
Generation of a Syngeneic Heterozygous ACVRL1(wt/mut) Knockout iPS Cell Line for the In Vitro Study of HHT2-Associated Angiogenesis

 , , ,
, , , 
 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. iPSCs
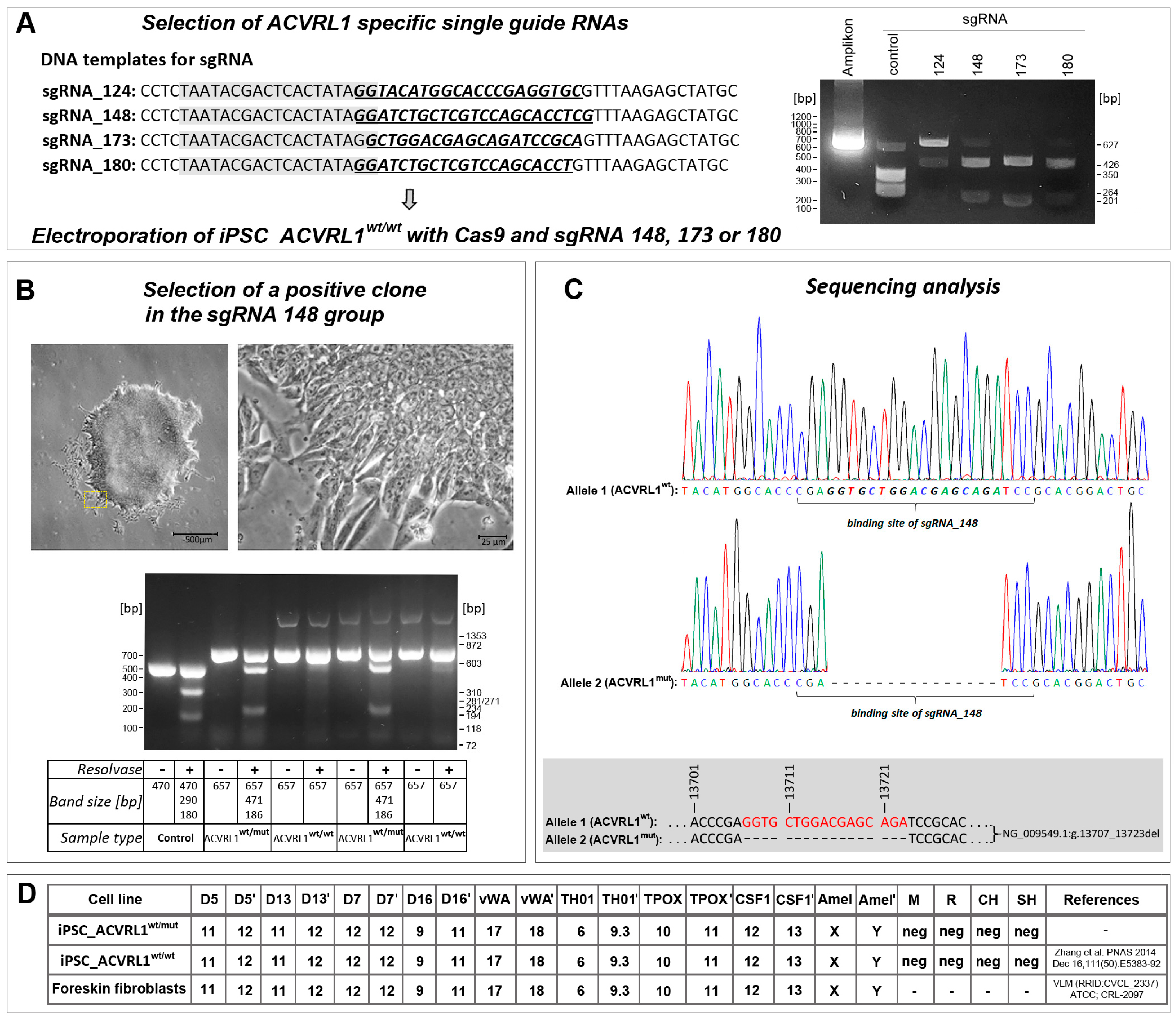
2.2. Design and Selection of sgRNAs
2.3. CRISPR/Cas9 ACVRL1 Knockout
2.4. Western Blot Analysis
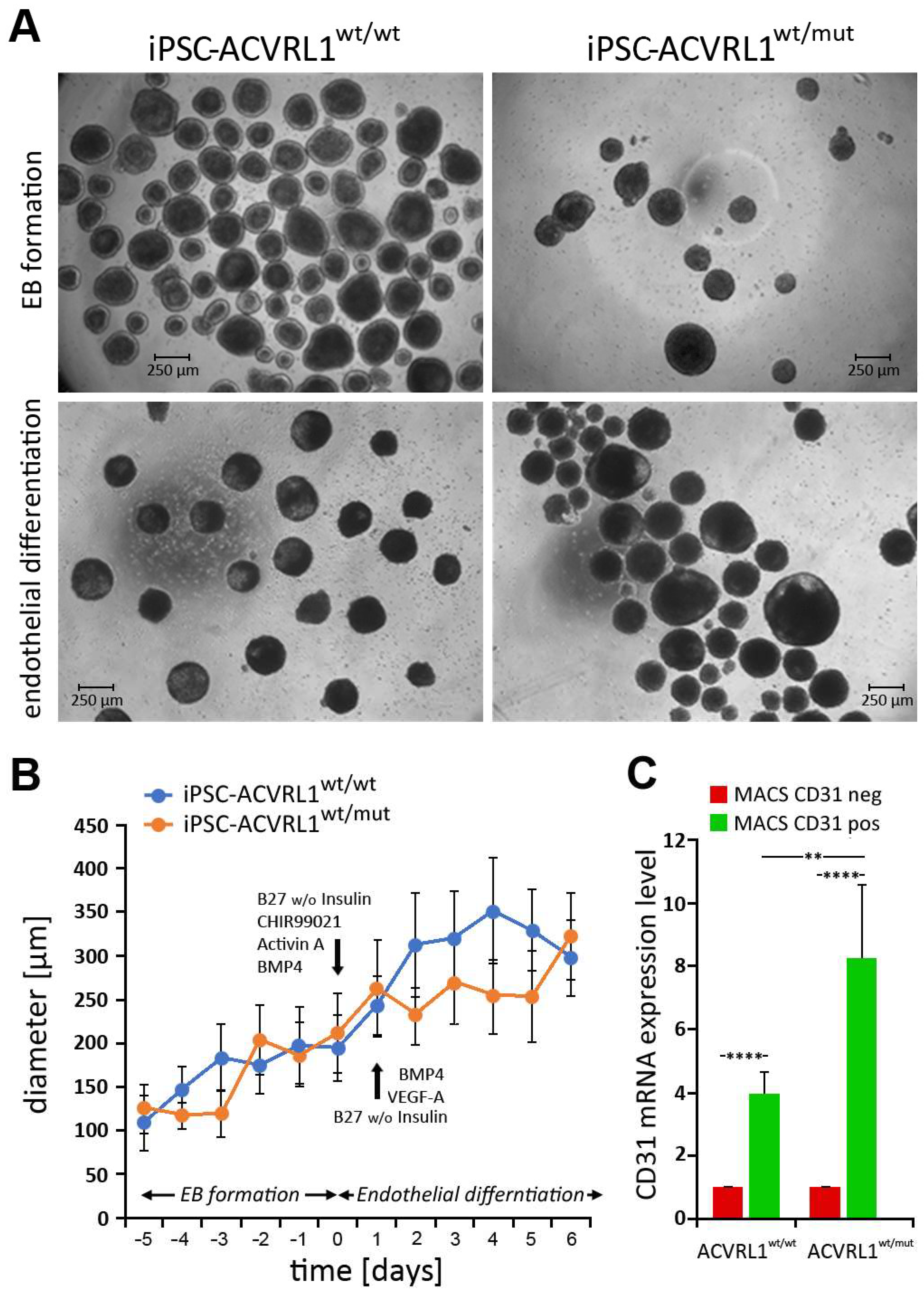
2.5. Generation of Embryoid Bodies (EBs)
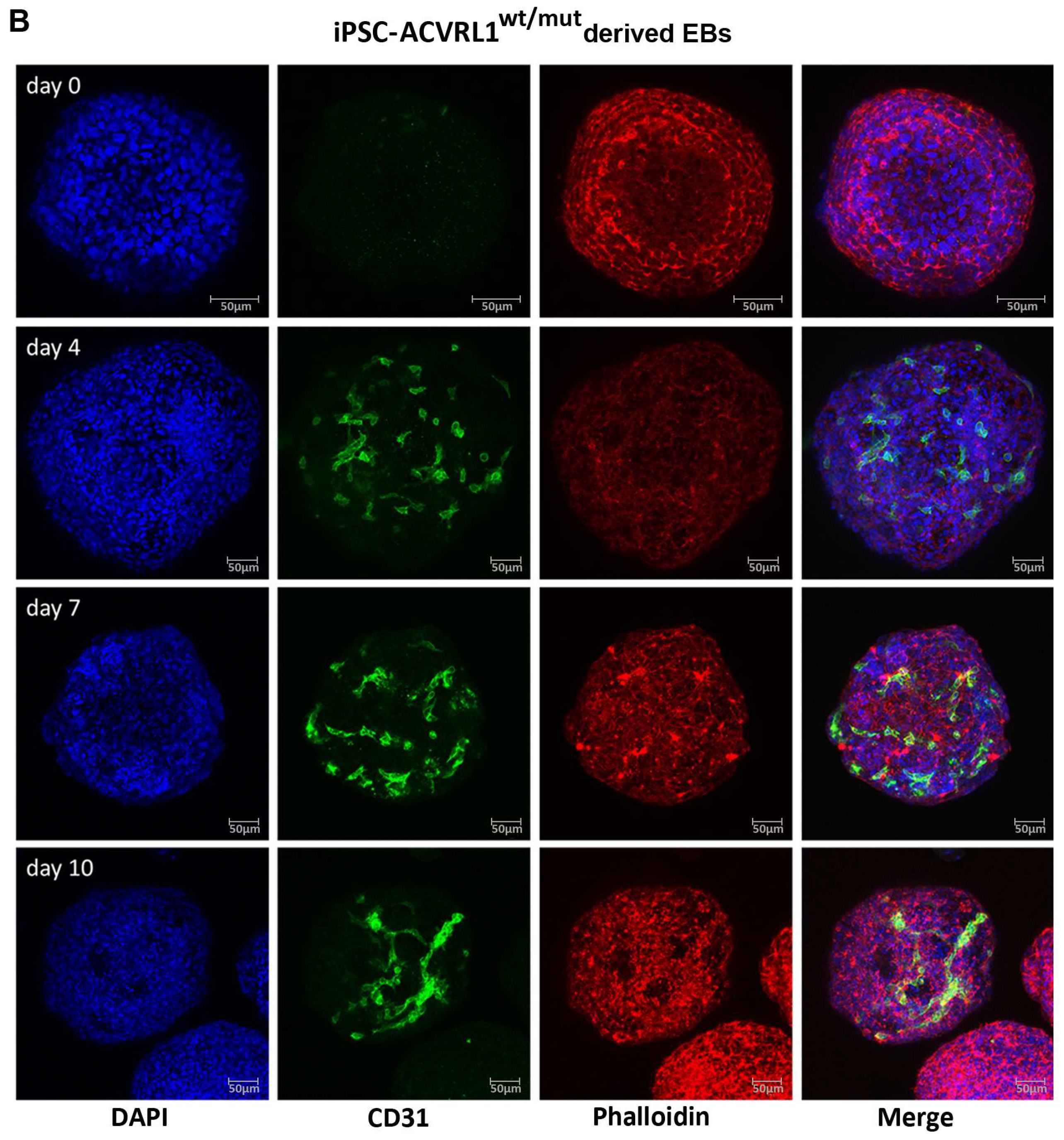
2.6. Endothelial Differentiation in EBs
2.7. Isolation of Endothelial Cells from EBs
2.8. Immunocytochemistry
2.9. Gene Expression Analysis
2.10. Statistical Analysis
3. Results
3.1. ACVRL1 Knockout Using CRISPR/Cas9
3.2. Effect of Heterozygous ACVRL1 Knockout on the Expression of TGF-Beta and BMP Signaling Molecules
3.3. Generation of Embryoid Bodies (EBs) from ACVRL1wt/wt and ACVRL1wt/mut iPSCs
3.4. Induction of Endothelial Differentiation in Embryoid Bodies (EBs) Derived from ACVRL1wt/wt and ACVRL1wt/mut iPSCs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kunimoto, K.; Yamamoto, Y.; Jinnin, M. ISSVA Classification of Vascular Anomalies and Molecular Biology. Int. J. Mol. Sci. 2022, 23, 2358. [Google Scholar] [CrossRef]
- Goumans, M.-J.; Liu, Z.; Dijke, P.T. TGF-β signaling in vascular biology and dysfunction. Cell Res. 2008, 19, 116–127. [Google Scholar] [CrossRef] [Green Version]
- Dijke, P.T.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol. 2007, 8, 857–869. [Google Scholar] [CrossRef]
- Schulte, C.; Geisthoff, U.; Lux, A.; Kupka, S.; Zenner, H.-P.; Blin, N.; Pfister, M. High frequency of ENG and ALK1/ACVRL1 mutations in German HHT patients. Hum. Mutat. 2005, 25, 595. [Google Scholar] [CrossRef]
- Letteboer, T.G.W.; Zewald, R.A.; Kamping, E.J.; De Haas, G.; Mager, J.J.; Snijder, R.J.; Lindhout, D.; Hennekam, F.A.M.; Westermann, C.J.J.; Van Amstel, J.K.P. Hereditary hemorrhagic telangiectasia: ENG and ALK-1 mutations in Dutch patients. Hum. Genet. 2005, 116, 8–16. [Google Scholar] [CrossRef]
- Fernandez-L, A.; Sanz-Rodriguez, F.; Zarrabeitia, R.; Pérez-Molino, A.; Hebbel, R.P.; Nguyen, J.; Bernabéu, C.; Botella, L.-M. Blood outgrowth endothelial cells from Hereditary Haemorrhagic Telangiectasia patients reveal abnormalities compatible with vascular lesions. Cardiovasc. Res. 2005, 68, 235–248. [Google Scholar] [CrossRef] [Green Version]
- Geisthoff, U.W.; Heckmann, K.; D'Amelio, R.; Grünewald, S.; Knöbber, D.; Falkai, P.; König, J. Health-Related Quality of Life in Hereditary Hemorrhagic Telangiectasia. Otolaryngol. Head Neck Surg. 2007, 136, 726–733. [Google Scholar] [CrossRef] [PubMed]
- Geisthoff, U.; Nguyen, H.-L.; Röth, A.; Seyfert, U. How to manage patients with hereditary haemorrhagic telangiectasia. Br. J. Haematol. 2015, 171, 443–452. [Google Scholar] [CrossRef]
- Faughnan, M.E.; Palda, V.A.; Garcia-Tsao, G.; Geisthoff, U.W.; McDonald, J.; Proctor, D.D.; Spears, J.; Brown, D.H.; Buscarini, E.; Chesnutt, M.S.; et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J. Med. Genet. 2011, 48, 73–87. [Google Scholar] [CrossRef] [PubMed]
- Abdalla, S.A.; Geisthoff, U.; Bonneau, D.; Plauchu, H.; McDonald, J.; Kennedy, S.; Faughnan, M.E.; Letarte, M. Visceral manifestations in hereditary haemorrhagic telangiectasia type 2. J. Med. Genet. 2003, 40, 494–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willemse, R.B.; Mager, J.J.; Westermann, C.J.J.; Overtoom, T.T.C.; Mauser, H.; Wolbers, J.G. Bleeding risk of cerebral vascular malformations in hereditary hemorrhagic telangiectasia. J. Neurosurg. 2000, 92, 779–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Droege, F.; Thangavelu, K.; Stuck, B.A.; Stang, A.; Lang, S.; Geisthoff, U. Life expectancy and comorbidities in patients with hereditary hemorrhagic telangiectasia. Vasc. Med. 2018, 23, 377–383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shovlin, C.L. Supermodels and disease: Insights from the HHT mice. J. Clin. Investig. 1999, 104, 1335–1336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bourdeau, A.; Faughnan, M.E.; Letarte, M. Endoglin-deficient Mice, a Unique Model to Study Hereditary Hemorrhagic Telangiectasia. Trends Cardiovasc. Med. 2000, 10, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Orlova, V.V.; Nahon, D.M.; Cochrane, A.; Cao, X.; Freund, C.; van den Hil, F.; Westermann, C.J.J.; Snijder, R.J.; Ploos van Amstel, J.K.; Ten Dijke, P.; et al. Vascular defects associated with hereditary hemorrhagic telangiectasia revealed in patient-derived isogenic iPSCs in 3D vessels on chip. Stem Cell Rep. 2022, 17, 1536–1545. [Google Scholar] [CrossRef]
- Freund, C.; Davis, R.P.; Gkatzis, K.; Oostwaard, D.W.-V.; Mummery, C.L. The first reported generation of human induced pluripotent stem cells (iPS cells) and iPS cell-derived cardiomyocytes in the Netherlands. Neth. Heart J. 2010, 18, 51–54. [Google Scholar]
- Bouma, M.J.; Orlova, V.; Hil, F.E.V.D.; Mager, H.-J.; Baas, F.; de Knijff, P.; Mummery, C.L.; Mikkers, H.; Freund, C. Generation and genetic repair of 2 iPSC clones from a patient bearing a heterozygous c.1120del18 mutation in the ACVRL1 gene leading to Hereditary Hemorrhagic Telangiectasia (HHT) type 2. Stem Cell Res. 2020, 46, 101786. [Google Scholar] [CrossRef]
- Zhang, M.; D’aniello, C.; Verkerk, A.O.; Wrobel, E.; Frank, S.; Oostwaard, D.W.-V.; Piccini, I.; Freund, C.; Rao, J.; Seebohm, G.; et al. Recessive cardiac phenotypes in induced pluripotent stem cell models of Jervell and Lange-Nielsen syndrome: Disease mechanisms and pharmacological rescue. Proc. Natl. Acad. Sci. USA 2014, 111, E5383–E5392. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Si-Tayeb, K.; Noto, F.K.; Sepac, A.; Sedlic, F.; Bosnjak, Z.J.; Lough, J.W.; Duncan, S.A. Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 2010, 10, 81. [Google Scholar] [CrossRef] [Green Version]
- Cirulli, A.; Liso, A.; D’ovidio, F.; Mestice, A.; Pasculli, G.; Gallitelli, M.; Rizzi, R.; Specchia, G.; Sabbà, C. Vascular Endothelial Growth Factor Serum Levels Are Elevated in Patients with Hereditary Hemorrhagic Telangiectasia. Acta Haematol. 2003, 110, 29–32. [Google Scholar] [CrossRef] [PubMed]
- Geisthoff, U.; Nguyen, H.-L.; Lefering, R.; Maune, S.; Thangavelu, K.; Droege, F. Trauma Can Induce Telangiectases in Hereditary Hemorrhagic Telangiectasia. J. Clin. Med. 2020, 9, 1507. [Google Scholar] [CrossRef] [PubMed]
- Itoh, F.; Itoh, S.; Carvalho, R.L.C.; Adachi, T.; Ema, M.; Goumans, M.-J.; Larsson, J.; Karlsson, S.; Takahashi, S.; Mummery, C.L.; et al. Poor vessel formation in embryos from knock-in mice expressing ALK5 with L45 loop mutation defective in Smad activation. Lab. Investig. 2009, 89, 800–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Zhang, D.; Zhou, F.; Zhou, M.; Li, Q.; Chen, J.; Yang, J. Whole-Mount In Situ Hybridization in Zebrafish Embryos and Tube Formation Assay in iPSC-ECs to Study the Role of Endoglin in Vascular Development. J. Vis. Exp. 2020, 159, e60498. [Google Scholar] [CrossRef]
- Ura, H.; Togi, S.; Iwata, Y.; Ozaki, M.; Niida, Y. Establishment of a human induced pluripotent stem cell line, KMUGMCi001-A, from a patient bearing a heterozygous c.772 + 3_772 + 4dup mutation in the ACVRL1 gene leading Telangiectasia, hereditary hemorrhagic, type 2 (HHT2). Stem Cell Res. 2022, 61, 102743. [Google Scholar] [CrossRef]
- McDonald, J.; Damjanovich, K.; Millson, A.; Wooderchak, W.; Chibuk, J.; Stevenson, D.A.; Gedge, F.; Bayrak-Toydemir, P. Molecular diagnosis in hereditary hemorrhagic telangiectasia: Findings in a series tested simultaneously by sequencing and deletion/duplication analysis. Clin. Genet. 2010, 79, 335–344. [Google Scholar] [CrossRef]
- Sadick, H.; Riedel, F.; Naim, R.; Goessler, U.; Hörmann, K.; Hafner, M.; Lux, A. Patients with hereditary hemorrhagic telangiectasia have increased plasma levels of vascular endothelial growth factor and transforming growth factor-beta1 as well as high ALK1 tissue expression. Haematologica 2005, 90, 818–828. [Google Scholar]
- Mitchell, A.; Adams, L.A.; MacQuillan, G.; Tibballs, J.; Driesen, R.V.; Delriviere, L. Bevacizumab reverses need for liver transplantation in hereditary hemorrhagic telangiectasia. Liver Transplant. 2008, 14, 210–213. [Google Scholar] [CrossRef]
- Vlachou, P.A.; Colak, E.; Koculym, A.; Kirpalani, A.; Kim, T.K.; Hirschfield, G.M.; Faughnan, M.E. Improvement of ischemic cholangiopathy in three patients with hereditary hemorrhagic telangiectasia following treatment with bevacizumab. J. Hepatol. 2013, 59, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Davidson, T.M.; Olitsky, S.E.; Wei, J.L. Hereditary hemorrhagic telangiectasia/avastin. Laryngoscope 2010, 120, 432–435. [Google Scholar] [CrossRef]
- Rohrmeier, C.; Sachs, H.G.; Kuehnel, T.S. A retrospective analysis of low dose, intranasal injected bevacizumab (Avastin) in hereditary haemorrhagic telangiectasia. Eur. Arch. Oto-Rhino-Laryngol. 2011, 269, 531–536. [Google Scholar] [CrossRef] [PubMed]
- Kanellopoulou, T.; Alexopoulou, A. Bevacizumab in the treatment of hereditary hemorrhagic telangiectasia. Expert Opin. Biol. Ther. 2013, 13, 1315–1323. [Google Scholar] [CrossRef]
- Dupuis-Girod, S.; Ambrun, A.; Decullier, E.; Fargeton, A.-E.; Roux, A.; Bréant, V.; Colombet, B.; Rivière, S.; Cartier, C.; Lacombe, P.; et al. Effect of Bevacizumab Nasal Spray on Epistaxis Duration in Hereditary Hemorrhagic Telangectasia: A Randomized Clinical Trial. JAMA 2016, 316, 934–942. [Google Scholar] [CrossRef] [Green Version]
- Khanwalkar, A.R.; Rathor, A.; Read, A.K.; Paknezhad, H.; Ma, Y.; Hwang, P.H. Randomized, controlled, double-blinded clinical trial of effect of bevacizumab injection in management of epistaxis in hereditary hemorrhagic telangiectasia patients undergoing surgical cauterization. Int. Forum Allergy Rhinol. 2022, 12, 1034–1042. [Google Scholar] [CrossRef] [PubMed]
- Alsina-Sanchís, E.; García-Ibáñez, Y.; Figueiredo, A.; Riera-Domingo, C.; Figueras, A.; Matias-Guiu, X.; Casanovas, O.; Botella, L.M.; Pujana, M.A.; Riera-Mestre, A.; et al. ALK1 Loss Results in Vascular Hyperplasia in Mice and Humans through PI3K Activation. Arter. Thromb. Vasc. Biol. 2018, 38, 1216–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, B.L.; Pham, V.N.; Lawson, N.D.; Kulik, M.; Childs, S.; Lekven, A.C.; Garrity, D.M.; Moon, R.T.; Fishman, M.C.; Lechleider, R.J.; et al. Disruption of acvrl1 increases endothelial cell number in zebrafish cranial vessels. Development 2002, 129, 3009–3019. [Google Scholar] [CrossRef]
- Lopez-Novoa, J.M. Angiogenic Stimuli and Endoglin Absence Induces Brain Arteriovenous Malformations: Are Local Endoglin Deletion and Angiogenesis the ‘Second Hit’ That Is Necessary for Arteriovenous Malformations Formation in HHT-1? Cerebrovasc. Dis. 2012, 33, 548. [Google Scholar] [CrossRef]
- Baeyens, N.; Larrivée, B.; Ola, R.; Hayward-Piatkowskyi, B.; Dubrac, A.; Huang, B.; Ross, T.D.; Coon, B.G.; Min, E.; Tsarfati, M.; et al. Defective fluid shear stress mechanotransduction mediates hereditary hemorrhagic telangiectasia. J. Cell Biol. 2016, 214, 807–816. [Google Scholar] [CrossRef] [Green Version]
- Arthur, H.M.; Roman, B.L. An update on preclinical models of hereditary haemorrhagic telangiectasia: Insights into disease mechanisms. Front. Med. 2022, 9, 973964. [Google Scholar] [CrossRef]
- Hiepen, C.; Jatzlau, J.; Knaus, P. Biomechanical stress provides a second hit in the establishment of BMP/TGFβ-related vascular disorders. Cell Stress 2020, 4, 44–47. [Google Scholar] [CrossRef] [Green Version]
- Snellings, D.A.; Gallione, C.J.; Clark, D.S.; Vozoris, N.T.; Faughnan, M.E.; Marchuk, D.A. Somatic Mutations in Vascular Malformations of Hereditary Hemorrhagic Telangiectasia Result in Bi-allelic Loss of ENG or ACVRL1. Am. J. Hum. Genet. 2019, 105, 894–906. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiang-Tischhauser, L.; Bette, M.; Rusche, J.R.; Roth, K.; Kasahara, N.; Stuck, B.A.; Bakowsky, U.; Wartenberg, M.; Sauer, H.; Geisthoff, U.W.; et al. Generation of a Syngeneic Heterozygous ACVRL1(wt/mut) Knockout iPS Cell Line for the In Vitro Study of HHT2-Associated Angiogenesis. Cells 2023, 12, 1600. https://doi.org/10.3390/cells12121600
Xiang-Tischhauser L, Bette M, Rusche JR, Roth K, Kasahara N, Stuck BA, Bakowsky U, Wartenberg M, Sauer H, Geisthoff UW, et al. Generation of a Syngeneic Heterozygous ACVRL1(wt/mut) Knockout iPS Cell Line for the In Vitro Study of HHT2-Associated Angiogenesis. Cells. 2023; 12(12):1600. https://doi.org/10.3390/cells12121600
Chicago/Turabian StyleXiang-Tischhauser, Li, Michael Bette, Johanna R. Rusche, Katrin Roth, Norio Kasahara, Boris A. Stuck, Udo Bakowsky, Maria Wartenberg, Heinrich Sauer, Urban W. Geisthoff, and et al. 2023. "Generation of a Syngeneic Heterozygous ACVRL1(wt/mut) Knockout iPS Cell Line for the In Vitro Study of HHT2-Associated Angiogenesis" Cells 12, no. 12: 1600. https://doi.org/10.3390/cells12121600
APA StyleXiang-Tischhauser, L., Bette, M., Rusche, J. R., Roth, K., Kasahara, N., Stuck, B. A., Bakowsky, U., Wartenberg, M., Sauer, H., Geisthoff, U. W., & Mandic, R. (2023). Generation of a Syngeneic Heterozygous ACVRL1(wt/mut) Knockout iPS Cell Line for the In Vitro Study of HHT2-Associated Angiogenesis. Cells, 12(12), 1600. https://doi.org/10.3390/cells12121600