1. Introduction
Usher syndrome is an autosomal recessively inherited disorder characterized by hearing impairment and a progressive loss of visual function due to retinitis pigmentosa (RP). Depending on the clinical type, patients may also experience balance problems as a consequence of vestibular dysfunction. Currently, four types of Usher syndrome (USH1-4) are distinguished based on the onset, the severity and the progression of the clinical features [
1]. Pathogenic variants in the
ADGRV1 gene, previously named
MASS1,
VLGR1 or
GPR98, have been identified as the underlying cause for Usher syndrome type 2C (USH2C) [
2]. This type of Usher syndrome is characterized by congenital hearing impairment and a progressive loss of vision in the absence of vestibular dysfunction [
3]. Worldwide, around 40,000 individuals develop USH2C, and these patients may benefit from hearing aids or cochlear implants to alleviate their hearing loss. However, there are currently no treatment options available to compensate for
ADGRV1-associated loss of vision.
The
ADGRV1 gene encodes the largest cell surface protein known in humans: the adhesion G-protein-coupled receptor v1 (ADGRV1) [
4]. The protein consists of a very large extracellular tail containing a signal peptide, multiple calx-beta domains, an epilepsy-associated repeat (EAR) domain, a thrombospondin/pentraxin/laminin G-like domain and a GPCR proteolytic site (GPS). A seven transmembrane region anchors the protein in the cell membrane, followed by a short cytoplasmic region containing a C-terminal class I PDZ binding motif [
2,
4,
5].
In the retina, ADGRV1 co-localizes with usherin (USH2A) and whirlin (USH2D), and together these proteins shape the USH2 complex at the periciliary membrane of the photoreceptor cells [
6]. Two types of photoreceptor cells can be distinguished, i.e., rods and cones, which are both built up by an outer segment (OS), an inner segment (IS), a cell body and a synaptic terminal. The OS contains the proteins responsible for phototransduction, and is separated from the IS by a connecting cilium. The membrane of the periciliary region, where the USH2 complex is localized, forms a collar-like extension of the IS that surrounds the connecting cilium. It has been shown that the USH2 complex forms molecular links between the periciliary membrane and the membrane of the connecting cilium [
6]. Moreover, it has been proposed that the USH2 complex participates in the trafficking and docking of trans-Golgi-derived vesicles that contain components essential for OS function and maintenance, from the IS towards the OS [
6,
7,
8,
9]. Furthermore, for ADGRV1 specifically, it has been postulated that the protein is involved in the regulation of the calcium homeostasis at mitochondria-associated ER membranes, and that it is involved in cell spreading and migration by mechanosensing at focal adhesions [
8,
9]. However, the exact pathophysiological mechanism underlying
ADGRV1-associated RP remains elusive.
To further unravel the pathophysiological mechanism underlying
ADGRV1-associated RP, and to aid the development of future therapeutic strategies, suitable cellular or animal models that mimic the human phenotype are essential. Patient-derived cellular models such as fibroblasts or induced pluripotent stem cell (iPSC)-derived organoids provide the opportunity to perform studies in the patient’s own genetic and molecular context [
10,
11,
12]. However, vision is a complex process that relies both on the conversion of light into electrical stimuli in the retina, and the integration and interpretation of these signals by the central nervous system. As such, animal models are still essential to evaluate the effect of therapies at the level of visual function. Multiple
Adgrv1 mouse models, either naturally occurring or genetically modified, have been reported. These models, however, only partially resemble the human phenotype since they do present with hearing loss, but do not recapitulate the retinal phenotype observed in patients [
13,
14,
15,
16,
17,
18]. This may be explained by interspecies anatomical differences in the photoreceptor periciliary region where Adgrv1 is expressed. It has been previously reported that the photoreceptor periciliary region is largely underdeveloped in rodents when compared to humans [
19], making rodents less suitable to study
ADGRV1-associated RP. As an alternative, zebrafish have been recognized as an attractive model to study retinal dysfunction [
20,
21,
22]. Zebrafish have a retinal structure comparable to humans and also the photoreceptor periciliary region has been highly conserved. Moreover, the advantages of zebrafish such as their high fecundity, ex utero fertilization, rapid development and transparency of embryos and larvae allow for easy genetic manipulation and experimentation.
The introduction of CRISPR/Cas-based genome editing technologies significantly improved the possibilities of generating mutant zebrafish models in a time- and cost-effective manner [
23]. In recent years several CRISPR/Cas9-based zebrafish mutants have been generated to study retinal dysfunction caused by pathogenic variants in the
USH2A gene—the gene encoding usherin. In contrast to murine models, these mutant zebrafish models (
ush2armc1,
ush2ab1245,
ush2ahzu6 and
ush2au507) present with retinal dysfunction, already observed in larval stages, although in the absence of an obvious audiologic phenotype [
20,
21,
22].
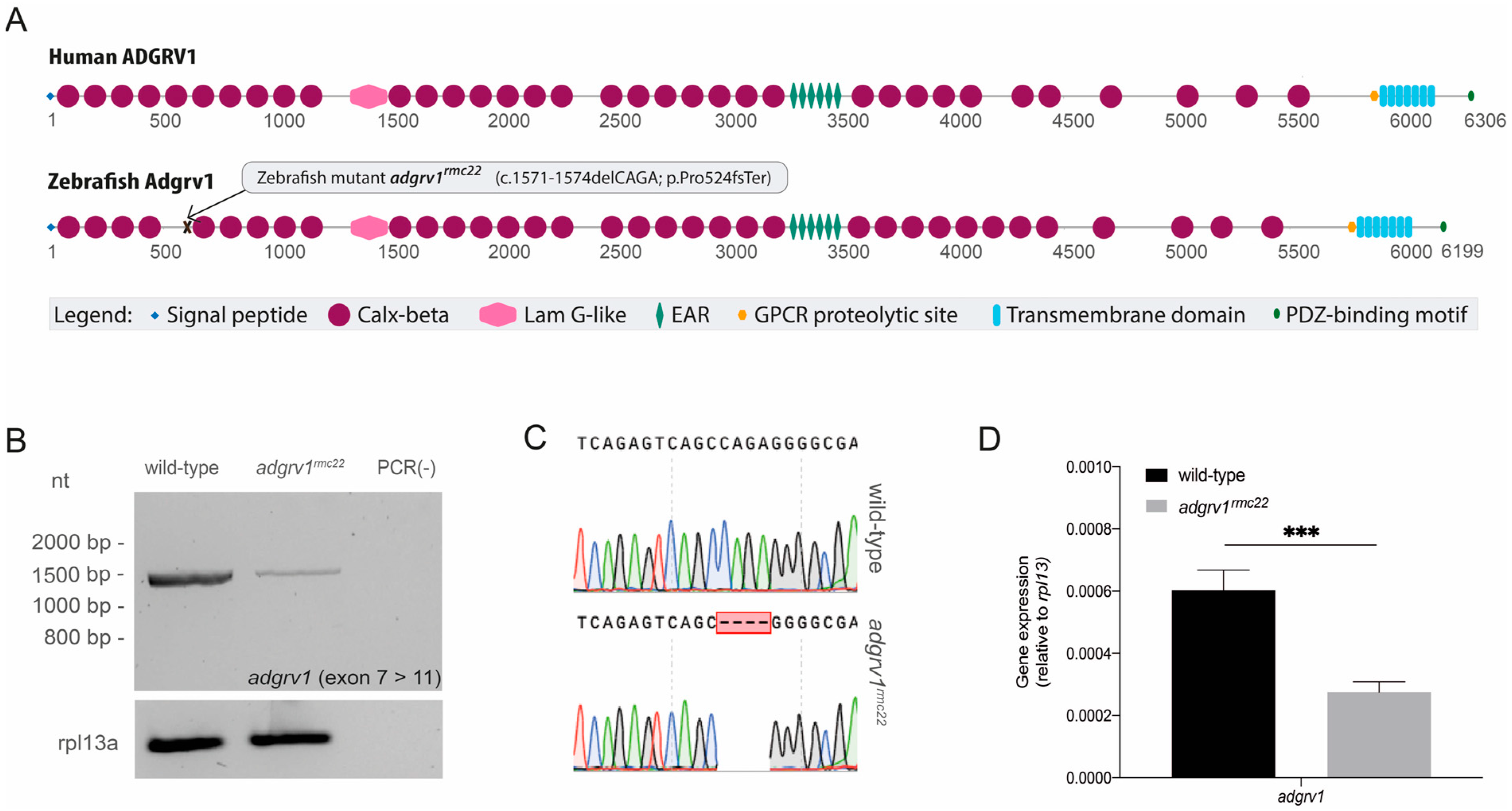
With the need for an animal model to study ADGRV1-associated RP, and with zebrafish being recognized as an attractive model to study retinal dysfunction, we here generated and characterized the adgrv1rmc22 zebrafish model. In the zebrafish genome, adgrv1 is present as a single copy that, as with the human ADGRV1 gene, consists of 90 exons. By employing CRISPR/Cas9-based genome editing technology, we introduced protein-truncating lesions into adgrv1, resulting in the absence of Adgrv1 and the reduced expression of the USH2 complex members at the periciliary region of the adgrv1rmc22 photoreceptors. Functional analyses revealed that knocking out adgrv1 resulted in an early-onset retinal phenotype. Therefore, we present the adgrv1rmc22 mutant as the first animal model that can be used to further unravel the molecular function of ADGRV1 in the retina, and to evaluate future therapeutic strategies for ADGRV1-associated RP.
2. Materials and Methods
2.1. Zebrafish Maintenance and Husbandry
Wild-type AB-strain zebrafish and
adgrv1rmc22 mutant zebrafish were bred and maintained by standard methods at the Radboud Zebrafish Facility Nijmegen [
24]. Zebrafish eggs were obtained from pair-wise matings of wild-type and mutant zebrafish. Experimental procedures were conducted in accordance with international and institutional guidelines (protocol #RU-DEC, AVD10300202215892).
2.2. CRISPR/Cas9 Genome Editing Design and Microinjections
For the generation of
adgrv1rmc22 mutant zebrafish, CRISPR targets were identified and guide RNAs with settings for Cas9 were designed using the web tool CRISPRscan [
25]. A cut-off value of at least 4 mismatches for potential off-target regions was used for the selection of an sgRNA. Based on the CRISPRscan predictions, an Alt-R
TM CRISPR/Cas9 sgRNA was ordered from Integrated DNA Technologies (IDT) for the following 20 bp genomic target sequence in
adgrv1 exon 9: 5′-GGGTATTCAGAGTCAGCCAG-3′.
The sgRNA and commercial Alt-R
® S.p. Cas9 Nuclease V3 (IDT, Coralville, IA, USA, #1081059) were co-injected in wild-type zebrafish embryos at the single cell stage. For this purpose, an injection mixture of 100 ng/μL sgRNA, 800 ng/μL Cas9 Nuclease, 300 mM KCl and 20% (
v/
v) phenol red solution (#P0290, Sigma Aldrich, Amsterdam, The Netherlands) was prepared and incubated at 37
for 5 min to allow sgRNA-Cas9 ribonucleoprotein complex formation. An amount of 1 nL of this mixture was injected into single-cell-stage zebrafish embryos using a Pneumatic PicoPump pv280 (World Precision Instruments, Friedberg, Germany). Following the injection, the embryos were raised at 28.5 °C in E3 embryo medium consisting of 5 mM NaCl, 0.17 mM KCl, 0.33 mM CaCl
2, 0.33 mM MgSO
4 and 0.1% (
v/
v) methylene blue. At 1 day post fertilization (dpf), 16 out of 200 injected embryos were analyzed for the presence of genome editing events using High-Resolution Melting Analysis (HRM) [
26]. Upon detection of genome-editing HRM signatures, the remainder of the injected embryos were raised to adulthood (F0 fish). Founder fish that transmitted germline mutations were outcrossed twice with wild-type zebrafish to further minimize the possible presence of unforeseen CRISPR/Cas9-induced off-target modifications. After the second round of outcrossing, heterozygous fish were incrossed to produce progeny of which the homozygous fish and their wild-type siblings were used for subsequent breeding and phenotypic analysis.
2.3. High-Resolution Melting Analysis
At 1 dpf, injected and uninjected zebrafish embryos were sampled individually in 25 μL hotshot lysis buffer (25 mM NaOH, 0.2 mM EDTA), and incubated at 95 °C for 20 min. The lysates were neutralized with 2.5 μL 1 M Tris pH8, diluted 5–10× and used as input for high-resolution melting (HRM) PCR analysis, which was performed in a QuantStudio™ 3 Real-Time PCR system (Applied Biosystems, Waltham, MA, USA) using Q5
® High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA, USA, #M0491L) and 1× EvaGreen Dye (Biotium, Fremont, CA, USA, #31000). Primers that were used for the PCR amplification are listed in
Table S1. The HRM procedure was initiated after the PCR amplification by rapid cooling (4 °C/s) to 10 °C and a melt curve procedure (0.1 °C/s) during which data were collected. When typical HRM heteroduplex peaks were observed in >25% of the injected embryos (
n = 16), the remainder of the injected embryos were raised to adulthood. Sanger sequencing was used to verify genome editing events in samples indicating heteroduplex HRM peaks.
2.4. Genotyping
Zebrafish samples, either individual 1 dpf embryos or adult tail fins, were lysed in 25 μL (embryos) or 75 μL (adult tail fins) lysis buffer (25 mM NaOH, 0.2 mM EDTA), and incubated at 95 °C for 20 min. The lysates were neutralized with 2.5 μL or 7.5 μL 1 M Tris pH 8, respectively, diluted 5–10× and used as input for PCR. The genomic region of
adgrv1 exon 9 surrounding the introduced lesion was amplified using standard PCR cycling conditions. Primers that were used for the PCR amplification are listed in
Table S1. Amplicons and genotypes were confirmed using Sanger Sequencing.
2.5. Transcript Analysis
The total RNA was isolated from three pooled (in liquid nitrogen snap-frozen) larval heads of the same genotype. Larval heads were homogenized prior to lysis and RNA isolation was performed using the RNeasy Micro kit according to the manufacturer’s instructions (Qiagen, Hilden, Germany). Subsequently, 100 μg of the total RNA was used as a template for cDNA synthesis using the Superscript
TM IV Reverse Transcriptase kit (Thermo Fisher Scientific, Waltham, MA, USA, #18090200). An
adgrv1 fragment spanning exon 7 to exon 11 (1500 bp) was amplified using Q5
® High-Fidelity DNA Polymerase kit (New England Biolabs, Ipswich, MA, USA, #M0491L). An amplicon of
rpl13 was used as a reference. Cycling conditions were as follows: 98 °C for 1 min, 35 cycles of 98 °C for 10 s, 68 °C for 20 s and 72 °C for 2 min, followed by a final 72 °C for 5 min. Afterwards, amplicons were separated on a 2% agarose gel and analyzed using Sanger Sequencing. All primers used in transcript analyses are listed in
Table S1.
2.6. Gene Expression Analysis
Following the same procedure as described in paragraph 2.5, the total RNA was isolated and cDNA was generated from four pools of five larvae (5 dpf), and from two juvenile zebrafish retinas (3 months post fertilization (3 mpf)) per genotype. Quantitative PCR was performed using GoTaq qPCR Master Mix (Promega) according to the manufacturer’s protocol. Transcript-specific primers have been designed targeted against
adgrv1 spanning from exon 84 to exon 85,
ush2a exon 55 to exon 56, and
whrnb exon 2 to exon 4. A target against the reference gene
rpl13 was also included. Amplifications were performed with the QuantStudio
TM 3 Real-Time PCR system (Applied Biosystems, Waltham, MA, USA). PCR reactions were performed in duplo and relative gene expression levels compared to the reference gene
rpl13 were determined with the 2
−∆Ct method. Primers that were used are listed in
Table S1.
2.7. Antibodies, Immunohistochemistry and Histology
Zebrafish larvae (5 dpf) and the eyes of juvenile zebrafish (3 mpf) (adgrv1rmc22 mutants and strain-matched wild-types) were cryoprotected with 10% sucrose in PBS for 5 min prior to embedding in an OCT compound (Sakura, Alphen aan den Rijn, The Netherlands, Tissue-Tek, #4583). After the embedding procedure, samples were slowly frozen using liquid-nitrogen-cooled isopentane, and cryosections (7 μm) were prepared following standard protocols. Unfixed cryosections were permeabilized for 20 min with 0.01% Tween-20 in PBS and blocked for 30 min with blocking buffer (10% normal goat serum and 2% bovine serum albumin in PBS). Both primary and secondary antibodies were diluted in blocking buffer, and sections were incubated overnight at 4 °C with the primary antibodies, followed by rinsing 3 times for 10 min with PBS, and a 1 h incubation with the secondary antibodies together with DAPI (1:800; D1306; Thermo Fisher, Waltham, MA, USA). After a second round of rinsing (3 times for 10 min with PBS), cryosections were post-fixed using 4% paraformaldehyde for 10 min, followed by rinsing 3 times for 5 min with PBS. Finally, the cryosections were mounted with Prolong Gold Anti-fade (P36930; Thermo Fisher, Waltham, MA, USA). The following primary antibodies and dilutions were used: rabbit anti-usherin (1:1000; #DZ01481, Boster Bio, Pleasanton, CA, USA), rabbit anti-Adgrv1 (1:100; #DZ41032; Boster Bio), rabbit anti-Whrnb (1:750; #42690002, Cip98a; Novus Biologicals, Centennial, CO, USA) and mouse anti-centrin (1:500; #04-1624; Millipore, Darmstadt, Germany). Secondary antibodies (Alexa Fluor 568 goat anti-rabbit (Thermo Fisher, Waltham, MA, USA, #A-11011) and Alexa Fluor 488 goat anti-mouse (Thermo Fisher, Waltham, MA, USA, #A-11029)) were used in a 1:800 dilution. The slides were imaged using a Zeiss Axio Imager fluorescence microscope equipped with an AxioCam MCR5 camera (Zeiss, Jena, Germany). The intensity of the usherin and Whrnb immunofluorescence was measured using Fiji (v.1.51n). Based on the centrin immunostaining, the outer segment layer was isolated. Next, a mask was generated based on the centrin staining using the “Find Maxima” option (noise = 25) and dilated five times. Next, the centrin mask and the usherin/Whrnb layer were combined to locate the exact position of the usherin or the Whrnb staining. Subsequently, the “Find Maxima” (noise = 10) was used to identify the usherin/Whrnb staining within the centrin mask, and touching objects were separated using the watershed option. The resulting mask was dilated six times to measure the area surrounding the usherin and Whrnb immunofluorescence signal. Subsequently, this enables a correction for background noise when the maximum and minimum gray values of the identified regions were measured on the original image of the usherin/Whrnb immunofluorescence (Analyze Particles option: size = 0–180, pixel circularity = 0.00–1.00).
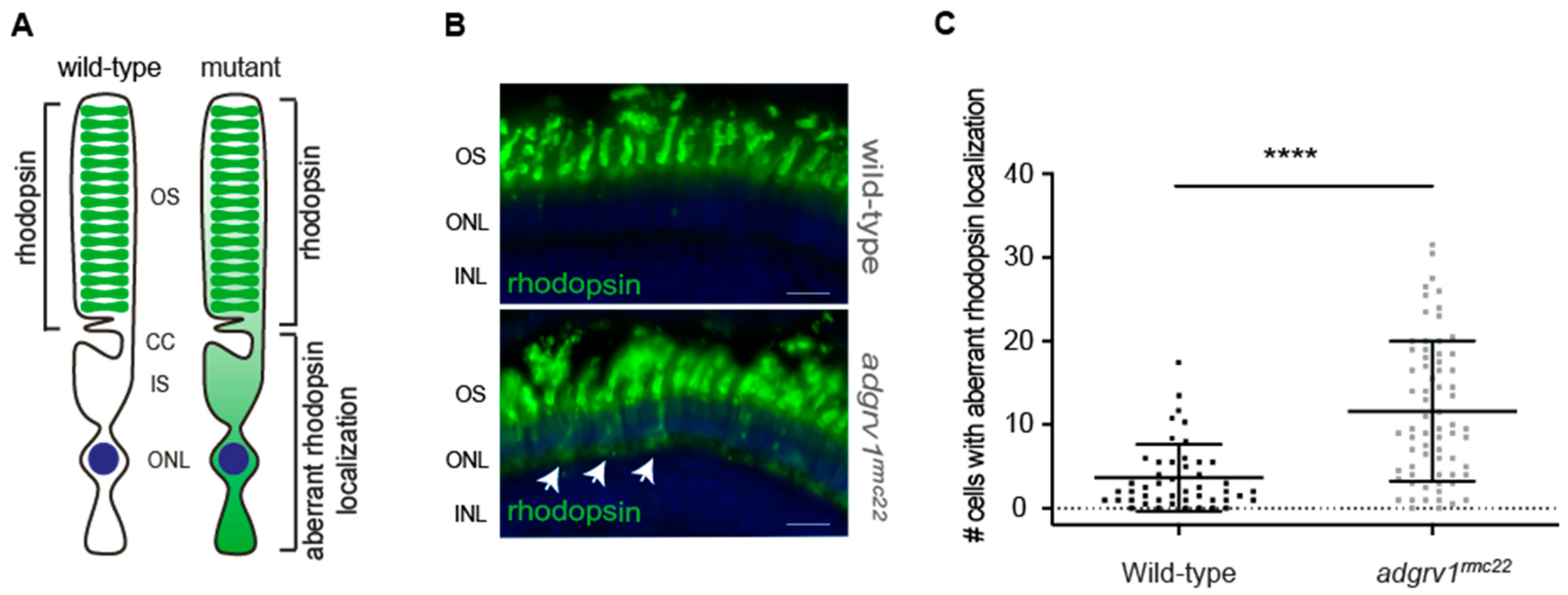
For the rhodopsin labeling,
adgrv1rmc22 zebrafish and strain-matched wild-type larvae were raised in transparent 10 cm Petri dishes under normal light conditions (300 lux white light in a 14/10 h day/night rhythm), and larvae were sampled at the morning of 6 dpf exactly 100 min after light exposure. Cryosections (7 μm) of 4% PFA fixed larvae were stained and imaged as published previously [
27]. As a primary antibody mouse anti-rhodopsin (1:4000, Clone 4D2; NBP2-59690, Novus Biologicals, Centennial, CO, USA) was used with secondary Alexa Fluor 488 goat anti-mouse (Thermo Fisher, Waltham, MA, USA, #A-11029) combined with DAPI (1:8000; D1306; Thermo Fisher, Waltham, MA, USA). The slides were imaged using a Zeiss Axio Imager fluorescence microscope equipped with an AxioCam MCR5 camera (Zeiss, Jena, Germany). Subsequently, images were blinded and randomized, and rhodopsin mislocalization was quantified independently by two researchers, by manually scoring the number of photoreceptor cells with a clear rhodopsin signal in the photoreceptor cell body per retinal section.
To assess the morphology of juvenile (3 mpf) and adult (6 mpf) zebrafish retinas, the eyes of adgrv1rmc22 mutants and strain-matched wild-types were dissected. Using 4% paraformaldehyde (PFA) in PBS, the eyes were fixed overnight at 4 °C, dehydrated in ascending methanol series and incubated overnight in 100% methanol. Afterwards the eyes were rehydrated in a descending methanol series to 0.1% PBS-Tween-20, followed by cryoprotecting with 10% sucrose in 0.1% PBS-Tween-20 for 15 min, and 30% sucrose in 0.1% PBS-Tween-20 for 1 h at room temperature. Next, the eyes were embedded in the OCT compound (Sakura, Alphen aan den Rijn, The Netherlands, Tissue-Tek, #4583), slowly frozen using liquid-nitrogen-cooled isopentane, and cryosections (7 μm) were prepared and stained with hematoxylin and eosin and analyzed using a Zeiss Axioscope light microscope (Zeiss, Oberkochen, Germany).
2.8. Electroretinogram (ERG) Recordings
ERG recordings were performed on juvenile (6–8 weeks post fertilization (wpf))
adgrv1rmc22 zebrafish and wild-type siblings. Before performing the ERG measurements, fish were dark-adapted for at least 30 min. Fish were anesthetized using 0.016% tricaine methane sulfonate solution and the spinal cord was severed to stop the heartbeat. A Petri dish was filled with agarose gel, and a reference electrode was placed into the gel. Next, the zebrafish was placed in this Petri dish with the right eye facing the light source. Using a 25-gauge syringe needle, a small incision was made at the edge of the cornea of the right eye, in which the recording electrode filled with E3 medium (5 mM NaCL, 0.17 mM KCl, 0.33 mM CaCl
2 and 0.33 mM MgSO
4) was placed [
28]. Two consecutive 100 ms white light stimuli with an intensity of ~550 lux were applied at an 8 s interval [
20,
28,
29]. Subsequently, the response was amplified 10,000 times with a band-pass of 700-0.1 Hz and recorded with the Signal6.03 software (Cambridge Electronic Design Limited, Cambridge, England). A baseline correction was performed on the recordings, with the baseline signal being determined during a 50 ms timespan, as the average signal before the stimulus was given. The maximum B wave amplitude was calculated after taking the average response to the two light stimuli. All steps were performed under dim, red light.
2.9. Statistical Analyses
All statistical analyses were performed using PRISM software (v9.0.0). The software was used to check for normality, calculate average scores and perform unpaired two-tailed Student’s t-tests (for the usherin and Whrnb quantification, rhodopsin localization and ERG recordings).
4. Discussion
As a consequence of mutations in the ADGRV1 gene, patients with Usher syndrome type 2C present with congenital hearing impairment and a progressive loss of vision due to RP. In the absence of a therapeutic strategy to prevent ADGRV1-associated RP, a suitable model that resembles the human retinal phenotype is essential to unravel the exact pathophysiological mechanism and to evaluate future therapeutic strategies. In this study, we generated and characterized the adgrv1rmc22 zebrafish as a model for ADGRV1-associated retinal dysfunction. By employing CRISPR/Cas9 genome editing technology, we introduced a 4bp deletion in zebrafish adgrv1 exon 9. We have shown that the adgrv1rmc22 allele results in the absence of Adgrv1 at the photoreceptor periciliary region where its absence causes the disintegration of the USH2 protein complex. Moreover, an increase in the aberrant localization of rhodopsin was observed in adgrv1rmc22 mutant larvae, and ERG recordings indicate reduced retinal function in juvenile adgrv1rmc22 zebrafish. Based on these findings, we present the adgrv1rmc22 zebrafish as a suitable model for studying ADGRV1-associated retinal dysfunction.
Until now, only
Adgrv1 mutant mouse models were reported, which in contrast to the
adgrv1rmc22 mutant zebrafish presented here, displayed an audiologic phenotype in the absence of a clear retinal phenotype [
13,
14,
15,
16,
17,
18]. In recent years, however, zebrafish have been recognized as suitable models to study
USH2A-associated retinal dysfunction [
20,
21,
22]. Due to the ex utero fertilization, zebrafish can be easily subjected to genetic manipulation and the introduction of CRISPR/Cas9 technology has radically reduced the efforts to generate targeted knock-out models when compared to ZFN- and TALEN-based genome editing [
23,
32,
33]. To fill the gap in animal models to study
ADGRV1-associated retinal dysfunction, we generated the CRISPR/Cas9-induced
adgrv1rmc22 mutant zebrafish.
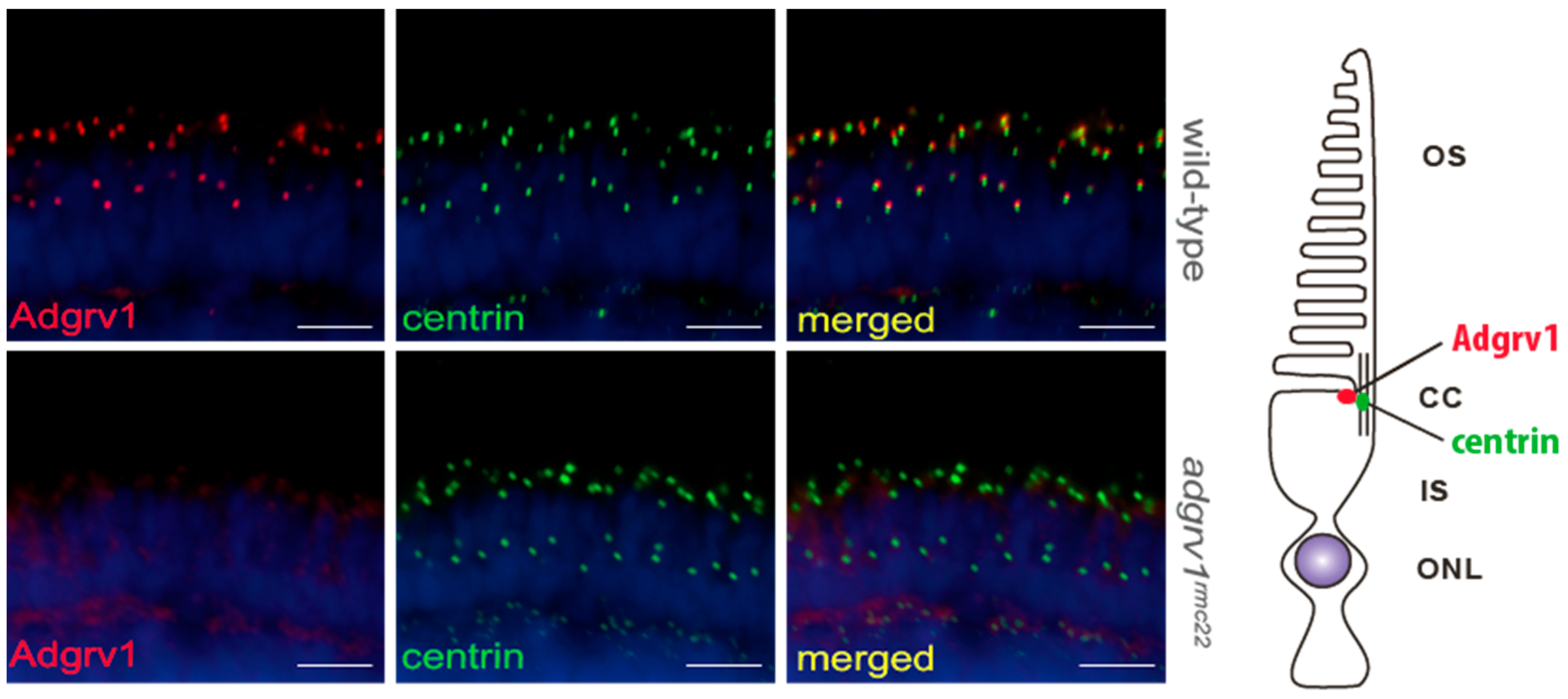
Immunohistochemical analysis confirmed the presence of the USH2 proteins Adgrv1, usherin and Whrnb at the region of the connecting cilium in the wild-type zebrafish retina, which is in line with previous studies [
20,
21,
31]. We showed that the introduction of a protein-truncating mutation at the N-terminal region of
adgrv1 resulted in the absence of the protein at the periciliary region of photoreceptor cells. Thus far, the effect of Adgrv1 depletion in the zebrafish retina has only been studied after morpholino antisense oligonucleotide-induced knock-down. In their study, however, Ebermann, et al. [
34] did not report any retinal defects after the knock-down of
adgrv1. When comparing our
adgrv1rmc22 zebrafish with the
adgrv1 knock-down larvae, the differences observed in retinal phenotype may be attributed to the transient and partial knock-down induced by morpholino antisense oligonucleotides versus the permanent protein-truncating mutation induced by CRISPR/Cas9. Additional immunohistochemical analyses revealed that the absence of Adgrv1 in the
adgrv1rmc22 zebrafish retina also results in a strong reduction in the usherin and Whrnb fluorescence signal. These results confirm the interdependency of these proteins in the correct formation of the periciliary USH2 protein complex that we previously proposed based on similar results in the
ush2armc1 and ush2ab1245 zebrafish [
20]. Therefore, our findings confirm the proposed model in which Adgrv1 interacts in a protein complex with the other USH2 proteins usherin and Whrnb at the zebrafish photoreceptor periciliary region. To unravel if the reduced immunoreactivity of usherin and Whrnb at this region is the result of the disassembly of the USH2 complex, or caused by altered gene expression levels, we analyzed the
ush2a and
whrnb gene expression levels. In
adgrv1rmc22 mutant larvae, the relative gene expression levels show a statistically significant reduction of 34% and 26% for
ush2a and
whrnb, respectively. It has been described that as little as 20% of functional
ush2a transcripts can be sufficient to result in usherin localization at the zebrafish photoreceptor periciliary region [
35]. Therefore, it is likely that the reduced localization of usherin and Whrnb in the
adgrv1rmc22 zebrafish retina is the consequence of the absence of Adgrv1 resulting in the disintegration of the USH2 complex.
The USH2 protein network is hypothesized to contribute to the ciliary trafficking of cargo, such as rhodopsin, towards the photoreceptor outer segments. It is also well accepted that the accumulation of proteins in the inner segments due to the dysfunction of ciliary transport may underlie the photoreceptor degeneration through the activation of cell stress pathways [
6,
36]. Moreover, it was reported that mislocalized rhodopsin results in ectopic phototransduction [
37], a process which has been found to accelerate photoreceptor cell death [
38]. The significant increase in the number of photoreceptors with the aberrant localization of rhodopsin in the retinal sections of
adgrv1rmc22 larvae indeed supports the notion that defective ciliary protein transport may lie at the origin of
ADGRV1-associated retinal degeneration. Interestingly, histological examination of the juvenile (3 mpf) and adult (6 mpf)
adgrv1rmc22 zebrafish retina did not reveal any signs of progressive retinal degeneration (
Figure S3), as the
adgrv1rmc22 zebrafish retinal layers were morphologically indistinguishable from wild-type retinas. The absence of a clear progressive phenotype, however, is in line with the previously published
ush2a mutant zebrafish models, where also no morphological differences were observed when compared to wild-types [
20]. A plausible explanation for the absence of the progressive degeneration of photoreceptor cells lies in the regenerative capacity of the zebrafish retina, since in contrast to the human retina, the zebrafish retina is able to replace lost retinal neurons [
39]. The phototoxic effect of mislocalized rhodopsin resulting in photoreceptor cell death may thus be masked by the continuous photoreceptor regeneration. Lacking a progressive phenotype, the restoration of the USH2 protein complex at the periciliary region, and normal rhodopsin trafficking, provide valuable biomarkers to evaluate future therapeutic strategies for
ADGRV1-associated retinal degeneration in the
adgrv1rmc22 zebrafish.
In the current study, we assessed the function of Adgrv1 localized at the photoreceptor periciliary membrane. Interestingly, a recent paper identified ADGRV1 at the ER membrane and at the mitochondria-associated ER membranes (MAMs) in cells derived from VLGR1-deficient mouse models, suggesting a novel function for ADGRV1 [
9]. The absence of ADGRV1 at these internal membranes results in increased distances between the ER and mitochondria, and in reduced Ca
2+ release from the ER, thereby disrupting the bioenergetics of cells [
9]. Due to the high energy demand of photoreceptor cells, it is tempting to speculate that these mechanisms could also underlie photoreceptor cell dysfunction [
40]. Based on our immunohistochemical analysis, however, we obtained no indication that Adgrv1 is present at the ER membranes and MAMs in photoreceptor cells, as this requires high-resolution microscopy techniques.
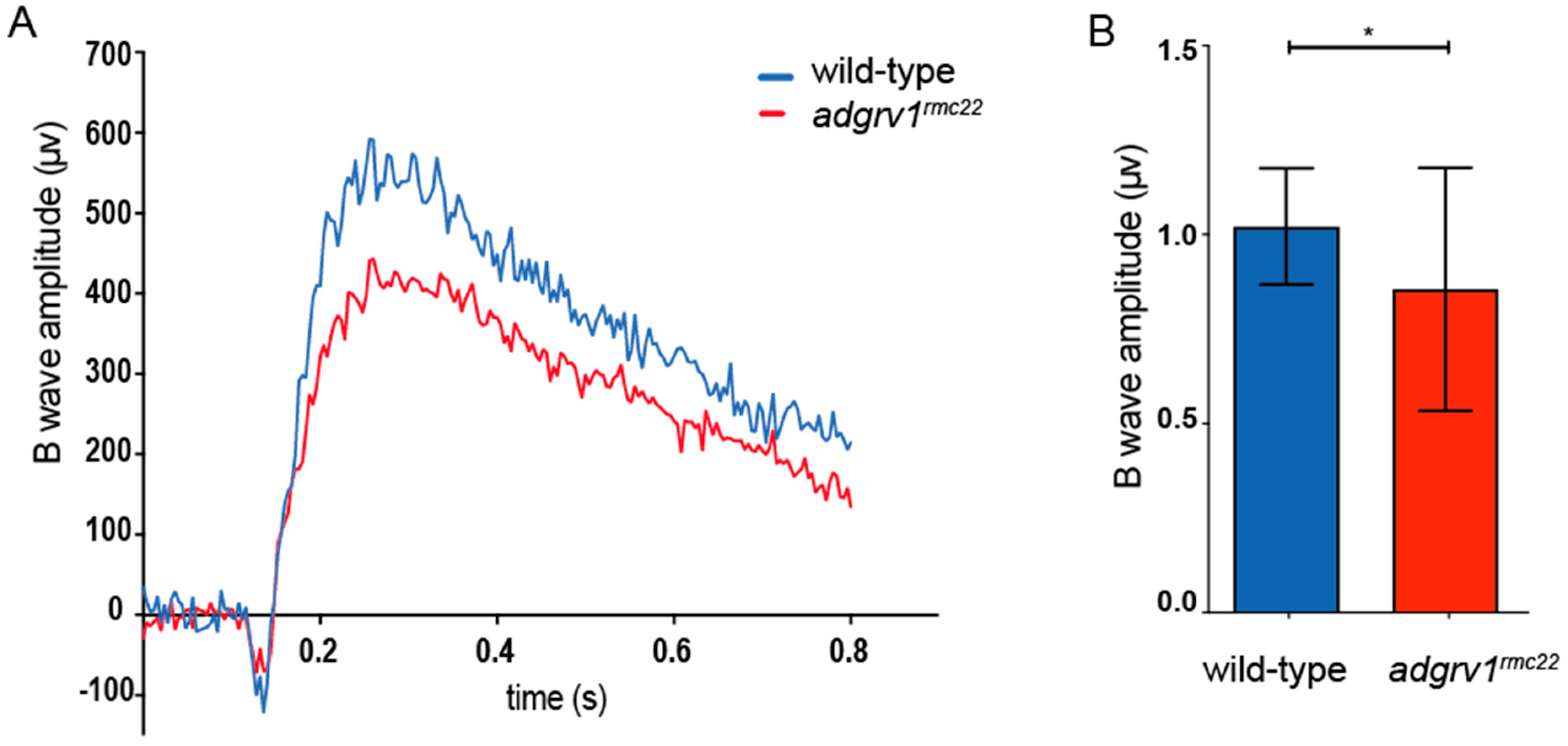
Finally, ERGs were recorded in the eyes of juvenile
adgrv1rmc22 (6–8 wpf) mutants and wild-type siblings. At this age, zebrafish rod and cone photoreceptors are considered to be mature, and both contribute to the visual function [
41]. When examining ERG traces, first a small initial A-wave is observed that represents the hyperpolarization of photoreceptor cells. The A-wave is largely obscured by the thereafter following B-wave that represents the depolarization of the ON bipolar cells [
42]. ERG recordings demonstrate a significant decrease in B-wave amplitude in the
adgrv1rmc22 zebrafish when compared to wild-types. The decrease in B-wave amplitude could be a result of a defect in phototransduction or in signal transduction towards the ON bipolar cells. Either way, the significant decrease in B-wave amplitude in the
adgrv1rmc22 zebrafish can be considered as an indication of impaired retinal function. These results are in line with the previously published ERGs recorded on the eyes of
ush2a mutant zebrafish larvae, where similar reductions in B-wave amplitudes were recorded [
20,
21,
22,
31].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}