

A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. iPSC Lines
2.2. iPSC Culture
2.3. SOSR-COS Differentiation
2.4. SOSR-CO Drug Treatment
2.5. SOSR-CO Fixing & Staining
2.6. Manual SOSR-CO Imaging and Analysis
2.7. Automated Imaging and Analysis
2.8. Random Forest Predictive Modeling
2.9. RNA Sequencing Experiment
2.10. Quantifying Apical Surface Areas
3. Results
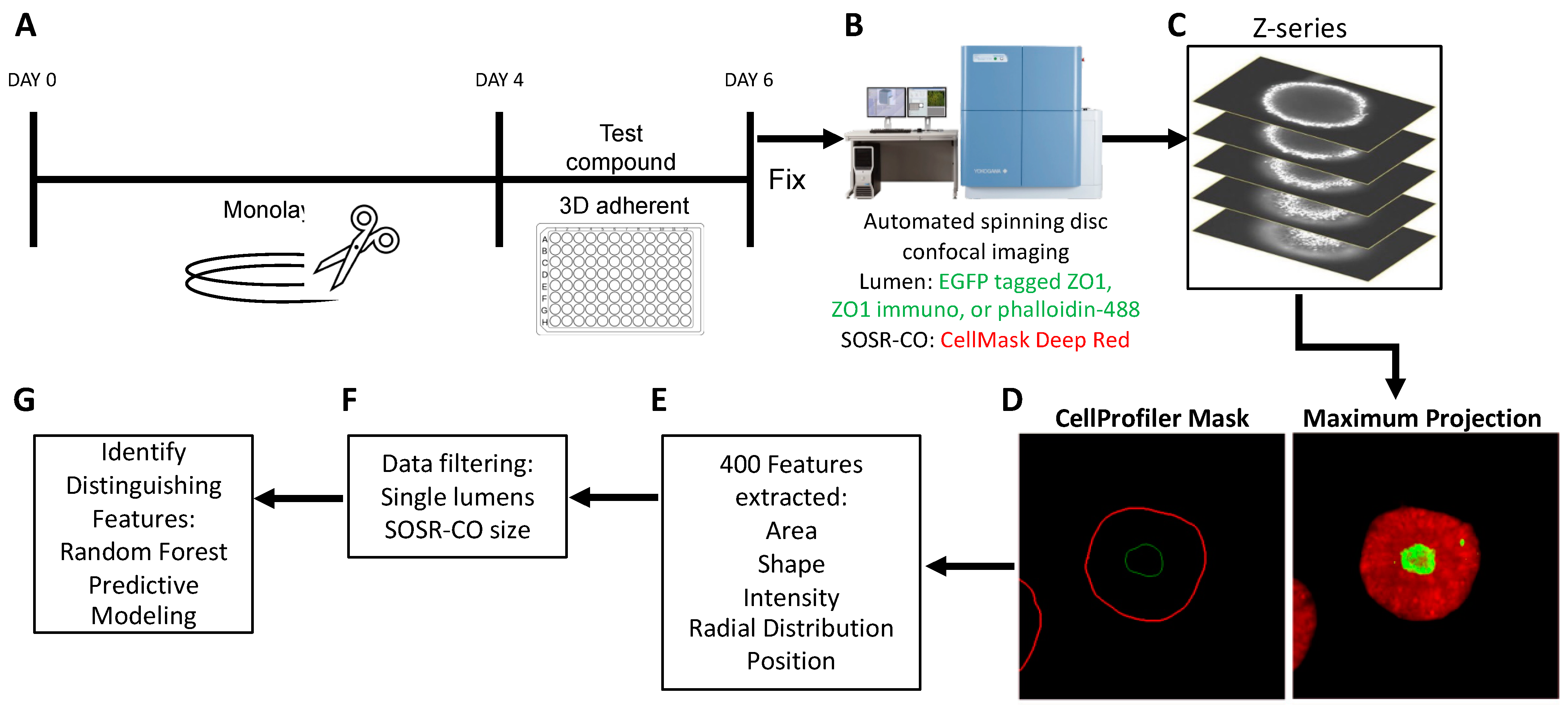
3.1. High Content Imaging Pipeline for SOSR-CO NTD Screening
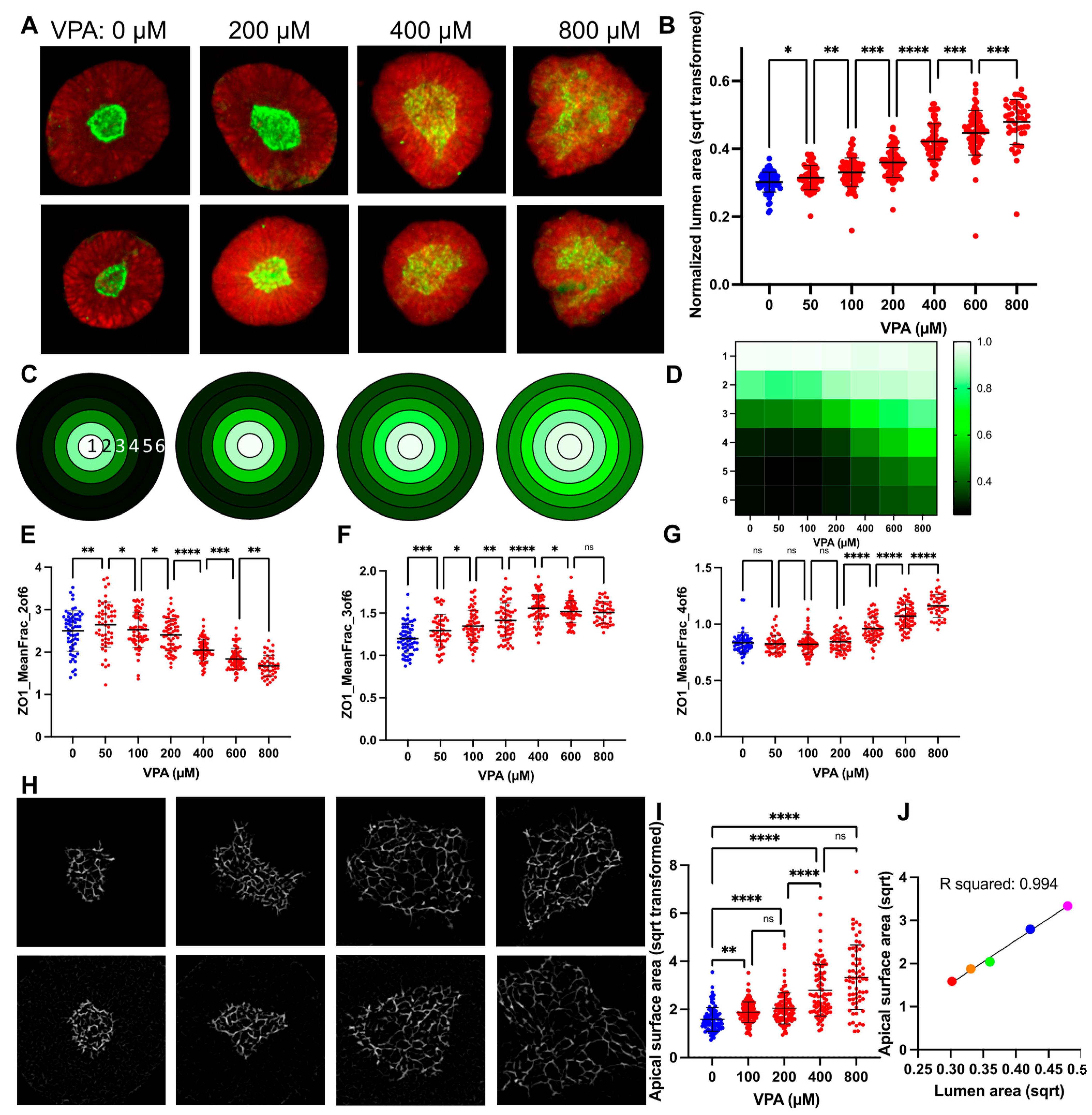
3.2. VPA Dose Response Shows SOSR-CO Lumen Size as Most Defining Feature
3.3. Lumen Enlargement Is Directly Related to Reduced Apical Constriction
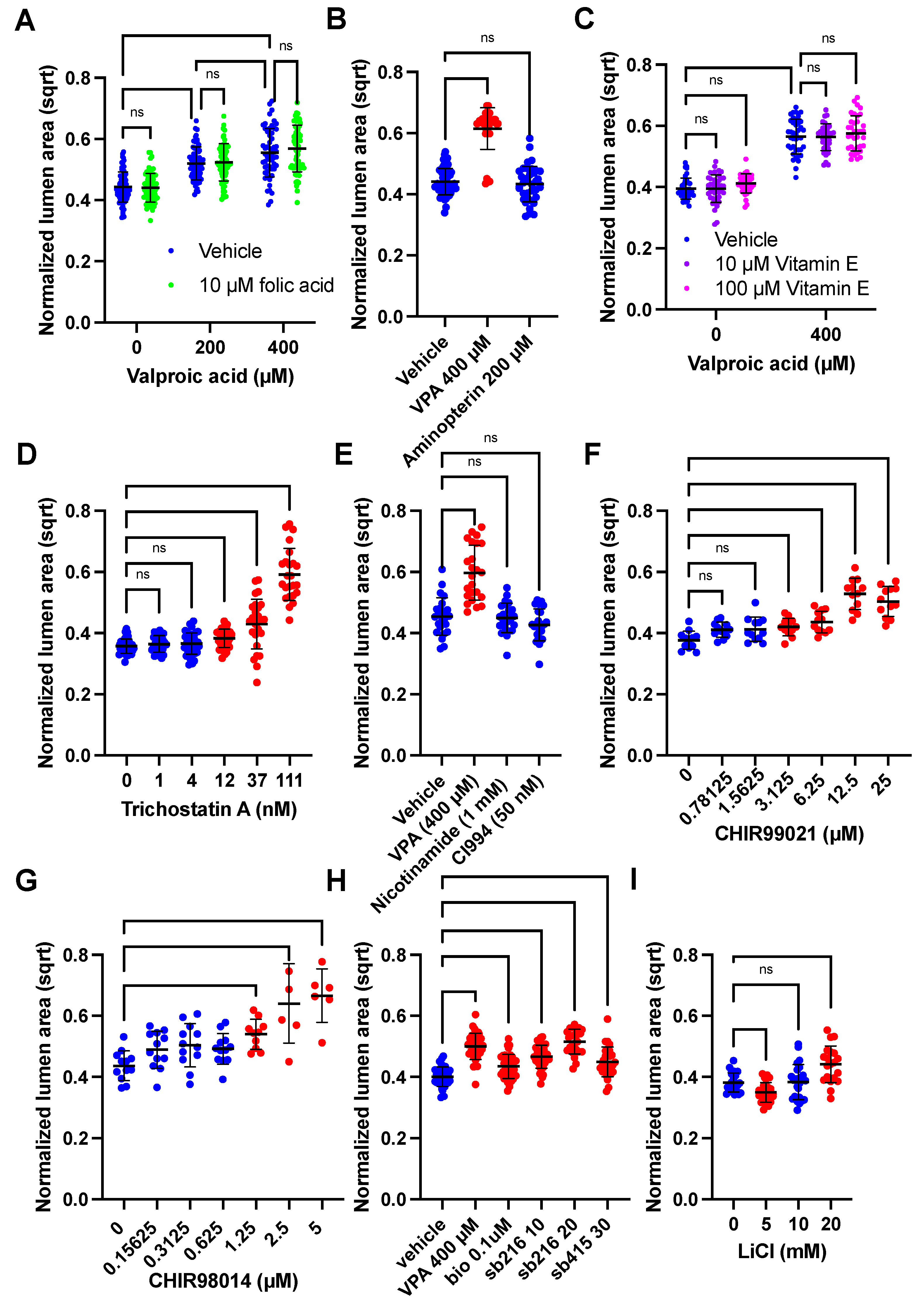
3.4. GSK3β or HDAC Inhibition Cause VPA-like Phenotypes, but No Evidence for Folic Acid or Oxidative Stress Involvement
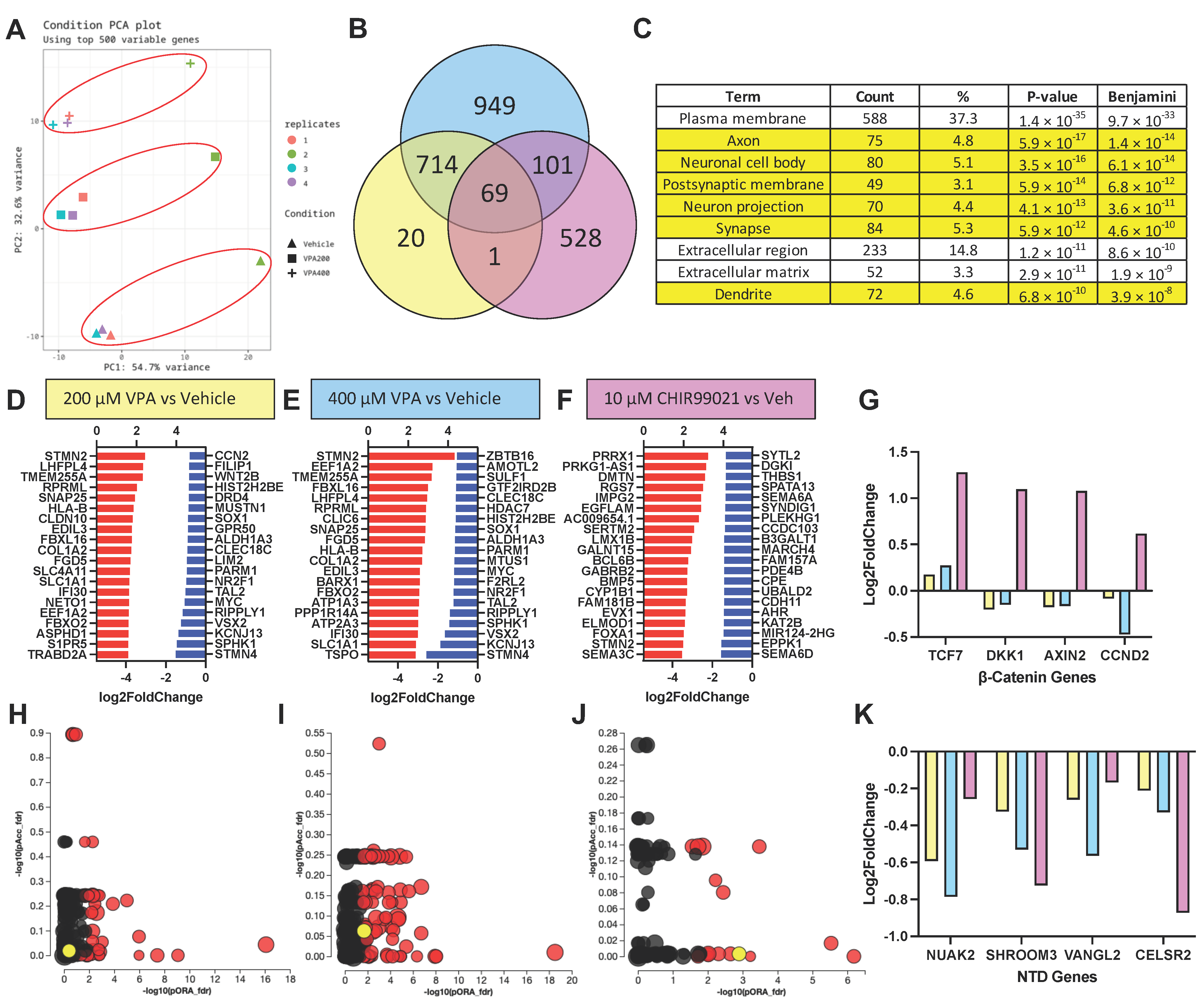
3.5. RNA Sequencing Results Suggest VPA Inhibits HDAC but Not GSK3β at Concentrations Causing NTD Phenotypes
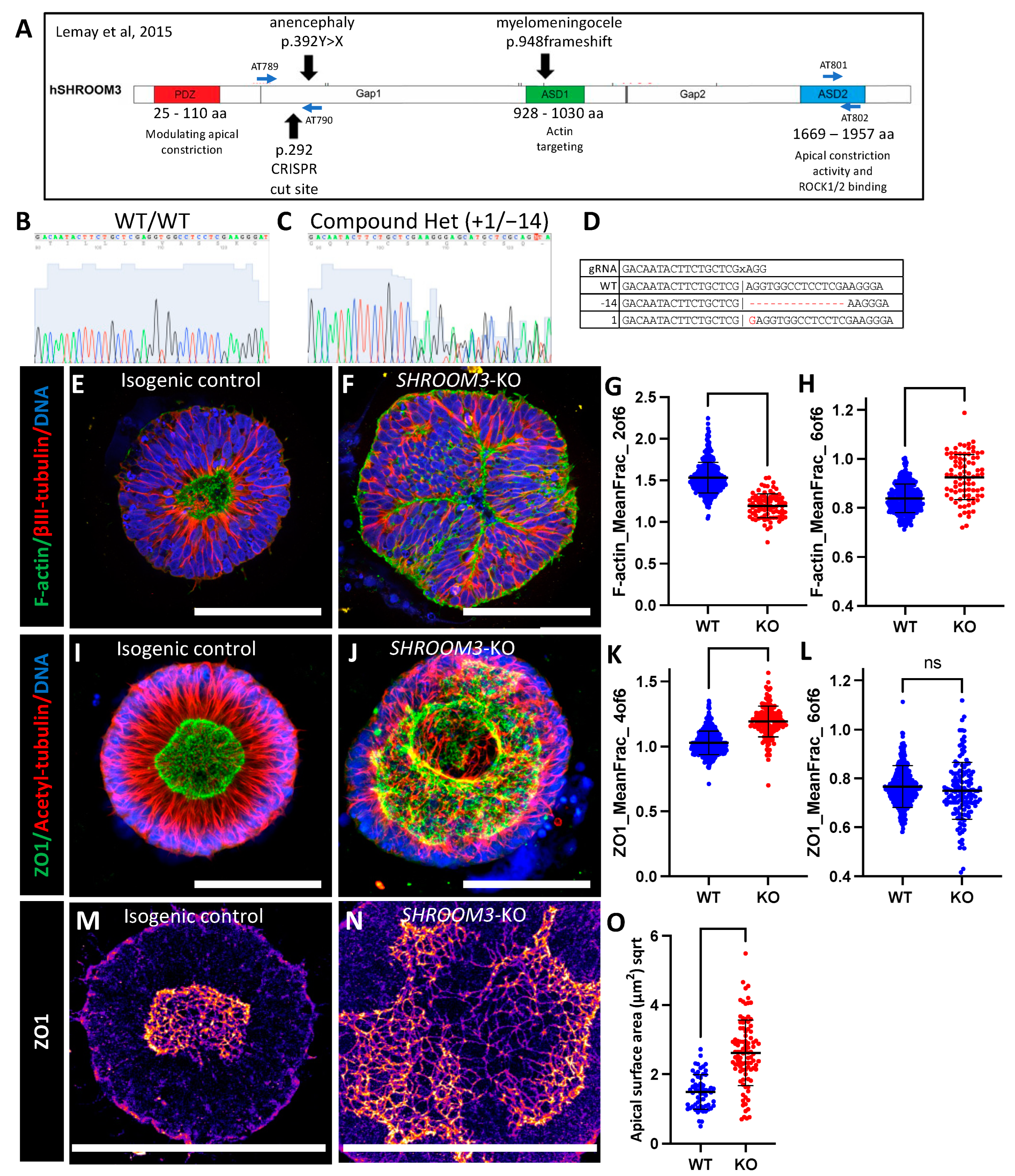
3.6. SHROOM3 Knockout Results in Enlarged, Dysmorphic Lumens and Reduced Apical Constriction
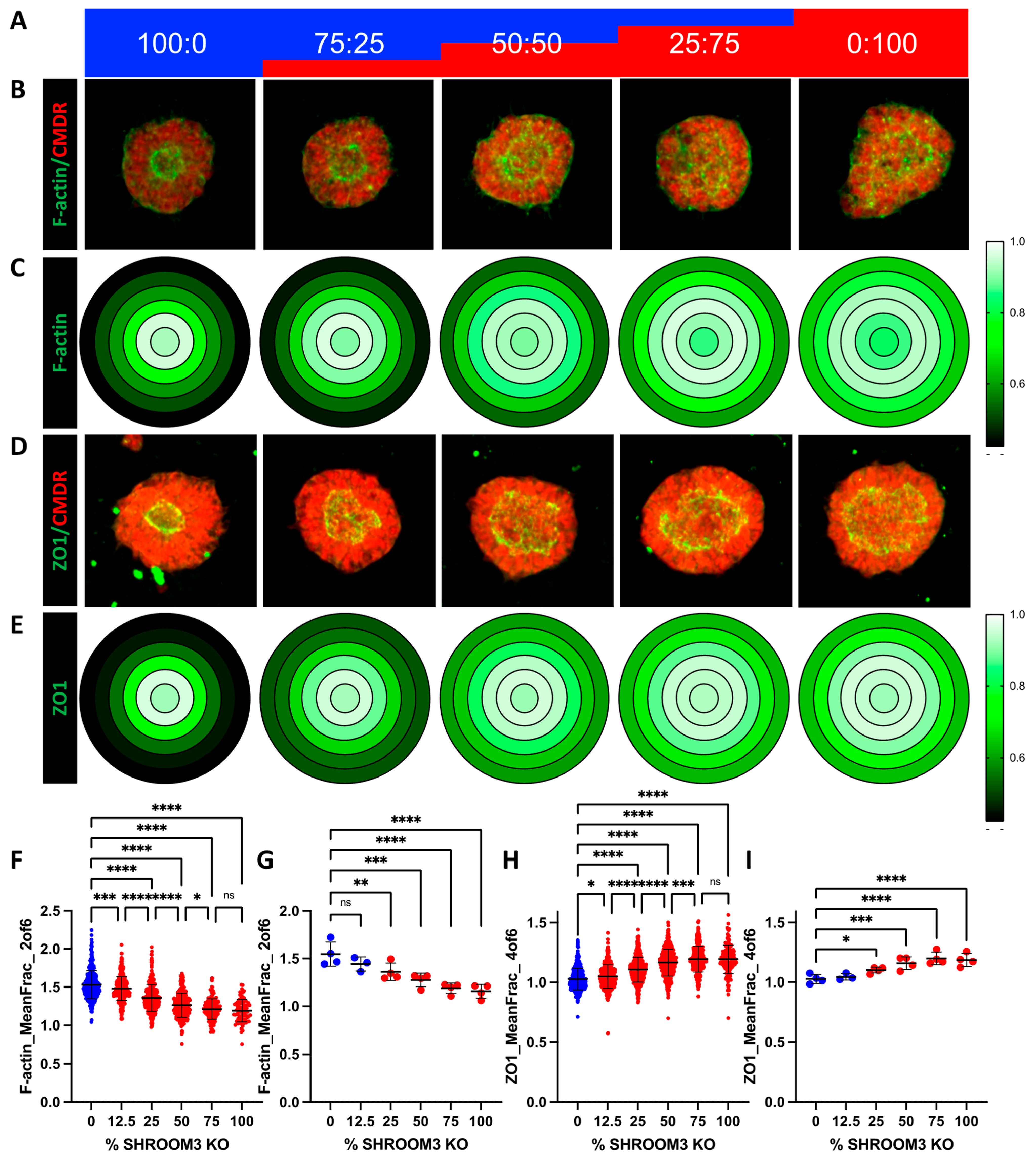
3.7. Mosaic SHROOM3 Knockout Shows Gene-Dose Responsive Lumen Size
4. Discussion
5. Conclusions
6. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mai, C.T.; Isenburg, J.; Langlois, P.H.; Alverson, C.; Gilboa, S.M.; Rickard, R.; Canfield, M.A.; Anjohrin, S.B.; Lupo, P.J.; Jackson, D.R. Population-based birth defects data in the United States, 2008 to 2012: Presentation of state-specific data and descriptive brief on variability of prevalence. Birth Defects Res. Part A Clin. Mol. Teratol. 2015, 103, 972–993. [Google Scholar] [CrossRef] [PubMed]
- Vargesson, N. Thalidomide-induced teratogenesis: History and mechanisms. Birth Defects Res. Part C Embryo Today Rev. 2015, 105, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Olson, H.; Betton, G.; Robinson, D.; Thomas, K.; Monro, A.; Kolaja, G.; Lilly, P.; Sanders, J.; Sipes, G.; Bracken, W. Concordance of the toxicity of pharmaceuticals in humans and in animals. Regul. Toxicol. Pharmacol. 2000, 32, 56–67. [Google Scholar] [CrossRef] [PubMed]
- Augustine-Rauch, K.; Zhang, C.X.; Panzica-Kelly, J.M. A developmental toxicology assay platform for screening teratogenic liability of pharmaceutical compounds. Birth Defects Res. Part B Dev. Reprod. Toxicol. 2016, 107, 4–20. [Google Scholar] [CrossRef] [PubMed]
- Van Norman, G.A. Limitations of animal studies for predicting toxicity in clinical trials: Is it time to rethink our current approach? JACC Basic Transl. Sci. 2019, 4, 845–854. [Google Scholar] [CrossRef]
- Warkany, J.; BEAUDRY, P.H.; Hornstein, S. Attempted Abortion with Aminopterin (4-Amino-Pteroylglutamic Acid): Malformations of the Child. AMA J. Dis. Child. 1959, 97, 274–281. [Google Scholar] [CrossRef]
- Nelson, M.M.; Asling, C.W.; Evans, H.M. Production of Multiple Congenital Abnormalities in Young by Maternal Pteroylglutamic Acid Deficiency during Gestation: Thirteen Figures. J. Nutr. 1952, 48, 61–79. [Google Scholar] [CrossRef]
- Wyszynski, D.; Nambisan, M.; Surve, T.; Alsdorf, R.; Smith, C.; Holmes, L. Increased rate of major malformations in offspring exposed to valproate during pregnancy. Neurology 2005, 64, 961–965. [Google Scholar] [CrossRef]
- Li, S.; Luo, D.; Yue, H.; Lyu, J.; Yang, Y.; Gao, T.; Liu, Y.; Qin, J.; Wang, X.; Guan, Z. Neural tube defects: Role of lithium carbonate exposure in embryonic neural development in a murine model. Pediatr. Res. 2021, 90, 82–92. [Google Scholar] [CrossRef]
- Zash, R.; Holmes, L.; Diseko, M.; Jacobson, D.L.; Brummel, S.; Mayondi, G.; Isaacson, A.; Davey, S.; Mabuta, J.; Mmalane, M. Neural-tube defects and antiretroviral treatment regimens in Botswana. N. Engl. J. Med. 2019, 381, 827–840. [Google Scholar] [CrossRef]
- Gentile, S. Lithium in pregnancy: The need to treat, the duty to ensure safety. Expert Opin. Drug Saf. 2012, 11, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Nau, H.; Hauck, R.S.; Ehlers, K. Valproic acid-induced neural tube defects in mouse and human: Aspects of chirality, alternative drug development, pharmacokinetics and possible mechanisms. Pharmacol. Toxicol. 1991, 69, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, E.H.; Green, R. Valproate and folate: Congenital and developmental risks. Epilepsy Behav. 2020, 108, 107068. [Google Scholar] [CrossRef] [PubMed]
- Göttlicher, M.; Minucci, S.; Zhu, P.; Krämer, O.H.; Schimpf, A.; Giavara, S.; Sleeman, J.P.; Coco, F.L.; Nervi, C.; Pelicci, P.G. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 2001, 20, 6969–6978. [Google Scholar] [CrossRef]
- Chanda, S.; Ang, C.E.; Lee, Q.Y.; Ghebrial, M.; Haag, D.; Shibuya, Y.; Wernig, M.; Südhof, T.C. Direct reprogramming of human neurons identifies MARCKSL1 as a pathogenic mediator of valproic acid-induced teratogenicity. Cell Stem Cell 2019, 25, 103–119.e106. [Google Scholar] [CrossRef]
- Tung, E.W.; Winn, L.M. Valproic acid increases formation of reactive oxygen species and induces apoptosis in postimplantation embryos: A role for oxidative stress in valproic acid-induced neural tube defects. Mol. Pharmacol. 2011, 80, 979–987. [Google Scholar] [CrossRef]
- Deak, K.L.; Siegel, D.G.; George, T.M.; Gregory, S.; Ashley-Koch, A.; Speer, M.C.; Group, N.C. Further evidence for a maternal genetic effect and a sex-influenced effect contributing to risk for human neural tube defects. Birth Defects Res. Part A Clin. Mol. Teratol. 2008, 82, 662–669. [Google Scholar] [CrossRef]
- Windham, G.C.; Sever, L.E. Neural tube defects among twin births. Am. J. Hum. Genet. 1982, 34, 988. [Google Scholar]
- Harris, M.J.; Juriloff, D.M. An update to the list of mouse mutants with neural tube closure defects and advances toward a complete genetic perspective of neural tube closure. Birth Defects Res. Part A Clin. Mol. Teratol. 2010, 88, 653–669. [Google Scholar] [CrossRef]
- Hildebrand, J.D.; Soriano, P. Shroom, a PDZ domain–containing actin-binding protein, is required for neural tube morphogenesis in mice. Cell 1999, 99, 485–497. [Google Scholar] [CrossRef]
- Curtin, J.A.; Quint, E.; Tsipouri, V.; Arkell, R.M.; Cattanach, B.; Copp, A.J.; Henderson, D.J.; Spurr, N.; Stanier, P.; Fisher, E.M. Mutation of Celsr1 disrupts planar polarity of inner ear hair cells and causes severe neural tube defects in the mouse. Curr. Biol. 2003, 13, 1129–1133. [Google Scholar] [CrossRef]
- Lemay, P.; Guyot, M.-C.; Tremblay, É.; Dionne-Laporte, A.; Spiegelman, D.; Henrion, É.; Diallo, O.; De Marco, P.; Merello, E.; Massicotte, C. Loss-of-function de novo mutations play an important role in severe human neural tube defects. J. Med. Genet. 2015, 52, 493–497. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.; Escuin, S.; Doudney, K.; Vekemans, M.; Stevenson, R.E.; Greene, N.D.; Copp, A.J.; Stanier, P. Mutations in the planar cell polarity genes CELSR1 and SCRIB are associated with the severe neural tube defect craniorachischisis. Hum. Mutat. 2012, 33, 440–447. [Google Scholar] [CrossRef]
- Lei, Y.; Zhu, H.; Yang, W.; Ross, M.E.; Shaw, G.M.; Finnell, R.H. Identification of novel CELSR1 mutations in spina bifida. PLoS ONE 2014, 9, e92207. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, J.D. Shroom regulates epithelial cell shape via the apical positioning of an actomyosin network. J. Cell Sci. 2005, 118, 5191–5203. [Google Scholar] [CrossRef] [PubMed]
- Galea, G.L.; Maniou, E.; Edwards, T.J.; Marshall, A.R.; Ampartzidis, I.; Greene, N.D.; Copp, A.J. Cell non-autonomy amplifies disruption of neurulation by mosaic Vangl2 deletion in mice. Nat. Commun. 2021, 12, 1159. [Google Scholar] [CrossRef]
- del Álamo, J.C.; Lemons, D.; Serrano, R.; Savchenko, A.; Cerignoli, F.; Bodmer, R.; Mercola, M. High throughput physiological screening of iPSC-derived cardiomyocytes for drug development. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2016, 1863, 1717–1727. [Google Scholar] [CrossRef]
- Worley, K.E.; Rico-Varela, J.; Ho, D.; Wan, L.Q. Teratogen screening with human pluripotent stem cells. Integr. Biol. 2018, 10, 491–501. [Google Scholar] [CrossRef]
- Shinde, V.; Hoelting, L.; Srinivasan, S.P.; Meisig, J.; Meganathan, K.; Jagtap, S.; Grinberg, M.; Liebing, J.; Bluethgen, N.; Rahnenführer, J. Definition of transcriptome-based indices for quantitative characterization of chemically disturbed stem cell development: Introduction of the STOP-Tox ukn and STOP-Tox ukk tests. Arch. Toxicol. 2017, 91, 839–864. [Google Scholar] [CrossRef]
- Dreser, N.; Madjar, K.; Holzer, A.-K.; Kapitza, M.; Scholz, C.; Kranaster, P.; Gutbier, S.; Klima, S.; Kolb, D.; Dietz, C. Development of a neural rosette formation assay (RoFA) to identify neurodevelopmental toxicants and to characterize their transcriptome disturbances. Arch. Toxicol. 2020, 94, 151–171. [Google Scholar] [CrossRef]
- Cao, X.; Tian, T.; Steele, J.W.; Cabrera, R.M.; Aguiar-Pulido, V.; Wadhwa, S.; Bhavani, N.; Bi, P.; Gargurevich, N.H.; Hoffman, E.N. Loss of RAD9B impairs early neural development and contributes to the risk for human spina bifida. Hum. Mutat. 2020, 41, 786–799. [Google Scholar] [CrossRef] [PubMed]
- Sahakyan, V.; Duelen, R.; Tam, W.L.; Roberts, S.J.; Grosemans, H.; Berckmans, P.; Ceccarelli, G.; Pelizzo, G.; Broccoli, V.; Deprest, J. Folic acid exposure rescues Spina bifida Aperta phenotypes in human induced pluripotent stem cell model. Sci. Rep. 2018, 8, 2942. [Google Scholar] [CrossRef]
- Bonnard, C.; Navaratnam, N.; Ghosh, K.; Chan, P.W.; Tan, T.T.; Pomp, O.; Ng, A.Y.J.; Tohari, S.; Changede, R.; Carling, D. A loss-of-function NUAK2 mutation in humans causes anencephaly due to impaired Hippo-YAP signaling. J. Exp. Med. 2020, 217, e20191561. [Google Scholar] [CrossRef]
- Knight, G.T.; Lundin, B.F.; Iyer, N.; Ashton, L.M.; Sethares, W.A.; Willett, R.M.; Ashton, R.S. Engineering induction of singular neural rosette emergence within hPSC-derived tissues. Elife 2018, 7, e37549. [Google Scholar] [CrossRef] [PubMed]
- Karzbrun, E.; Khankhel, A.H.; Megale, H.C.; Glasauer, S.M.; Wyle, Y.; Britton, G.; Warmflash, A.; Kosik, K.S.; Siggia, E.D.; Shraiman, B.I. Human neural tube morphogenesis in vitro by geometric constraints. Nature 2021, 599, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Zhang, X.; Huo, H.-Q.; Zhu, Y.-Q.; Feng, H.-Y.; Jiao, J.; Tan, J.-X.; Wang, Y.; Hu, P. Folic acid rescues valproic acid-induced morphogenesis inhibition in neural rosettes derived from human pluripotent stem cells. Front. Cell. Neurosci. 2022, 16, 244. [Google Scholar]
- Tidball, A.M.; Niu, W.; Ma, Q.; Takla, T.N.; Walker, J.C.; Margolis, J.L.; Mojica-Perez, S.P.; Sudyk, R.; Moore, S.J.; Chopra, R.; et al. Self-organizing Single-Rosette Brain Organoids from Human Pluripotent Stem Cells. bioRxiv 2022. [Google Scholar] [CrossRef]
- Wang, Y.; Chiola, S.; Yang, G.; Russell, C.; Armstrong, C.J.; Wu, Y.; Spampanato, J.; Tarboton, P.; Ullah, H.; Edgar, N.U. Modeling human telencephalic development and autism-associated SHANK3 deficiency using organoids generated from single neural rosettes. Nat. Commun. 2022, 13, 5688. [Google Scholar] [CrossRef]
- Tidball, A.M.; Dang, L.T.; Glenn, T.W.; Kilbane, E.G.; Klarr, D.J.; Margolis, J.L.; Uhler, M.D.; Parent, J.M. Rapid generation of human genetic loss-of-function iPSC lines by simultaneous reprogramming and gene editing. Stem Cell Rep. 2017, 9, 725–731. [Google Scholar] [CrossRef]
- Okita, K.; Matsumura, Y.; Sato, Y.; Okada, A.; Morizane, A.; Okamoto, S.; Hong, H.; Nakagawa, M.; Tanabe, K.; Tezuka, K.-i. A more efficient method to generate integration-free human iPS cells. Nat. Methods 2011, 8, 409. [Google Scholar] [CrossRef]
- Nie, Y.; Walsh, P.; Clarke, D.L.; Rowley, J.A.; Fellner, T. Scalable passaging of adherent human pluripotent stem cells. PLoS ONE 2014, 9, e88012. [Google Scholar] [CrossRef] [PubMed]
- Yu, I.T.; Park, J.-Y.; Kim, S.H.; Lee, J.-s.; Kim, Y.-S.; Son, H. Valproic acid promotes neuronal differentiation by induction of proneural factors in association with H4 acetylation. Neuropharmacology 2009, 56, 473–480. [Google Scholar] [CrossRef]
- Plageman, T.F., Jr.; Chung, M.-I.; Lou, M.; Smith, A.N.; Hildebrand, J.D.; Wallingford, J.B.; Lang, R.A. Pax6-dependent Shroom3 expression regulates apical constriction during lens placode invagination. Development 2010, 137, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.B.; Short, N.E.; Maniou, E.; Alexandre, P.; Greene, N.D.; Copp, A.J.; Galea, G.L. Rho kinase-dependent apical constriction counteracts M-phase apical expansion to enable mouse neural tube closure. J. Cell Sci. 2019, 132, jcs230300. [Google Scholar] [CrossRef]
- Haigo, S.L.; Hildebrand, J.D.; Harland, R.M.; Wallingford, J.B. Shroom induces apical constriction and is required for hingepoint formation during neural tube closure. Curr. Biol. 2003, 13, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Erol, S.A.; Tanacan, A.; Firat Oguz, E.; Anuk, A.T.; Goncu Ayhan, S.; Neselioglu, S.; Sahin, D. A comparison of the maternal levels of serum proprotein convertase subtilisin/kexin type 9 in pregnant women with the complication of fetal open neural tube defects. Congenit. Anom. 2021, 61, 169–176. [Google Scholar] [CrossRef]
- Holmes, L.B.; Harvey, E.A.; Coull, B.A.; Huntington, K.B.; Khoshbin, S.; Hayes, A.M.; Ryan, L.M. The teratogenicity of anticonvulsant drugs. N. Engl. J. Med. 2001, 344, 1132–1138. [Google Scholar] [CrossRef]
- Lei, Y.-P.; Zhang, T.; Li, H.; Wu, B.-L.; Jin, L.; Wang, H.-Y. VANGL2 mutations in human cranial neural-tube defects. N. Engl. J. Med. 2010, 362, 2232–2235. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Takla, T.N.; Luo, J.; Sudyk, R.; Huang, J.; Walker, J.C.; Vora, N.L.; Sexton, J.Z.; Parent, J.M.; Tidball, A.M. A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects. Cells 2023, 12, 1697. https://doi.org/10.3390/cells12131697
Takla TN, Luo J, Sudyk R, Huang J, Walker JC, Vora NL, Sexton JZ, Parent JM, Tidball AM. A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects. Cells. 2023; 12(13):1697. https://doi.org/10.3390/cells12131697
Chicago/Turabian StyleTakla, Taylor N., Jinghui Luo, Roksolana Sudyk, Joy Huang, John Clayton Walker, Neeta L. Vora, Jonathan Z. Sexton, Jack M. Parent, and Andrew M. Tidball. 2023. "A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects" Cells 12, no. 13: 1697. https://doi.org/10.3390/cells12131697
APA StyleTakla, T. N., Luo, J., Sudyk, R., Huang, J., Walker, J. C., Vora, N. L., Sexton, J. Z., Parent, J. M., & Tidball, A. M. (2023). A Shared Pathogenic Mechanism for Valproic Acid and SHROOM3 Knockout in a Brain Organoid Model of Neural Tube Defects. Cells, 12(13), 1697. https://doi.org/10.3390/cells12131697