


Astrocytes Differentiated from LRRK2-I1371V Parkinson’s-Disease-Induced Pluripotent Stem Cells Exhibit Similar Yield but Cell-Intrinsic Dysfunction in Glutamate Uptake and Metabolism, ATP Generation, and Nrf2-Mediated Glutathione Machinery
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Ethics Clearance
2.3. Generation of Neural Progenitors (NPs) from Induced Pluripotent Stem Cells (iPSCs)
2.4. Differentiation of Astrocytes from NPs
2.5. U87 Astrocyte Cell Line Maintenance
2.6. Plasmid Constructs and Transfection
2.7. Phase Contrast Microscopy
2.8. Immunocytochemistry
2.9. Flow Cytometry
2.10. Quantitative Polymerase Chain Reaction (qPCR)
2.11. Glutathione Content
2.12. Glutamine and Glutamate Contents
2.13. Glutamate Uptake
2.14. ATP Determination
2.15. Time-Lapse Live-Cell Fluorescence Imaging for Glutamate Dehydrogenase (GDH) Enzyme Kinetics
2.16. Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blot
2.17. Statistical Analysis
3. Results
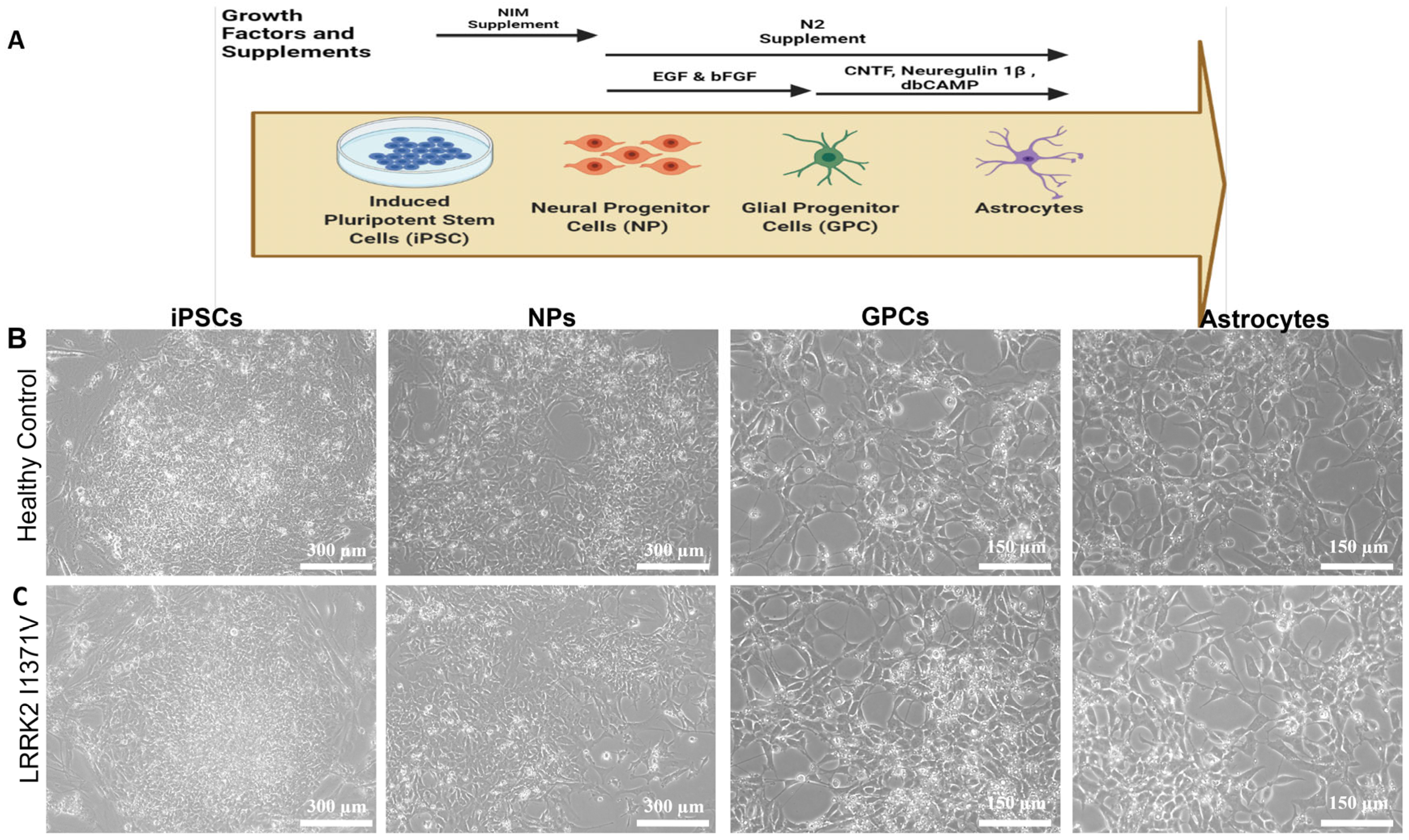
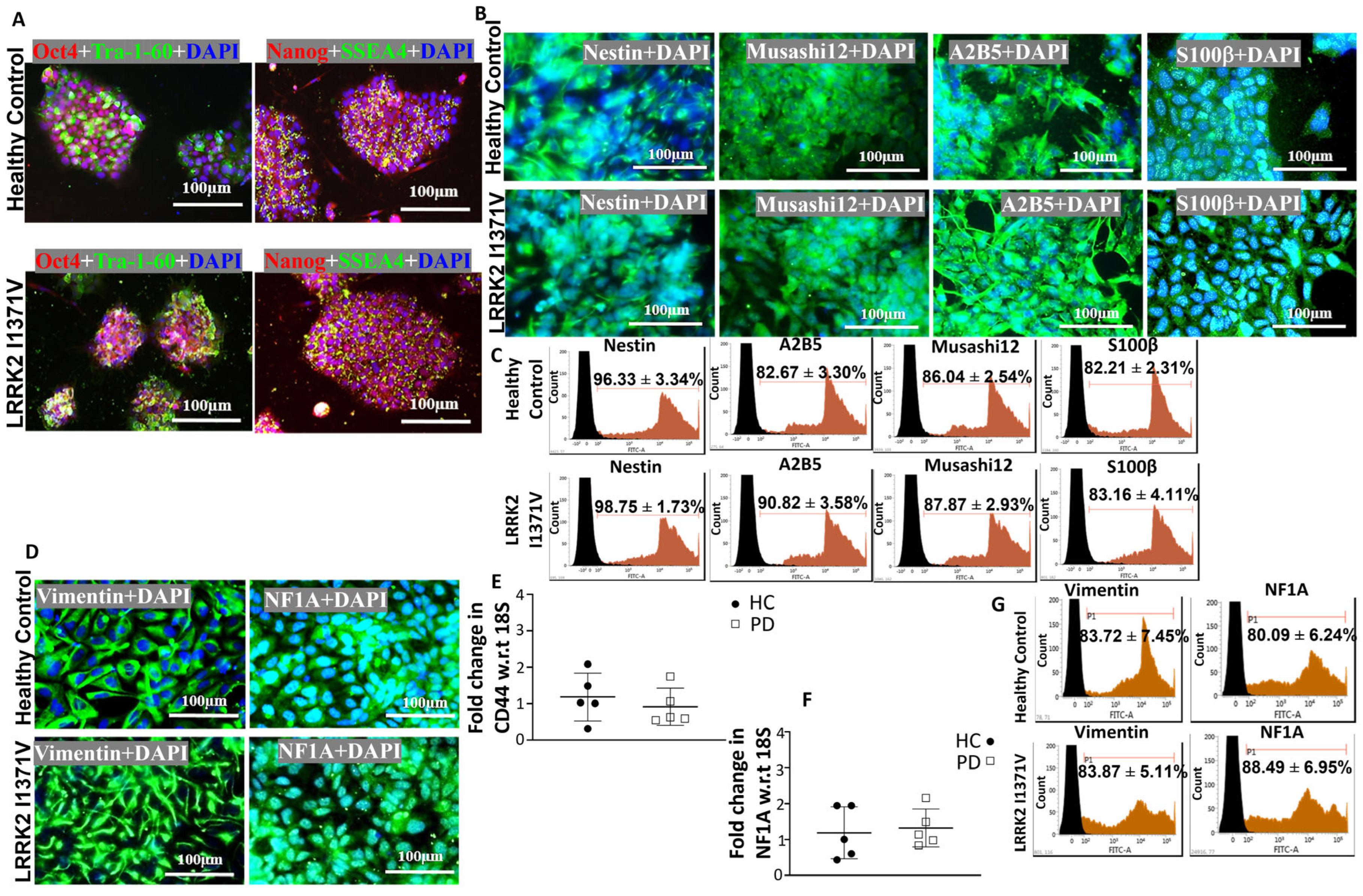
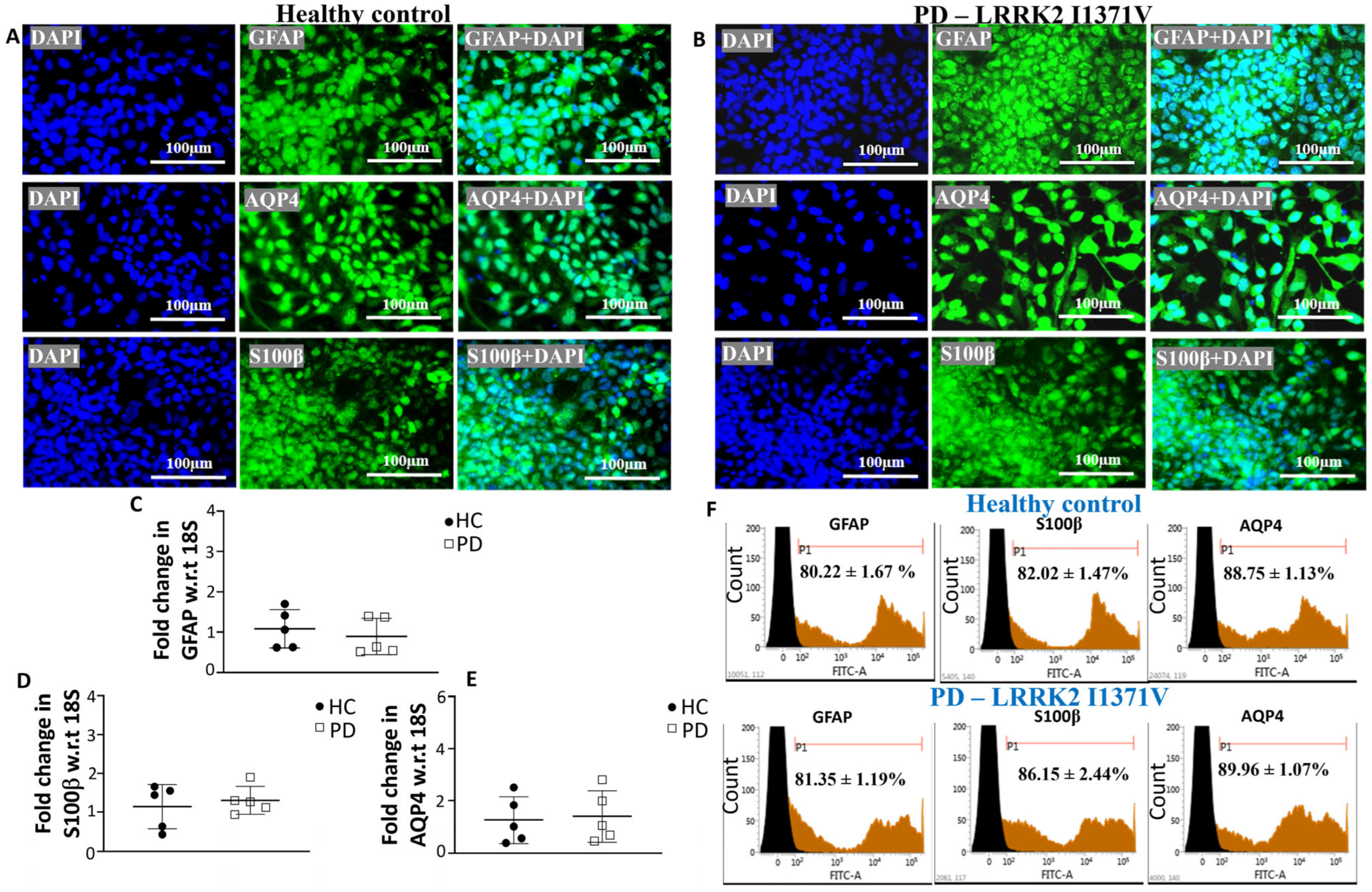
3.1. Generating Terminally Differentiated Astrocyte Populations from iPSC Lines
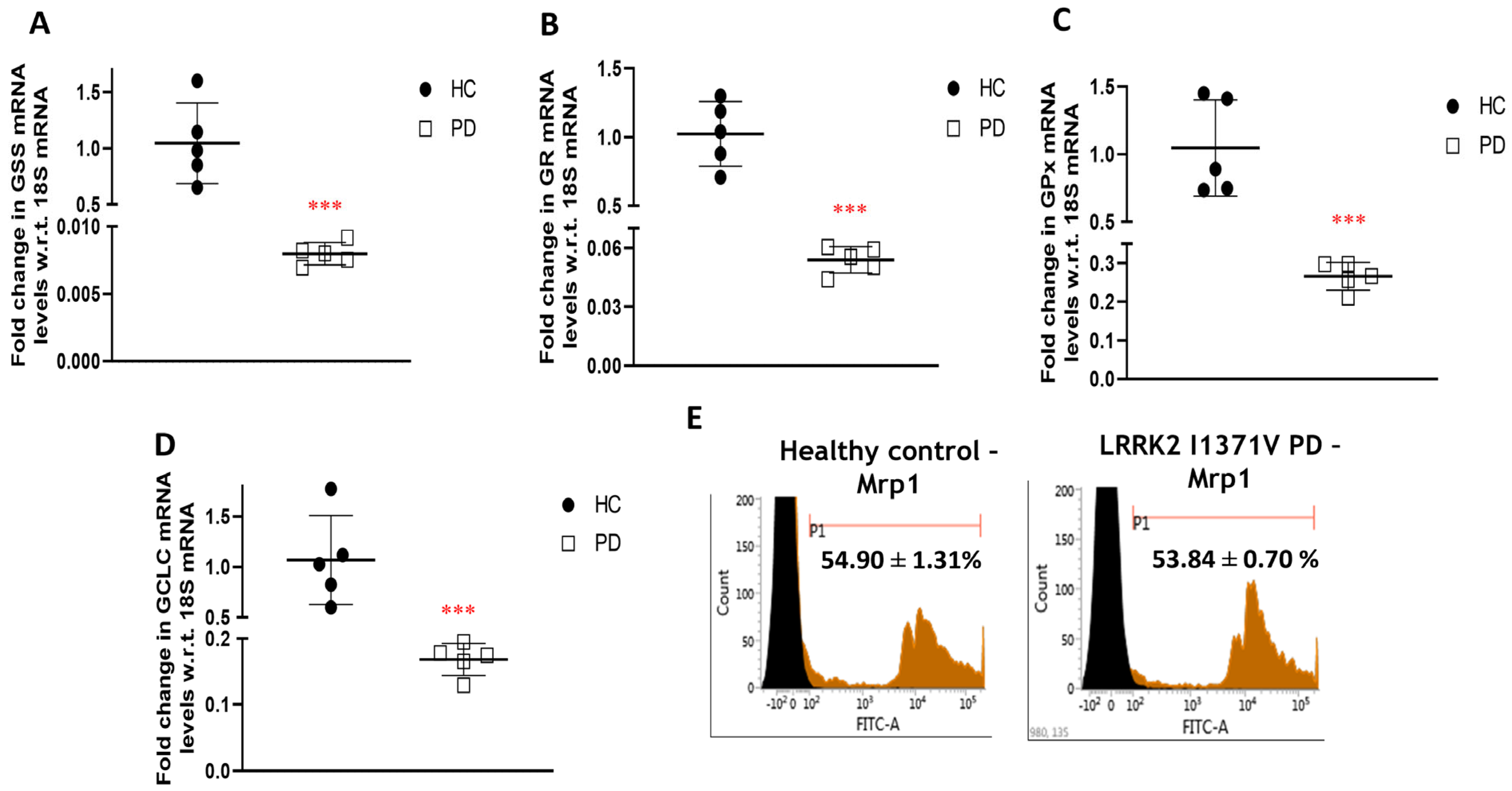
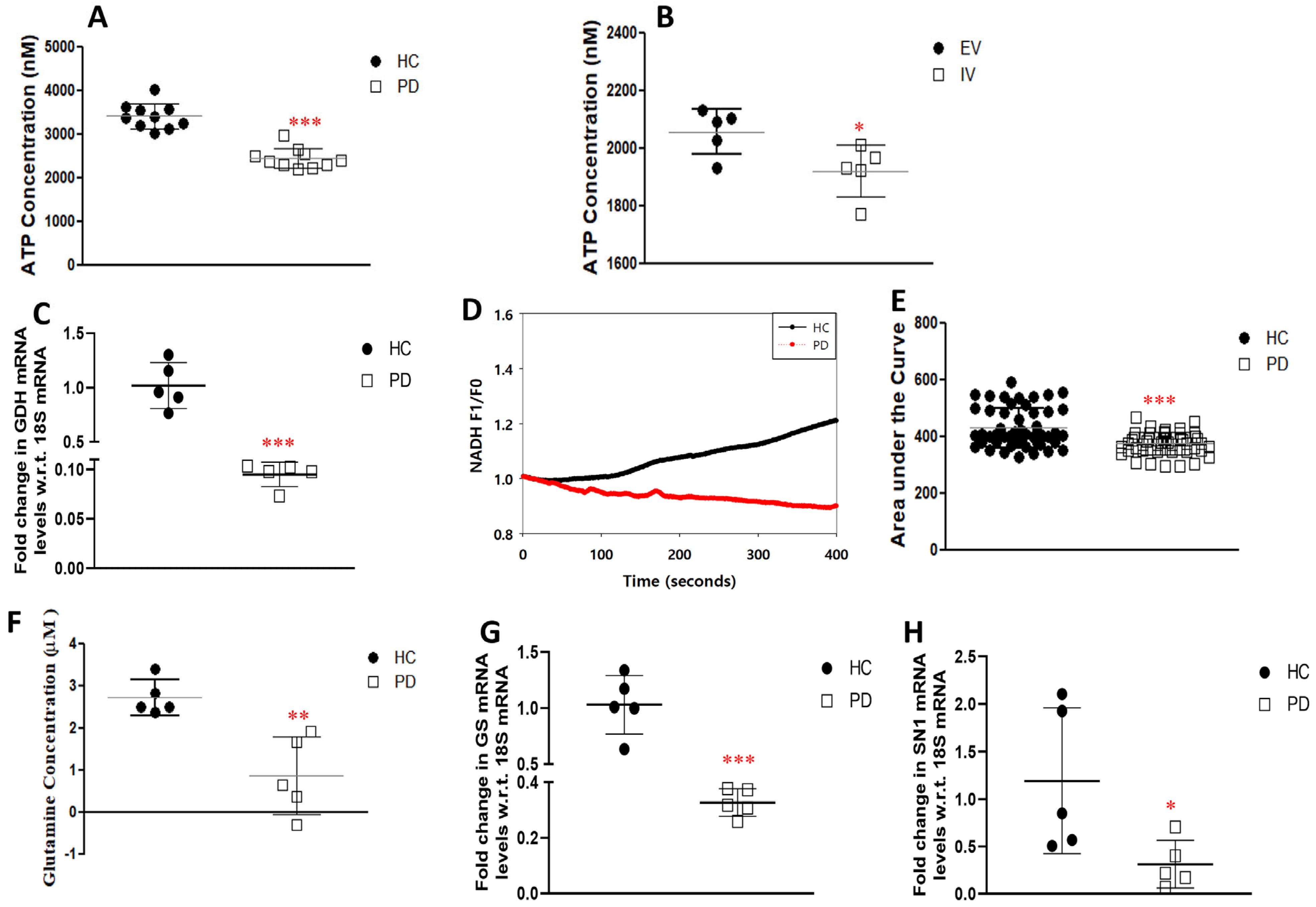
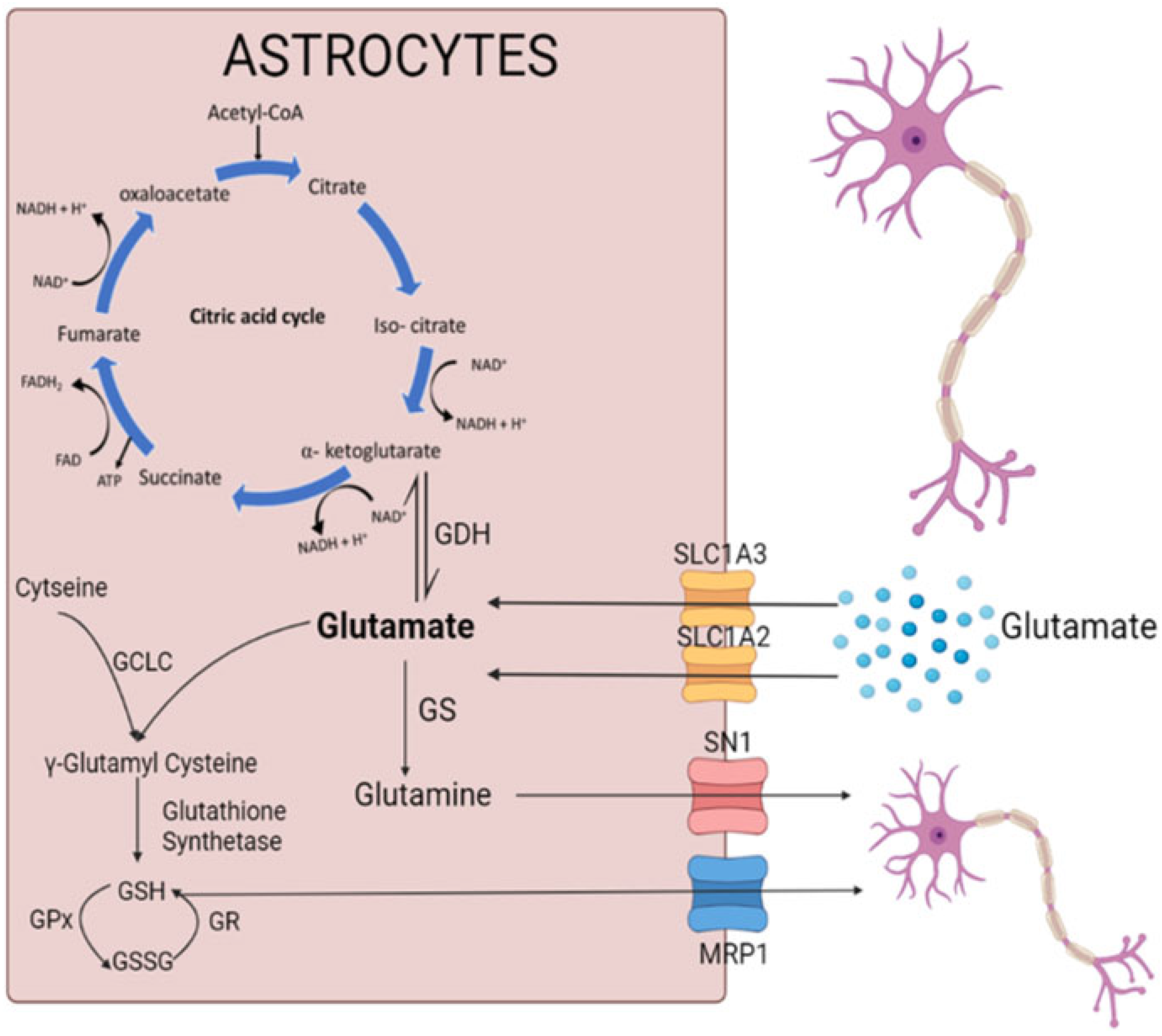
3.2. Glutathione Machinery
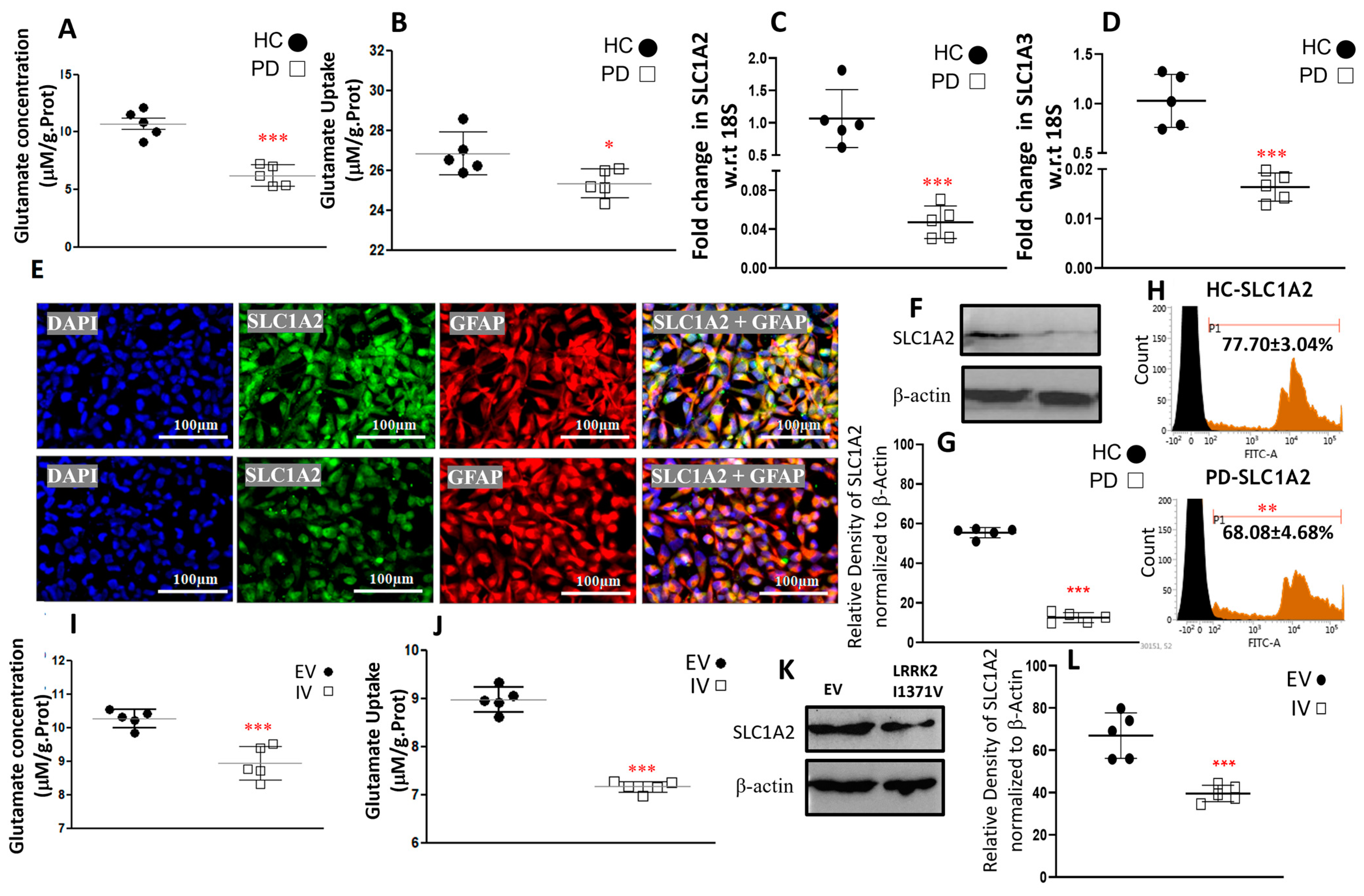
3.3. Glutamate Uptake and Metabolism
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Guaitoli, G.; Raimondi, F.; Gilsbach, B.K.; Gómez-Llorente, Y.; Deyaert, E.; Renzi, F.; Li, X.; Schaffner, A.; Jagtap, P.K.A.; Boldt, K.; et al. Structural model of the dimeric Parkinson’s protein LRRK2 reveals a compact architecture involving distant interdomain contacts. Proc. Natl. Acad. Sci. USA 2016, 113, E4357–E4366. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.S.; Kaila-Sharma, P.; Weng, J.-H.; Aoto, P.; Schmidt, S.H.; Knapp, S.; Mathea, S.; Herberg, F.W. Kinase Domain Is a Dynamic Hub for Driving LRRK2 Allostery. Front. Mol. Neurosci. 2020, 13, 538219. [Google Scholar] [CrossRef]
- Deng, J.; Lewis, P.A.; Greggio, E.; Sluch, E.; Beilina, A.; Cookson, M.R. Structure of the ROC domain from the Parkinson’s disease-associated leucine-rich repeat kinase 2 reveals a dimeric GTPase. Proc. Natl. Acad. Sci. USA 2008, 105, 1499–1504. [Google Scholar] [CrossRef]
- Giasson, B.I.; Van Deerlin, V.M. Mutations in LRRK2 as a Cause of Parkinson’s Disease. Neurosignals 2008, 16, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Paisan-Ruiz, C.; Lang, A.E.; Kawarai, T.; Sato, C.; Salehi-Rad, S.; Fisman, G.K.; Al-Khairallah, T.; George-Hyslop, P.S.; Singleton, A.; Rogaeva, E. LRRK2 gene in Parkinson disease: Mutation analysis and case control association study. Neurology 2005, 65, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, T.; Vishal, M.; Das, G.; Sharma, A.; Mukhopadhyay, A.; Das, S.K.; Ray, K.; Ray, J. Evaluation of the role of LRRK2 gene in Parkinson’s disease in an East Indian cohort. Dis. Markers 2012, 32, 355–362. [Google Scholar] [CrossRef]
- Cheng, Y.-C.; Huang, C.-Y.; Ho, M.-C.; Hsu, Y.-H.; Syu, S.-H.; Lu, H.-E.; Lin, H.-I.; Lin, C.-H.; Hsieh, P.C.H.S. Generation of 2 induced pluripotent stem cell lines derived from patients with Parkinson’s disease carrying LRRK2 G2385R variant. Stem Cell Res. 2018, 28, 1–5. [Google Scholar] [CrossRef]
- Ho, D.H.; Jang, J.; Joe, E.; Son, I.; Seo, H.; Seol, W. G2385R and I2020T Mutations Increase LRRK2 GTPase Activity. Biomed. Res. Int. 2016, 2016, 7917128. [Google Scholar] [CrossRef]
- Datta, I.; Sowmithra Jagtap, S.; Potdar, C.; Yadav, R.; Pal, P. Generation of induced pluripotent stem cells (NIMHi001-A) from a Parkinson’s disease patient of East Indian ethnicity carrying LRRK2 I1371V variant. Stem Cell Res. 2020, 44, 101768. [Google Scholar] [CrossRef]
- Rideout, H.; Stefanis, L. The neurobiology of LRRK2 and its role in the pathogenesis of Parkinson’s disease. Neurochem. Res. 2014, 39, 576–592. [Google Scholar] [CrossRef]
- Liao, J.; Wu, C.; Burlak, C.; Zhang, S.; Sahm, H.; Wang, M.; Zhang, Z.-Y.; Vogel, K.W.; Federici, M.; Riddle, S.M.; et al. Parkinson disease-associated mutation R1441H in LRRK2 prolongs the “active state” of its GTPase domain. Proc. Natl. Acad. Sci. USA 2014, 111, 4055–4060. [Google Scholar] [CrossRef]
- Xu, L.; Nagai, Y.; Kajihara, Y.; Ito, G.; Tomita, T. The Regulation of Rab GTPases by Phosphorylation. Biomolecules 2021, 11, 1340. [Google Scholar] [CrossRef]
- Iannotta, L.; Biosa, A.; Kluss, J.H.; Tombesi, G.; Kaganovich, A.; Cogo, S.; Plotegher, N.; Civiero, L.; Lobbestael, E.; Baekelandt, V.; et al. Divergent Effects of G2019S and R1441C LRRK2 Mutations on LRRK2 and Rab10 Phosphorylations in Mouse Tissues. Cells 2020, 9, 2344. [Google Scholar] [CrossRef]
- Fan, Y.; Nirujogi, R.S.; Garrido, A.; Ruiz-Martínez, J.; Bergareche-Yarza, A.; Mondragón-Rezola, E.; Vinagre-Aragón, A.; Croitoru, I.; Pagola, A.G.; Markinez, L.P.; et al. R1441G but not G2019S mutation enhances LRRK2 mediated Rab10 phosphorylation in human peripheral blood neutrophils. Acta Neuropathol. 2021, 142, 475–494. [Google Scholar] [CrossRef]
- Cooper, O.; Seo, H.; Andrabi, S.; Guardia-Laguarta, C.; Graziotto, J.; Sundberg, M.; McLean, J.R.; Carrillo-Reid, L.; Xie, Z.; Osborn, T.; et al. Pharmacological rescue of mitochondrial deficits in iPSC-derived neural cells from patients with familial Parkinson’s disease. Sci. Transl. Med. 2012, 4, 141ra90. [Google Scholar] [CrossRef]
- Chan, D.; Citro, A.; Cordy, J.M.; Shen, G.C.; Wolozin, B. Rac1 protein rescues neurite retraction caused by G2019S leucine-rich repeat kinase 2 (LRRK2). J. Biol. Chem. 2011, 286, 16140–16149. [Google Scholar] [CrossRef]
- Sánchez-Danés, A.; Richaud-Patin, Y.; Carballo-Carbajal, I.; Jiménez-Delgado, S.; Caig, C.; Mora, S.; Di Guglielmo, C.; Ezquerra, M.; Patel, B.; Giralt, A.; et al. Disease-specific phenotypes in dopamine neurons from human iPS-based models of genetic and sporadic Parkinson’s disease. EMBO Mol. Med. 2012, 4, 380–395. [Google Scholar] [CrossRef]
- Jagtap, S.; Potdar, C.; Yadav, R.; Pal, P.K.; Datta, I. Dopaminergic Neurons Differentiated from LRRK2 I1371V-Induced Pluripotent Stem Cells Display a Lower Yield, α-Synuclein Pathology, and Functional Impairment. ACS Chem. Neurosci. 2022, 13, 2632–2645. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J.; Arai, T.; Guo, J.-P.; Klegeris, A.; Yu, S.; McGeer, E.G.; McGeer, P.L. LRRK2 expression in normal and pathologic human brain and in human cell lines. J. Neuropathol. Exp. Neurol. 2006, 65, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Hirst, W.D.; Wade-martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Lindström, V.; Fagerqvist, T.; Nordström, E.; Eriksson, F.; Lord, A.; Tucker, S.; Andersson, J.; Johannesson, M.; Schell, H.; Kahle, P.J.; et al. Immunotherapy targeting α-synuclein protofibrils reduced pathology in (Thy-1)-h[A30P] α-synuclein mice. Neurobiol. Dis. 2014, 69, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Neumann, M.; Kahle, P.J.; Giasson, B.I.; Ozmen, L.; Borroni, E.; Spooren, W.; Müller, V.; Odoy, S.; Fujiwara, H.; Hasegawa, M.; et al. Misfolded proteinase K–resistant hyperphosphorylated α-synuclein in aged transgenic mice with locomotor deterioration and in human α-synucleinopathies. J. Clin. Investig. 2002, 110, 1429–1439. [Google Scholar] [CrossRef]
- Raj, A.; Kaushal, A.; Datta, I. Impact of monomeric and aggregated wild-type and A30P/A53T double-mutant α-synuclein on antioxidant mechanism and glutamate metabolic profile of cultured astrocytes. J. Neurosci. Res. 2022, 100, 681–706. [Google Scholar] [CrossRef]
- Takeuchi, H.; Jin, S.; Wang, J.; Zhang, G.; Kawanokuchi, J.; Kuno, R.; Sonobe, Y.; Mizuno, T.; Suzumura, A. Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J. Biol. Chem. 2006, 281, 21362–21368. [Google Scholar] [CrossRef]
- Wetherington, J.; Serrano, G.; Dingledine, R. Astrocytes in the Epileptic Brain. Neuron 2008, 58, 168–178. [Google Scholar] [CrossRef]
- Sonsalla, P.K.; Albers, D.S.; Zeevalk, G.D. Role of glutamate in neurodegeneration of dopamine neurons in several animal models of parkinsonism. Amino Acids 1998, 14, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Meredith, G.E.; Totterdell, S.; Beales, M.; Meshul, C.K. Impaired glutamate homeostasis and programmed cell death in a chronic MPTP mouse model of Parkinson’s disease. Exp. Neurol. 2009, 219, 334–340. [Google Scholar] [CrossRef]
- Meiser, J.; Weindl, D.; Hiller, K. Complexity of dopamine metabolism. Cell Commun. Signal 2013, 11, 34. [Google Scholar] [CrossRef]
- Baxter, P.S.; Hardingham, G.E. Adaptive regulation of the brain’s antioxidant defences by neurons and astrocytes. Free Radic. Biol. Med. 2016, 100, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.A.; Johnson, D.A.; Kraft, A.D.; Calkins, M.J.; Jakel, R.J.; Vargas, M.R.; Chen, P.-C. The Nrf2-ARE pathway: An indicator and modulator of oxidative stress in neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 61–69. [Google Scholar] [CrossRef]
- Dringen, R.; Hirrlinger, J. Glutathione Pathways in the Brain. Biol. Chem. 2003, 384, 505–516. [Google Scholar] [CrossRef]
- Sian, J.; Dexter, D.T.; Lees, A.J.; Daniel, S.; Agid, Y.; Javoy-Agid, F.; Jenner, P.; Marsden, C.D. Alterations in glutathione levels in Parkinson’s disease and other neurodegenerative disorders affecting basal ganglia. Ann. Neurol. 1994, 36, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Volterra, A.; Trotti, D.; Tromba, C.; Floridi, S.; Racagni, G. Glutamate uptake inhibition by oxygen free radicals in rat cortical astrocytes. J. Neurosci. 1994, 14, 2924–2932. [Google Scholar] [CrossRef]
- Di Domenico, A.; Carola, G.; Calatayud, C.; Pons-Espinal, M.; Muñoz, J.P.; Richaud-Patin, Y.; Fernandez-Carasa, I.; Gut, M.; Faella, A.; Parameswaran, J.; et al. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Rep. 2019, 12, 213–229. [Google Scholar] [CrossRef] [PubMed]
- Booth, H.D.E.; Wessely, F.; Connor-Robson, N.; Rinaldi, F.; Vowles, J.; Browne, C.; Evetts, S.G.; Hu, M.T.; Cowley, S.A.; Webber, C.; et al. RNA sequencing reveals MMP2 and TGFB1 downregulation in LRRK2 G2019S Parkinson’s iPSC-derived astrocytes. Neurobiol. Dis. 2019, 129, 56–66. [Google Scholar] [CrossRef]
- De Rus Jacquet, A.; Tancredi, J.L.; Lemire, A.L.; DeSantis, M.C.; Li, W.-P.; O’Shea, E.K. The LRRK2 G2019S mutation alters astrocyte-to-neuron communication via extracellular vesicles and induces neuron atrophy in a human iPSC-derived model of Parkinson’s disease. Elife 2021, 30, 10. [Google Scholar]
- Sonninen, T.-M.; Hämäläinen, R.H.; Koskuvi, M.; Oksanen, M.; Shakirzyanova, A.; Wojciechowski, S.; Puttonen, K.; Naumenko, N.; Goldsteins, G.; Laham-Karam, N.; et al. Metabolic alterations in Parkinson’s disease astrocytes. Sci. Rep. 2020, 10, 14474. [Google Scholar] [CrossRef]
- Von Linstow, C.; Gan-Or, Z.; Brundin, P. Precision medicine in Parkinson’s disease patients with LRRK2 and GBA risk variants–Let’s get even more personal. Transl. Neurodegener. 2020, 9, 39. [Google Scholar] [CrossRef] [PubMed]
- Burroughs, V.J.; Maxey, R.W.; Levy, R.A. Racial and ethnic differences in response to medicines: Towards individualized pharmaceutical treatment. J. Natl. Med. Assoc. 2002, 94 (Suppl. 10), 1–26. [Google Scholar]
- Lewis, P.; Rack, P.H.; Vaddadi, K.S.; Allen, J.J. Ethnic differences in drug response. Postgrad. Med. J. 1980, 56 (Suppl. 1), 46–49. [Google Scholar]
- Sauerbier, A.; Aris, A.; Lim, E.; Bhattacharya, K.; Ray, C.K. Impact of ethnicity on the natural history of Parkinson disease. Med. J. Aust. 2018, 208, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Titova, N.; Chaudhuri, K.R. Nonmotor Parkinson’s and Future Directions. Int. Rev. Neurobiol. 2017, 134, 1493–1505. [Google Scholar]
- Hertz, L.; Rothman, D. Glutamine-Glutamate Cycle Flux Is Similar in Cultured Astrocytes and Brain and Both Glutamate Production and Oxidation Are Mainly Catalyzed by Aspartate Aminotransferase. Biology 2017, 6, 17. [Google Scholar] [CrossRef] [PubMed]
- Hertz, L.; Chen, Y. Integration between Glycolysis and Glutamate-Glutamine Cycle Flux May Explain Preferential Glycolytic Increase during Brain Activation, Requiring Glutamate. Front. Integr. Neurosci. 2017, 11, 18. [Google Scholar] [CrossRef]
- Jagtap, S.; Sowmithra Yadav, R.; Pal, P.K.; Datta, I. Generation of induced pluripotent stem cells (NIMHi004-A, NIMHi005-A and NIMHi006-A) from healthy individuals of Indian ethnicity with no mutation for Parkinson’s disease related genes. Stem Cell Res. 2022, 60, 102716. [Google Scholar] [CrossRef] [PubMed]
- Sowmithra, S.; Jain, N.K.; Datta, I. Evaluating in vitro neonatal hypoxic-ischemic injury using neural progenitors derived from human embryonic stem cells. Stem Cells Dev. 2020, 29, 929–951. [Google Scholar] [CrossRef]
- Serio, A.; Bilican, B.; Barmada, S.J.; Ando, D.M.; Zhao, C.; Siller, R.; Burr, K.; Haghi, G.; Story, D.; Nishimura, A.L.; et al. Astrocyte pathology and the absence of non-cell autonomy in an induced pluripotent stem cell model of TDP-43 proteinopathy. Proc. Natl. Acad. Sci. USA 2013, 110, 4697–4702. [Google Scholar] [CrossRef]
- Ganapathy, K.; Datta, I.; Bhonde, R. Astrocyte-like Cells Differentiated from Dental Pulp Stem Cells Protect Dopaminergic Neurons Against 6-Hydroxydopamine Toxicity. Mol. Neurobiol. 2018, 56, 4395–4413. [Google Scholar] [CrossRef]
- Cho, I.K.; Yang, B.; Forest, C.; Qian, L.; Chan, A.W.S. Amelioration of Huntington’s disease phenotype in astrocytes derived from iPSC-derived neural progenitor cells of Huntington’s disease monkeys. PLoS ONE 2019, 14, 14474. [Google Scholar] [CrossRef]
- Malarkey, E.B.; Ni, Y.; Parpura, V. Ca2+ entry through TRPC1 channels contributes to intracellular Ca2+ dynamics and consequent glutamate release from rat astrocytes. Glia 2008, 56, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Ganapathy, K.; Datta, I.; Sowmithra, S.; Joshi, P.; Bhonde, R. Influence of 6-Hydroxydopamine toxicity on α-synuclein phosphorylation, resting vesicle expression, and vesicular dopamine release. J. Cell Biochem. 2016, 117, 2719–2736. [Google Scholar] [CrossRef] [PubMed]
- Schousboe, A.; Scafidi, S.; Bak, L.K.; Waagepetersen, H.S.; McKenna, M.C. Glutamate Metabolism in the Brain Focusing on Astrocytes. Adv. Neurobiol. 2014, 11, 13–30. [Google Scholar] [PubMed]
- Lu, C.-S.; Simons, E.J.; Wu-Chou, Y.-H.; Fonzo ADi Chang, H.-C.; Chen, R.-S.; Wen, Y.-H.; Rohé, C.F.; Breedveld, G.J.; Hattori, N.; Gasser, T.; et al. The LRRK2 I2012T, G2019S, and I2020T mutations are rare in Taiwanese patients with sporadic Parkinson’s disease. Park. Relat. Disord. 2005, 11, 521–522. [Google Scholar] [CrossRef] [PubMed]
- Luzon-Toro, B.; de la Torre, E.R.; Delgado, A.; Perez-Tur, J.; Hilfiker, S. Mechanistic insight into the dominant mode of the Parkinson’s disease-associated G2019S LRRK2 mutation. Hum. Mol. Genet. 2007, 16, 2031–2039. [Google Scholar] [CrossRef]
- Chittoor-Vinod, V.G.; Nichols, R.J.; Schüle, B. Genetic and Environmental Factors Influence the Pleomorphy of LRRK2 Parkinsonism. Int. J. Mol. Sci. 2021, 22, 1045. [Google Scholar] [CrossRef]
- Shu, L.; Zhang, Y.; Sun, Q.; Pan, H.; Tang, B. A Comprehensive Analysis of Population Differences in LRRK2 Variant Distribution in Parkinson’s Disease. Front. Aging Neurosci. 2019, 11, 13. [Google Scholar] [CrossRef]
- Trinh, J.; Zeldenrust, F.M.J.; Huang, J.; Kasten, M.; Schaake, S.; Petkovic, S.; Madoev, H.; Grünewald, A.; Almuammar, S.; König, I.R.; et al. Genotype-phenotype relations for the Parkinson’s disease genes SNCA, LRRK2, VPS35: MDSGene systematic review. Mov. Disord. 2018, 33, 1857–1870. [Google Scholar] [CrossRef]
- Mata, I.F.; Jang, Y.; Kim, C.-H.; Hanna, D.S.; Dorschner, M.O.; Samii, A.; Agarwal, P.; Roberts, J.W.; Klepitskaya, O.; Shprecher, D.R.; et al. The RAB39B p.G192R mutation causes X-linked dominant Parkinson’s disease. Mol. Neurodegener. 2015, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- An, X.-K.; Peng, R.; Li, T.; Burgunder, J.-M.; Wu, Y.; Chen, W.-J.; Zhang, J.-H.; Wang, Y.-C.; Xu, Y.-M.; Gou, Y.-R.; et al. LRRK2 Gly2385Arg variant is a risk factor of Parkinson’s disease among Han-Chinese from mainland China. Eur. J. Neurol. 2008, 15, 301–305. [Google Scholar] [CrossRef]
- Zimprich, A.; Biskup, S.; Leitner, P.; Lichtner, P.; Farrer, M.; Lincoln, S.; Kachergus, J.; Hulihan, M.; Uitti, R.J.; Calne, D.B.; et al. Mutations in LRRK2 Cause Autosomal-Dominant Parkinsonism with Pleomorphic Pathology. Neuron 2004, 44, 601–607. [Google Scholar] [CrossRef]
- Schneider, S.A.; Alcalay, R.N. Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov. Disord. 2017, 32, 1504–1523. [Google Scholar] [CrossRef]
- Henderson, M.X.; Sengupta, M.; Trojanowski, J.Q.; Lee, V.M.Y. Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 2019, 7, 183. [Google Scholar] [CrossRef]
- Li, Y.; Liu, W.; Oo, T.F.; Wang, L.; Tang, Y.; Jackson-Lewis, V.; Zhou, C.; Geghman, K.; Bogdanov, M.; Przedborski, S.; et al. Mutant LRRK2R1441G BAC transgenic mice recapitulate cardinal features of Parkinson’s disease. Nat. Neurosci. 2009, 12, 826–828. [Google Scholar] [CrossRef]
- Li, J.-Q.; Tan, L.; Yu, J.-T. The role of the LRRK2 gene in Parkinsonism. Mol. Neurodegener. 2014, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Steger, M.; Diez, F.; Dhekne, H.S.; Lis, P.; Nirujogi, R.S.; Karayel, O.; Tonelli, F.; Martinez, T.N.; Lorentzen, E.; Pfeffer, A.R.; et al. Systematic proteomic analysis of LRRK2-mediated Rab GTPase phosphorylation establishes a connection to ciliogenesis. Elife 2017, 6, e31012. [Google Scholar] [CrossRef]
- Steger, M.; Tonelli, F.; Ito, G.; Davies, P.; Trost, M.; Vetter, M.; Wachter, S.; Lorentzen, E.; Duddy, G.; Wilson, S.; et al. Phosphoproteomics reveals that Parkinson’s disease kinase LRRK2 regulates a subset of Rab GTPases. Elife 2016, 29, 5. [Google Scholar] [CrossRef] [PubMed]
- Datta, I.; Ganapathy, K.; Razdan, R.; Bhonde, R. Location and Number of Astrocytes Determine Dopaminergic Neuron Survival and Function under 6-OHDA Stress Mediated through Differential BDNF Release. Mol. Neurobiol. 2017, 55, 5505–5525. [Google Scholar] [CrossRef] [PubMed]
- Angeles, D.C.; Gan, B.-H.; Onstead, L.; Zhao, Y.; Lim, K.-L.; Dachsel, J.; Melrose, H.; Farrer, M.; Wszolek, Z.K.; Dickson, D.W.; et al. Mutations in LRRK2 increase phosphorylation of peroxiredoxin 3 exacerbating oxidative stress-induced neuronal death. Hum. Mutat. 2011, 32, 1390–1397. [Google Scholar] [CrossRef]
- Loeffler, D.A.; Klaver, A.C.; Coffey, M.P.; Aasly, J.O.; LeWitt, P.A. Increased Oxidative Stress Markers in Cerebrospinal Fluid from Healthy Subjects with Parkinson’s Disease-Associated LRRK2 Gene Mutations. Front. Aging Neurosci. 2017, 9, 89. [Google Scholar] [CrossRef]
- Kawakami, F.; Imai, M.; Tamaki, S.; Ohta, E.; Kawashima, R.; Maekawa, T.; Kurosaki, Y.; Ohba, K.; Ichikawa, T. Nrf2 Expression Is Decreased in LRRK2 Transgenic Mouse Brain and LRRK2 Overexpressing SH-SY5Y Cells. Biol. Pharm. Bull. 2023, 46, b22-00356. [Google Scholar] [CrossRef]
- Melrose, H. Update on the functional biology ofLrrk2. Future Neurol. 2008, 3, 669–681. [Google Scholar] [CrossRef]
- Thakur, G.; Kumar, V.; Lee, K.W.; Won, C. Structural Insights and Development of LRRK2 Inhibitors for Parkinson’s Disease in the Last Decade. Genes 2022, 13, 1426. [Google Scholar] [CrossRef]
- Lin, Y.-E.; Lin, C.-H.; Ho, E.-P.; Ke, Y.-C.; Petridi, S.; Elliott, C.J.; Sheen, L.-Y.; Chien, C.-T. Glial Nrf2 signaling mediates the neuroprotection exerted by Gastrodia elata Blume in Lrrk2-G2019S Parkinson’s disease. Elife 2021, 10, e73753. [Google Scholar] [CrossRef] [PubMed]
- Delcambre, S.; Ghelfi, J.; Ouzren, N.; Grandmougin, L.; Delbrouck, C.; Seibler, P.; Wasner, K.; Aasly, J.O.; Klein, C.; Trinh, J.; et al. Mitochondrial Mechanisms of LRRK2 G2019S Penetrance. Front. Neurol. 2020, 11, 881. [Google Scholar] [CrossRef]
- Muriel, P.; Gordillo, K.R. Role of Oxidative Stress in Liver Health and Disease. Oxidative Med. Cell. Longev. 2016, 2016, 9037051. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Glutathione synthesis. Biochim. Biophys. Acta-Gen. Subj. 2013, 1830, 3143–3153. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-K.; Na, K.-S. Role of glutamate receptors and glial cells in the pathophysiology of treatment-resistant depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2016, 70, 117–126. [Google Scholar] [CrossRef]
- Maragakis, N.J.; Dietrich, J.; Wong, V.; Xue, H.; Mayer-Proschel, M.; Rao, M.S.; Rothstein, J.D. Glutamate transporter expression and function in human glial progenitors. Glia 2004, 45, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Lauriat, T.L.; McInnes, L.A. EAAT2 regulation and splicing: Relevance to psychiatric and neurological disorders. Mol. Psychiatry 2007, 12, 1065–1078. [Google Scholar] [CrossRef]
- Sheldon, A.L.; Robinson, M.B. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem. Int. 2007, 51, 333–355. [Google Scholar] [CrossRef]
- Figura, M.; Kuśmierska, K.; Bucior, E.; Szlufik, S.; Koziorowski, D.; Jamrozik, Z.; Janik, P. Serum amino acid profile in patients with Parkinson’s disease. PLoS ONE 2018, 13, e0191670. [Google Scholar] [CrossRef] [PubMed]
- Gröger, A.; Kolb, R.; Schäfer, R.; Klose, U. Dopamine Reduction in the Substantia Nigra of Parkinson’s Disease Patients Confirmed by In Vivo Magnetic Resonance Spectroscopic Imaging. PLoS ONE 2014, 9, e84081. [Google Scholar] [CrossRef]
- Chua, C.E.L.; Tang, B.L. Rab 10—A traffic controller in multiple cellular pathways and locations. J. Cell. Physiol. 2018, 233, 6483–6494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; He, X.; Meng, X.; Wu, X.; Tong, H.; Zhang, X.; Qu, S. Regulation of glutamate transporter trafficking by Nedd4-2 in a Parkinson’s disease model. Cell Death Dis. 2017, 8, e2574. [Google Scholar] [CrossRef] [PubMed]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The Glutamate Dehydrogenase Pathway and Its Roles in Cell and Tissue Biology in Health and Disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef]
- Giordana, M.; D’Agostino, C.; Albani, G.; Mauro, A.; Di Fonzo, A.; Antonini, A.; Bonifati, V. Neuropathology of Parkinson’s disease associated with the LRRK2 Ile1371Val mutation. Mov. Disord. 2007, 22, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dunn, L.; Greggio, E.; Krumm, B.; Jackson, G.; Cookson, M.; Lewis, P.A.; Deng, J. The R1441C mutation alters the folding properties of the ROC domain of LRRK2. Biochim. Biophys Acta BBA-Mol. Basis Dis. 2009, 1792, 1194–1197. [Google Scholar] [CrossRef]
- Pajarillo, E.; Rizor, A.; Lee, J.; Aschner, M.; Lee, E. The role of astrocytic glutamate transporters GLT-1 and GLAST in neurological disorders: Potential targets for neurotherapeutics. Neuropharmacology 2019, 161, 107559. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Banerjee, R.; Raj, A.; Potdar, C.; Pal, P.K.; Yadav, R.; Kamble, N.; Holla, V.; Datta, I. Astrocytes Differentiated from LRRK2-I1371V Parkinson’s-Disease-Induced Pluripotent Stem Cells Exhibit Similar Yield but Cell-Intrinsic Dysfunction in Glutamate Uptake and Metabolism, ATP Generation, and Nrf2-Mediated Glutathione Machinery. Cells 2023, 12, 1592. https://doi.org/10.3390/cells12121592
Banerjee R, Raj A, Potdar C, Pal PK, Yadav R, Kamble N, Holla V, Datta I. Astrocytes Differentiated from LRRK2-I1371V Parkinson’s-Disease-Induced Pluripotent Stem Cells Exhibit Similar Yield but Cell-Intrinsic Dysfunction in Glutamate Uptake and Metabolism, ATP Generation, and Nrf2-Mediated Glutathione Machinery. Cells. 2023; 12(12):1592. https://doi.org/10.3390/cells12121592
Chicago/Turabian StyleBanerjee, Roon, Aishwarya Raj, Chandrakanta Potdar, Pramod Kumar Pal, Ravi Yadav, Nitish Kamble, Vikram Holla, and Indrani Datta. 2023. "Astrocytes Differentiated from LRRK2-I1371V Parkinson’s-Disease-Induced Pluripotent Stem Cells Exhibit Similar Yield but Cell-Intrinsic Dysfunction in Glutamate Uptake and Metabolism, ATP Generation, and Nrf2-Mediated Glutathione Machinery" Cells 12, no. 12: 1592. https://doi.org/10.3390/cells12121592
APA StyleBanerjee, R., Raj, A., Potdar, C., Pal, P. K., Yadav, R., Kamble, N., Holla, V., & Datta, I. (2023). Astrocytes Differentiated from LRRK2-I1371V Parkinson’s-Disease-Induced Pluripotent Stem Cells Exhibit Similar Yield but Cell-Intrinsic Dysfunction in Glutamate Uptake and Metabolism, ATP Generation, and Nrf2-Mediated Glutathione Machinery. Cells, 12(12), 1592. https://doi.org/10.3390/cells12121592