.png)

Neuronal p58IPK Protects Retinal Ganglion Cells Independently of Macrophage/Microglia Activation in Ocular Hypertension

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Microbead-Induced OHT Model
2.3. Immunohistochemistry and Analyses
2.4. Dark- and Light-Adapted Electroretinography (ERG)
2.5. Isolation and Culture of Primary Mouse BMDMs
2.6. RNA Isolation and Quantitative Real-Time PCR (qPCR)
2.7. Western Blot Analysis
2.8. Intravitreal Injection of AAVs
2.9. Statistical Data Analysis
3. Results
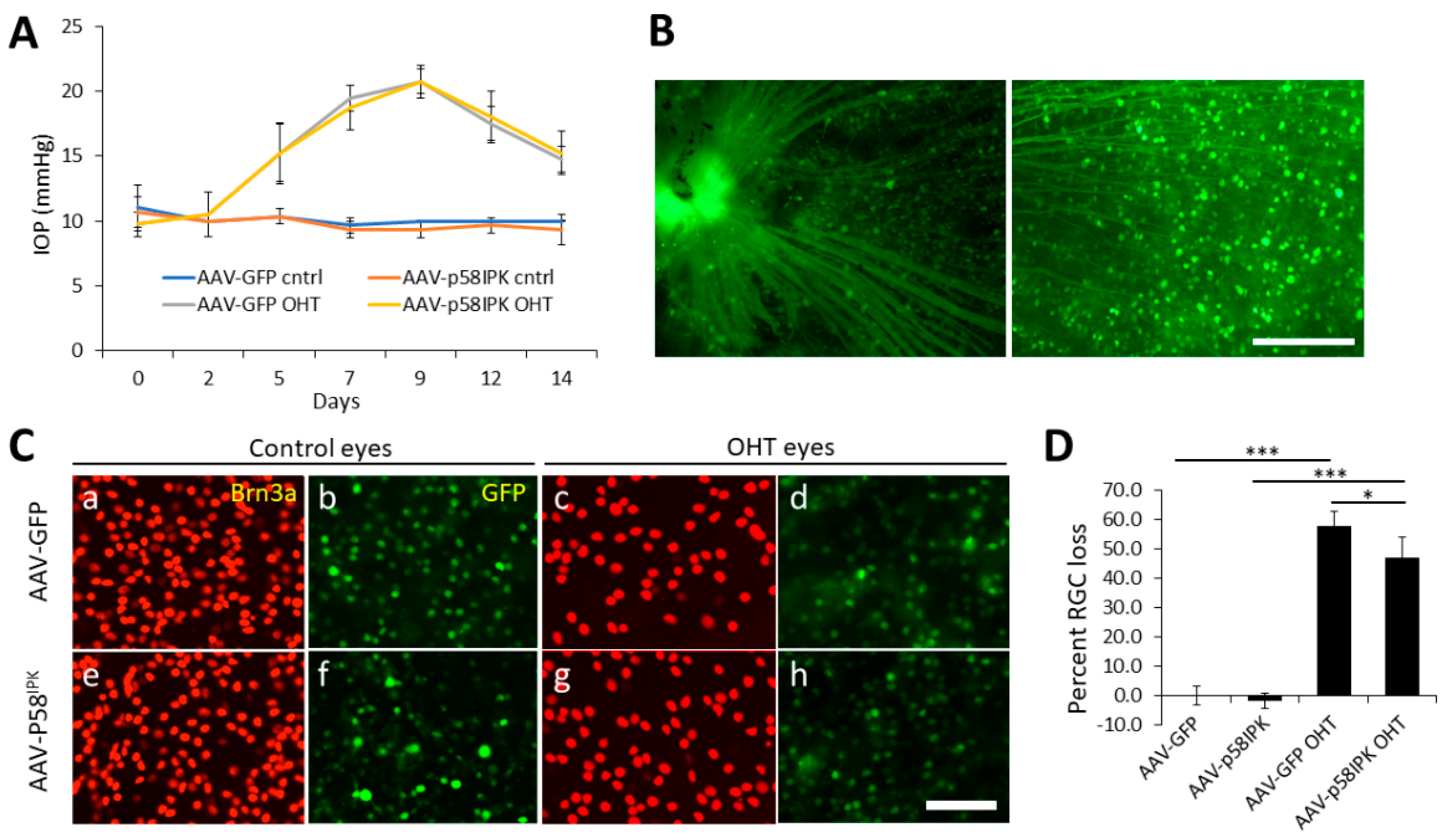
3.1. In Vivo Overexpression of p58IPK by AAV in the Retina Reduces RGC Loss in OHT
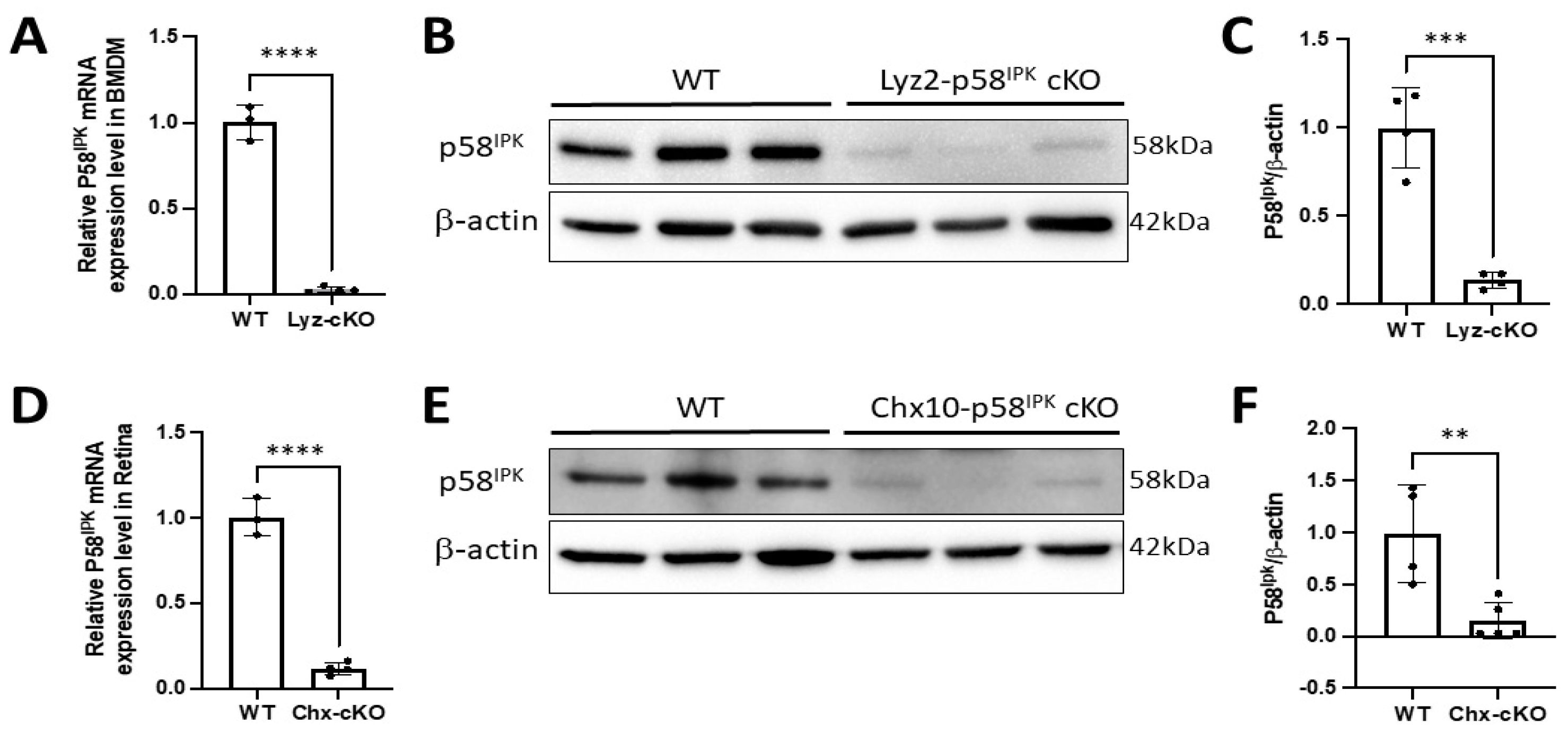
3.2. Generation of Retina-Specific and Myeloid Cell-Specific p58IPK cKO Mouse Lines
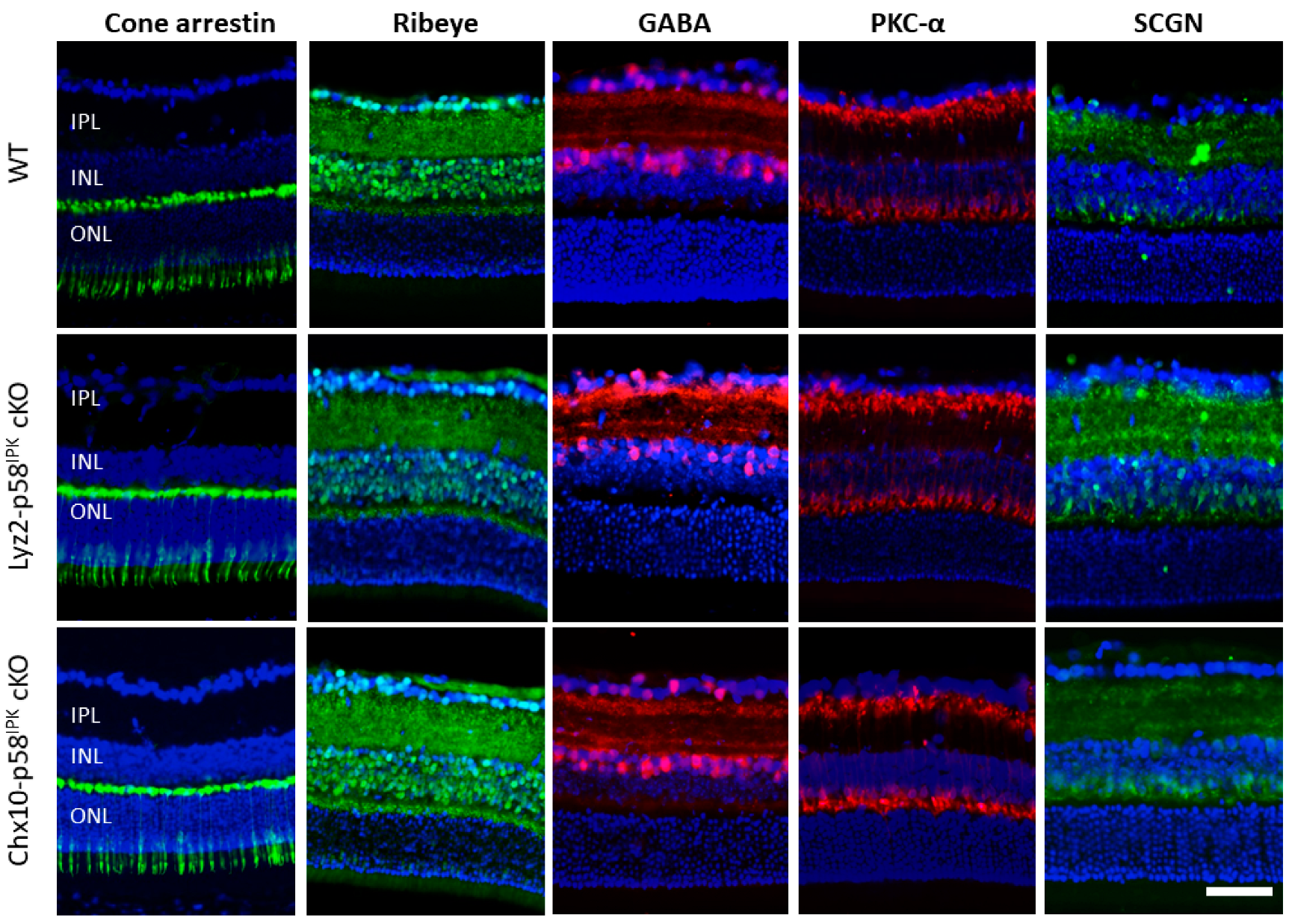
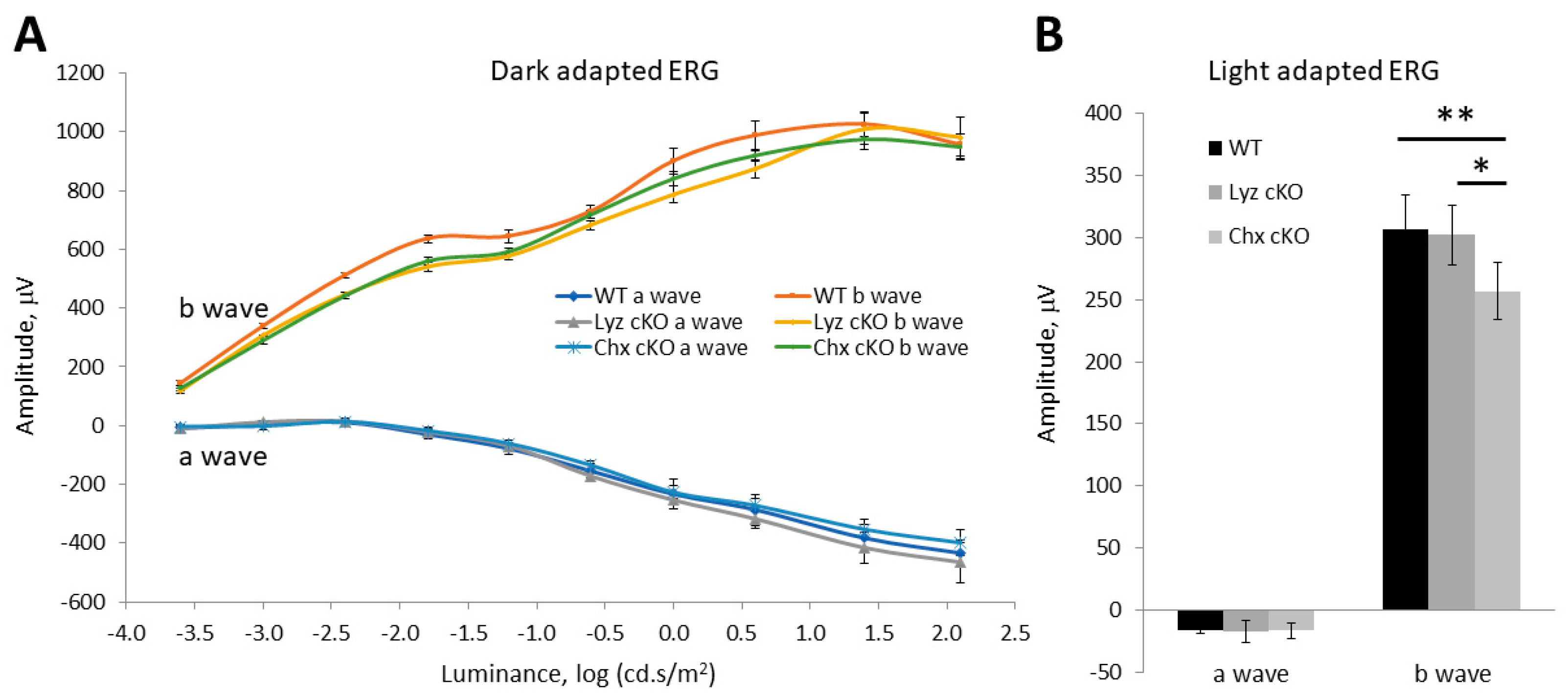
3.3. Impacts of p58IPK Deficiency in Macrophages or Retinal Neurons on Retinal Structure or Function
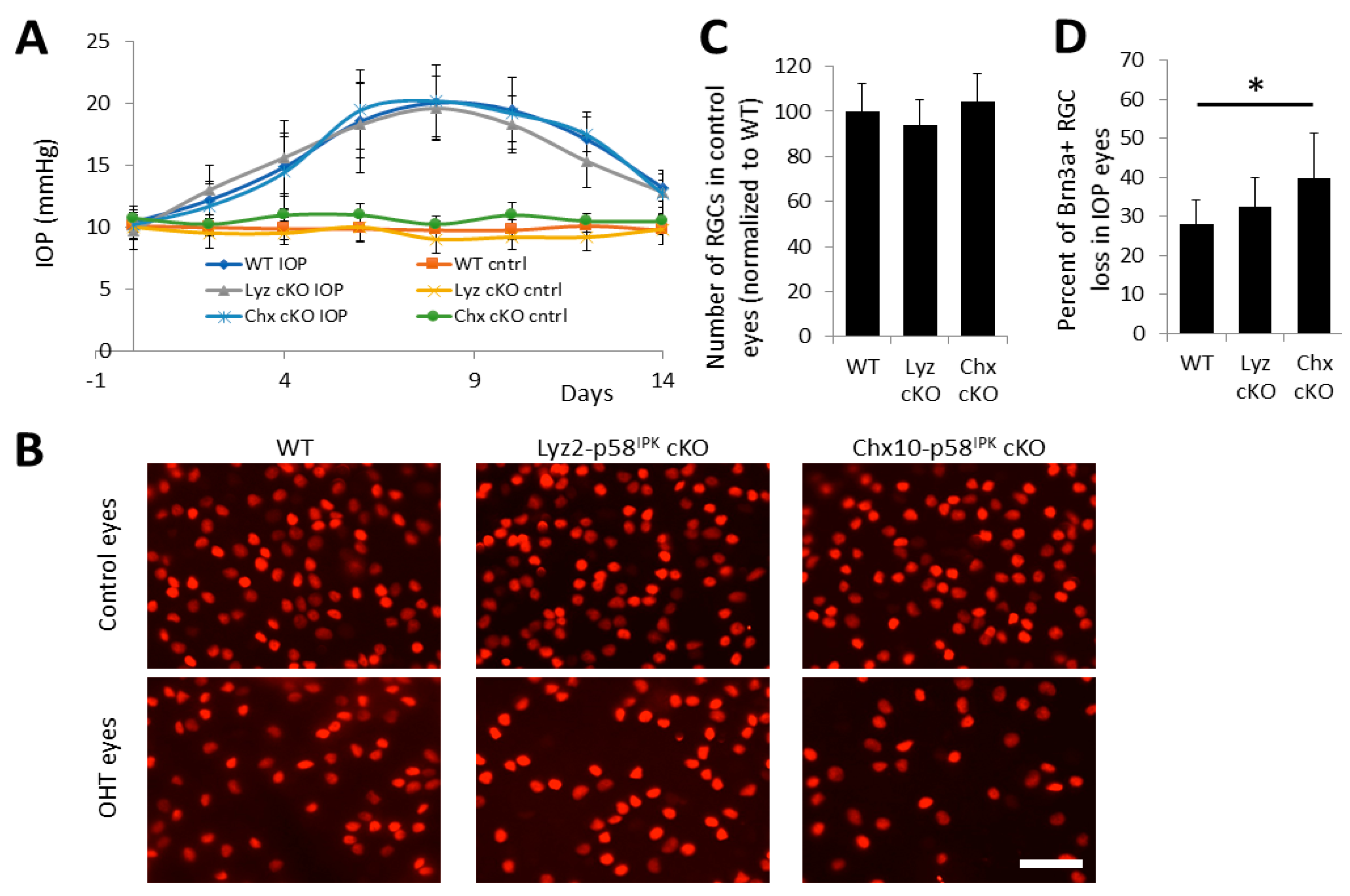
3.4. Increased RGC Loss after OHT in Chx10-p58IPK cKO Mice, but Not in Lyz2-p58IPK cKO Mice
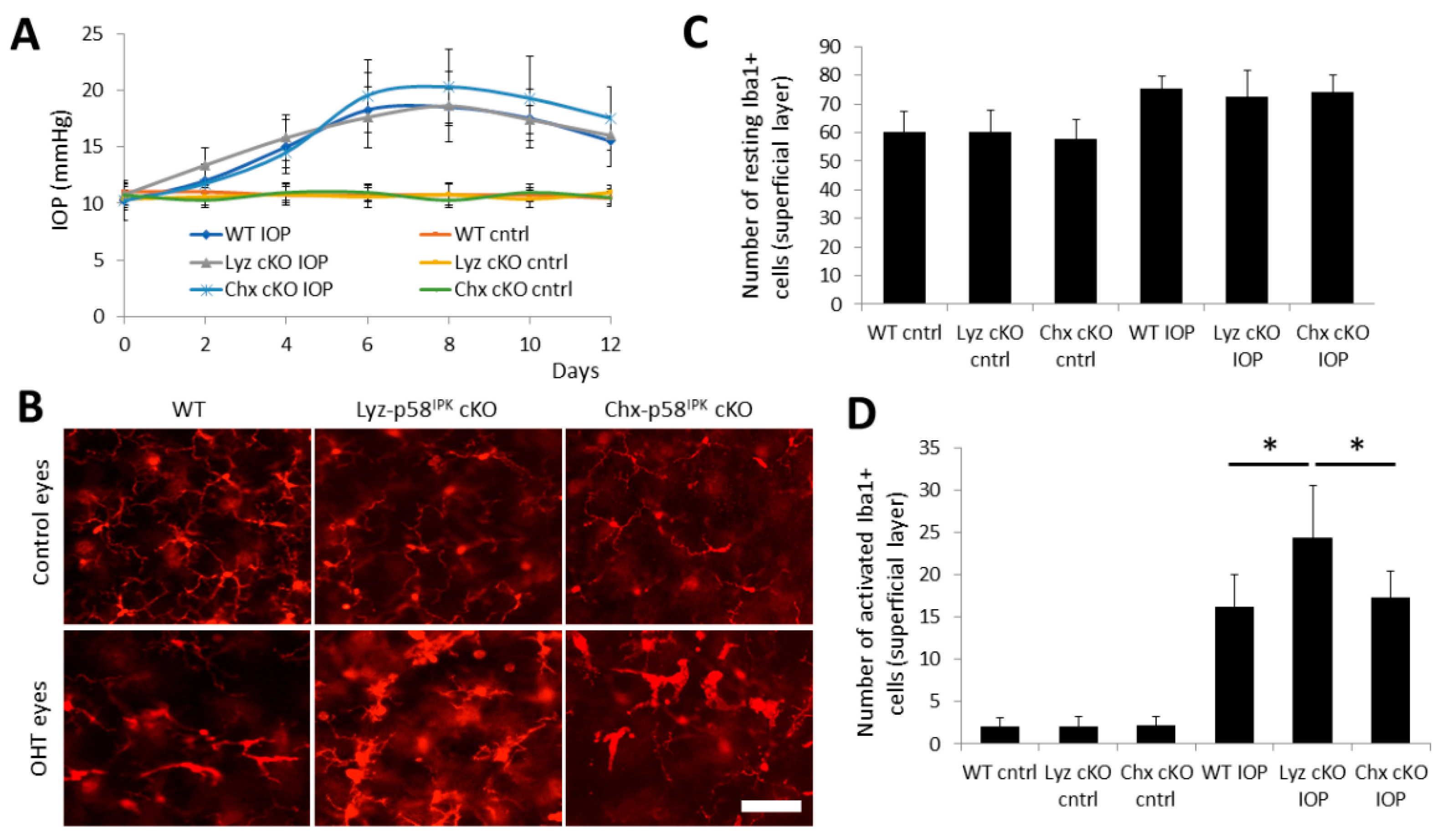
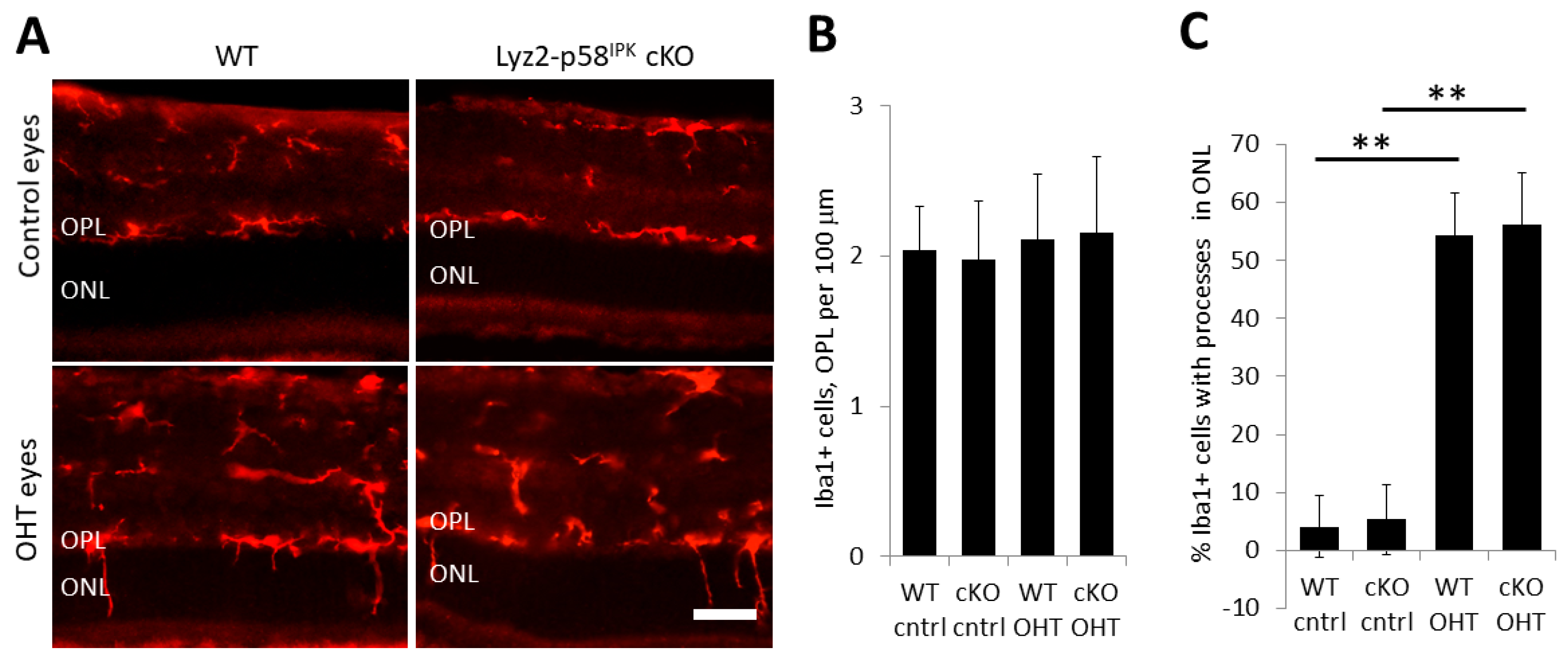
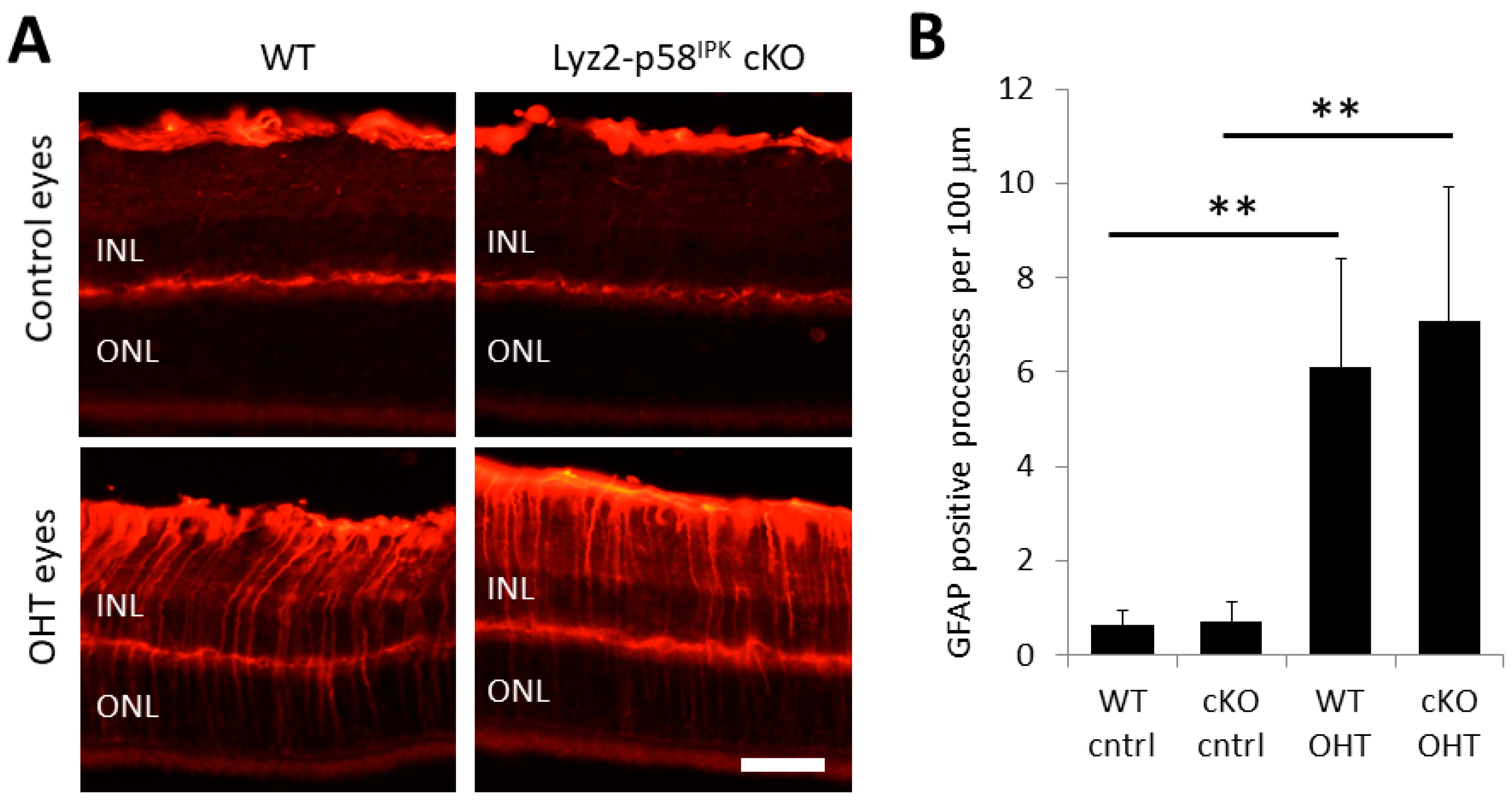
3.5. Increased Macrophage/Microglial Activation in the Retina of Lyz2-p58IPK cKO Mice after OHT
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Almasieh, M.; Levin, L.A. Neuroprotection in Glaucoma: Animal Models and Clinical Trials. Annu. Rev. Vis. Sci. 2017, 3, 91–120. [Google Scholar] [CrossRef] [PubMed]
- Storgaard, L.; Tran, T.L.; Freiberg, J.C.; Hauser, A.S.; Kolko, M. Glaucoma Clinical Research: Trends in Treatment Strategies and Drug Development. Front. Med. 2021, 8, 733080. [Google Scholar] [CrossRef] [PubMed]
- Alqawlaq, S.; Flanagan, J.G.; Sivak, J.M. All roads lead to glaucoma: Induced retinal injury cascades contribute to a common neurodegenerative outcome. Exp. Eye Res. 2019, 183, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Cho, K.S.; Thee, E.F.; Jager, M.J.; Chen, D.F. Neuroinflammation and microglia in glaucoma: Time for a paradigm shift. J. Neurosci. Res. 2019, 97, 70–76. [Google Scholar] [CrossRef]
- Coyle, S.; Khan, M.N.; Chemaly, M.; Callaghan, B.; Doyle, C.; Willoughby, C.E.; Atkinson, S.D.; Gregory-Ksander, M.; McGilligan, V. Targeting the NLRP3 Inflammasome in Glaucoma. Biomolecules 2021, 11, 1239. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Medina, A.; Perkins, J.; Yera, M.; Wang, J.J.; Zhang, S.X. Cellular stress signaling and the unfolded protein response in retinal degeneration: Mechanisms and therapeutic implications. Mol. Neurodegener. 2022, 17, 25. [Google Scholar] [CrossRef]
- Van Huizen, R.; Martindale, J.L.; Gorospe, M.; Holbrook, N.J. P58IPK, a Novel Endoplasmic Reticulum Stress-inducible Protein and Potential Negative Regulator of eIF2α Signaling. J. Biol. Chem. 2003, 278, 15558–15564. [Google Scholar] [CrossRef]
- Yan, W.; Frank, C.L.; Korth, M.J.; Sopher, B.L.; Novoa, I.; Ron, D.; Katze, M.G. Control of PERK eIF2α kinase activity by the endoplasmic reticulum stress-induced molecular chaperone P58IPK. Proc. Natl. Acad. Sci. USA 2002, 99, 15920–15925. [Google Scholar] [CrossRef]
- Daverkausen-Fischer, L.; Pröls, F. Dual topology of co-chaperones at the membrane of the endoplasmic reticulum. Cell Death Discov. 2021, 7, 203. [Google Scholar] [CrossRef]
- Rutkowski, D.T.; Kang, S.-W.; Goodman, A.G.; Garrison, J.L.; Taunton, J.; Katze, M.G.; Kaufman, R.J.; Hegde, R.S.; Gilmore, R. The Role of p58IPK in Protecting the Stressed Endoplasmic Reticulum. Mol. Biol. Cell 2007, 18, 3681–3691. [Google Scholar] [CrossRef]
- Gale, M., Jr.; Tan, S.L.; Wambach, M.; Katze, M.G. Interaction of the interferon-induced PKR protein kinase with inhibitory proteins P58IPK and vaccinia virus K3L is mediated by unique domains: Implications for kinase regulation. Mol. Cell. Biol. 1996, 16, 4172–4181. [Google Scholar] [CrossRef] [PubMed]
- Roobol, A.; Roobol, J.; Bastide, A.; Knight, J.R.; Willis, A.E.; Smales, C.M. p58IPK is an inhibitor of the eIF2alpha kinase GCN2 and its localization and expression underpin protein synthesis and ER processing capacity. Biochem. J. 2015, 465, 213–225. [Google Scholar] [CrossRef] [PubMed]
- Boriushkin, E.; Wang, J.J.; Li, J.; Bhatta, M.; Zhang, S.X. p58(IPK) suppresses NLRP3 inflammasome activation and IL-1beta production via inhibition of PKR in macrophages. Sci. Rep. 2016, 6, 25013. [Google Scholar] [CrossRef] [PubMed]
- Boriushkin, E.; Wang, J.J.; Li, J.; Jing, G.; Seigel, G.M.; Zhang, S.X. Identification of p58IPK as a Novel Neuroprotective Factor for Retinal Neurons. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1374–1386. [Google Scholar] [CrossRef]
- McLaughlin, T.; Dhimal, N.; Li, J.; Wang, J.J.; Zhang, S.X. p58IPK Is an Endogenous Neuroprotectant for Retinal Ganglion Cells. Front. Aging Neurosci. 2018, 10, 267. [Google Scholar] [CrossRef]
- George, S.H.; Gertsenstein, M.; Vintersten, K.; Korets-Smith, E.; Murphy, J.; Stevens, M.E.; Haigh, J.J.; Nagy, A. Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 4455–4460. [Google Scholar] [CrossRef]
- Clausen, B.E.; Burkhardt, C.; Reith, W.; Renkawitz, R.; Förster, I. Conditional gene targeting in macrophages and granulocytes using LysMcre mice. Transgenic Res. 1999, 8, 265–277. [Google Scholar] [CrossRef]
- Rowan, S.; Cepko, C.L. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev. Biol. 2004, 271, 388–402. [Google Scholar] [CrossRef]
- McLaughlin, T.; Falkowski, M.; Park, J.W.; Keegan, S.; Elliott, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 2018, 13, 16. [Google Scholar] [CrossRef]
- Zhang, X.; Goncalves, R.; Mosser, D.M. The isolation and characterization of murine macrophages. Curr. Protoc. Immunol. 2008, 83, 14-1. [Google Scholar] [CrossRef]
- Muzumdar, M.D.; Tasic, B.; Miyamichi, K.; Li, L.; Luo, L. A global double-fluorescent Cre reporter mouse. Genesis 2007, 45, 593–605. [Google Scholar] [CrossRef]
- Chen, H.; Wei, X.; Cho, K.-S.; Chen, G.; Sappington, R.; Calkins, D.J.; Chen, D.F. Optic Neuropathy Due to Microbead-Induced Elevated Intraocular Pressure in the Mouse. Investig. Ophthalmol. Vis. Sci. 2011, 52, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.G.; Tang, N.; Thompson, S.; Miller, J.; Katze, M.G. The 58,000-dalton cellular inhibitor of the interferon-induced double-stranded RNA-activated protein kinase (PKR) is a member of the tetratricopeptide repeat family of proteins. Mol. Cell. Biol. 1994, 14, 2331–2342. [Google Scholar] [CrossRef] [PubMed]
- Ocansey, S.; Pullen, D.; Atkinson, P.; Clarke, A.; Hadonou, M.; Crosby, C.; Short, J.; Lloyd, I.C.; Smedley, D.; Assunta, A.; et al. Biallelic DNAJC3 variants in a neuroendocrine developmental disorder with insulin dysregulation. Clin. Dysmorphol. 2022, 31, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Synofzik, M.; Tobias, H.B.; Kopajtich, R.; Gorza, M.; Rapaport, D.; Greiner, M.; Schönfeld, C.; Freiberg, C.; Schorr, S.; Reinhard, H.W.; et al. Absence of BiP Co-chaperone DNAJC3 Causes Diabetes Mellitus and Multisystemic Neurodegeneration. Am. J. Hum. Genet. 2014, 95, 689–697. [Google Scholar] [CrossRef]
- Ozon, Z.A.; Alikasifoglu, A.; Kandemir, N.; Aydin, B.; Gonc, E.N.; Karaosmanoglu, B.; Celik, N.B.; Eroglu-Ertugrul, N.G.; Taskiran, E.Z.; Haliloglu, G.; et al. Novel insights into diabetes mellitus due to DNAJC3-defect: Evolution of neurological and endocrine phenotype in the pediatric age group. Pediatr. Diabetes 2020, 21, 1176–1182. [Google Scholar] [CrossRef] [PubMed]
- Ladiges, W.C.; Knoblaugh, S.E.; Morton, J.F.; Korth, M.J.; Sopher, B.L.; Baskin, C.R.; MacAuley, A.; Goodman, A.G.; LeBoeuf, R.C.; Katze, M.G. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005, 54, 1074–1081. [Google Scholar] [CrossRef]
- Han, J.; Song, B.; Kim, J.; Kodali, V.K.; Pottekat, A.; Wang, M.; Hassler, J.; Wang, S.; Pennathur, S.; Back, S.H.; et al. Antioxidants Complement the Requirement for Protein Chaperone Function to Maintain β-Cell Function and Glucose Homeostasis. Diabetes 2015, 64, 2892–2904. [Google Scholar] [CrossRef]
- Lytrivi, M.; Senée, V.; Salpea, P.; Fantuzzi, F.; Philippi, A.; Abdulkarim, B.; Sawatani, T.; Marín-Cañas, S.; Pachera, N.; Degavre, A.; et al. DNAJC3 deficiency induces β-cell mitochondrial apoptosis and causes syndromic young-onset diabetes. Eur. J. Endocrinol. 2021, 184, 455–468. [Google Scholar] [CrossRef]
- Jennings, M.J.; Hathazi, D.; Nguyen, C.D.L.; Munro, B.; Münchberg, U.; Ahrends, R.; Schenck, A.; Eidhof, I.; Freier, E.; Synofzik, M.; et al. Intracellular Lipid Accumulation and Mitochondrial Dysfunction Accompanies Endoplasmic Reticulum Stress Caused by Loss of the Co-chaperone DNAJC3. Front. Cell Dev. Biol. 2021, 9, 710247. [Google Scholar] [CrossRef]
- Kroeger, H.; Grandjean, J.M.D.; Chiang, W.-C.J.; Bindels, D.D.; Mastey, R.; Okalova, J.; Nguyen, A.; Powers, E.T.; Kelly, J.W.; Grimsey, N.J.; et al. ATF6 is essential for human cone photoreceptor development. Proc. Natl. Acad. Sci. USA 2021, 118, e2103196118. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Zobor, D.; Chiang, W.C.; Weisschuh, N.; Staller, J.; Menendez, G.I.; Chang, S.; Beck, S.C.; Garrido, G.M.; Sothilingam, V.; et al. Mutations in the unfolded protein response regulator ATF6 cause the cone dysfunction disorder achromatopsia. Nat. Genet. 2015, 47, 757–765. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Struebing, F.L.; Wang, J.; King, R.; Geisert, E.E. Different Effect of Sox11 in Retinal Ganglion Cells Survival and Axon Regeneration. Front. Genet. 2018, 9, 633. [Google Scholar] [CrossRef]
- Pupo, A.; Fernández, A.; Low, S.H.; François, A.; Suárez-Amarán, L.; Samulski, R.J. AAV vectors: The Rubik’s cube of human gene therapy. Mol. Ther. 2022, 30, 3515–3541. [Google Scholar] [CrossRef]
- Williams, P.A.; Marsh-Armstrong, N.; Howell, G.R. Neuroinflammation in glaucoma: A new opportunity. Exp. Eye Res. 2017, 157, 20–27. [Google Scholar] [CrossRef] [PubMed]
- Russo, R.; Varano, G.P.; Adornetto, A.; Nucci, C.; Corasaniti, M.T.; Bagetta, G.; Morrone, L.A. Retinal ganglion cell death in glaucoma: Exploring the role of neuroinflammation. Eur. J. Pharmacol. 2016, 787, 134–142. [Google Scholar] [CrossRef]
- Tezel, G. Molecular regulation of neuroinflammation in glaucoma: Current knowledge and the ongoing search for new treatment targets. Prog. Retin. Eye Res. 2022, 87, 100998. [Google Scholar] [CrossRef]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLaughlin, T.; Wang, J.; Jia, L.; Wu, F.; Wang, Y.; Wang, J.J.; Mu, X.; Zhang, S.X. Neuronal p58IPK Protects Retinal Ganglion Cells Independently of Macrophage/Microglia Activation in Ocular Hypertension. Cells 2023, 12, 1558. https://doi.org/10.3390/cells12121558
McLaughlin T, Wang J, Jia L, Wu F, Wang Y, Wang JJ, Mu X, Zhang SX. Neuronal p58IPK Protects Retinal Ganglion Cells Independently of Macrophage/Microglia Activation in Ocular Hypertension. Cells. 2023; 12(12):1558. https://doi.org/10.3390/cells12121558
Chicago/Turabian StyleMcLaughlin, Todd, Jinli Wang, Liyun Jia, Fuguo Wu, Yaqin Wang, Joshua J. Wang, Xiuqian Mu, and Sarah X. Zhang. 2023. "Neuronal p58IPK Protects Retinal Ganglion Cells Independently of Macrophage/Microglia Activation in Ocular Hypertension" Cells 12, no. 12: 1558. https://doi.org/10.3390/cells12121558
APA StyleMcLaughlin, T., Wang, J., Jia, L., Wu, F., Wang, Y., Wang, J. J., Mu, X., & Zhang, S. X. (2023). Neuronal p58IPK Protects Retinal Ganglion Cells Independently of Macrophage/Microglia Activation in Ocular Hypertension. Cells, 12(12), 1558. https://doi.org/10.3390/cells12121558