

Cell-Penetrating Peptides as Valuable Tools for Nose-to-Brain Delivery of Biological Drugs


Abstract
1. Introduction
2. CPPs: Chemical Helpers to Improve Membrane Permeability of Biologics
3. CPP Classification Based on Physical–Chemical Properties
4. CPP Classification Based on the Type of Coupling to the Cargo
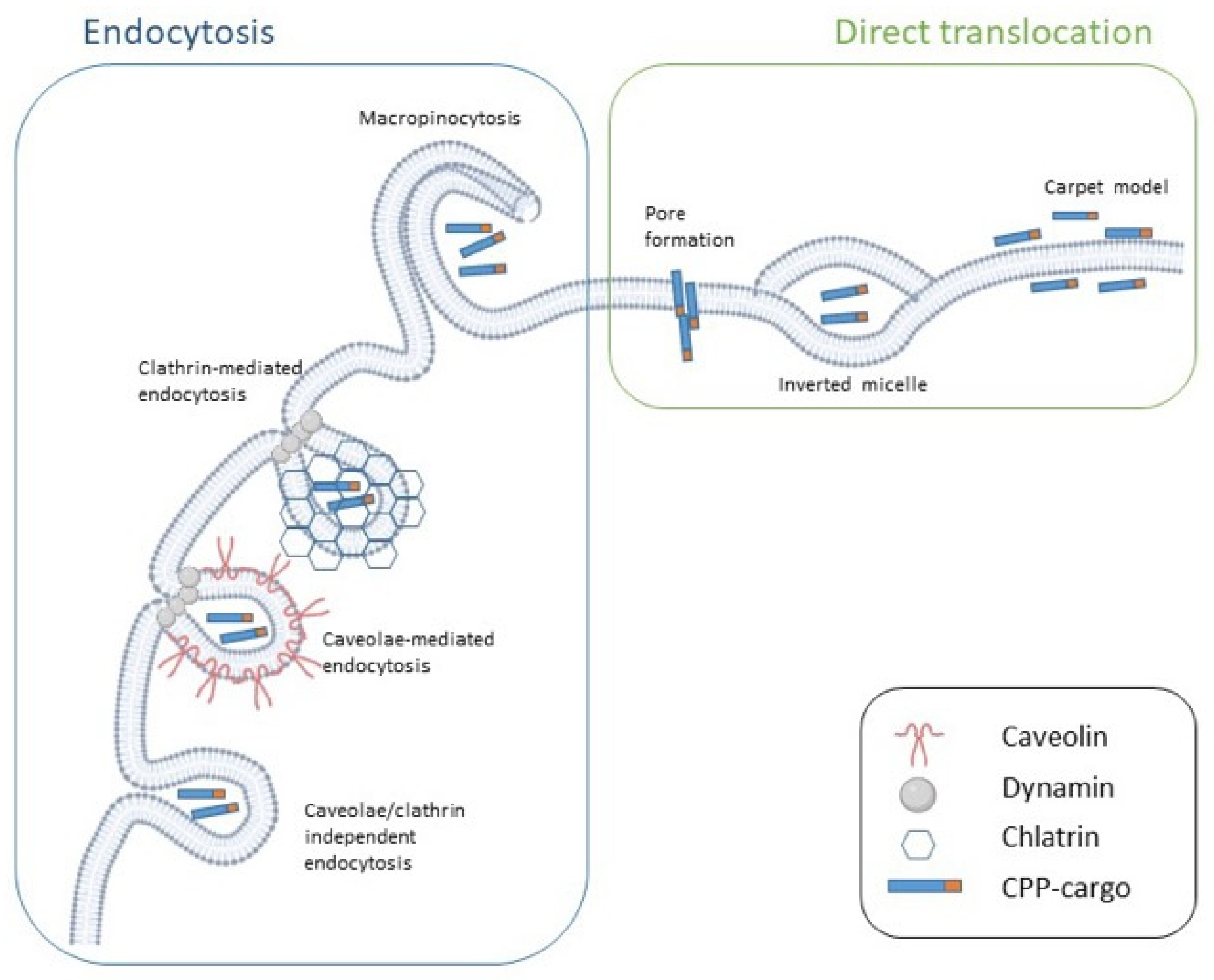
5. CPP Classification Based on Uptake Mechanism
6. Overcoming the Current Limitations of CPPs
7. Attempts to Deliver Drugs through the BBB
8. Nose-to-Brain Delivery
9. Transport of Therapeutics from Nose-to-Brain
10. Delivering Biotherapeutics through the Intranasal Route of Administration
11. Challenges and Strategies to Achieve Efficient Intranasal Delivery
12. CPPs to Improve N2B Delivery of Biologics
13. CPP-Functionalized Nanocarriers for IN Delivery
14. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhong, X.; Neumann, P.; Corbo, M.; Loh, E. Recent Advances in Biotherapeutics Drug Discovery and Development. In Drug Discovery and Development—Present and Future; Kapetanovi, I., Ed.; InTech: London, UK, 2011. [Google Scholar] [CrossRef]
- Oo, C.; Kalbag, S.S. Leveraging the attributes of biologics and small molecules, and releasing the bottlenecks: A new wave of revolution in drug development. Expert Rev. Clin. Pharmacol. 2016, 9, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef] [PubMed]
- Guidotti, G.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides: From Basic Research to Clinics. Trends Pharmacol. Sci. 2017, 38, 406–424. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, A.M.; Godinho, B.M.D.C.; Cryan, J.F.; O’Driscoll, C.M. Non-Viral Nanosystems for Gene and Small Interfering RNA Delivery to the Central Nervous System: Formulating the Solution. J. Pharm. Sci. 2013, 102, 3469–3484. [Google Scholar] [CrossRef] [PubMed]
- Fominaya, J.; Bravo, J.; Rebollo, A. Strategies to stabilize cell penetrating peptides for in vivo applications. Ther. Deliv. 2015, 6, 1171–1194. [Google Scholar] [CrossRef]
- Cohen, S.; Myneni, S.; Batt, A.; Guerrero, J.; Brumm, J.; Chung, S. Immunogenicity risk assessment for biotherapeutics through in vitro detection of CD134 and CD137 on T helper cells. mAbs 2021, 13, 1898831. [Google Scholar] [CrossRef]
- Kurano, T.; Kanazawa, T.; Ooba, A.; Masuyama, Y.; Maruhana, N.; Yamada, M.; Iioka, S.; Ibaraki, H.; Kosuge, Y.; Kondo, H.; et al. Nose-to-brain/spinal cord delivery kinetics of liposomes with different surface properties. J. Control. Release 2022, 344, 225–234. [Google Scholar] [CrossRef]
- Lochhead, J.J.; Thorne, R.G. Intranasal delivery of biologics to the central nervous system. Adv. Drug Deliv. Rev. 2012, 64, 614–628. [Google Scholar] [CrossRef]
- Ross, C.; Taylor, M.; Fullwood, N.; Allsop, D. Liposome delivery systems for the treatment of Alzheimer′s disease. Int. J. Nanomed. 2018, 13, 8507–8522. [Google Scholar] [CrossRef]
- Pardridge, W.M. The blood-brain barrier: Bottleneck in brain drug development. NeuroRx 2005, 2, 12. [Google Scholar] [CrossRef]
- Chiriboga, C.A.; Swoboda, K.J.; Darras, B.T.; Iannaccone, S.T.; Montes, J.; De Vivo, D.C.; Norris, D.A.; Bennett, C.F.; Bishop, K.M. Results from a phase 1 study of nusinersen (ISIS-SMNRx) in children with spinal muscular atrophy. Neurology 2016, 86, 890–897. [Google Scholar] [CrossRef]
- Finkel, R.S.; Chiriboga, C.A.; Vajsar, J.; Day, J.W.; Montes, J.; De Vivo, D.C.; Yamashita, M.; Rigo, F.; Hung, G.; Schneider, E.; et al. Treatment of infantile-onset spinal muscular atrophy with nusinersen: A phase 2, open-label, dose-escalation study. Lancet 2016, 388, 3017–3026. [Google Scholar] [CrossRef]
- Haché, M.; Swoboda, K.J.; Sethna, N.; Farrow-Gillespie, A.; Khandji, A.; Xia, S.; Bishop, K.M. Intrathecal Injections in Children with Spinal Muscular Atrophy: Nusinersen Clinical Trial Experience. J. Child Neurol. 2016, 31, 899–906. [Google Scholar] [CrossRef]
- Glascock, J.J.; Osman, E.Y.; Coady, T.H.; Rose, F.F.; Shababi, M.; Lorson, C.L. Delivery of Therapeutic Agents Through Intracerebroventricular (ICV) and Intravenous (IV) Injection in Mice. J. Vis. Exp. JoVE 2011, 56, 2968. [Google Scholar] [CrossRef]
- Spetea, M.; Eans, S.O.; Ganno, M.L.; Lantero, A.; Mairegger, M.; Toll, L.; Schmidhammer, H.; McLaughlin, J.P. Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice. Br. J. Pharmacol. 2017, 174, 2444–2456. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, H.; Wang, Y.; Elhussini, M.Z.; Tran, P.V.; Haraguchi, S.; Cockrem, J.F.; Bungo, T.; Furuse, M.; Chowdhury, V.S. Central administration of neuropeptide Y reduces the cellular heat stress response and may enhance spleen antioxidative functions in heat-exposed chicks. Neurosci. Lett. 2022, 784, 136749. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Frey, W.H. Intranasal delivery bypasses the blood-brain barrier to target therapeutic agents to the central nervous system and treat neurodegenerative disease. BMC Neurosci. 2008, 9, S5. [Google Scholar] [CrossRef] [PubMed]
- Sharma, G.; Lakkadwala, S.; Modgil, A.; Singh, J. The Role of Cell-Penetrating Peptide and Transferrin on Enhanced Delivery of Drug to Brain. Int. J. Mol. Sci. 2016, 17, 806. [Google Scholar] [CrossRef]
- Kamei, N.; Takeda-Morishita, M. Brain delivery of insulin boosted by intranasal coadministration with cell-penetrating peptides. J. Control. Release 2015, 197, 105–110. [Google Scholar] [CrossRef]
- Zou, L.-L.; Ma, J.-L.; Wang, T.; Yang, T.-B.; Liu, C.-B. Cell-Penetrating Peptide-Mediated Therapeutic Molecule Delivery into the Central Nervous System. Curr. Neuropharmacol. 2013, 11, 197–208. [Google Scholar] [CrossRef] [PubMed]
- Szabó, I.; Yousef, M.; Soltétz, D.; Bató, C.; Mezó, G.; Bánóczi, Z. Redesigning of Cell-Penetrating Peptides to Improve Their Efficacy as a Drug Delivery System. Pharmaceutics 2022, 14, 907. [Google Scholar] [CrossRef]
- Illum, L. Nasal drug delivery—Possibilities, problems and solutions. J. Control. Release 2003, 87, 187–198. [Google Scholar] [CrossRef]
- Reissmann, S. Cell penetration: Scope and limitations by the application of cell-penetrating peptides: CPPS, TYPES, UPTAKE, TRAFFICKING, SELECTIVITY, AND CLINICAL STUDIES. J. Pept. Sci. 2014, 20, 760–784. [Google Scholar] [CrossRef]
- Ruseska, I.; Zimmer, A. Internalization mechanisms of cell-penetrating peptides. Beilstein J. Nanotechnol. 2020, 11, 101–123. [Google Scholar] [CrossRef]
- Frankel, A.D.; Pabo, C.O. Cellular uptake of the tat protein from human immunodeficiency virus. Cell 1988, 55, 1189–1193. [Google Scholar] [CrossRef]
- Vivès, E.; Brodin, P.; Lebleu, B. A Truncated HIV-1 Tat Protein Basic Domain Rapidly Translocates through the Plasma Membrane and Accumulates in the Cell Nucleus. J. Biol. Chem. 1997, 272, 16010–16017. [Google Scholar] [CrossRef] [PubMed]
- Derossi, D.; Joliot, A.H.; Chassaing, G.; Prochiantz, A. The third helix of the Antennapedia homeodomain translocates through biological membranes. J. Biol. Chem. 1994, 269, 10444–10450. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.C.; Cheon, D.H.; Lee, Y. Challenge to overcome current limitations of cell-penetrating peptides. Biochim. Biophys. Acta BBA-Proteins Proteom. 2021, 1869, 140604. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S.; Filatova, M.P. New generation of cell-penetrating peptides: Functionality and potential clinical application. J. Pept. Sci. 2021, 27, e3300. [Google Scholar] [CrossRef] [PubMed]
- Pujals, S.; Giralt, E. Proline-rich, amphipathic cell-penetrating peptides. Adv. Drug Deliv. Rev. 2008, 60, 473–484. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, Y.; Liu, E.; Gong, J.; Shin, M.C.; Huang, Y. CPP-mediated Protein Delivery in a Noncovalent Form: Proof-of-Concept for Percutaneous and Intranasal Delivery. Protein Pept. Lett. 2014, 21, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Morris, M.C.; Divita, G.; Heitz, F. Interactions of amphipathic CPPs with model membranes. Biochim. Biophys. Acta BBA-Biomembr. 2006, 1758, 328–335. [Google Scholar] [CrossRef] [PubMed]
- Deshayes, S.; Gerbal-Chaloin, S.; Morris, M.C.; Aldrian-Herrada, G.; Charnet, P.; Divita, G.; Heitz, F. On the mechanism of non-endosomial peptide-mediated cellular delivery of nucleic acids. Biochim. Biophys. Acta BBA-Biomembr. 2004, 1667, 141–147. [Google Scholar] [CrossRef]
- Conner, S.D.; Schmid, S.L. Regulated portals of entry into the cell. Nature 2003, 422, 37–44. [Google Scholar] [CrossRef]
- Wadia, J.S.; Stan, R.V.; Dowdy, S.F. Transducible TAT-HA fusogenic peptide enhances escape of TAT-fusion proteins after lipid raft macropinocytosis. Nat. Med. 2004, 10, 310–315. [Google Scholar] [CrossRef]
- Nakase, I.; Hirose, H.; Tanaka, G.; Tadokoro, A.; Kobayashi, S.; Tateuchi, T.; Futaki, S. Cell-surface Accumulation of Flock House Virus-derived Peptide Leads to Efficient Internalization via Macropinocytosis. Mol. Ther. 2009, 17, 1868–1876. [Google Scholar] [CrossRef]
- Nakase, I.; Tadokoro, A.; Kawabata, N.; Tateuchi, T.; Katoh, H.; Hiramoto, K.; Negishi, M.; Nomizu, M.; Sugiura, Y.; Futaki, S. Interaction of Arginine-Rich Peptides with Membrane-Associated Proteoglycans Is Crucial for Induction of Actin Organization and Macropinocytosis. Biochemistry 2007, 46, 492–501. [Google Scholar] [CrossRef]
- Trofimenko, E.; Grasso, G.; Heulot, M.; Chevalier, N.; Deriu, M.A.; Dubuis, G.; Arribat, Y.; Serulla, M.; Michel, S.; Vantomme, G.; et al. Genetic, cellular, and structural characterization of the membrane potential-dependent cell-penetrating peptide translocation pore. eLife 2021, 10, e69832. [Google Scholar] [CrossRef]
- Futaki, S. Oligoarginine vectors for intracellular delivery: Design and cellular-uptake mechanisms. Pept. Sci. 2006, 84, 241–249. [Google Scholar] [CrossRef]
- Pang, H.-B.; Braun, G.B.; Ruoslahti, E. Neuropilin-1 and heparan sulfate proteoglycans cooperate in cellular uptake of nanoparticles functionalized by cationic cell-penetrating peptides. Sci. Adv. 2015, 1, e1500821. [Google Scholar] [CrossRef] [PubMed]
- Richard, J.P.; Melikov, K.; Brooks, H.; Prevot, P.; Lebleu, B.; Chernomordick, L.V. Cellular Uptake of Unconjugated TAT Peptide Involves Clathrin-dependent Endocytosis and Heparan Sulfate Receptors. J. Biol. Chem. 2005, 280, 15300–15306. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, Y.; Takeuchi, T.; Kuwata, K.; Chiba, J.; Hatanaka, Y.; Nakase, I.; Futaki, S. Syndecan-4 Is a Receptor for Clathrin-Mediated Endocytosis of Arginine-Rich Cell-Penetrating Peptides. Bioconjug. Chem. 2016, 27, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Arukuusk, P.; Pärnaste, L.; Oskolkov, N.; Copolovici, D.-A.; Margus, H.; Padari, K.; Möll, K.; Maslovskaja, J.; Tegova, R.; Kivi, G.; et al. New generation of efficient peptide-based vectors, NickFects, for the delivery of nucleic acids. Biochim. Biophys. Acta BBA-Biomembr. 2013, 1828, 1365–1373. [Google Scholar] [CrossRef]
- Taylor, B.N.; Mehta, R.R.; Yamada, T.; Lekmine, F.; Christov, K.; Chakrabarty, A.M.; Green, A.; Bratescu, L.; Shilkaitis, A.; Beattie, C.W.; et al. Noncationic Peptides Obtained from Azurin Preferentially Enter Cancer Cells. Cancer Res. 2009, 69, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.; Pellegrini, V.; Arcangeli, C.; Fittipaldi, A.; Giacca, M.; Beltram, F. Caveolae-Mediated internalization of extracellular HIV-1 tat fusion proteins visualized in real time. Mol. Ther. 2003, 8, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Säälik, P.; Padari, K.; Niinep, A.; Lorents, A.; Hansen, M.; Jokitalo, E.; Langel, U.; Pooga, M. Protein Delivery with Transportans Is Mediated by Caveolae Rather Than Flotillin-Dependent Pathways. Bioconjug. Chem. 2009, 20, 877–887. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Liu, E.; Yu, Z.; Pei, X.; Chen, S.; Zhang, P.; Shin, M.C.; Gong, J.; He, H.; Yang, V.C. CPP-Assisted Intracellular Drug Delivery, What Is Next? Int. J. Mol. Sci. 2016, 17, 1892. [Google Scholar] [CrossRef]
- Guidotti, G.; Brambilla, L.; Rossi, D. Peptides in clinical development for the treatment of brain tumors. Curr. Opin. Pharmacol. 2019, 47, 102–109. [Google Scholar] [CrossRef]
- Purkayastha, N.; Eyer, K.; Robinson, T.; Dittrich, P.S.; Beck, A.K.; Seebach, D.; Kolesinska, B.; Cadalbert, R. Enantiomeric and Diastereoisomeric (Mixed) L/ D-Octaarginine Derivatives—A Simple Way of Modulating the Properties of Cell-Penetrating Peptides. Chem. Biodivers. 2013, 10, 1165–1184. [Google Scholar] [CrossRef]
- Nielsen, E.J.B.; Yoshida, S.; Kamei, N.; Iwamae, R.; Khafagy, E.-S.; Olsen, J.; Rahbek, U.L.; Pedersen, B.L.; Takayama, K.; Takeda-Morishita, M. In vivo proof of concept of oral insulin delivery based on a co-administration strategy with the cell-penetrating peptide penetratin. J. Control. Release 2014, 189, 19–24. [Google Scholar] [CrossRef]
- Yamada, T.; Signorelli, S.; Cannistraro, S.; Beattie, C.W.; Bizzarri, A.R. Chirality Switching within an Anionic Cell-Penetrating Peptide Inhibits Translocation without Affecting Preferential Entry. Mol. Pharm. 2015, 12, 140–149. [Google Scholar] [CrossRef]
- Park, S.E.; Sajid, M.I.; Parang, K.; Tiwari, R.K. Cyclic Cell-Penetrating Peptides as Efficient Intracellular Drug Delivery Tools. Mol. Pharm. 2019, 16, 3727–3743. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Nakase, I.; Michiue, H.; Tateuchi, T.; Tomizawa, K.; Matsui, H.; Futaki, S. Enhanced intracellular delivery using arginine-rich peptides by the addition of penetration accelerating sequences (Pas). J. Control. Release 2009, 138, 128–133. [Google Scholar] [CrossRef]
- Mäe, M.; Andaloussi, S.E.; Lundin, P.; Oskolkov, N.; Johansson, H.J.; Guterstam, P.; Langel, U. A stearylated CPP for delivery of splice correcting oligonucleotides using a non-covalent co-incubation strategy. J. Control. Release 2009, 134, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Kosuge, M.; Tadokoro, A.; Sugiura, Y.; Nishi, M.; Kawata, M.; Sakai, N.; Matile, S.; Futaki, S. Direct and Rapid Cytosolic Delivery Using Cell-Penetrating Peptides Mediated by Pyrenebutyrate. ACS Chem. Biol. 2006, 1, 299–303. [Google Scholar] [CrossRef] [PubMed]
- Katayama, S.; Hirose, H.; Takayama, K.; Nakase, I.; Futaki, S. Acylation of octaarginine: Implication to the use of intracellular delivery vectors. J. Control. Release 2011, 149, 29–35. [Google Scholar] [CrossRef]
- Swiecicki, J.-M.; Di Pisa, M.; Lippi, F.; Chwetzoff, S.; Mansuy, C.; Trugnan, G.; Chassaing, G.; Lavielle, S.; Burlina, F. Unsaturated acyl chains dramatically enhanced cellular uptake by direct translocation of a minimalist oligo-arginine lipopeptide. Chem. Commun. 2015, 51, 14656–14659. [Google Scholar] [CrossRef]
- Marks, J.R.; Placone, J.; Hristova, K.; Wimley, W.C. Spontaneous Membrane-Translocating Peptides by Orthogonal High-Throughput Screening. J. Am. Chem. Soc. 2011, 133, 8995–9004. [Google Scholar] [CrossRef]
- de Jong, H.; Bonger, K.M.; Löwik, D.W.P.M. Activatable cell-penetrating peptides: 15 years of research. RSC Chem. Biol. 2020, 1, 192–203. [Google Scholar] [CrossRef]
- Jiang, T.; Olson, E.S.; Nguyen, Q.T.; Roy, M.; Jennings, P.A.; Tsien, R.Y. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc. Natl. Acad. Sci. USA 2004, 101, 17867–17872. [Google Scholar] [CrossRef]
- Schwarze, S.R.; Ho, A.; Vocero-Akbani, A.; Dowdy, S.F. In Vivo Protein Transduction: Delivery of a Biologically Active Protein into the Mouse. Science 1999, 285, 1569–1572. [Google Scholar] [CrossRef] [PubMed]
- Fu, A.; Wang, Y.; Zhan, L.; Zhou, R. Targeted Delivery of Proteins into the Central Nervous System Mediated by Rabies Virus Glycoprotein-Derived Peptide. Pharm. Res. 2012, 29, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.; Liu, D.; Yao, S.; Collins, R.D.; Hawiger, J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat. Med. 2005, 11, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- Caillé, I.; Allinquant, B.; Dupont, E.; Bouillot, C.; Langer, A.; Müller, U.; Prochiantz, A. Soluble form of amyloid precursor protein regulates proliferation of progenitors in the adult subventricular zone. Development 2004, 131, 2173–2181. [Google Scholar] [CrossRef]
- Kurzrock, R.; Gabrail, N.; Chandhasin, C.; Moulder, S.; Smith, C.; Brenner, A.; Sankhala, K.; Mita, A.; Elian, K.; Bouchard, D.; et al. Safety, pharmacokinetics, and activity of GRN1005, a novel conjugate of angiopep-2, a peptide facilitating brain penetration, and paclitaxel, in patients with advanced solid tumors. Mol. Cancer Ther. 2012, 11, 308–316. [Google Scholar] [CrossRef]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro-Oncology 2016, 18, 1319–1325. [Google Scholar] [CrossRef]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef]
- Gänger, S.; Schindowski, K. Tailoring Formulations for Intranasal Nose-to-Brain Delivery: A Review on Architecture, Physico-Chemical Characteristics and Mucociliary Clearance of the Nasal Olfactory Mucosa. Pharmaceutics 2018, 10, 116. [Google Scholar] [CrossRef]
- Frey William, H. Neurologic Agents for Nasal Administration to the Brain. U.S. Patent WO1991007947A1, 4 December 1990. [Google Scholar]
- Pires, P.C.; Rodrigues, M.; Alves, G.; Santos, A.O. Strategies to Improve Drug Strength in Nasal Preparations for Brain Delivery of Low Aqueous Solubility Drugs. Pharmaceutics 2022, 14, 588. [Google Scholar] [CrossRef]
- Merkus, F.V.; Verhoef, J.C.; Schipper, N.G.; Marttin, E. Nasal mucociliary clearance as a factor in nasal drug delivery. Adv. Drug Deliv. Rev. 1998, 29, 13–38. [Google Scholar] [CrossRef] [PubMed]
- Crowe, T.P.; Greenlee, M.H.W.; Kanthasamy, A.G.; Hsu, W.H. Mechanism of intranasal drug delivery directly to the brain. Life Sci. 2018, 195, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal drug targeting of hypocretin-1 (orexin-A) to the central nervous system. J. Pharm. Sci. 2009, 98, 2501–2515. [Google Scholar] [CrossRef]
- Thorne, R.G.; Pronk, G.J.; Padmanabhan, V.; Frey, W.H. Delivery of insulin-like growth factor-I to the rat brain and spinal cord along olfactory and trigeminal pathways following intranasal administration. Neuroscience 2004, 127, 481–496. [Google Scholar] [CrossRef]
- Thorne, R.G.; Hanson, L.R.; Ross, T.M.; Tung, D.; Frey, W.H. Delivery of interferon-beta to the monkey nervous system following intranasal administration. Neuroscience 2008, 152, 785–797. [Google Scholar] [CrossRef]
- Selvaraj, K.; Gowthamarajan, K.; Karri, V.V.S.R. Nose to brain transport pathways an overview: Potential of nanostructured lipid carriers in nose to brain targeting. Artif. Cells Nanomed. Biotechnol. 2018, 46, 2088–2095. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Intranasal delivery to the central nervous system: Mechanisms and experimental considerations. J. Pharm. Sci. 2010, 99, 1654–1673. [Google Scholar] [CrossRef]
- Crowe, T.P.; Hsu, W.H. Evaluation of Recent Intranasal Drug Delivery Systems to the Central Nervous System. Pharmaceutics 2022, 14, 629. [Google Scholar] [CrossRef] [PubMed]
- Keller, L.-A.; Merkel, O.; Popp, A. Intranasal drug delivery: Opportunities and toxicologic challenges during drug development. Drug Deliv. Transl. Res. 2022, 12, 735–757. [Google Scholar] [CrossRef] [PubMed]
- Dihydroergotamine Mesylate (DHE-45), Migranal®. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=020148 (accessed on 21 March 2023).
- Desmopressin Acetate, Stimate®. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=020355 (accessed on 21 March 2023).
- SYNAREL® (NAFARELIN Acetate)|Pfizer Medical Information–US. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/nda/98/019886S013%20.cfm (accessed on 21 March 2023).
- Butorphanol Tartrate, Stadol®. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=019890 (accessed on 21 March 2023).
- Zolmitriptan, Zomig®, Nasal Spray. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=021450 (accessed on 21 March 2023).
- Desmopressin Acetate, DDAVP®. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=017922 (accessed on 21 March 2023).
- Esketamine, SPRAVATO®. Available online: https://www.spravato.com/home-1 (accessed on 21 March 2023).
- Midazolam, Nayzilam. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=211321 (accessed on 21 March 2023).
- Information for Patients|NAYZILAM® (Midazolam) Nasal Spray, CIV. Available online: https://www.nayzilam.com/ (accessed on 21 March 2023).
- Diazepam Nasal Spray, Valtoco®. Available online: https://www.neurelis.com/our-product (accessed on 21 March 2023).
- Lochhead, J.J.; Kellohen, K.L.; Ronaldson, P.T.; Davis, T.P. Distribution of insulin in trigeminal nerve and brain after intranasal administration. Sci. Rep. 2019, 9, 2621. [Google Scholar] [CrossRef]
- Brabazon, F.; Wilson, C.M.; Jaiswal, S.; Reed, J.; Frey, W.H.; Byrnes, K.R. Intranasal insulin treatment of an experimental model of moderate traumatic brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 3203–3218. [Google Scholar] [CrossRef] [PubMed]
- Renner, D.B.; Svitak, A.L.; Gallus, N.J.; Ericson, M.E.; Frey, W.H.; Hanson, L.R. Intranasal delivery of insulin via the olfactory nerve pathway. J. Pharm. Pharmacol. 2012, 64, 1709–1714. [Google Scholar] [CrossRef] [PubMed]
- Nedelcovych, M.T.; Gadiano, A.J.; Wu, Y.; Manning, A.A.; Thomas, A.G.; Khuder, S.S.; Yoo, S.-W.; Xu, J.; McArthur, J.C.; Haughey, N.J.; et al. Pharmacokinetics of Intranasal versus Subcutaneous Insulin in the Mouse. ACS Chem. Neurosci. 2018, 9, 809–816. [Google Scholar] [CrossRef]
- Banks, W.A.; During, M.J.; Niehoff, M.L. Brain Uptake of the Glucagon-Like Peptide-1 Antagonist Exendin(9-39) after Intranasal Administration. J. Pharmacol. Exp. Ther. 2004, 309, 469–475. [Google Scholar] [CrossRef]
- Banks, W.A.; Farrell, C.L. Impaired transport of leptin across the blood-brain barrier in obesity is acquired and reversible. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E10–E15. [Google Scholar] [CrossRef]
- Fliedner, S.; Schulz, C.; Lehnert, H. Brain Uptake of Intranasally Applied Radioiodinated Leptin in Wistar Rats. Endocrinology 2006, 147, 2088–2094. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Paulus, K.; Jöhren, O.; Lehnert, H. Intranasal Leptin Reduces Appetite and Induces Weight Loss in Rats with Diet-Induced Obesity (DIO). Endocrinology 2012, 153, 143–153. [Google Scholar] [CrossRef]
- Berger, S.; Pho, H.; Fleury-Curado, T.; Bevans-Fonti, S.; Younas, H.; Shin, M.-K.; Jun, J.C.; Anokye-Danso, F.; Ahima, R.S.; Enquist, L.W.; et al. Intranasal Leptin Relieves Sleep-disordered Breathing in Mice with Diet-induced Obesity. Am. J. Respir. Crit. Care Med. 2019, 199, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Erdő, F.; Bors, L.A.; Farkas, D.; Bajza, Á.; Gizurarson, S. Evaluation of intranasal delivery route of drug administration for brain targeting. Brain Res. Bull. 2018, 143, 155–170. [Google Scholar] [CrossRef]
- Chavoshinezhad, S.; Kouchesfahani, H.M.; Salehi, M.S.; Pandamooz, S.; Ahmadiani, A.; Darghai, L. Intranasal interferon beta improves memory and modulates inflammatory responses in a mutant APP-overexpressing rat model of Alzheimer’s disease. Brain Res. Bull. 2019, 150, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Ross, T.M.; Martinez, P.M.; Renner, J.C.; Thorne, R.G.; Hanson, L.R.; Frey, W.H. Intranasal administration of interferon beta bypasses the blood-brain barrier to target the central nervous system and cervical lymph nodes: A non-invasive treatment strategy for multiple sclerosis. J. Neuroimmunol. 2004, 151, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Pietrowsky, R.; Strüben, C.; Mölle, M.; Fehm, H.L.; Born, J. Brain potential changes after intranasal vs. intravenous administration of vasopressin: Evidence for a direct nose-brain pathway for peptide effects in humans. Biol. Psychiatry 1996, 39, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Derad, I.; Sayk, F.; Lehnert, H.; Marshall, L.; Born, J.; Nitschke, M. Intranasal Angiotensin II in Humans Reduces Blood Pressure When Angiotensin II Type 1 Receptors Are Blocked. Hypertension 2014, 63, 762–767. [Google Scholar] [CrossRef] [PubMed]
- Kern, W.; Born, J.; Schreiber, H.; Fehm, H.L. Central nervous system effects of intranasally administered insulin during euglycemia in men. Diabetes 1999, 48, 557–563. [Google Scholar] [CrossRef]
- Benedict, C.; Hallschmid, M.; Hatke, A.; Schultes, B.; Fehm, H.L.; Born, J.; Kern, W. Intranasal insulin improves memory in humans. Psychoneuroendocrinology 2004, 29, 1326–1334. [Google Scholar] [CrossRef]
- Kang, Y.S.; Park, J.H. Brain uptake and the analgesic effect of oxytocin—Its usefulness as an analgesic agent. Arch. Pharm. Res. 2000, 23, 391–395. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.C.S.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates beta-amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal Insulin Therapy for Alzheimer Disease and Amnestic Mild Cognitive Impairment. Arch. Neurol. 2012, 69, 29–38. [Google Scholar] [CrossRef]
- Maggio, E.T. IntravailTM: Highly effective intranasal delivery of peptide and protein drugs. Expert Opin. Drug Deliv. 2006, 3, 529–539. [Google Scholar] [CrossRef]
- Nonaka, N.; Farr, S.A.; Kageyama, H.; Shioda, S.; Banks, W.A. Delivery of Galanin-Like Peptide to the Brain: Targeting with Intranasal Delivery and Cyclodextrins. J. Pharmacol. Exp. Ther. 2008, 325, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Zolkowska, D.; Wu, C.-Y.; Rogawski, M.A. Intranasal Allopregnanolone Confers Rapid Seizure Protection: Evidence for Direct Nose-to-Brain Delivery. Neurotherapeutics 2021, 18, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Hanson, L.R.; Ii, W.H.F.; Hoekman, J.D.; Pohl, J.D. Lipid growth factor formulations. U.S. Patent 9,480,649, 1 November 2016. [Google Scholar]
- Khan, S.A.; Patil, K.S.; Yeole, P.G. Intranasal mucoadhesive buspirone formulation: In vitro characterization and nasal clearance studies. J. Pharm. Sci. 2008, 63, 348–351. [Google Scholar] [CrossRef]
- Khan, S.; Patil, K.; Yeole, P.; Gaikwad, R. Brain targeting studies on buspirone hydrochloride after intranasal administration of mucoadhesive formulation in rats. J. Pharm. Pharmacol. 2009, 61, 669–675. [Google Scholar] [CrossRef]
- Dalvi, A.; Ravi, P.R.; Uppuluri, C.T. Rufinamide-Loaded Chitosan Nanoparticles in Xyloglucan-Based Thermoresponsive in Situ Gel for Direct Nose to Brain Delivery. Front. Pharmacol. 2021, 12, 691936. [Google Scholar] [CrossRef]
- Graff, C.L.; Pollack, G.M. P-Glycoprotein Attenuates Brain Uptake of Substrates After Nasal Instillation. Pharm. Res. 2003, 20, 1225–1230. [Google Scholar] [CrossRef] [PubMed]
- Graff, C.L.; Zhao, R.; Pollack, G.M. Pharmacokinetics of Substrate Uptake and Distribution in Murine Brain After Nasal Instillation. Pharm. Res. 2005, 22, 235–244. [Google Scholar] [CrossRef]
- Dhuria, S.V.; Hanson, L.R.; Frey, W.H. Novel Vasoconstrictor Formulation to Enhance Intranasal Targeting of Neuropeptide Therapeutics to the Central Nervous System. J. Pharmacol. Exp. Ther. 2009, 328, 312–320. [Google Scholar] [CrossRef]
- Abouhussein, D.M.N.; Khattab, A.; Bayoumi, N.A.; Mahmoud, A.F.; Sakr, T.M. Brain targeted rivastigmine mucoadhesive thermosensitive in situ gel: Optimization, in vitro evaluation, radiolabeling, in vivo pharmacokinetics and biodistribution. J. Drug Deliv. Sci. Technol. 2018, 43, 129–140. [Google Scholar] [CrossRef]
- Ravi, P.R.; Aditya, N.; Patil, S.; Cherian, L. Nasal in-situ gels for delivery of rasagiline mesylate: Improvement in bioavailability and brain localization. Drug Deliv. 2015, 22, 903–910. [Google Scholar] [CrossRef]
- Bhandwalkar, M.J.; Avachat, A.M. Thermoreversible Nasal in situ Gel of Venlafaxine Hydrochloride: Formulation, Characterization, and Pharmacodynamic Evaluation. AAPS PharmSciTech 2013, 14, 101–110. [Google Scholar] [CrossRef]
- Lin, T.; Liu, E.; He, H.; Shin, M.C.; Moon, C.; Yang, V.C.; Huang, Y. Nose-to-brain delivery of macromolecules mediated by cell-penetrating peptides. Acta Pharm. Sin. B 2016, 6, 352–358. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Akiyama, F.; Kakizaki, S.; Takashima, Y.; Seta, Y. Delivery of siRNA to the brain using a combination of nose-to-brain delivery and cell-penetrating peptide-modified nano-micelles. Biomaterials 2013, 34, 9220–9226. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Xiang, Q.; Su, J.; Yang, P.; Zhang, Q.; Su, Z.; Xiao, F.; Huang, Y. Evaluation of the Safety and Brain-Related Tissues Distribution Characteristics of TAT-HaFGF via Intranasal Administration. Biol. Pharm. Bull. 2014, 37, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Khafagy, E.-S.; Kamei, N.; Nielsen, E.J.B.; Nishio, R.; Takeda-Morishita, M. One-month subchronic toxicity study of cell-penetrating peptides for insulin nasal delivery in rats. Eur. J. Pharm. Biopharm. 2013, 85, 736–743. [Google Scholar] [CrossRef]
- Iwase, Y.; Kamei, N.; Khafagy, E.-S.; Miyamoto, M.; Takeda-Morishita, M. Use of a non-covalent cell-penetrating peptide strategy to enhance the nasal delivery of interferon beta and its PEGylated form. Int. J. Pharm. 2016, 510, 304–310. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, X.Y.; Wu, S.W.; Zhang, R.; Zhou, B.L.; Tang, L.; Tian, Y.; Men, K.; Yang, L. Enhanced nose-to-brain delivery of siRNA using hyaluronan-enveloped nanomicelles for glioma therapy. J. Control. Release 2022, 342, 66–80. [Google Scholar] [CrossRef]
- Kamei, N.; Tanaka, M.; Choi, H.; Okada, N.; Ikeda, T.; Itokazu, R.; Takeda-Morishita, M. Effect of an Enhanced Nose-to-Brain Delivery of Insulin on Mild and Progressive Memory Loss in the Senescence-Accelerated Mouse. Mol. Pharm. 2017, 14, 916–927. [Google Scholar] [CrossRef]
- Kamei, N.; Shingaki, T.; Kanayama, Y.; Tanaka, M.; Zochi, R.; Hasegawa, K.; Watanabe, Y.; Takeda-Morishita, M. Visualization and Quantitative Assessment of the Brain Distribution of Insulin through Nose-to-Brain Delivery Based on the Cell-Penetrating Peptide Noncovalent Strategy. Mol. Pharm. 2016, 13, 1004–1011. [Google Scholar] [CrossRef]
- Kamei, N.; Okada, N.; Ikeda, T.; Choi, H.; Fujiwara, Y.; Okumura, H.; Takeda-Morishita, M. Effective nose-to-brain delivery of exendin-4 via coadministration with cell-penetrating peptides for improving progressive cognitive dysfunction. Sci. Rep. 2018, 8, 17641. [Google Scholar] [CrossRef]
- Sasaki-Hamada, S.; Nakamura, R.; Nakao, Y.; Akimoto, T.; Sanai, E.; Nagai, M.; Horiguchi, M.; Yamashita, C.; Oka, J.-I. Antidepressant-like effects exerted by the intranasal administration of a glucagon-like peptide-2 derivative containing cell-penetrating peptides and a penetration-accelerating sequence in mice. Peptides 2017, 87, 64–70. [Google Scholar] [CrossRef]
- Akita, T.; Kimura, R.; Akaguma, S.; Nagai, M.; Nakao, Y.; Tsugane, M.; Suzuki, H.; Oka, J.-I.; Yamashita, C. Usefulness of cell-penetrating peptides and penetration accelerating sequence for nose-to-brain delivery of glucagon-like peptide-2. J. Control. Release 2021, 335, 575–583. [Google Scholar] [CrossRef]
- Khafagy, E.-S.; Kamei, N.; Fujiwara, Y.; Okumura, H.; Yuasa, T.; Kato, M.; Arime, K.; Nonomura, A.; Ogino, H.; Hirano, S.; et al. Systemic and brain delivery of leptin via intranasal coadministration with cell-penetrating peptides and its therapeutic potential for obesity. J. Control. Release 2020, 319, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Sasaki-Hamada, S.; Funane, T.; Nakao, Y.; Sasaki, R.; Nagai, M.; Ueta, Y.; Yoshizawa, K.; Horiguchi, M.; Yamashita, C.; Oka, J.-I. Intranasal administration of neuromedin U derivatives containing cell-penetrating peptides and a penetration-accelerating sequence induced memory improvements in mice. Peptides 2018, 99, 241–246. [Google Scholar] [CrossRef]
- Lou, G.; Zhang, Q.; Xiao, F.; Xiang, Q.; Su, Z.; Zhang, L.; Yang, P.; Yang, Y.; Zheng, Q.; Huang, Y. Intranasal administration of TAT-haFGF14–154 attenuates disease progression in a mouse model of Alzheimer’s disease. Neuroscience 2012, 223, 225–237. [Google Scholar] [CrossRef]
- Lou, G.; Zhang, Q.; Xiao, F.; Xiang, Q.; Su, Z.; Huang, Y. Intranasal TAT-haFGF Improves Cognition and Amyloid-β Pathology in an AβPP/PS1 Mouse Model of Alzheimer’s Disease. J. Alzheimers Dis. 2016, 51, 985–990. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Suh, J.S.; Lee, H.J.; Num, H.; Jo, B.S.; Lee, D.W.; Kim, J.-H.; Lee, J.Y.; Chung, C.P.; Lee, G.; Park, Y.J. Control of cancer stem cell like population by intracellular target identification followed by the treatment with peptide-siRNA complex. Biochem. Biophys. Res. Commun. 2017, 491, 827–833. [Google Scholar] [CrossRef]
- He, H.; Sheng, J.; David, A.E.; Kwom, Y.M.; Zhang, J.; Huang, Y.; Wang, J.; Yang, V.C. The Use of Low Molecular Weight Protamine Chemical Chimera to Enhance Monomeric Insulin Intestinal Absorption. Biomaterials 2013, 34, 7733–7743. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T. Brain delivery of small interfering ribonucleic acid and drugs through intranasal administration with nano-sized polymer micelles. Med. Devices Evid. Res. 2015, 8, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, A.; Carreiró, F.; Oliveira, A.M.; Neves, A.; Pires, B.; Venkatesh, D.N.; Durazzo, A.; Lucarini, M.; Eder, P.; Silva, A.M.; et al. Polymeric Nanoparticles: Production, Characterization, Toxicology and Ecotoxicology. Molecules 2020, 25, 3731. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Wong, L.R.; Xie, H.; Ho, P.C.-L. In Vitro and In Vivo Comparison of Curcumin-Encapsulated Chitosan-Coated Poly(lactic-co-glycolic acid) Nanoparticles and Curcumin/Hydroxypropyl-β-Cyclodextrin Inclusion Complexes Administered Intranasally as Therapeutic Strategies for Alzheimer’s Disease. Mol. Pharm. 2020, 17, 4256–4269. [Google Scholar] [CrossRef]
- Gao, X.; Tao, W.; Lu, W.; Zhang, Q.; Jiang, X.; Fu, S. Lectin-conjugated PEG–PLA nanoparticles: Preparation and brain delivery after intranasal administration. Biomaterials 2006, 27, 3482–3490. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, S.; Huang, L.; Ho, P.C.-L. Poly (ethylene glycol)-block-poly (D, L-lactide) (PEG-PLA) micelles for brain delivery of baicalein through nasal route for potential treatment of neurodegenerative diseases due to oxidative stress and inflammation: An in vitro and in vivo study. Int. J. Pharm. 2020, 591, 119981. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Chavhan, S.; Soni, H.; Babbar, A.K.; Mathur, R.; Mishra, A.K.; Sawant, K. Brain targeting of risperidone-loaded solid lipid nanoparticles by intranasal route. J. Drug Target. 2011, 19, 468–474. [Google Scholar] [CrossRef]
- Zheng, X.; Shao, X.; Zhang, C.; Tan, Y.; Liu, Q.; Wan, X.; Zhang, Q.; Xu, S.; Jiang, X. Intranasal H102 Peptide-Loaded Liposomes for Brain Delivery to Treat Alzheimer’s Disease. Pharm. Res. 2015, 32, 3837–3849. [Google Scholar] [CrossRef]
- Kubek, M.J.; Domb, A.J.; Veronesi, M.C. Attenuation of kindled seizures by intranasal delivery of neuropeptide-loaded nanoparticles. Neurotherapeutics 2009, 6, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Taki, H.; Tanaka, K.; Takashima, Y.; Okada, H. Cell-Penetrating Peptide-Modified Block Copolymer Micelles Promote Direct Brain Delivery via Intranasal Administration. Pharm. Res. 2011, 28, 2130–2139. [Google Scholar] [CrossRef] [PubMed]
- Taki, H.; Kanazawa, T.; Akiyama, F.; Takashima, Y.; Okada, H. Intranasal Delivery of Camptothecin-Loaded Tat-Modified Nanomicells for Treatment of Intracranial Brain Tumors. Pharmaceuticals 2012, 5, 1092–1103. [Google Scholar] [CrossRef]
- Kanazawa, T.; Morisaki, K.; Suzuki, S.; Takashima, Y. Prolongation of Life in Rats with Malignant Glioma by Intranasal siRNA/Drug Codelivery to the Brain with Cell-Penetrating Peptide-Modified Micelles. Mol. Pharm. 2014, 11, 1471–1478. [Google Scholar] [CrossRef] [PubMed]
- Kanazawa, T.; Kurano, T.; Ibaraki, H.; Takashima, Y.; Suzuki, T.; Seta, Y. Therapeutic Effects in a Transient Middle Cerebral Artery Occlusion Rat Model by Nose-To-Brain Delivery of Anti-TNF-Alpha siRNA with Cell-Penetrating Peptide-Modified Polymer Micelles. Pharmaceutics 2019, 11, 478. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Gao, X.; Gu, G.; Liu, Z.; Zeng, N.; Hu, Q.; Song, Q.; Yao, L.; Pang, Z.; Jiang, X.; et al. Low molecular weight protamine-functionalized nanoparticles for drug delivery to the brain after intranasal administration. Biomaterials 2011, 32, 9888–9898. [Google Scholar] [CrossRef]
- Rassu, G.; Soddu, E.; Posadino, A.M.; Pintus, G.; Sarmento, B.; Giunchedi, P.; Gavini, E. Nose-to-brain delivery of BACE1 siRNA loaded in solid lipid nanoparticles for Alzheimer’s therapy. Colloids Surf. B Biointerfaces 2017, 152, 296–301. [Google Scholar] [CrossRef] [PubMed]
- Samaridou, E.; Walgrave, H.; Salta, E.; Moriera Alvarez, D.; Castro-López, V.; Loza, M.; Alonso, M.J. Nose-to-brain delivery of enveloped RNA-cell permeating peptide nanocomplexes for the treatment of neurodegenerative diseases. Biomaterials 2020, 230, 119657. [Google Scholar] [CrossRef]
- Kanazawa, T.; Kaneko, M.; Niide, T.; Akiyama, M.; Kakizaki, S.; Ibaraki, H.; Shiraishi, S.; Takashima, Y.; Suzuki, T.; Seta, Y. Enhancement of nose-to-brain delivery of hydrophilic macromolecules with stearate- or polyethylene glycol-modified arginine-rich peptide. Int. J. Pharm. 2017, 530, 195–200. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Type of Classification | Categories | Examples | Refs. |
|---|---|---|---|
| Based on physical–chemical properties | Cationic peptides | TAT-derived moieties, penetratin, polyarginines, LMWP, crotamine | [4,24,25] |
| Amphipathic peptides | MPG, Pep-1, MAP, transportan, azurin-derived p28 peptide, proline-rich CPPs | [4] | |
| Hydrophobic peptides | C105Y and derivatives, Pep-7 | [4] | |
| Based on type of coupling to the cargo | Covalently bound | TAT derivatives, penetratin, and polyarginines | [24,26] |
| Non-covalently bound | Pep, MPG, KALA, KLA and PepFect types | [24,26] | |
| Based on uptake mechanism | Membrane translocation | Transportan analogs, MPG, Pep-1, dermaseptin | [4,22,26,31] |
| Endocytosis | TAT derivatives, polyarginines, transportan-based complexes, NickFect1, proline-rich CPPs, azurin-derived peptides, LMWP | [32,33,34,35,36,37,38] |
| Drug | Brand Name | Indications | Manufacturer | Status | Refs. |
|---|---|---|---|---|---|
| Dihydroergotamine mesylate (DHE-45) | Migranal® | Migraine | BAUSCH | Available | [83] |
| Desmopressin acetate | Stimate® | Hemophilia A | Ferring Pharmaceuticals US | Voluntary recall * | [84] |
| Nafarelin acetate | Synarel® | Central precocious puberty | Pfizer | Available | [85] |
| Butorphanol tartrate | Stadol® | Migraine and pain | Bristol Myers Squibb | Discontinued * | [86] |
| Zolmitriptan | Zomig® | Migraine | Astra Zeneca | Available | [87] |
| Desmopressin acetate | DDAVP® | Prevention of polydipsia and polyurea, head trauma | Ferring Pharmaceuticals US | Voluntary recall * | [88] |
| Esketamine | Spravato® | Resistant depression | Janssen-Cilag S.p.a. | Available | [89] |
| Midazolam | Nayzilam® | Seizure clusters | UCB pharmaceutics | Available (only in USA) | [90,91] |
| Diazepam | Valtoco® | Seizure clusters | Neurelis INC | Available (only in USA) | [92] |
| Strategies to Increase Brain Bioavailability | Examples | Refs. | |
|---|---|---|---|
| Enhancing drug solubility in the nasal cavity | Encapsulation complexes | IN delivery to the brain of GALP improved to threefold by encapsulation in cyclodextrins. | [113] |
| Microemulsion and nanoemulsion formulations | IN emulsion-like formulation improved delivery of GDF5 to all regions of the CNS and to trigeminal nerve. | [80,115] | |
| Reducing clearance, prolonging the residence time of the formulation at the delivery site. | Mucoadhesive and viscosity enhancing agents | Retention time and cell permeability of buspirone significantly increased in the rat nasal compartment, when formulated with 1% chitosan. | [116] |
| Mucoadhesive and viscosity enhancing agents and encapsulating agents | IN delivery of buspirone resulted in twofold higher brain AUC when formulated with 1% chitosan and 5% hydroxypropyl β-cyclodextrin compared to the simple solution. | [117] | |
| Reversible and irreversible ciliostatics and ciliotoxic drugs | Ciliostatics impair ciliary movement and decrease mucus clearance. Ciliotoxic drugs cause damage to the cilia or epithelium by destroying their structure or integrity. | [81] | |
| Biogels | IN administration of rufinamide in xyloglucan-based, heat triggered biogels resulted in higher brain AUC compared to the simple suspension. | [118] | |
| Reducing clearance due to efflux transporters or absorption by the nasal vasculature. | Transporter inhibitors | The use of transporter inhibitors such as rifampin resulted in greater brain uptake of several drugs. | [119,120] |
| Vasoconstrictors | The addition of phenylephrine to IN formulations of hypocretin-1 or L-Tyr-D Arg resulted in a reduction in the amount of drug adsorbed into the blood and an increase in the amount delivered to the OB. | [121] | |
| Reducing degradation by enzymes and proteases in the nasal cavity | Enzyme inhibitors | The use of P-glycoprotein inhibitors, CYP450 inhibitors. or acetazolamide reduced degradation and increased the amount of drug transported from the nasal compartment to the brain. | [81] |
| Biotherapeutic/Pathology | CPP Conjugated | Results | Refs. |
|---|---|---|---|
| Insulin/Alzheimer’s disease | Non-covalently conjugated L-/D-penetratin | -Both peptides enhanced insulin levels in OB. -L-penetratin increased insulin concentrations in the hypothalamus, cerebral cortex, cerebellum, and brain stem. -L-penetratin increased insulin levels in plasma (systemic side effects). -D-penetratin maintained a more favorable AUCbrain/AUCblood ratio | [20,132] |
| Insulin/Dementia | Non-covalently conjugated L-/D-penetratin | -L-penetratin enhanced IN delivery of insulin. -In early stage dementia, L-penetratin contributed to ameliorate the therapeutic action of insulin, although causing an undesirable hypoglycemic effect. | [131] |
| Exendin-4/Dementia | Non-covalently conjugated L-penetratin | -L-penetratin facilitated N2B delivery of exendin-4, increasing its concentration in hypothalamus, hippocampus, cerebral cortex, cerebellum, and brain stem. -Co-administration of exendin-4 and L-penetratin only slightly improved progressive cognitive dysfunction in a senescence-accelerated mouse model (SAMP8 mice). | [133] |
| Exendin-4 + Insulin/Dementia | Non-covalently conjugated L-penetratin | -Co-administration of exendin-4 and low-dose insulin with L-penetratin improved cognitive and spatial functions in SAMP8 mice. | [133] |
| haFGF/Alzheimer’s disease | Covalently-bound TAT | -TAT conjugation enhanced delivery and distribution of haFGF to the brain. -Favorable brain to blood ratio is maintained. -IN TAT-haFGF was more effective than the IV administration in increasing ACh levels, improving learning and memory abilities, and reducing the number and size of Aβ plaques in the AβPP/PS1 AD mouse brain. | [138,139] |
| haFGF/Dementia | Covalently-bound TAT | -TAT-haFGF treatment corrected cholinergic deficits, decreased the amount of Aβ deposits, and decreased the number of apoptotic neurons and oxidative stress in SAMP8 mice. -Improved learning and memory abilities of SAMP8 mice. | [138] |
| Leptin/Obesity | Non-covalently conjugated L-penetratin | -The treatment causes stimulation of leptin receptors and activation of Stat3 effector, with consequent reduction in the plasma triglycerides and decreased body weight. | [136] |
| NMU/Inflammation-mediated amnesia | PAS-R8 F4-R8 | -PAS-R8-NMU is more stable and more efficiently delivered to the CNS than F4-R8-NMU. -While both IN-administered NMU derivatives prevented or reduced LPS-induced memory impairment, IN NMU alone did not. | [137] |
| GLP-2/Major depression | PAS-R8 | -Both the CPP and the PAS portions are essential to ensure GLP-2 access to the brain upon IN administration and to induce an antidepressant-like effect. -The systemic absorption of PAS-CPP-GLP-2 was extremely low. | [134,135] |
| BSA | Covalently-bound LMWP | -Conjugation with LMWP gives BSA the ability to reach the CNS (OB) and achieve deep inward penetration. | [125] |
| HRP β-gal | Covalently-bound LMWP | -Conjugation with LMWP enhanced the retention of enzymatic activity of the two proteins in the CNS. | [125] |
| Nanocarrier | Pharmacological Agent | Results | Ref. |
|---|---|---|---|
| MPEG-PLC-TAT polymeric micelles | Coumarin | -Brain distribution of coumarin, 4 h after IN administration, was higher with MPEG-PLC-TAT micelles than with the MPEG-PLC micelles. -Concentration of coumarin in non-targeted tissues was lower when administered with MPEG-PLC-TAT compared to the solution alone. | [55] |
| Camptothecin | -MPEG-PLC-TAT micelles were more effective than the MPEG-PLC micelles in ensuring an interaction with glioma cells and delivering the agent to the rat brain. | [152] | |
| siRNAs | -MPEG-PLC-TAT guaranteed a better mucosa permeability and a higher transfer to the OB and trigeminal nerve when compared to the naked siRNA. | [126] | |
| Camptothecin+siRaf-1 | -MPEG-PLC-TAT micelles enhanced siRaf-1 and camptothecin delivery to the brain both individually or in combination | [153] | |
| siTNF-α | -This system provided shrinkage of the infarcted area, suppression of TNF-α production, and improvement in the neurology scores in the t-MCAO rats | [154] | |
| HA/DP7-C polymeric micelles | siVEGF siPLK1 | -In glioma cells, these systems provided the downregulation of VEGF and PLK1 mRNA and protein levels, suppression of angiogenesis for the siVEGF system and induction of apoptosis for the siPLK1 system. -In mice, the IN administration of these systems downregulated the VEGF and PLK1 protein levels and prolonged survival. | [130] |
| PEG-PLA nanoparticles functionalized with LMWP | Coumarin | -These systems enhanced cellular accumulation in vitro when compared to non-CPP-coupled nanoparticles. -These nanoparticles increased the amount of drug delivered to different brain areas in vivo, exhibited higher Cmax and AUC0–8h, and showed enhanced AUCbrain/AUCblood ratio than non-functionalized nanoparticles. | [155] |
| RVG-9R- chitosan coated/uncoated solid lipid nanoparticles | BACE1 siRNA | -Both formulations enhanced the penetration of the siRNA through an in vitro model of the nasal epithelium. | [156] |
| C12-R8 PEG-PGA nanocomplexes | miR-132 | -These nanocomplexes increased the rate of internalization of the therapeutic agent in vitro. -These systems were capable of delivering miR-132 to the hippocampus, increasing the level of miRNA-132, and downregulating the target mRNAs. | [157] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Martini, L.B.; Sulmona, C.; Brambilla, L.; Rossi, D. Cell-Penetrating Peptides as Valuable Tools for Nose-to-Brain Delivery of Biological Drugs. Cells 2023, 12, 1643. https://doi.org/10.3390/cells12121643
De Martini LB, Sulmona C, Brambilla L, Rossi D. Cell-Penetrating Peptides as Valuable Tools for Nose-to-Brain Delivery of Biological Drugs. Cells. 2023; 12(12):1643. https://doi.org/10.3390/cells12121643
Chicago/Turabian StyleDe Martini, Lisa Benedetta, Claudia Sulmona, Liliana Brambilla, and Daniela Rossi. 2023. "Cell-Penetrating Peptides as Valuable Tools for Nose-to-Brain Delivery of Biological Drugs" Cells 12, no. 12: 1643. https://doi.org/10.3390/cells12121643
APA StyleDe Martini, L. B., Sulmona, C., Brambilla, L., & Rossi, D. (2023). Cell-Penetrating Peptides as Valuable Tools for Nose-to-Brain Delivery of Biological Drugs. Cells, 12(12), 1643. https://doi.org/10.3390/cells12121643