

Identification of Small Molecules Affecting the Secretion of Therapeutic Antibodies with the Retention Using Selective Hook (RUSH) System

 ,
,  , ,
, ,  and
and 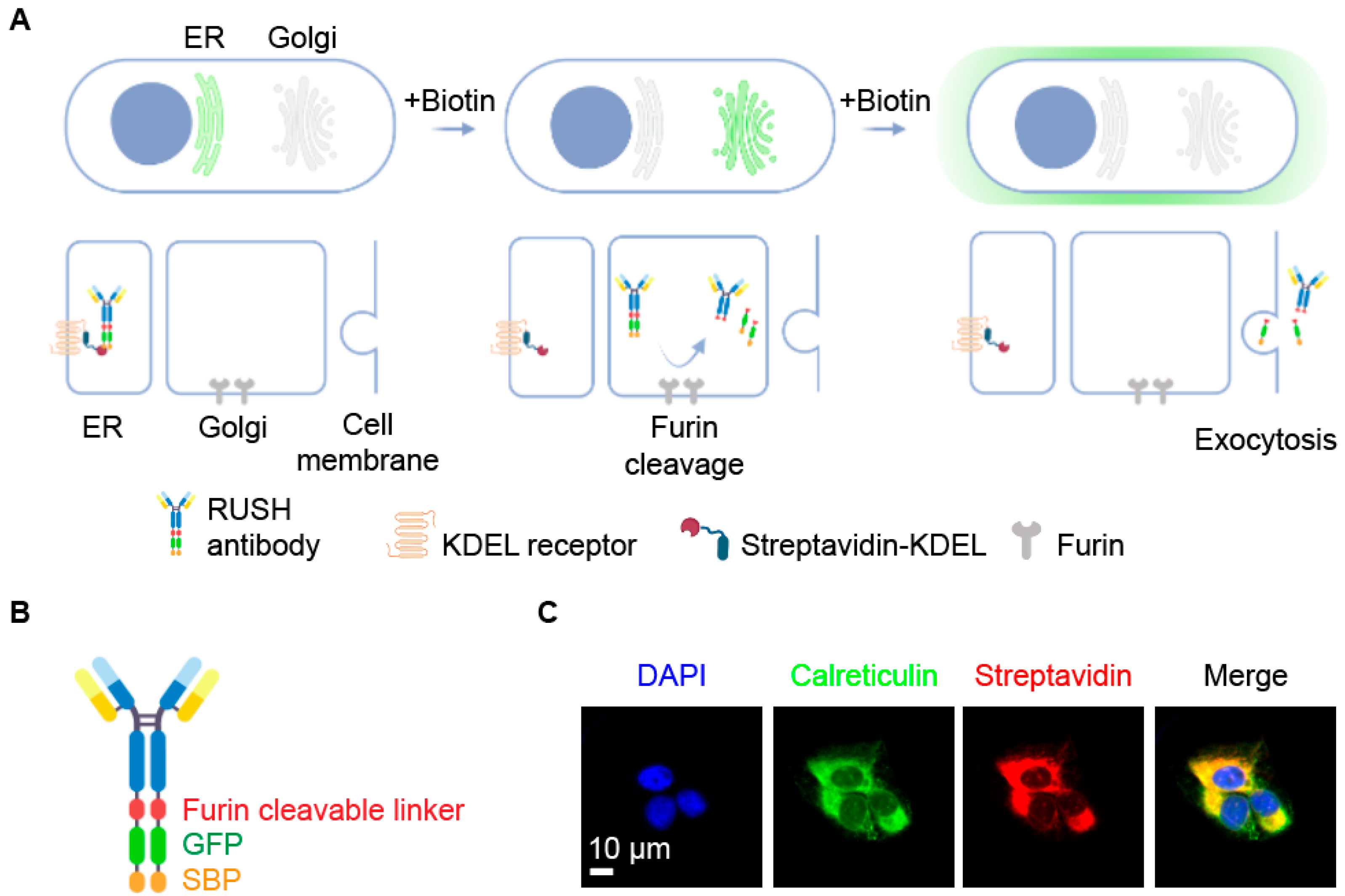{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture, Chemicals, and Antibodies
2.2. Constructs Used for the RUSH Antibody Secretion System
2.3. Establishment of the RUSH-Antibody Cell Line
2.4. Immunofluorescence
2.5. Antibody Expression and Secretion Characterization by Western Blot
2.6. Flow Cytometric Validation of Antibody Reactivity
2.7. High-Throughput Compound Screening for Modulators of Protein Secretion
2.8. ELISA
2.9. Statistics
3. Results
3.1. Design of an Antibody RUSH System
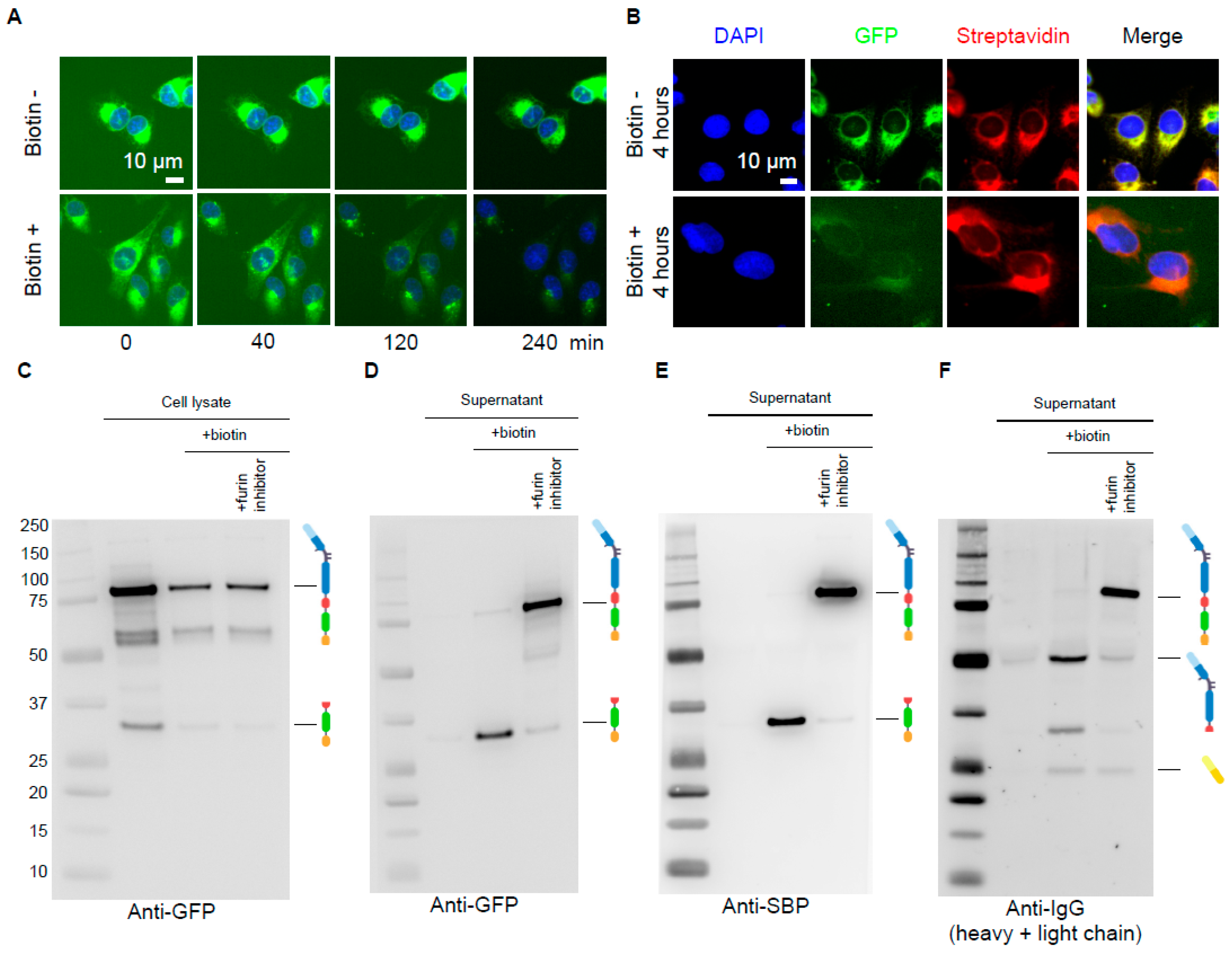
3.2. Biochemical Validation of the Antibody RUSH System
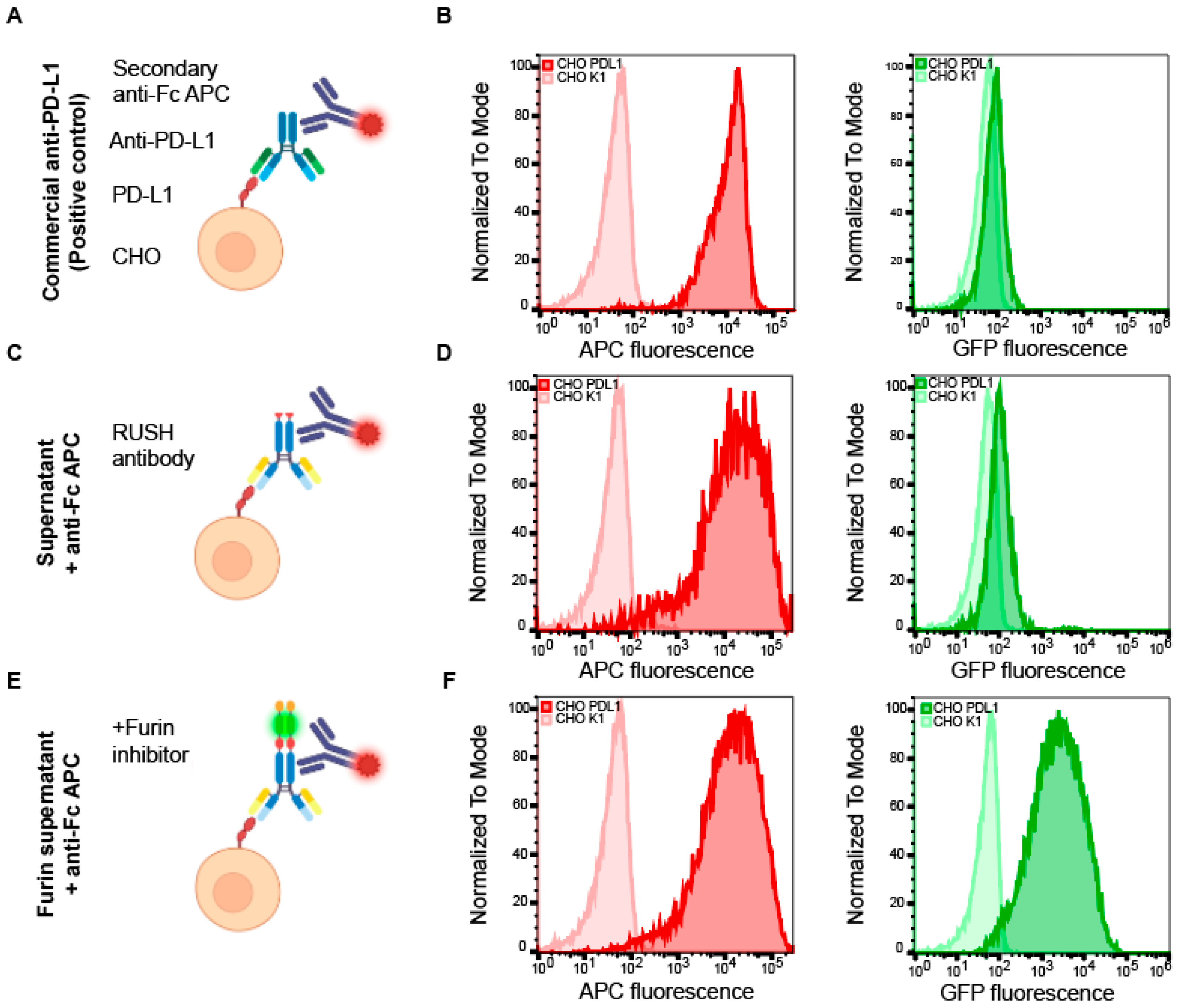
3.3. Immunological Validation of the Antibody RUSH System
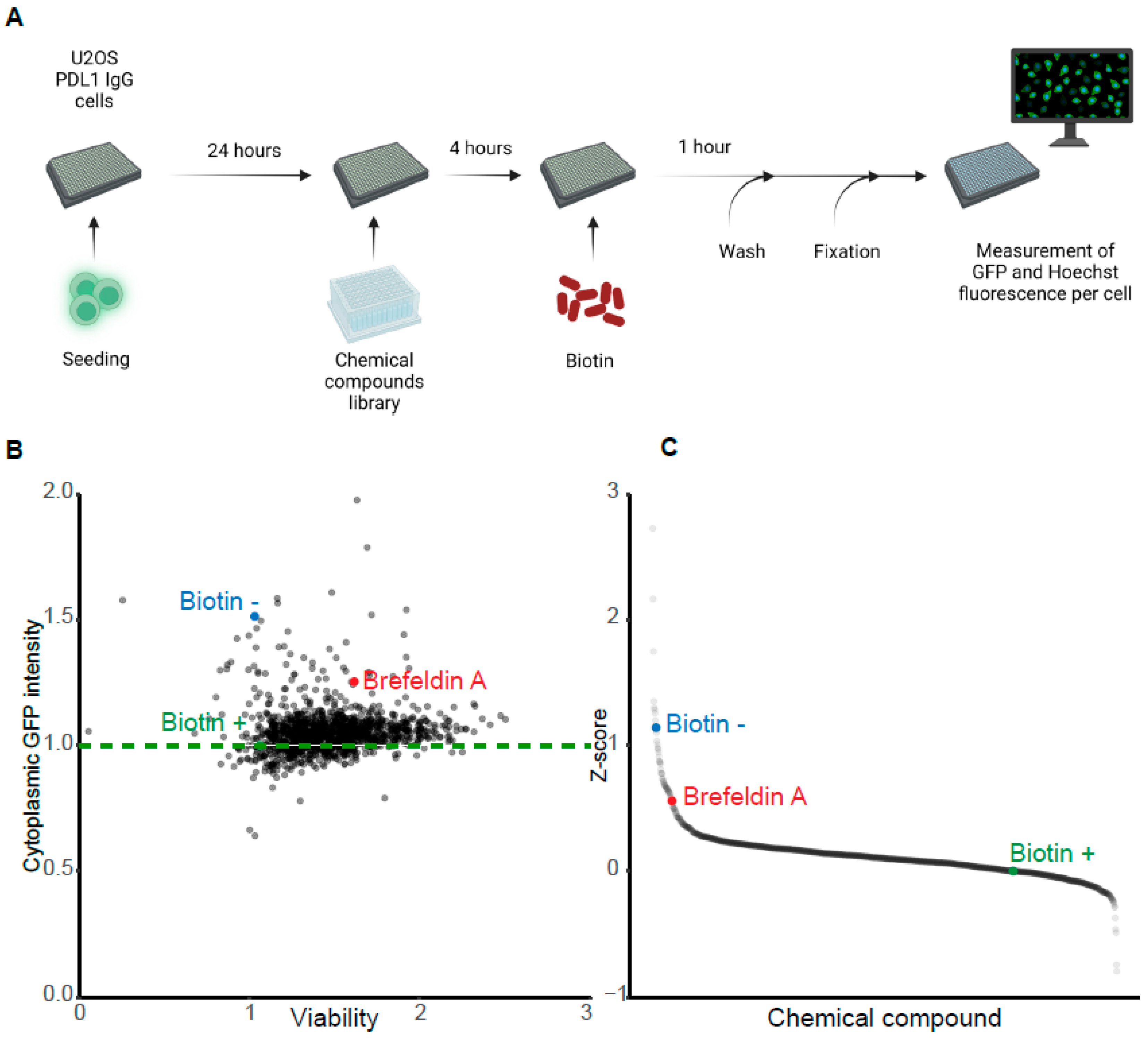
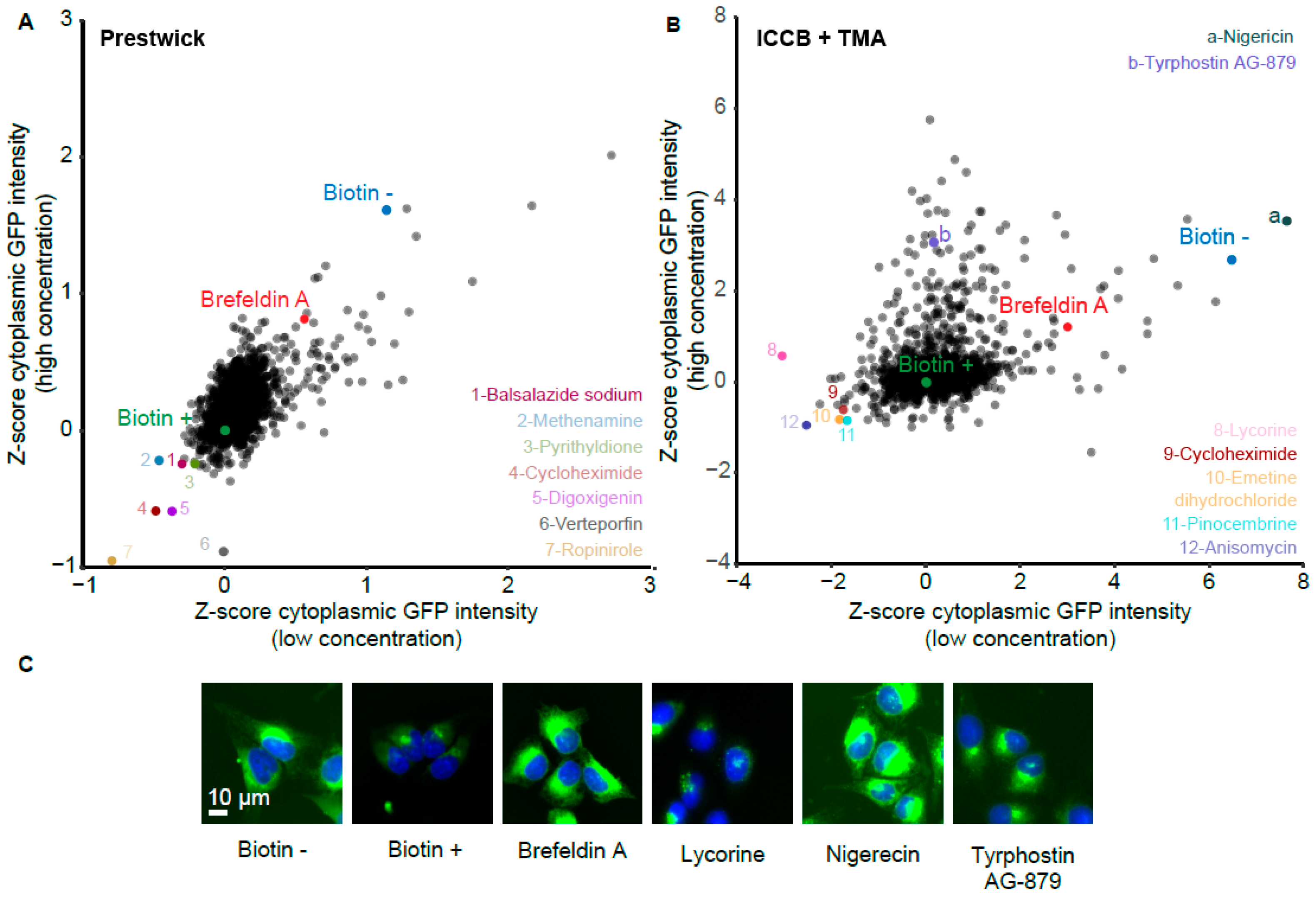
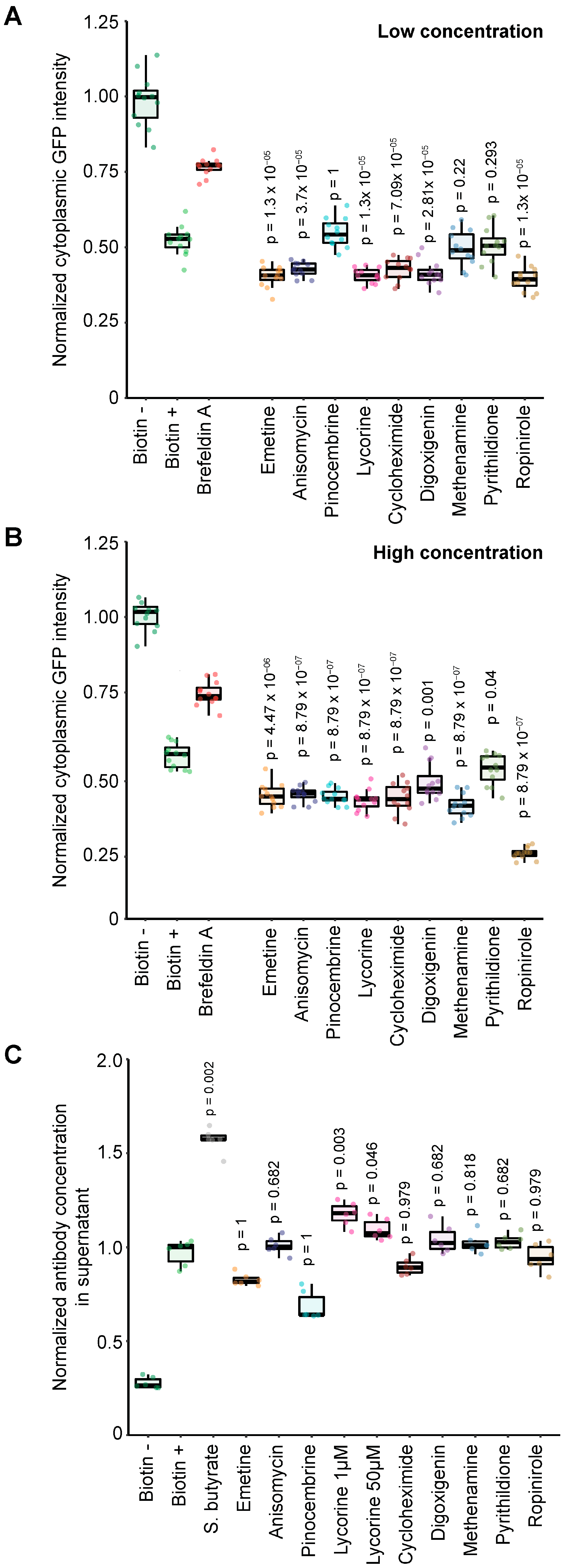
3.4. Pharmacological Screen for Modulators of Biotin-Induced Antibody Release
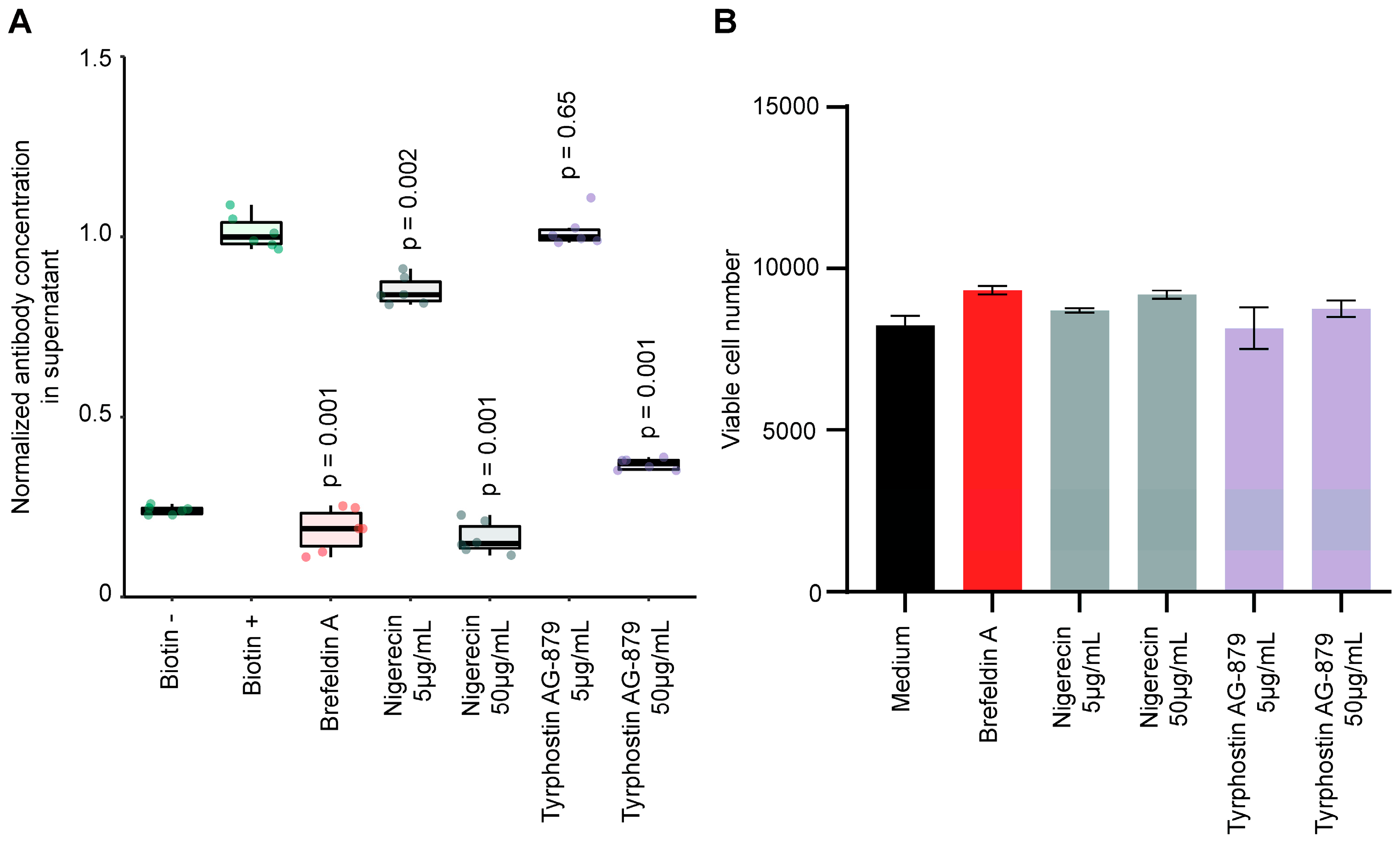
3.5. Validation of Stimulators and Inhibitors of Biotin-Induced Antibody Release
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Viotti, C. ER to Golgi-Dependent Protein Secretion: The Conventional Pathway. Methods Mol. Biol. 2016, 1459, 3–29. [Google Scholar] [CrossRef]
- Torres, M.; Hussain, H.; Dickson, A.J. The secretory pathway—The key for unlocking the potential of Chinese hamster ovary cell factories for manufacturing therapeutic proteins. Crit. Rev. Biotechnol. 2022, 43, 628–645. [Google Scholar] [CrossRef]
- Jena, B.P. Molecular machinery and mechanism of cell secretion. Exp. Biol. Med. 2005, 230, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Paavilainen, V.O. Natural products as modulators of eukaryotic protein secretion. Nat. Prod. Rep. 2020, 37, 717–736. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Kaufman, R.J. Protein misfolding in the endoplasmic reticulum as a conduit to human disease. Nature 2016, 529, 326–335. [Google Scholar] [CrossRef]
- Dejeans, N.; Manié, S.; Hetz, C.; Bard, F.; Hupp, T.; Agostinis, P.; Samali, A.; Chevet, E. Addicted to secrete—Novel concepts and targets in cancer therapy. Trends Mol. Med. 2014, 20, 242–250. [Google Scholar] [CrossRef]
- Walsh, G.; Walsh, E. Biopharmaceutical benchmarks 2022. Nat. Biotechnol. 2022, 40, 1722–1760. [Google Scholar] [CrossRef] [PubMed]
- Wurm, F.M. Production of recombinant protein therapeutics in cultivated mammalian cells. Nat. Biotechnol. 2004, 22, 1393–1398. [Google Scholar] [CrossRef]
- Chiu, M.L.; Goulet, D.R.; Teplyakov, A.; Gilliland, G.L. Antibody Structure and Function: The Basis for Engineering Therapeutics. Antibodies 2019, 8, 55. [Google Scholar] [CrossRef]
- Pelham, H.R. Multiple targets for brefeldin A. Cell 1991, 67, 449–451. [Google Scholar] [CrossRef]
- Badr, C.E.; Hewett, J.W.; Breakefield, X.O.; Tannous, B.A. A highly sensitive assay for monitoring the secretory pathway and ER stress. PLoS ONE 2007, 2, e571. [Google Scholar] [CrossRef]
- Aowicki, D.; Huczyński, A. Structure and antimicrobial properties of monensin A and its derivatives: Summary of the achievements. BioMed Res. Int. 2013, 2013, 742149. [Google Scholar] [CrossRef]
- Fussenegger, M.; Mazur, X.; Bailey, J.E. A novel cytostatic process enhances the productivity of Chinese hamster ovary cells. Biotechnol. Bioeng. 1997, 55, 927–939. [Google Scholar] [CrossRef]
- Sunley, K.; Butler, M. Strategies for the enhancement of recombinant protein production from mammalian cells by growth arrest. Biotechnol. Adv. 2010, 28, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Chung, J.Y.; Lim, S.W.; Hong, Y.J.; Hwang, S.O.; Lee, G.M. Effect of doxycycline-regulated calnexin and calreticulin expression on specific thrombopoietin productivity of recombinant Chinese hamster ovary cells. Biotechnol. Bioeng. 2004, 85, 539–546. [Google Scholar] [CrossRef]
- Berger, A.; Le Fourn, V.; Masternak, J.; Regamey, A.; Bodenmann, I.; Girod, P.A.; Mermod, N. Overexpression of transcription factor Foxa1 and target genes remediate therapeutic protein production bottlenecks in Chinese hamster ovary cells. Biotechnol. Bioeng. 2020, 117, 1101–1116. [Google Scholar] [CrossRef]
- Chandrawanshi, V.; Kulkarni, R.; Prabhu, A.; Mehra, S. Enhancing titers and productivity of rCHO clones with a combination of an optimized fed-batch process and ER-stress adaptation. J. Biotechnol. 2020, 311, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Peng, R.W.; Abellan, E.; Fussenegger, M. Differential effect of exocytic SNAREs on the production of recombinant proteins in mammalian cells. Biotechnol. Bioeng. 2011, 108, 611–620. [Google Scholar] [CrossRef]
- Rahimpour, A.; Vaziri, B.; Moazzami, R.; Nematollahi, L.; Barkhordari, F.; Kokabee, L.; Adeli, A.; Mahboudi, F. Engineering the cellular protein secretory pathway for enhancement of recombinant tissue plasminogen activator expression in Chinese hamster ovary cells: Effects of CERT and XBP1s genes. J. Microbiol. Biotechnol. 2013, 23, 1116–1122. [Google Scholar] [CrossRef]
- Hammond, S.; Lee, K.H. RNA interference of cofilin in Chinese hamster ovary cells improves recombinant protein productivity. Biotechnol. Bioeng. 2012, 109, 528–535. [Google Scholar] [CrossRef]
- Pourcel, L.; Buron, F.; Garcia, F.; Delaloix, M.S.; Le Fourn, V.; Girod, P.A.; Mermod, N. Transient vitamin B5 starving improves mammalian cell homeostasis and protein production. Metab. Eng. 2020, 60, 77–86. [Google Scholar] [CrossRef]
- Pourcel, L.; Buron, F.; Arib, G.; Le Fourn, V.; Regamey, A.; Bodenmann, I.; Girod, P.A.; Mermod, N. Influence of cytoskeleton organization on recombinant protein expression by CHO cells. Biotechnol. Bioeng. 2020, 117, 1117–1126. [Google Scholar] [CrossRef]
- Boncompain, G.; Divoux, S.; Gareil, N.; de Forges, H.; Lescure, A.; Latreche, L.; Mercanti, V.; Jollivet, F.; Raposo, G.; Perez, F. Synchronization of secretory protein traffic in populations of cells. Nat. Methods 2012, 9, 493–498. [Google Scholar] [CrossRef]
- Shapiro, J.; Sciaky, N.; Lee, J.; Bosshart, H.; Angeletti, R.H.; Bonifacino, J.S. Localization of endogenous furin in cultured cell lines. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1997, 45, 3–12. [Google Scholar] [CrossRef]
- Lefranc, M.P.; Ehrenmann, F.; Kossida, S.; Giudicelli, V.; Duroux, P. Use of IMGT(®) Databases and Tools for Antibody Engineering and Humanization. Methods Mol. Biol. 2018, 1827, 35–69. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Liu, P.; Boncompain, G.; Loos, F.; Lachkar, S.; Bezu, L.; Chen, G.; Zhou, H.; Perez, F.; Kepp, O.; et al. Identification of pharmacological inhibitors of conventional protein secretion. Sci. Rep. 2018, 8, 14966. [Google Scholar] [CrossRef] [PubMed]
- Barrette-Ng, I.H.; Wu, S.C.; Tjia, W.M.; Wong, S.L.; Ng, K.K. The structure of the SBP-Tag-streptavidin complex reveals a novel helical scaffold bridging binding pockets on separate subunits. Acta Crystallographica. Sect. D Biol. Crystallogr. 2013, 69, 879–887. [Google Scholar] [CrossRef]
- Delgadillo, R.F.; Mueser, T.C.; Zaleta-Rivera, K.; Carnes, K.A.; González-Valdez, J.; Parkhurst, L.J. Detailed characterization of the solution kinetics and thermodynamics of biotin, biocytin and HABA binding to avidin and streptavidin. PLoS ONE 2019, 14, e0204194. [Google Scholar] [CrossRef] [PubMed]
- Capitani, M.; Sallese, M. The KDEL receptor: New functions for an old protein. FEBS Lett. 2009, 583, 3863–3871. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Qi, X.; Wang, X.; Wei, D.; Wu, J.; Feng, L.; Cai, H.; Wang, Y.; Zeng, N.; Xu, T.; et al. Structural basis of the therapeutic anti-PD-L1 antibody atezolizumab. Oncotarget 2017, 8, 90215–90224. [Google Scholar] [CrossRef]
- Nomura, R.; Orii, M.; Senda, T. Calreticulin-2 is localized in the lumen of the endoplasmic reticulum but is not a Ca2+ -binding protein. Histochem. Cell Biol. 2011, 135, 531–538. [Google Scholar] [CrossRef]
- Misumi, Y.; Misumi, Y.; Miki, K.; Takatsuki, A.; Tamura, G.; Ikehara, Y. Novel blockade by brefeldin A of intracellular transport of secretory proteins in cultured rat hepatocytes. J. Biol. Chem. 1986, 261, 11398–11403. [Google Scholar] [CrossRef] [PubMed]
- Coulet, M.; (Sanofi R&D, Vitry-sur-Seine, France). Personal communication, 2023.
- Jiang, Z.; Sharfstein, S.T. Sodium butyrate stimulates monoclonal antibody over-expression in CHO cells by improving gene accessibility. Biotechnol. Bioeng. 2008, 100, 189–194. [Google Scholar] [CrossRef]
- Mimura, Y.; Lund, J.; Church, S.; Dong, S.; Li, J.; Goodall, M.; Jefferis, R. Butyrate increases production of human chimeric IgG in CHO-K1 cells whilst maintaining function and glycoform profile. J. Immunol. Methods 2001, 247, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Zhao, L.; Loos, F.; Marty, C.; Xie, W.; Martins, I.; Lachkar, S.; Qu, B.; Waeckel-Énée, E.; Plo, I.; et al. Immunosuppression by Mutated Calreticulin Released from Malignant Cells. Mol. Cell 2020, 77, 748–760.e9. [Google Scholar] [CrossRef]
- Fourriere, L.; Divoux, S.; Roceri, M.; Perez, F.; Boncompain, G. Microtubule-independent secretion requires functional maturation of Golgi elements. J. Cell Sci. 2016, 129, 3238–3250. [Google Scholar] [CrossRef] [PubMed]
- Deffieu, M.S.; Cesonyte, I.; Delalande, F.; Boncompain, G.; Dorobantu, C.; Song, E.; Lucansky, V.; Hirschler, A.; Cianferani, S.; Perez, F.; et al. Rab7-harboring vesicles are carriers of the transferrin receptor through the biosynthetic secretory pathway. Sci. Adv. 2021, 7, eaba7803. [Google Scholar] [CrossRef]
- Haas, A.K.; Mayer, K.; Brinkmann, U. Generation of fluorescent IgG fusion proteins in mammalian cells. Methods Mol. Biol. 2012, 901, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Luria, Y.; Raichlin, D.; Benhar, I. Fluorescent IgG fusion proteins made in E. coli. MAbs 2012, 4, 373–384. [Google Scholar] [CrossRef]
- Leitzgen, K.; Knittler, M.R.; Haas, I.G. Assembly of immunoglobulin light chains as a prerequisite for secretion. A model for oligomerization-dependent subunit folding. J. Biol. Chem. 1997, 272, 3117–3123. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Helenius, A. Quality control in the secretory pathway. Curr. Opin. Cell Biol. 1995, 7, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Dul, J.L.; Aviel, S.; Melnick, J.; Argon, Y. Ig light chains are secreted predominantly as monomers. J. Immunol. 1996, 157, 2969–2975. [Google Scholar] [CrossRef] [PubMed]
- Hopper, J.E.; Papagiannes, E. Evidence by radioimmunoassay that mitogen-activated human blood mononuclear cells secrete significant amounts of light chain Ig unassociated with heavy chain. Cell. Immunol. 1986, 101, 122–131. [Google Scholar] [CrossRef]
- Sreelatha, A.; Kinch, L.N.; Tagliabracci, V.S. The secretory pathway kinases. Biochim. Et Biophys. Acta 2015, 1854, 1687–1693. [Google Scholar] [CrossRef]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Gao, G.; Liu, F.; Xu, Z.; Wan, D.; Han, Y.; Kuang, Y.; Wang, Q.; Zhi, Q. Evidence of nigericin as a potential therapeutic candidate for cancers: A review. Biomed. Pharmacother. 2021, 137, 111262. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.; Liang, L.; Xiao, X.; Feng, P.; Ye, M.; Liu, J. Lycorine: A prospective natural lead for anticancer drug discovery. Biomed. Pharmacother. 2018, 107, 615–624. [Google Scholar] [CrossRef]
- Zhao, S.; Guo, Y.; Wang, Q.; An, B. Antifungal effects of lycorine on Botrytis cinerea and possible mechanisms. Biotechnol. Lett. 2021, 43, 1503–1512. [Google Scholar] [CrossRef]
- Kola, A.; Lamponi, S.; Currò, F.; Valensin, D. A Comparative Study between Lycorine and Galantamine Abilities to Interact with AMYLOID β and Reduce In Vitro Neurotoxicity. Int. J. Mol. Sci. 2023, 24, 2500. [Google Scholar] [CrossRef]
- Li, F.; Vijayasankaran, N.; Shen, A.Y.; Kiss, R.; Amanullah, A. Cell culture processes for monoclonal antibody production. MAbs 2010, 2, 466–479. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coulet, M.; Lachkar, S.; Leduc, M.; Trombe, M.; Gouveia, Z.; Perez, F.; Kepp, O.; Kroemer, G.; Basmaciogullari, S. Identification of Small Molecules Affecting the Secretion of Therapeutic Antibodies with the Retention Using Selective Hook (RUSH) System. Cells 2023, 12, 1642. https://doi.org/10.3390/cells12121642
Coulet M, Lachkar S, Leduc M, Trombe M, Gouveia Z, Perez F, Kepp O, Kroemer G, Basmaciogullari S. Identification of Small Molecules Affecting the Secretion of Therapeutic Antibodies with the Retention Using Selective Hook (RUSH) System. Cells. 2023; 12(12):1642. https://doi.org/10.3390/cells12121642
Chicago/Turabian StyleCoulet, Mathilde, Sylvie Lachkar, Marion Leduc, Marc Trombe, Zelia Gouveia, Franck Perez, Oliver Kepp, Guido Kroemer, and Stéphane Basmaciogullari. 2023. "Identification of Small Molecules Affecting the Secretion of Therapeutic Antibodies with the Retention Using Selective Hook (RUSH) System" Cells 12, no. 12: 1642. https://doi.org/10.3390/cells12121642
APA StyleCoulet, M., Lachkar, S., Leduc, M., Trombe, M., Gouveia, Z., Perez, F., Kepp, O., Kroemer, G., & Basmaciogullari, S. (2023). Identification of Small Molecules Affecting the Secretion of Therapeutic Antibodies with the Retention Using Selective Hook (RUSH) System. Cells, 12(12), 1642. https://doi.org/10.3390/cells12121642