1. Introduction
Niemann–Pick diseases are a subgroup of lysosomal storage disorders (LSDs) first described by Albert Niemann and Ludwig Pick in the early 20th century that can be further categorised by their respective lysosomal protein deficiency. Niemann–Pick Type A (NP-A) and B (NP-B) are caused by the loss of acid sphingomyelinase (ASM) function, whereas Type C1 and C2 result from the loss of either
NPC1 or
NPC2 function, respectively [
1,
2]. Niemann–Pick disease type C1 (NP-C) (OMIM 257220) results from mutations in the
NPC1 gene and is a prematurely lethal genetic lysosomal storage disorder (LSD). Although visceral organs including the liver and spleen are affected, the clinical course is dominated by progressive neurodegeneration in the brain leading to premature death [
1,
2]. The most prominent and earliest pathology is seen in the brain, resulting in neuron loss, especially that of the Purkinje cells in the cerebellum. The clinical onset of symptoms varies, but can be broadly classified into early infantile, late infantile, juvenile, and adult cases [
1]. Progressive neurological symptoms include ataxia, cognitive decline, dementia, vertical gaze palsy, epilepsy, and dysphagia with subsequent aspiration pneumonia as a leading cause of death [
3].
The
NPC1 gene encodes a 13-domain transmembrane protein that is localised to the membrane of the late endosomes and lysosomes and plays a central role in transporting cholesterol from these vesicular organelles into the membranes [
4]. Mutations in
NPC1 lead to an accumulation of glycosphingolipids, sphingosine, sphingomyelin, and cholesterol in cells of the body. However, which of these metabolites leads to pathology is yet to be determined [
5,
6].
Various mouse models of NP-C are available for the characterisation of pathology and pre-clinical assessment of therapeutic modalities. The most extensively studied is the NPC1 null model (
Npc1nih or
Npc1m1n) that carries a spontaneous mutation and accumulates the hallmark cholesterol and sphingolipids [
7] (
https://www.informatics.jax.org/allele/summary?markerId=MGI:1097712, accessed on 11 June 2023). Neurodegeneration is observed in the cerebellum but also the thalamus, cortex, and substantia nigra and is accompanied by microglial and astrocyte activation [
8]. The neurological symptoms that develop at 6 weeks of age (tremor and decline in locomotor function) reflect those seen in NP-C patients and the mice reach their humane endpoint at 9–10 weeks of age. An alternative mouse model of NP-C is the
Npc1nmf164 which carries a point mutation in a region of the
NPC1 gene where a high proportion of mutations are found in NP-C patients [
9]. The biochemical, locomotor, and pathological phenotype is similar to that observed in
Npc1nih but has a slower progression.
While there are ongoing efforts such as enzyme replacement therapy [
10] and autophagic modulation [
11] for NP-A and NP-B, the availability of reliable mouse models has led to the successful pre-clinical testing of different therapeutic modalities and their subsequent clinical trials. These include the heat shock protein arimoclomol (ClinicalTrials.gov Identifier: NCT04316637), hydroxypropyl-beta-cyclodextrin (NCT04860960, NCT03887533), N-Acetyl-L-Leucine (NCT03759639), HDAC inhibitor vorinostat (NCT02124083), and substrate reduction therapy using miglustat (NCT00517153). To date, miglustat is currently the only licensed drug for treating NP-C in some territories and is generally considered to have a disease modifying effect [
12,
13].
Following the recent regulatory approval and licensing of the adeno-associated viral vector (AAV)-based gene therapy Zolgensma for the treatment of spinal muscular atrophy [
14,
15], there is hope for this approach in other neurological diseases including NP-C. A number of pre-clinical studies, including those conducted by our group, have shown that the AAV9 vector is able to deliver a therapeutic copy of the
NPC1 gene to the brain of the
Npc1nih mouse model leading to extension of lifespan, improved locomotor function, an amelioration of neuronal loss and neuroinflammation, and a reduction in cholesterol and glycosphingolipid accumulation [
16,
17,
18,
19]. This is encouraging given that there is no therapeutic advantage from the cross-correction of untransduced neighbouring cells, as NPC1 is membrane-bound [
20]. Furthermore, the
NPC1 cDNA sequence is relatively large (3.8Kb final coding sequence, RefSeq: CCDS11878.1) in the context of the limited packaging capacity of AAV vectors (4.7 Kb) and restricts the selection of genetic regulatory sequences such as promoters and polyA signals, leading to possible packaging and truncation errors [
21]. Therefore, despite the promising results of these preclinical studies, further optimisation of the vectors is required to rise to the challenge of having a therapeutic effect in the larger and more complex human brain.
The promoter sequence is a crucial DNA regulatory element in the viral vector genome that determines the level of transgene expression as well as tissue specificity. Often, a strong, constitutively expressed promoter is desired for the high-level expression of NPC1, as there is no possible cross-correction of this membrane-bound protein [
20]. Commonly used promoters of this type include the CMV (cytomegalovirus) promoter/enhancer, EFS (elongation factor 1a), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), mPGK (phosphoglycerate kinase), CBA (chicken β-actin), and CAG (chicken β-actin promoter with CMV enhancer). All these promoters provide constitutively active gene expression in most cell types [
22,
23]. We have previously described the efficacy of the neuron-selective human Synapsin1 (SYN) promoter in treating mouse models of neurodegenerative LSDs, including neuronopathic Gaucher disease [
24] and NP-C [
16]. However, as many of these promoters have sizes larger than 400 bp that may lead to packaging and truncation issues during AAV vector production, it is crucial to find a promoter that drives strong expression but is small in length for developing an optimal gene therapy strategy for NP-C.
In this study, we compared a broad range of promoters to identify one that is small enough while maintaining high-level gene expression of the large NPC1 gene and remain within the optimal 4.7 Kb packaging capacity of AAV9. Ideally, this small promoter would also outperform other sequences commonly used in gene therapy studies and clinical trials to provide maximum levels of NPC1 expression and subsequent enhanced therapeutic efficacy. We found that a small, truncated version of the endogenous human NPC1 promoter fulfilled these criteria and significantly enhanced the therapeutic efficacy in both the Npc1nih and Npc1nmf164 mouse models. This optimised AAV vector provides a major step forward towards the effective clinical translation of gene therapy for NP-C.
2. Materials and Methods
2.1. Plasmid and Vector Production
Human
NPC1 cDNA (RefSeq CCDS11878.1) was cloned into an AAV construct containing the various promoter sequences (
Supplementary File S2) and SV40 late polyadenylation signal sequence, flanked by AAV2 inverted terminal repeats.
Human Embryonic Kidney 293T (HEK293T) cells were triple transfected with the ITR-containing plasmid carrying the respective therapeutic transgene cassette, a helper plasmid expressing AAV2 Rep and AAV9 Capsid, and a third construct containing the adenovirus helper functions (HGTI) using 3.5 mg/mL DNA polyethylenimine MAX (Polysciences, Inc. Warrington, PA, USA) and a 1:1:3 ratio of the respective plasmids [
25]. Cells were harvested by centrifugation 72 h post-transfection and lysed by three freeze–thaw cycles (−80 °C to 37 °C) with vortexing in a lysis buffer (150 mM NaCl, 50 mM Tris, pH 8.5). Subsequent benzonase treatment (Sigma, Dorset, UK) was followed by lysate, and was cleared by centrifugation at 3200 g for 30 min. Subsequently, iodixanol gradient purification was performed in ultracentrifuge tubes (Beckman Instruments, High Wycombe, UK) with the lysate overlaid on increasing layers of 15, 25, 40, and 60% iodixanol (OptiPrep; Sigma). The tubes were centrifuged (Sorvall Discovery 90SE) for 3 h at 200,000 g in a TH641 (ThermoScientific, Paisley, UK) rotor. The vector was extracted from the 40% fraction with a 19-gauge needle, diluted in sterile phosphate-buffered saline, filtered at 0.22 mm, and concentrated in a Vivaspin 20 with a 100,000 molecular weight cut-off (Sartorious Stedim Biotech, Epsom, UK) centrifugal concentrator.
The concentrated vector genome was determined using quantitative PCR, using primers to the SV40 polyA sequence. The integrity of the vector genome was assessed using alkaline agarose gel electrophoresis [
26]. Briefly, alkaline gels were run with 0.05 M NaOH as the running buffer, stained post-electrophoresis with 4× GelRed stain (Biotium, Fremont, CA, USA), and quantified against a HyperLadder 1 kb (Bioline Reagents, London, UK). Further analysis of concentrated AAV particle titre and vector purity was also assessed by visualization of the capsid proteins VP1, VP2, and VP3 via a SYPRO Ruby (ThermoScientific) protein stain, after sodium dodecyl sulfate–polyacrylamide gel electrophoresis [
27].
2.2. Cell Culture and Plasmid Transfection
HEK 293T cells were grown in a standard growth medium of Dulbecco’s modified Eagle’s medium (DMEM; Gibco, ThermoFisher Scientific, Waltham, MA, USA) with high glucose (4.5 mg/mL), GlutaMAX (0.8 mg/mL), and pyruvate (0.1 mg/mL) and supplemented with 10% heat-inactivated foetal bovine serum (FBS; Sigma-Aldrich), penicillin (100 units/mL), and streptomycin (100 µg/mL). All cells were grown on poly-D-lysine coated six-well plates, T75 and T175 flasks, and 100 mm dishes to maintain adherence (Nunc, ThermoFisher Scientific). Cells were incubated at 37 °C in 5% CO2 for an average of 48–72 h between passaging.
For transfecting plasmids, cells were seeded in either a six-well plate (Nunc) at a ratio of 200,000 cells/well or a twelve-well plate at 10,000 cells/well with Poly-D-Lysine (PDL) coated coverslips (Thermofisher) along with standard growth media and incubated for 24 h. Plasmid DNA was mixed with the transfection agent linear polyethylenimine MAX (PEI MAX Mw ~ 40,000, Polysciences Inc., Warrington, PA, USA) at a ratio of 1:3 in reduced serum media OptiMEM (Gibco). Transfection mix was incubated at room temperature for 15 min to allow DNA-PEI complex formation and was subsequently added to the cells in the standard growth media. After 24 h, the media was replaced with fresh standard growth media and incubated for a further 24–48 h, at which point cells were either imaged or collected for downstream protein analysis.
2.3. Primary Cortical Cell Culture of E15 Mouse Embryos
Pregnant CD1 mice were sacrificed by schedule 1 methods at 15 days of gestation and embryonic cortices prepared as described previously [
28]. Briefly, cortices from embryos in a single litter were dissected, meninges were removed, and the tissue was pooled. Cortices were roughly chopped before incubation in 0.25% trypsin/EDTA followed by trituration. Cells were pelleted by centrifugation, resuspended, and plated in a Neurobasal medium supplemented with B27, 100 units/mL of penicillin, 100 μg/mL of streptomycin, 0.25 μg/mL of amphotericin B, 300 μM of glutamine, and 25 μM of 2-mercaptoethanol at a density of 2.106 cells/6 cm plate. Transfections with plasmids were carried out on cells grown for a minimum of 7 days in vitro.
2.4. Immunocytochemistry and Cellular Imaging
For imaging cells, cells grown and transfected on PDL-coated coverslips were fixed using 4% Paraformaldehyde added to culture media for 15 min at room temperature. Coverslips were washed five times with Phosphate Buffered Saline (PBS) and blocked with 10% Normal serum solution in 0.1% PBS with Triton-X (PBS-T). Coverslips were then incubated overnight in an anti-GFP antibody (Abcam, Cambridge, UK), ab290, 1:200) in 10% normal serum solution in PBS-T followed by three washes in PBS and 2 h incubation in a secondary antibody (goat anti-rabbit Alexa Fluor 488, 1:200, Life Technologies, Paisley, UK) and three washes in PBS. Coverslips were then counterstained with DAPI for nuclear visualization, mounted, and coverslipped with Fluoromount G (SouthernBiotech, Birmingham, AL, USA). Sections were visualized with a laser scanning confocal microscope (Zeiss LSM 710, Carl Zeiss AG, Cambridge, UK).
2.5. Animals
All animal studies were approved by the UK Home Office for the conduct of regulated procedures under license (Animal Scientific Procedures Act, 1986) and according to ARRIVE guidelines and recommendations. Wild-type CD-1 mice, as well as wild-type and Npc1nih/nih mice on Npc1nih (BALB/c) and Npc1nmf164 (C57Bl6/J) were maintained as individual colonies in approved biological service units at University College London under a 12 h light/dark cycle and provided food and water ad libitum. For survival, any mice that lost 15% of their body weight after a 24 h period were considered to have reached their humane endpoint and sacrificed.
2.6. Intracerebroventricular (ICV) Injections
Neonatal injections were carried out as previously described [
27]. Briefly, viral vector preparations were injected into 1-day post-gestation (P1) neonatal mice via bilateral ICV injection targeting the anterior horn of the lateral ventricle using a 33-gauge needle (Hamilton, Reno, NV, USA). Injected neonates were subsequently returned to the dam. After appropriate duration post-injection, the mice underwent terminal exsanguination by trans-cardiac perfusion with phosphate-buffered saline. Brains and organs were subsequently extracted and either fixed in 4% paraformaldehyde for immunohistochemistry or snap frozen on dry ice and stored at −80 °C for protein analysis.
2.7. IVIS Imaging
Imaging for luciferase expression in vivo was performed as previously described [
29]. Briefly, animals were anesthetized with isoflurane (Abbott Laboratories), injected intraperitoneally (I.P.) with firefly D-luciferin (15 mg/mL in PBS; Gold Biotechnology, St. Louis, MO, USA), and imaged 5 min later with a cooled charge-coupled device (CCD) camera (IVIS; PerkinElmer, Waltham, MA, USA). The luciferin dose was 150 mg/kg; the volume varied with the age/size of the animal. Grey-scale photographs were acquired with a 24 cm field of view and a bioluminescence image was obtained using a binning/resolution factor of 4, a 1.2/f stop, and open filter. Regions of interest (ROIs) were defined manually using a standard area for each organ under investigation. Signal intensities were calculated with Living Image software v4.0(Perkin Elmer) and expressed as photons/second/cm
2/steradian.
2.8. Western Blotting
For cell culture experiments, 48–72 h post-transfection cells were collected for protein extraction. A total of 100 µL of cold RIPA lysis buffer (ThermoFisher Scientific) with a 1× protease inhibitor cocktail (ThermoFisher Scientific) was added per well and incubated on ice for 5 min, swirling the plate every minute. Cells were then scraped and collected at the bottom of the well and the lysate was transferred to 1.5 mL microcentrifuge tubes. Resulting lysates were incubated on ice for 30 min, after which debris was pelleted by centrifugation at 14,000× g, 4 °C for 20 min. Lysate was carefully extracted and the overall protein concentration was determined by Pierce BCA Protein Assay (LifeTechnologies (Carlsbad, CA, USA), ThermoFisher Scientific). Sample protein concentrations were standardised to 1 µg/µL to standardise the loading volumes.
For brain tissues, tissues were homogenized (Ultra-Turrax TP, IKA Labortechnik, Wasserburg, Germany) on ice in 300 mL of RIPA lysis buffer (Thermo) per 100 mg of tissue with a 1× protease inhibitor cocktail (Thermo) and incubated for 30 min. Lysates were centrifuged at 14,000× g, 4 °C for 20 min and the overall protein concentrations of the supernatant was determined by Pierce BCA Protein Assay (Life Technologies).
Samples were incubated at 37 °C for 30 min in a 1× LDS sample buffer (Life Technologies) and a 1× sample reducing agent (Life Technologies), after which 40 mg of protein were loaded per well in a NuPAGE Bis–Tris 4–12% polyacrylamide gel for protein separation via SDS-PAGE electrophoresis. Proteins were transferred to a PDVF membrane at 400 mA for 1 h and the membrane was blocked for 1 h at 4 °C with 5% BSA in Tris Buffered Saline (TBS) with 3% Tween-20. Membranes were subsequently incubated overnight at 4 °C with primary antibodies for NPC1 (1:10,000, ab134113, Abcam), b-tubulin (1:2000, ab6161, Abcam), or b-Actin (1:100, ab8227, Abcam) with 3% BSA in TBS with 3% Tween-20. After three washes in TBS, antibody staining was revealed using HRP-conjugated goat anti-rabbit IgG (1:2000, ab6721, Abcam) and goat anti-rat IgG (1:10,000, ab97057, Abcam) incubated for 2 h at room temperature in TBS 3% Tween-20 with 3% BSA. Blots were developed with the ECL system (SuperSignal West Pico, Life Technologies) and imaged using a Genegnome imager (Syngene, Cambridge, UK).
2.9. Behavioural Analysis
2.9.1. Tremor
Npc1nih/nih mice demonstrate an age-dependent increase in 32–55 Hz high frequency tremors [
5]. Tremor analysis was carried out on control, untreated, and treated
Npc1nih/nih at 10 weeks of age, using a commercial tremor monitor (San Diego Instruments, San Diego, CA, USA) as previously described [
30]. Individual mice were placed inside the apparatus on an anti-vibration table and monitored for 256 s, after 30 s of acclimatisation time. The output (amplitude/time) was analysed using LabView software, to give a measurement of power at each monitored frequency (0–64 Hz).
2.9.2. Gait
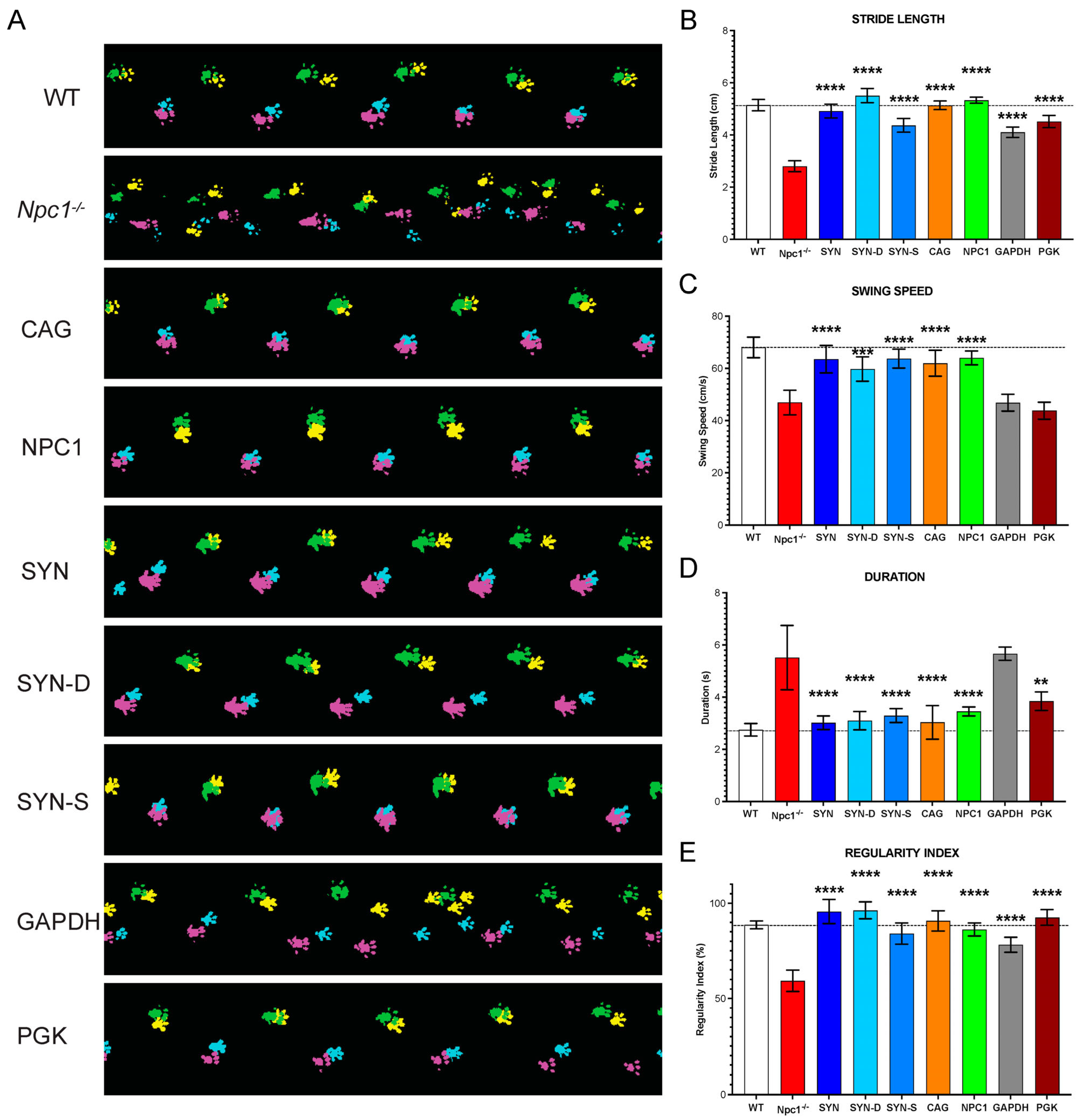
Gait monitoring was performed on control, untreated, and treated
Npc1nih/nih at 10 weeks of age by using the automated gait analysis CatWalk system (Noldus, Wageningen, The Netherlands). The mice were monitored by being individually placed at one end of the CatWalk and were filmed freely walking across a filmed section with a backlit stage. A minimum of five successful runs were recorded per session, where a run across the stage was deemed successful if standard run criteria were met. Paw prints during successful runs were checked, classified, and analysed using the CatWalk XT software v10.6 (Noldus) to produce overall run measurements. Parameters recorded included the duration of the run (seconds), stride length (the distance between successive paw placement of the same paw in cm), swing speed (speed of the paw between successive paw placement, cm/sec), and regularity index (% index for the degree of interlimb coordination during gait) [
16].
2.10. Histological Tissue Processing and Analysis
After 48 h fixation in 4% PFA, the brains were transferred into 30% sucrose in phosphate-buffered saline for cryoprotection. The brains were then cryosectioned at 20 °C using a Leica CM3050 cryostat microtome to 20 µm thickness.
Briefly, a one in twelve series of coronal brain sections and one in six sagittal cerebellum sections from each mouse were stained on slides using a modified immunofluorescence protocol [
31] for the following antibodies–astrocytes (mouse anti-GFAP, 1:200, Millipore MAB3402), microglia (rat anti-mouse CD68, 1:400, Bio-Rad MCA1957), LAMP1 (rabbit anti-LAMP1, 1:200, Abcam ab24170), and calbindin (Rabbit anti-calbindin, 1:2000, Swant CB38a). A total of 20 µm coronal sections were mounted on superfrost plus slides (Fisher Scientific) and air-dried for 30 min, and the slides were then blocked in a 15% serum solution (Normal goat serum, S-1000 Vector Laboratories) in 4% TBS-T (1× Tris Buffered Saline, pH 7.6 with 4% Triton-X100, Fisher Scientific) for 1 h. The slides were then incubated in a primary antibody in 10% serum solution in 4% TBS-T for 2 h. Slides were washed three times in 1× TBS and incubated in fluorescent Alexa-Fluor-labelled IgG secondary antibodies (Alexa-Fluor goat anti-rabbit 488 Invitrogen A-11008, goat anti-rat 546 Invitrogen A-11081, Alexa-Fluor goat anti-mouse 546 Invitrogen A-11003) in 10% serum solution in 4% TBS-T for 2 h, washed three times in 1× TBS, and incubated in a 1× solution of TrueBlack lipofuscin autofluoresence quencher (Biotium, Fremont, CA, USA) in 70% ethanol for 2 min before rinsing in 1× TBS. Slides were coverslipped in a fluoromount-G mounting medium with DAPI (Southern Biotech, Birmingham, AL, USA).
A 3,3′-diaminobenzidine (DAB, Sigma-Aldrich) mediated immunohistochemistry (IHC) was used for neuron counts with NeuN (mouse anti-NeuN, 1:500, Millipore MAB377) and cortical analyses. Free-floating brain sections were incubated in a 1% H2O2 solution for 30 min to block endogenous peroxidase activity followed by three washes with TBS. A block of non-specific binding was then performed by a 30 min incubation in 15% normal serum with 0.3% TBS-T. The solution was then changed to 10% normal serum in 0.3% TBS-T with the primary antibody and left to incubate overnight at 4 °C. The sections were washed three times with TBS followed by incubation in a biotinylated secondary antibody (anti-mouse, 1:1000, Vector Laboratories) in 10% normal serum with 0.3% TBS-T for 2 h. After another three washes with TBS, visualisation of the staining was achieved with a Vectastain avidin-biotin solution (Vector Laboratories) and DAB (Sigma). The sections were then mounted, dehydrated, cleaned in histoclear, and coverslipped with DPX (VWR).
To analyse the degree of immunofluoresence in cellular images as well as the brain sections, a semiautomated thresholding image analysis was used with Image-Pro Premier software v9.1 (Media Cybernetics, Rockville, MD, USA) [
31]. For in vitro cultures, 30 non-overlapping images were collected for
n = 3 samples, as described above, at a 40× magnification. For brain sections, slide-scanned images at 10× magnification for stained sections were collected for all animals using a Zeiss Axioscan Z1 (Zeiss Microscopy Deutschland GmbH, Oberkochen, Germany) followed by demarcation of the anatomical regions of interest. Images were subsequently analyzed using Image-Pro Premier (Media Cybernetics) using appropriate thresholds that selected the foreground immunoreactivity above the background. Separate thresholds were used for each antigen and each anatomical region analysed.
Cortical thickness was measured in brain sections stained with Anti-NeuN antibody using Stereo Investigator software (MBF Bioscience, Williston, VT, USA). This was done by drawing 10 individual contours over the entire thickness of the cortex across three separate sections and collecting the average value in µm.
Estimates of neuron populations in the cerebellum for Calbindin-positive Purkinje neurons were performed by manual stereological counts and normalised per 1000 µm. Counts in the barrel field of the primary somatosensory cortex (S1BF) and Ventral posteromedial/lateral nuclei (VPM/VPL) of the thalamus were performed using a design-based optical fractionator method in a 1 in 12 series of NeuN-stained sections using Stereo Investigator software (MBF Bioscience) [
31]. Cells were sampled with counting frames (90 × 100 μm) distributed over a sampling grid (VPM/VPL, 350 × 350 μm; S1BF, 450 × 450 μm) that was superimposed over the region of interest at 100× magnification.
2.11. Statistical Analysis
All statistical analyses were carried out with GraphPad Prism software (Version 9.0e). Multiple comparisons were analysed by one-way analysis of variance (ANOVA) followed by post hoc Bonferroni’s correction, or a two-way ANOVA post hoc Bonferroni’s correction. All graphs are plotted as the mean ± the standard deviation (SD), unless stated otherwise, and statistical significance was compared to controls and assumed for
p < 0.05; ns—non-significant, *
p < 0.05, **
p < 0.01, ***
p < 0.001, and ****
p < 0.0001. Full details of all tests are reported in
Supplementary File S1.
4. Discussion
NP-C is a prematurely fatal neurodegenerative lysosomal storage disorder. Despite a plethora of preclinical therapies being tested for NP-C, only one drug, miglustat, has been approved for clinical therapy. However, miglustat slows disease progression and extends life span, but it does not halt disease progression [
12,
13]. Therefore, there is a significant need for readily translatable preclinical therapies for NP-C. With the recent regulatory approval of Zolgensma for spinal muscular atrophy [
14,
15], there is greater hope for a similar approach to be successful for other neurological disorders, including NP-C. While gene therapy has previously been tested in preclinical models of NP-C [
16,
17,
18,
19], these approaches are yet to be successfully translated into the clinic. A major hurdle in designing an AAV vector for NP-C is the size of the human
NPC1 gene. The large size of the gene means less space for other crucial elements such as the promoter, polyA signal, or regulatory sequences. Exceeding the limited packaging capacity of AAV vectors (4.7 Kb) could lead to truncation and packaging defects during vector manufacturing [
21].
While techniques using spilt vectors, overlapping dual vectors [
36], or development of minigenes [
37] exist to reduce the size of the transgene, another avenue to be pursued would be the choice of promoter. The promoter sequence not only determines the level of transgene expression as well as tissue specificity but may also significantly influence the overall size of the gene product to be packaged [
24,
38,
39,
40].
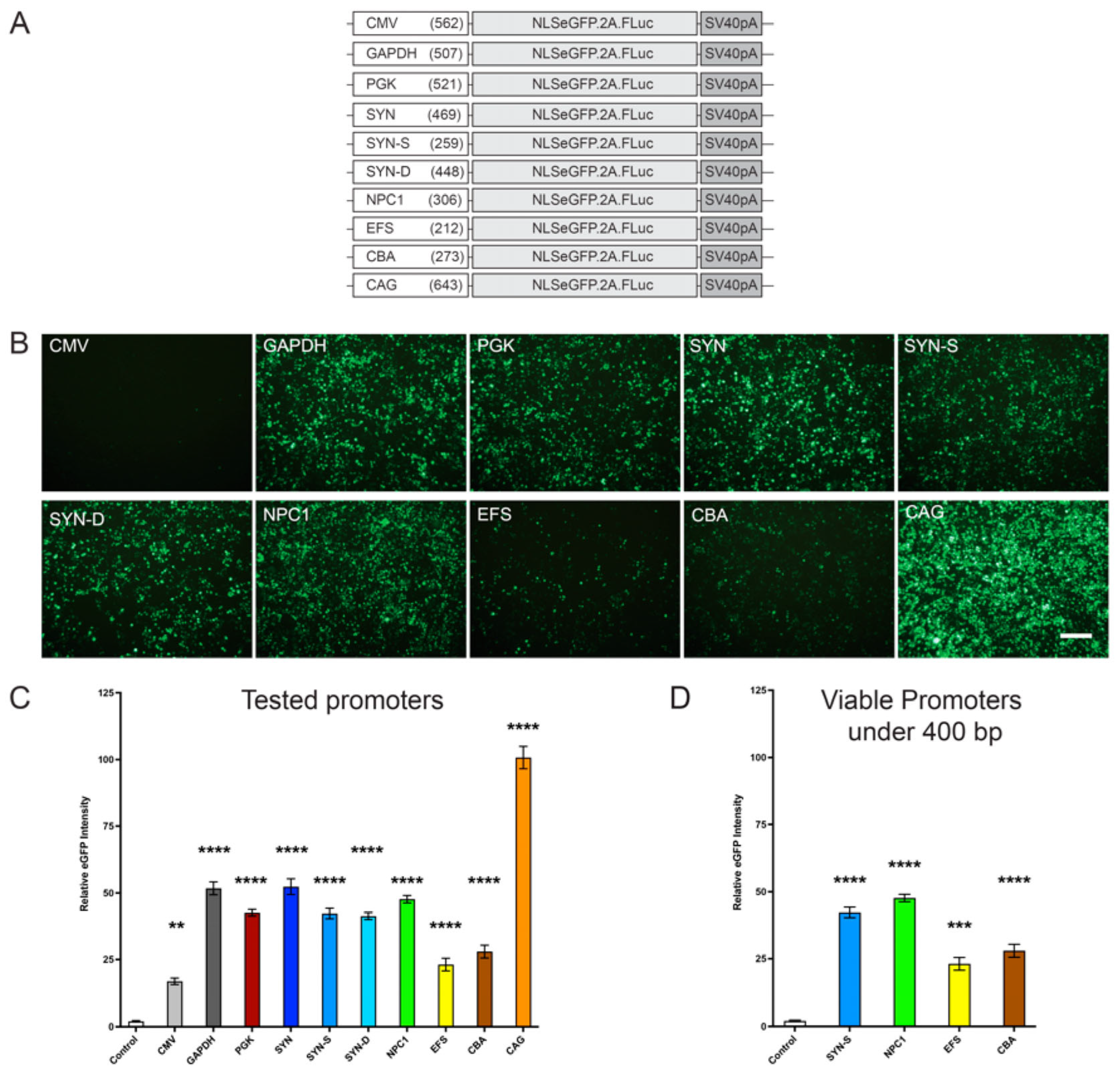
Therefore, given the need for a small, ubiquitous, and efficient promoter for NPC1, we compared various promoters, including CMV (cytomegalovirus) promoter/enhancer, EFS (elongation factor 1a), GAPDH (glyceraldehyde-3-phosphate dehydrogenase), mPGK (phosphoglycerate kinase), CBA (chicken β-actin), CAG (chicken β-actin promoter with CMV enhancer), Synapsin-1, and a truncated NPC1 promoter. Of these, the EFS, CBA, and NPC1 promoters were the only promoters under the desired 400 bp size. Further, we also tested a neuron de-targeted Synapsin-1 (SYN-D) promoter and a shortened Synapsin-1 (SYN-S) promoter under 400 bp. While the focus was to find an efficient promoter under 400 bp, it was also crucial to compare these with more commonly used, but larger promoters, to see if this efficacy could be matched.
Our data revealed that when tested in vitro, the small truncated
NPC1 promoter showed the highest level of expression of both a reported eGFP as well as the h
NPC1 gene, among the promoters under 400 bp. While this expression was lower than other larger promoters including CAG, PGK, and GAPDH, these were still unexpectedly significant results for this novel promoter (
Figure 1 and
Figure 3). Furthermore, unlike Synapsin-1, which is a predominantly neuronal promoter, the
NPC1 promoter was also expressed in astrocytes in culture, similar to the CAG promoter, indicating its ubiquitous expression in the CNS (
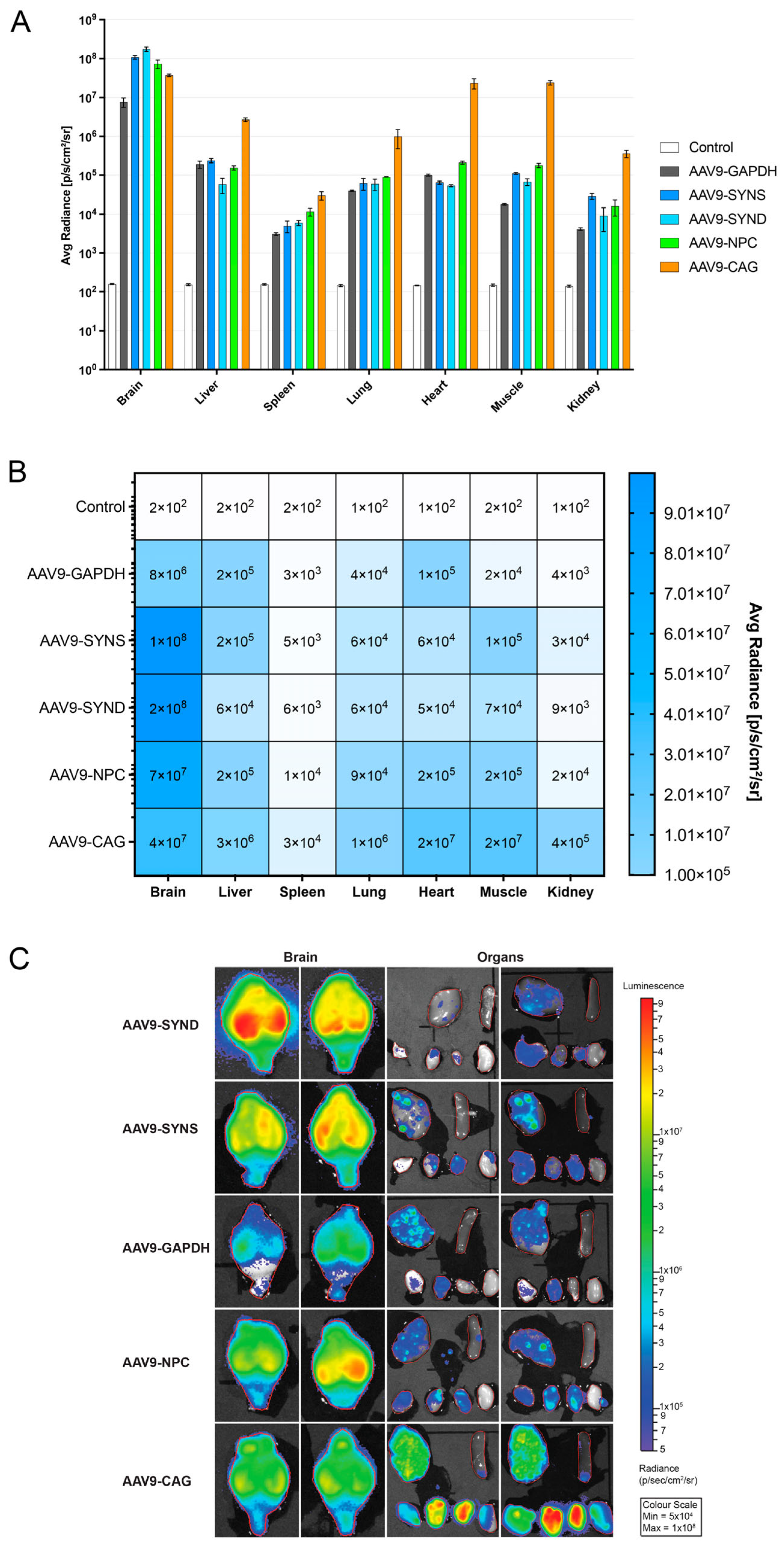
Supplementary Figures S1 and S2). In vivo injections of viral preparations into the ventricles of the neonatal wild-type mice also showed high levels of brain expression, even when compared to a strong neuronal promoter—synapsin-1 (
Figure 2).
To test therapeutic efficacy, we then injected the various viral preparations into the neonatal
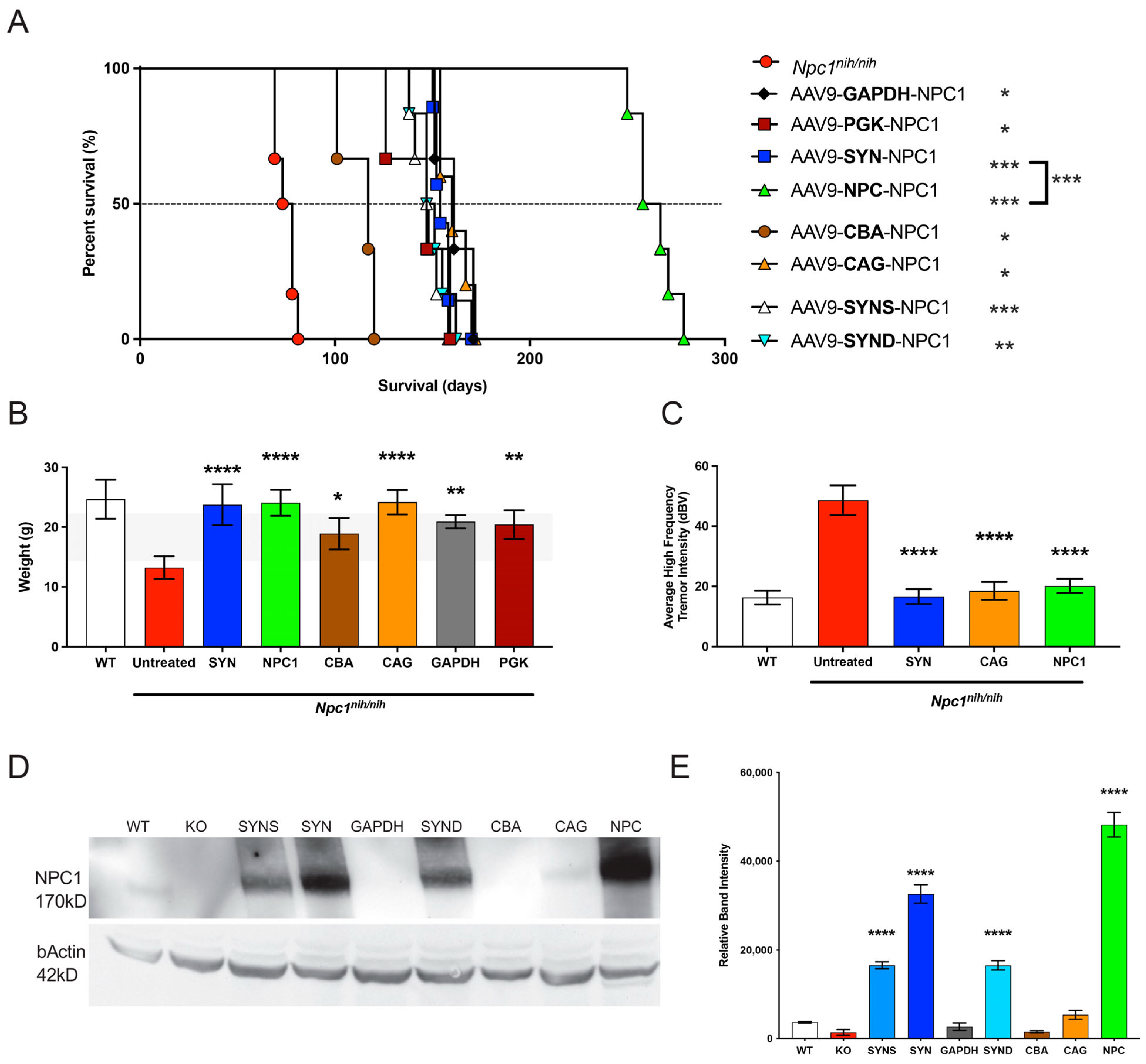
Npc1nih mice, and here, the
NPC1 promoter showed a comparable effect in attenuating weight loss and the tremor phenotype at 10 weeks, as compared to most promoters. However, in terms of overall survival and NPC1 protein expression, there were significantly improved outcomes with the
NPC1 promoter, as compared to any other promoter tested (
Figure 4 and
Figure 5). The significant result of this novel, small promoter is perhaps explained by the differential expression observed when AAV viral preparations were injected into the wild-type versus the
Npc1-deficient mice (
Figure 7), indicating that the
NPC1 promoter drives greater gene expression in the deficient model. However, the mechanism underlying such differential expression awaits further investigation. Moreover, the possibility of additional gene interactions arising from the expression of NPC1 from this truncated endogenous promoter and their downstream epigenetic and translational consequences must also be elucidated.
Lastly, we investigated whether the
NPC1 promoter is effective in reducing neuropathological phenotypes in a more slowly progressing mouse model, the
Npc1nmf164 mouse [
9], comparing it to the previously described Synapsin-1 (SYN) promoter [
16]. In contrast to the NPC1 null
Npc1nih model, the
Npc1nmf164 mice express low levels of the misfolded NPC1 protein, similar to the majority of NPC patients. Our data show a significantly higher level of NPC1 protein expression driven by the
NPC1 promoter compared to the SYN promoter, consistent with the previous western blot analysis (
Figure 4). Further, there was an overall comparable attenuation of microglial activation, astrocytosis, and lysosomal burden between NPC1 and SYN promoters. However, while the SYN.hNPC1-treated mice did not show a significant rescue of neurons in the deeper-lying thalamus, the NPC.h
NPC1-treated mice showed a significant rescue in this region, possibly due to the overall higher level of NPC1 expression in the brain. These findings, along with significant neuron rescue in the S1BF and Purkinje cells in the cerebellum, show that the novel
NPC1 promoter is overall more effective in preventing the onset of neuropathological features, as compared to the SYN promoter.
Taken together, our data for the first time describe a novel small, truncated endogenous NPC1 promoter that is efficient in driving gene expression both in vitro and in vivo, with significantly improved survival and NPC1 expression in the knockout Npc1nih mouse and overall effective reduction in neuropathology in the Npc1nmf164 mouse models of NP-C that carry a missense point mutation. Unexpectedly, this promoter drives gene expression at a higher level in the NPC1-deficient mice, implying a potential feedback loop, whose mechanism awaits further validation.
Taken together, our studies corroborate our previous data, demonstrating that AAV-mediated gene therapy administered ICV has significant therapeutic potential for NP-C, and that an optimised AAV design strategy must also take into account the DNA regulatory elements as well as the overall packaging restrictions depending on the gene of interest. The translation of pre-clinical gene therapy studies to successful clinical trials for other neurodegenerative conditions means that this is an opportune moment to further pursue the development of gene therapy for NP-C.



 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






