. Cell 2019, 73, 1174–1190.e1112. [Google Scholar] [CrossRef] [PubMed]
- Yashiro-Ohtani, Y.; Wang, H.; Zang, C.; Arnett, K.L.; Bailis, W.; Ho, Y.; Knoechel, B.; Lanauze, C.; Louis, L.; Forsyth, K.S.; et al. Long-range enhancer activity determines Myc sensitivity to Notch inhibitors in T cell leukemia. Proc. Natl. Acad. Sci. USA 2014, 111, E4946–E4953. [Google Scholar] [CrossRef]
- Shi, J.; Whyte, W.A.; Zepeda-Mendoza, C.J.; Milazzo, J.P.; Shen, C.; Roe, J.-S.; Minder, J.L.; Mercan, F.; Wang, E.; Eckersley-Maslin, M.A.; et al. Role of SWI/SNF in acute leukemia maintenance and enhancer-mediated Myc regulation. Genes Dev. 2013, 27, 2648–2662. [Google Scholar] [CrossRef]
- Hung, K.L.; Luebeck, J.; Dehkordi, S.R.; Colón, C.I.; Li, R.; Wong, I.T.-L.; Coruh, C.; Dharanipragada, P.; Lomeli, S.H.; Weiser, N.E.; et al. Targeted profiling of human extrachromosomal DNA by CRISPR-CATCH. Nat. Genet. 2022, 54, 1746–1754. [Google Scholar] [CrossRef]
- Kim, H.; Nguyen, N.-P.; Turner, K.; Wu, S.; Gujar, A.D.; Luebeck, J.; Liu, J.; Deshpande, V.; Rajkumar, U.; Namburi, S.; et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nat. Genet. 2020, 52, 891–897. [Google Scholar] [CrossRef]
- Pongor, L.S.; Schultz, C.W.; Rinaldi, L.; Wangsa, D.; Redon, C.E.; Takahashi, N.; Fialkoff, G.; Desai, P.; Zhang, Y.; Burkett, S.; et al. Extrachromosomal DNA Amplification Contributes to Small Cell Lung Cancer Heterogeneity and is Associated with Worse Outcomes. Cancer Discov. 2023, 13, 928–949. [Google Scholar] [CrossRef]
- Turner, K.M.; Deshpande, V.; Beyter, D.; Koga, T.; Rusert, J.; Lee, C.; Li, B.; Arden, K.; Ren, B.; Nathanson, D.A.; et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 2017, 543, 122–125. [Google Scholar] [CrossRef]
- Storlazzi, C.T.; Lonoce, A.; Guastadisegni, M.C.; Trombetta, D.; D’Addabbo, P.; Daniele, G.; L’Abbate, A.; Macchia, G.; Surace, C.; Kok, K.; et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res. 2010, 20, 1198–1206. [Google Scholar] [CrossRef]
- Van Roy, N.; Vandesompele, J.; Menten, B.; Nilsson, H.; De Smet, E.; Rocchi, M.; De Paepe, A.; Påhlman, S.; Speleman, F. Translocation-excision-deletion-amplification mechanism leading to nonsyntenic coamplification of MYC and ATBF1. Genes Chromosom. Cancer 2006, 45, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Hung, K.L.; Yost, K.E.; Xie, L.; Shi, Q.; Helmsauer, K.; Luebeck, J.; Schöpflin, R.; Lange, J.T.; Chamorro González, R.; Weiser, N.E.; et al. ecDNA hubs drive cooperative intermolecular oncogene expression. Nature 2021, 600, 731–736. [Google Scholar] [CrossRef] [PubMed]
- Stasevich, E.M.; Uvarova, A.N.; Murashko, M.M.; Khabusheva, E.R.; Sheetikov, S.A.; Prassolov, V.S.; Kuprash, D.V.; Demin, D.E.; Schwartz, A.M. Enhancer RNA AL928768.3 from the IGH Locus Regulates MYC Expression and Controls the Proliferation and Chemoresistance of Burkitt Lymphoma Cells with IGH/MYC Translocation. Int. J. Mol. Sci. 2022, 23, 4624. [Google Scholar] [CrossRef]
- Ottema, S.; Mulet-Lazaro, R.; Erpelinck-Verschueren, C.; van Herk, S.; Havermans, M.; Arricibita Varea, A.; Vermeulen, M.; Beverloo, H.B.; Gröschel, S.; Haferlach, T.; et al. The leukemic oncogene EVI1 hijacks a MYC super-enhancer by CTCF-facilitated loops. Nat. Commun. 2021, 12, 5679. [Google Scholar] [CrossRef]
- Sastre, D.; Baiochi, J.; de Souza Lima, I.M.; Canto de Souza, F.; Corveloni, A.C.; Thomé, C.H.; Faca, V.M.; Schiavinato, J.L.D.S.; Covas, D.T.; Panepucci, R.A. Focused screening reveals functional effects of microRNAs differentially expressed in colorectal cancer. BMC Cancer 2019, 19, 1239. [Google Scholar] [CrossRef]
- Su, B.; Xu, T.; Bruce, J.P.; Yip, K.W.; Zhang, N.; Huang, Z.; Zhang, G.; Liu, F.-F.; Liang, J.; Yang, H.; et al. hsa-miR-24 suppresses metastasis in nasopharyngeal carcinoma by regulating the c-Myc/epithelial-mesenchymal transition axis. Oncol. Rep. 2018, 40, 2536–2546. [Google Scholar] [CrossRef]
- Li, Z.-Y.; Xie, Y.; Deng, M.; Zhu, L.; Wu, X.; Li, G.; Shi, N.-X.; Wen, C.; Huang, W.; Duan, Y.; et al. c-Myc-activated intronic miR-210 and lncRNA MIR210HG synergistically promote the metastasis of gastric cancer. Cancer Lett. 2022, 526, 322–334. [Google Scholar] [CrossRef]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.I.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef]
- Yao, W.; Li, S.; Liu, R.; Jiang, M.; Gao, L.; Lu, Y.; Liang, X.; Zhang, H. Long non-coding RNA PVT1: A promising chemotherapy and radiotherapy sensitizer. Front. Oncol. 2022, 12, 959208. [Google Scholar] [CrossRef]
- Takahashi, Y.; Sawada, G.; Kurashige, J.; Uchi, R.; Matsumura, T.; Ueo, H.; Takano, Y.; Eguchi, H.; Sudo, T.; Sugimachi, K.; et al. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br. J. Cancer 2014, 110, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Zamani, M.; Foroughmand, A.-M.; Hajjari, M.-R.; Bakhshinejad, B.; Johnson, R.; Galehdari, H. CASC11 and PVT1 spliced transcripts play an oncogenic role in colorectal carcinogenesis. Front. Oncol. 2022, 12, 954634. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Xu, J.; Sun, R.; Mumbach, M.R.; Carter, A.C.; Chen, Y.G.; Yost, K.E.; Kim, J.; He, J.; Nevins, S.A.; et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 2018, 173, 1398–1412.e22. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hong, S.; Hu, H.; Liu, T.; Yan, G.; Sun, D. MYC-Induced Upregulation of Lncrna ELFN1-AS1 Contributes to Tumor Growth in Colorectal Cancer via Epigenetically Silencing TPM1. Mol. Cancer Res. 2022, 20, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-X.; Li, W.; Li, J.-T.; Liu, F.; Zhou, L. Screening key long non-coding RNAs in early-stage colon adenocarcinoma by RNA-sequencing. Epigenomics 2018, 10, 1215–1228. [Google Scholar] [CrossRef]
- Qiu, X.; Boufaied, N.; Hallal, T.; Feit, A.; de Polo, A.; Luoma, A.M.; Alahmadi, W.; Larocque, J.; Zadra, G.; Xie, Y.; et al. MYC drives aggressive prostate cancer by disrupting transcriptional pause release at androgen receptor targets. Nat. Commun. 2022, 13, 2559. [Google Scholar] [CrossRef]
- Ganguly, K.; Bhatia, R.; Rauth, S.; Kisling, A.; Atri, P.; Thompson, C.; Vengoji, R.; Ram Krishn, S.; Shinde, D.; Thomas, V.; et al. Mucin 5AC Serves as the Nexus for β-Catenin/c-Myc Interplay to Promote Glutamine Dependency During Pancreatic Cancer Chemoresistance. Gastroenterology 2022, 162, 253–268.e13. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhou, B.; Hu, X.; Ying, S.; Zhou, Q.; Xu, W.; Feng, L.; Hou, T.; Wang, X.; Zhu, L.; et al. LncRNA LINC00942 promotes chemoresistance in gastric cancer by suppressing MSI2 degradation to enhance c-Myc mRNA stability. Clin. Transl. Med. 2022, 12, e703. [Google Scholar] [CrossRef]
- Zhu, P.; He, F.; Hou, Y.; Tu, G.; Li, Q.; Jin, T.; Zeng, H.; Qin, Y.; Wan, X.; Qiao, Y.; et al. A novel hypoxic long noncoding RNA KB-1980E6.3 maintains breast cancer stem cell stemness via interacting with IGF2BP1 to facilitate c-Myc mRNA stability. Oncogene 2021, 40, 1609–1627. [Google Scholar] [CrossRef]
- Tokheim, C.; Wang, X.; Timms, R.T.; Zhang, B.; Mena, E.L.; Wang, B.; Chen, C.; Ge, J.; Chu, J.; Zhang, W.; et al. Systematic characterization of mutations altering protein degradation in human cancers. Mol. Cell 2021, 81, 1292–1308.e11. [Google Scholar] [CrossRef]
- Bhatia, K.; Huppi, K.; Spangler, G.; Siwarski, D.; Iyer, R.; Magrath, I. Point mutations in the c-Myc transactivation domain are common in Burkitt’s lymphoma and mouse plasmacytomas. Nat. Genet. 1993, 5, 56–61. [Google Scholar] [CrossRef] [PubMed]
- Schaub, F.X.; Dhankani, V.; Berger, A.C.; Trivedi, M.; Richardson, A.B.; Shaw, R.; Zhao, W.; Zhang, X.; Ventura, A.; Liu, Y.; et al. Pan-cancer Alterations of the MYC Oncogene and Its Proximal Network across the Cancer Genome Atlas. Cell Syst. 2018, 6, 282–300.e2. [Google Scholar] [CrossRef] [PubMed]
- Takeda, D.Y.; Spisák, S.; Seo, J.-H.; Bell, C.; O’connor, E.; Korthauer, K.; Ribli, D.; Csabai, I.; Solymosi, N.; Szállási, Z.; et al. A Somatically Acquired Enhancer of the Androgen Receptor Is a Noncoding Driver in Advanced Prostate Cancer. Cell 2018, 174, 422–432.e13. [Google Scholar] [CrossRef]
- Morton, A.R.; Dogan-Artun, N.; Faber, Z.J.; MacLeod, G.; Bartels, C.F.; Piazza, M.S.; Allan, K.C.; Mack, S.C.; Wang, X.; Gimple, R.C.; et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 2019, 179, 1330–1341.e1313. [Google Scholar] [CrossRef]
- Nathanson, D.A.; Gini, B.; Mottahedeh, J.; Visnyei, K.; Koga, T.; Gomez, G.; Eskin, A.; Hwang, K.; Wang, J.; Masui, K.; et al. Targeted therapy resistance mediated by dynamic regulation of extrachromosomal mutant EGFR DNA. Science 2014, 343, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Møller, H.D.; Mohiyuddin, M.; Prada-Luengo, I.; Sailani, M.R.; Halling, J.F.; Plomgaard, P.; Maretty, L.; Hansen, A.J.; Snyder, M.P.; Pilegaard, H.; et al. Circular DNA elements of chromosomal origin are common in healthy human somatic tissue. Nat. Commun. 2018, 9, 1069. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Jia, R.; Ge, T.; Ge, S.; Zhuang, A.; Chai, P.; Fan, X. Extrachromosomal circular DNA: Biogenesis, structure, functions and diseases. Signal Transduct. Target. Ther. 2022, 7, 342. [Google Scholar] [CrossRef]
- Guérin, T.M.; Marcand, S. Breakage in breakage-fusion-bridge cycle: An 80-year-old mystery. Trends. Genet. 2022, 38, 641–645. [Google Scholar] [CrossRef]
- Ly, P.; Cleveland, D.W. Rebuilding Chromosomes After Catastrophe: Emerging Mechanisms of Chromothripsis. Trends Cell Biol. 2017, 27, 917–930. [Google Scholar] [CrossRef]
- Shoshani, O.; Brunner, S.F.; Yaeger, R.; Ly, P.; Nechemia-Arbely, Y.; Kim, D.H.; Fang, R.; Castillon, G.A.; Yu, M.; Li, J.S.Z.; et al. Chromothripsis drives the evolution of gene amplification in cancer. Nature 2021, 591, 137–141. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Nguyen-Khac, F. “Double-Hit” Chronic Lymphocytic Leukemia, Involving the TP53 and MYC Genes. Front. Oncol. 2021, 11, 826245. [Google Scholar] [CrossRef]
- Labbe, D.P.; Zadra, G.; Yang, M.; Reyes, J.M.; Lin, C.Y.; Cacciatore, S.; Ebot, E.M.; Creech, A.L.; Giunchi, F.; Fiorentino, M.; et al. High-fat diet fuels prostate cancer progression by rewiring the metabolome and amplifying the MYC program. Nat. Commun. 2019, 10, 4358. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.S.; Sears, R.C. MYC degradation. Cold Spring Harb. Perspect. Med. 2014, 4, a014365. [Google Scholar] [CrossRef] [PubMed]
- Sipol, A.; Hameister, E.; Xue, B.; Hofstetter, J.; Barenboim, M.; Öllinger, R.; Jain, G.; Prexler, C.; Rubio, R.A.; Baldauf, M.C.; et al. MondoA drives malignancy in B-ALL through enhanced adaptation to metabolic stress. Blood 2022, 139, 1184–1197. [Google Scholar] [CrossRef] [PubMed]
- Wilde, B.R.; Ayer, D.E. Interactions between Myc and MondoA transcription factors in metabolism and tumourigenesis. Br. J. Cancer 2015, 113, 1529–1533. [Google Scholar] [CrossRef]
- Lim, T.-Y.; Wilde, B.R.; Thomas, M.L.; Murphy, K.E.; Vahrenkamp, J.M.; Conway, M.E.; Varley, K.E.; Gertz, J.; Ayer, D.E. TXNIP loss expands Myc-dependent transcriptional programs by increasing Myc genomic binding. PLoS Biol. 2023, 21, e3001778. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; O’Shea, J.M.; Kaadige, M.R.; Cunha, S.; Wilde, B.R.; Cohen, A.L.; Welm, A.L.; Ayer, D.E. Metabolic reprogramming in triple-negative breast cancer through Myc suppression of TXNIP. Proc. Natl. Acad. Sci. USA 2015, 112, 5425–5430. [Google Scholar] [CrossRef]
- Parmenter, T.J.; Kleinschmidt, M.; Kinross, K.M.; Bond, S.T.; Li, J.; Kaadige, M.R.; Rao, A.; Sheppard, K.E.; Hugo, W.; Pupo, G.M.; et al. Response of BRAF-mutant melanoma to BRAF inhibition is mediated by a network of transcriptional regulators of glycolysis. Cancer Discov. 2014, 4, 423–433. [Google Scholar] [CrossRef]
- Qu, X.; Sun, J.; Zhang, Y.; Li, J.; Hu, J.; Li, K.; Gao, L.; Shen, L. c-Myc-driven glycolysis via TXNIP suppression is dependent on glutaminase-MondoA axis in prostate cancer. Biochem. Biophys. Res. Commun. 2018, 504, 415–421. [Google Scholar] [CrossRef]
- Carroll, P.A.; Diolaiti, D.; McFerrin, L.; Gu, H.; Djukovic, D.; Du, J.; Cheng, P.F.; Anderson, S.; Ulrich, M.; Hurley, J.B.; et al. Deregulated Myc requires MondoA/Mlx for metabolic reprogramming and tumorigenesis. Cancer Cell 2015, 27, 271–285. [Google Scholar] [CrossRef]
- Yang, G.; Hurlin, P.J. MNT and Emerging Concepts of MNT-MYC Antagonism. Genes 2017, 8, 83. [Google Scholar] [CrossRef] [PubMed]
- Walker, W.; Zhou, Z.-Q.; Ota, S.; Wynshaw-Boris, A.; Hurlin, P.J. Mnt-Max to Myc-Max complex switching regulates cell cycle entry. J. Cell Biol. 2005, 169, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Darnell, J.E., Jr. Transcription factors as targets for cancer therapy. Nat. Rev. Cancer 2002, 2, 740–749. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef]
- Sipos, F.; Firneisz, G.; Műzes, G. Therapeutic aspects of c-MYC signaling in inflammatory and cancerous colonic diseases. World J. Gastroenterol. 2016, 22, 7938–7950. [Google Scholar] [CrossRef] [PubMed]
- Tögel, L.; Nightingale, R.; Chueh, A.C.; Jayachandran, A.; Tran, H.; Phesse, T.; Wu, R.; Sieber, O.M.; Arango, D.; Dhillon, A.S.; et al. Dual Targeting of Bromodomain and Extraterminal Domain Proteins, and WNT or MAPK Signaling, Inhibits c-MYC Expression and Proliferation of Colorectal Cancer Cells. Mol. Cancer Ther. 2016, 15, 1217–1226. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Uyar, B.; Ozsamur, N.G.; Celik, F.S.; Ozbayram, I.; Erbas-Cakmak, S. Downregulation of gene expression in hypoxic cancer cells by an activatable G-quadruplex stabiliser. Chem. Commun. 2023, 59, 2247–2250. [Google Scholar] [CrossRef]
- Valiuska, S.; Psaras, A.M.; Noé, V.; Brooks, T.A.; Ciudad, C.J. Targeting MYC Regulation with Polypurine Reverse Hoogsteen Oligonucleotides. Int. J. Mol. Sci. 2022, 24, 378. [Google Scholar] [CrossRef]
- Ou, T.-M.; Lin, J.; Lu, Y.-J.; Hou, J.-Q.; Tan, J.-H.; Chen, S.-H.; Li, Z.; Li, Y.-P.; Li, D.; Gu, L.-Q.; et al. Inhibition of cell proliferation by quindoline derivative (SYUIQ-05) through its preferential interaction with c-myc promoter G-quadruplex. J. Med. Chem. 2011, 54, 5671–5679. [Google Scholar] [CrossRef] [PubMed]
- Chu, E.C.; Chai, J.; Ahluwalia, A.; Tarnawski, A.S. Mesalazine downregulates c-Myc in human colon cancer cells. A key to its chemopreventive action? Aliment. Pharmacol. Ther. 2007, 25, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Schwab, M.; Reynders, V.; Loitsch, S.; Shastri, Y.M.; Steinhilber, D.; Schroder, O.; Stein, J. PPARγ is involved in mesalazine-mediated induction of apoptosis and inhibition of cell growth in colon cancer cells. Carcinogenesis 2008, 29, 1407–1414. [Google Scholar] [CrossRef]
- Shimada, T.; Kojima, K.; Yoshiura, K.; Hiraishi, H.; Terano, A. Characteristics of the peroxisome proliferator activated receptor gamma (PPARgamma) ligand induced apoptosis in colon cancer cells. Gut 2002, 50, 658–664. [Google Scholar] [CrossRef]
- Yamakawa-Karakida, N.; Sugita, K.; Inukai, T.; Goi, K.; Nakamura, M.; Uno, K.; Sato, H.; Kagami, K.; Barker, N.; Nakazawa, S. Ligand activation of peroxisome proliferator-activated receptor γ induces apoptosis of leukemia cells by down-regulating the c-myc gene expression via blockade of the Tcf-4 activity. Cell Death Differ. 2002, 9, 513–526. [Google Scholar] [CrossRef]
- Park, J.; Kim, S.Y.; Kim, H.-J.; Kim, K.-M.; Choi, E.Y.; Kang, M.-S. A reciprocal regulatory circuit between CD44 and FGFR2 via c-myc controls gastric cancer cell growth. Oncotarget 2016, 7, 28670–28683. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.K.; Khan, J.; Arumugam, A.; Muzaffar, S.; Guroji, P.; Gorbatyuk, M.S.; Elmets, C.A.; Slominski, A.T.; Mukhtar, M.S.; Athar, M. 5′-Cap-Dependent Translation as a Potent Therapeutic Target for Lethal Human Squamous Cell Carcinoma. J. Investig. Dermatol. 2021, 141, 742–753.e10. [Google Scholar] [CrossRef]
- Sodir, N.M.; Swigart, L.B.; Karnezis, A.N.; Hanahan, D.; Evan, G.I.; Soucek, L. Endogenous Myc maintains the tumor microenvironment. Genes Dev. 2011, 25, 907–916. [Google Scholar] [CrossRef]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgün, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef]
- Beaulieu, M.-E.; Martínez-Martín, S.; Kaur, J.; Castillo Cano, V.; Massó-Vallés, D.; Felip, L.F.; López-Estévez, S.; del Pozo, E.S.; Thabussot, H.; Soucek, L. Pharmacokinetic Analysis of Omomyc Shows Lasting Structural Integrity and Long Terminal Half-Life in Tumor Tissue. Cancers 2023, 15, 826. [Google Scholar] [CrossRef]
- Han, H.; Jain, A.D.; Truica, M.I.; Izquierdo-Ferrer, J.; Anker, J.F.; Lysy, B.; Sagar, V.; Luan, Y.; Chalmers, Z.R.; Unno, K.; et al. Small-Molecule MYC Inhibitors Suppress Tumor Growth and Enhance Immunotherapy. Cancer Cell 2019, 36, 483–497.e15. [Google Scholar] [CrossRef] [PubMed]
- Urbanski, L.; Brugiolo, M.; Park, S.; Angarola, B.L.; Leclair, N.K.; Yurieva, M.; Palmer, P.; Sahu, S.K.; Anczuków, O. MYC regulates a pan-cancer network of co-expressed oncogenic splicing factors. Cell Rep. 2022, 41, 111704. [Google Scholar] [CrossRef] [PubMed]
- Iwai, K.; Yaguchi, M.; Nishimura, K.; Yamamoto, Y.; Tamura, T.; Nakata, D.; Dairiki, R.; Kawakita, Y.; Mizojiri, R.; Ito, Y.; et al. Anti-tumor efficacy of a novel CLK inhibitor via targeting RNA splicing and MYC-dependent vulnerability. EMBO Mol. Med. 2018, 10, e8289. [Google Scholar] [CrossRef] [PubMed]
- Bates, D.O.; Morris, J.C.; Oltean, S.; Donaldson, L.F. Pharmacology of Modulators of Alternative Splicing. Pharmacol. Rev. 2017, 69, 63–79. [Google Scholar] [CrossRef]
- Yin, Y.-W.; Jin, H.-J.; Zhao, W.; Gao, B.; Fang, J.; Wei, J.; Zhang, D.D.; Zhang, J.; Fang, D. The Histone Acetyltransferase GCN5 Expression Is Elevated and Regulated by c-Myc and E2F1 Transcription Factors in Human Colon Cancer. Gene Expr. 2015, 16, 187–196. [Google Scholar] [CrossRef]
- Chen, L.; Wei, T.; Si, X.; Wang, Q.; Li, Y.; Leng, Y.; Deng, A.; Chen, J.; Wang, G.; Zhu, S.; et al. Lysine acetyltransferase GCN5 potentiates the growth of non-small cell lung cancer via promotion of E2F1, cyclin D1, and cyclin E1 expression. J. Biol. Chem. 2013, 288, 14510–14521. [Google Scholar] [CrossRef]
- Xiong, J.; Du, Q.; Liang, Z. Tumor-suppressive microRNA-22 inhibits the transcription of E-box-containing c-Myc target genes by silencing c-Myc binding protein. Oncogene 2010, 29, 4980–4988. [Google Scholar] [CrossRef]
- Serrano, L.; Martínez-Redondo, P.; Marazuela-Duque, A.; Vazquez, B.N.; Dooley, S.J.; Voigt, P.; Beck, D.B.; Kane-Goldsmith, N.; Tong, Q.; Rabanal, R.M.; et al. The tumor suppressor SirT2 regulates cell cycle progression and genome stability by modulating the mitotic deposition of H4K20 methylation. Genes Dev. 2013, 27, 639–653. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Jing, H.; Hu, J.; He, B.; Negron Abril, Y.L.; Stupinski, J.; Weiser, K.; Carbonaro, M.; Chiang, Y.-L.; Southard, T.; Giannakakou, P.; et al. A SIRT2-Selective Inhibitor Promotes c-Myc Oncoprotein Degradation and Exhibits Broad Anticancer Activity. Cancer Cell 2016, 29, 297–310. [Google Scholar] [CrossRef]
- Seo, S.-K.; Jin, H.-O.; Woo, S.-H.; Kim, Y.-S.; An, S.; Lee, J.-H.; Hong, S.-I.; Lee, K.-H.; Choe, T.-B.; Park, I.-C. Histone deacetylase inhibitors sensitize human non-small cell lung cancer cells to ionizing radiation through acetyl p53-mediated c-myc down-regulation. J. Thorac. Oncol. 2011, 6, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Zeleke, T.Z.; Pan, Q.; Chiuzan, C.; Onishi, M.; Li, Y.; Tan, H.; Alvarez, M.J.; Honan, E.; Yang, M.; Chia, P.L.; et al. Network-based assessment of HDAC6 activity predicts preclinical and clinical responses to the HDAC6 inhibitor ricolinostat in breast cancer. Nat. Cancer 2023, 4, 257–275. [Google Scholar] [CrossRef] [PubMed]
- Kretzner, L.; Scuto, A.; Dino, P.M.; Kowolik, C.M.; Wu, J.; Ventura, P.; Jove, R.; Forman, S.J.; Yen, Y.; Kirschbaum, M.H. Combining histone deacetylase inhibitor vorinostat with aurora kinase inhibitors enhances lymphoma cell killing with repression of c-Myc, hTERT, and microRNA levels. Cancer Res. 2011, 71, 3912–3920. [Google Scholar] [CrossRef]
- Peiró-Jordán, R.; Krishna-Subramanian, S.; Hanski, M.-L.; Lüscher-Firzlaff, J.; Zeitz, M.; Hanski, C. The chemopreventive agent ursodeoxycholic acid inhibits proliferation of colon carcinoma cells by suppressing c-Myc expression. Eur. J. Cancer Prev. 2012, 21, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, F.; Saha, P.; Singh, V.; Wahid, M.; Mandal, R.K.; Nath Mishra, B.; Fagoonee, S.; Haque, S. Diet as a modifiable factor in tumorigenesis: Focus on microbiome-derived bile acid metabolites and short-chain fatty acids. Food Chem. 2023, 410, 135320. [Google Scholar] [CrossRef]
- Höglund, A.; Nilsson, L.M.; Muralidharan, S.V.; Hasvold, L.A.; Merta, P.; Rudelius, M.; Nikolova, V.; Keller, U.; Nilsson, J.A. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin. Cancer Res. 2011, 17, 7067–7079. [Google Scholar] [CrossRef]
- Thng, D.K.H.; Toh, T.B.; Pigini, P.; Hooi, L.; Dan, Y.Y.; Chow, P.K.; Bonney, G.K.; Rashid, M.B.M.A.; Guccione, E.; Wee, D.K.B.; et al. Splice-switch oligonucleotide-based combinatorial platform prioritizes synthetic lethal targets CHK1 and BRD4 against MYC-driven hepatocellular carcinoma. Bioeng. Transl. Med. 2022, 8, e10363. [Google Scholar] [CrossRef]
- Goga, A.; Yang, D.; Tward, A.D.; Morgan, D.O.; Bishop, J.M. Inhibition of CDK1 as a potential therapy for tumors over-expressing MYC. Nat. Med. 2007, 13, 820–827. [Google Scholar] [CrossRef]
- Kwiatkowski, N.; Zhang, T.; Rahl, P.B.; Abraham, B.J.; Reddy, J.; Ficarro, S.B.; Dastur, A.; Amzallag, A.; Ramaswamy, S.; Tesar, B.; et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature 2014, 511, 616–620. [Google Scholar] [CrossRef]
- Shroff, E.H.; Eberlin, L.S.; Dang, V.M.; Gouw, A.M.; Gabay, M.; Adam, S.J.; Bellovin, D.I.; Tran, P.T.; Philbrick, W.M.; Garcia-Ocana, A.; et al. MYC oncogene overexpression drives renal cell carcinoma in a mouse model through glutamine metabolism. Proc. Natl. Acad. Sci. USA 2015, 112, 6539–6544. [Google Scholar] [CrossRef]
- Yeung, C.; Gibson, A.E.; Issaq, S.H.; Oshima, N.; Baumgart, J.T.; Edessa, L.D.; Rai, G.; Urban, D.J.; Johnson, M.S.; Benavides, G.A.; et al. Targeting Glycolysis through Inhibition of Lactate Dehydrogenase Impairs Tumor Growth in Preclinical Models of Ewing Sarcoma. Cancer Res. 2019, 79, 5060–5073. [Google Scholar] [CrossRef]
- He, T.-L.; Zhang, Y.-J.; Jiang, H.; Li, X.-H.; Zhu, H.; Zheng, K.-L. The c-Myc-LDHA axis positively regulates aerobic glycolysis and promotes tumor progression in pancreatic cancer. Med. Oncol. 2015, 32, 187. [Google Scholar] [CrossRef]
- Zhang, Q.; Cui, K.; Yang, X.; He, Q.; Yu, J.; Yang, L.; Yao, G.; Guo, W.; Luo, Z.; Liu, Y.; et al. c-Myc-IMPDH1/2 axis promotes tumourigenesis by regulating GTP metabolic reprogramming. Clin. Transl. Med. 2023, 13, e1164. [Google Scholar] [CrossRef]
- Huang, F.; Ni, M.; Chalishazar, M.D.; Huffman, K.E.; Kim, J.; Cai, L.; Shi, X.; Cai, F.; Zacharias, L.G.; Ireland, A.S.; et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018, 28, 369–382.e5. [Google Scholar] [CrossRef] [PubMed]
- Mokgautsi, N.; Kuo, Y.-C.; Chen, C.-H.; Huang, Y.-J.; Wu, A.T.H.; Huang, H.-S. Multiomics Study of a Novel Naturally Derived Small Molecule, NSC772864, as a Potential Inhibitor of Proto-Oncogenes Regulating Cell Cycle Progression in Colorectal Cancer. Cells 2023, 12, 340. [Google Scholar] [CrossRef]
- Gil-Edo, R.; Espejo, S.; Falomir, E.; Carda, M. Synthesis and Biological Evaluation of Potential Oncoimmunomodulator Agents. Int. J. Mol. Sci. 2023, 24, 2614. [Google Scholar] [CrossRef] [PubMed]
- Li, C.S.; Nguyen, T.V.; Chai, O.H.; Park, B.H.; Lee, J.-S.; Kim, S.M. 3,3′-Diindolylmethane Augments 5-Fluorouracil-InducedGrowth Suppression in Gastric Cancer Cells through Suppression of the Akt/GSK-3β and WNT/Beta-Catenin. J. Oncol. 2023, 2023, 8268955. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Type of Alteration | Affected Molecule/Region | Cancer Types | References | |
|---|---|---|---|---|
| Germ-line genetic variants (SNPs) | rs6983267 (CCAT2) | PrC, CRC | [79,80,81,82,83,84,85,86] | |
| rs13281615 | BC | [84,87] | ||
| rs55705857 (CCDC26) | Glioma | [88,89] | ||
| Super-enhancer amplification with/without MYC | de novo, 3′ from MYC | PC | [90,91] | |
| focal amplification, 450kb downstream | LUAD | [92] | ||
| focal amplification, 800kb downstream | OvC | [92] | ||
| duplication, 1.47 Mb 3′ of MYC | T-ALL | [93,94,95] | ||
| focal amplification, 1.7 Mb downstream | AML | [96] | ||
| ecDNA | multiple | [97,98,99,100,101,102,103] | ||
| Translocation | MYC/IGH1 | Burkitt lymphoma | [104] | |
| MYC/EVI1 | AML | [105] | ||
| Gene expression | Transcription | miR-24 | CRC, NPC | [106,107] |
| let-7 | multiple | [37,38] | ||
| miR-138 | CRC, HCC | [39] | ||
| miR-3648 | GC | [45] | ||
| miR-210 (MIR210HG) | GC | [108] | ||
| miR-3622b-3p | PrC | [41] | ||
| miR-23a/b | B-cell lymohoma, PrC | [109] | ||
| CCAT1 | multiple | [31,35,110] | ||
| PVT1 | multiple | [31,35,110,111,112,113] | ||
| ELFN1-AS1 | CRC | [114,115] | ||
| Androgen receptor | PrC | [49,116] | ||
| MUC5AC | PC | [117] | ||
| (Post) translation | MNX1-AS1 | NSCLC, CRC | [40] | |
| LINC00942 | GC | [118] | ||
| KB-1980E6.3 | BC | [119] | ||
| CUL3 | multiple | [120] | ||
| USP29 | multiple | [51] | ||
| GLCC1 | CRC | [56] | ||
| Targeted Event | Targeted Molecule/Structure | Drug (If Available) | Cancer Type | References | |
|---|---|---|---|---|---|
| Targeting upstream features | Chromatin interactions | BRD4 | JQ1, OTX015 | multiple | [103,147] |
| G-quadruplex | Quindoline derivatives (SYUIQ-05) | multiple | [148,149,150,151] | ||
| MYC indirect inhibition | PPARγ | 5-ASA (mesalazine) | CRC | [152,153,154,155] | |
| FGFR2 | FGFR2 kinase inhibitors | GC | [156] | ||
| eIF4E | - | SCC | [157] | ||
| Targeting downstream (effector) features | MYC/MAX heterodimerization | Omomyc, MYCi361, MYCi975 | multiple | [158,159,160,161] | |
| CLK1/4 | CLK inhibitors (T-025) | multiple | [162,163,164] | ||
| GCN5 | HAT inhibitor (CPTH2) | CRC, NSCLC | [165,166] | ||
| Regulatory RNAs | miR-22 | - | multiple | [167] | |
| miR-24 | - | CRC, NPC | [36] | ||
| let-7 | - | multiple | [37] | ||
| PVT1 | - | multiple | [31,110] | ||
| PCGEM1 | - | PrC | [76] | ||
| Protein degradation | SIRT2 | TM | multiple | [168,169,170] | |
| HDAC | HDAC inhibitors | PC, NSCLC, BC, lymphoma | [53,171,172,173] | ||
| PP2A | SMAPs (FTY-720) | multiple | [53,134] | ||
| SET7 | OP449 | multiple | [53,133] | ||
| dephosphorylases | UDCA | CRC | [174,175] | ||
| USP29 | - | multiple | [51] | ||
| Synthetic lethal interactions | CHK1 | Chekin | HCC | [103,176,177] | |
| CDK1 | CDK1 inhibitors (purvalanol, roscovitine) | multiple | [178] | ||
| CDK7 | CDK7 inhibitors (THZ1) | T-ALL | [179] | ||
| GLS | GLS inhibitors (BPTES) | multiple | [180] | ||
| LDHA | LDHA inhibitors (NCI-737, NCI-006) | multiple | [181,182] | ||
| IMPDH2 | IMPDH inhibitors (mizoribine) | SCLC | [183,184] | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vízkeleti, L.; Spisák, S. Rewired Metabolism Caused by the Oncogenic Deregulation of MYC as an Attractive Therapeutic Target in Cancers. Cells 2023, 12, 1745. https://doi.org/10.3390/cells12131745
Vízkeleti L, Spisák S. Rewired Metabolism Caused by the Oncogenic Deregulation of MYC as an Attractive Therapeutic Target in Cancers. Cells. 2023; 12(13):1745. https://doi.org/10.3390/cells12131745
Chicago/Turabian StyleVízkeleti, Laura, and Sándor Spisák. 2023. "Rewired Metabolism Caused by the Oncogenic Deregulation of MYC as an Attractive Therapeutic Target in Cancers" Cells 12, no. 13: 1745. https://doi.org/10.3390/cells12131745
APA StyleVízkeleti, L., & Spisák, S. (2023). Rewired Metabolism Caused by the Oncogenic Deregulation of MYC as an Attractive Therapeutic Target in Cancers. Cells, 12(13), 1745. https://doi.org/10.3390/cells12131745