Abstract
Alt-EJ is an error-prone DNA double-strand break (DSBs) repair pathway coming to the fore when first-line repair pathways, c-NHEJ and HR, are defective or fail. It is thought to benefit from DNA end-resection—a process whereby 3′ single-stranded DNA-tails are generated—initiated by the CtIP/MRE11-RAD50-NBS1 (MRN) complex and extended by EXO1 or the BLM/DNA2 complex. The connection between alt-EJ and resection remains incompletely characterized. Alt-EJ depends on the cell cycle phase, is at maximum in G2-phase, substantially reduced in G1-phase and almost undetectable in quiescent, G0-phase cells. The mechanism underpinning this regulation remains uncharacterized. Here, we compare alt-EJ in G1- and G0-phase cells exposed to ionizing radiation (IR) and identify CtIP-dependent resection as the key regulator. Low levels of CtIP in G1-phase cells allow modest resection and alt-EJ, as compared to G2-phase cells. Strikingly, CtIP is undetectable in G0-phase cells owing to APC/C-mediated degradation. The suppression of CtIP degradation with bortezomib or CDH1-depletion rescues CtIP and alt-EJ in G0-phase cells. CtIP activation in G0-phase cells also requires CDK-dependent phosphorylation by any available CDK but is restricted to CDK4/6 at the early stages of the normal cell cycle. We suggest that suppression of mutagenic alt-EJ in G0-phase is a mechanism by which cells of higher eukaryotes maintain genomic stability in a large fraction of non-cycling cells in their organisms.
1. Introduction
Exposure of cells to ionizing radiation (IR) induces DNA double-strand breaks –(DSBs) that challenge genomic stability. In higher eukaryotes, DSBs are mainly processed by classical non-homologous end-joining (c-NHEJ) and homologous recombination (HR), while alternative end-joining (alt-EJ) and single-strand annealing (SSA) contribute variably depending on several parameters, including the cell cycle phase and growth state [1,2]. While HR is conceptually designed to faithfully restore the genome [2], c-NHEJ effectively rejoins DNA ends without template requirements and is, therefore, less faithful than HR. However, its simplicity and speed make it the most prominent route of DSB repair in higher eukaryotes, especially after high doses of IR exposure [3,4,5,6]. The failures of c-NHEJ or HR may be compensated by alt-EJ [6,7,8], which is slow, more error-prone than c-NHEJ, and, therefore, a frequent source of chromosomal translocations. This defines alt-EJ as a mutagenic route of DSB processing that promotes genomic instability induction [9].
How c-NHEJ and HR suppress error-prone alt-EJ to maintain genomic stability is a subject of intensive research. However, emphasis is presently placed on the regulation of DNA end-resection (henceforth here simply, resection). Resection processes the end of a DSB to expose a 3′ single-stranded DNA that can then invade a homologous DNA molecule or anneal with another resected end using homologies. Resection inhibits c-NHEJ and triggers homology-directed DSB repair pathways, including HR and SSA, that play key roles in the DSB repair pathway choice. Lower degrees of homology are utilized by alt-EJ [10]. Resection requires CtIP [11] to stimulate the endonuclease activity of MRE11 and proceeds bi-directionally [12,13]. The activity of the MRE11-RAD50-NBS1 (MRN) complex processes the DNA end in the 3ʹ–5ʹ direction, while Exonuclease 1 (EXO1) or Bloom helicase (BLM)/DNA Replication Helicase/Nuclease 2 (DNA2) catalyze long-range 5ʹ–3ʹ resection [14]. Resection is regulated in a cell-cycle-dependent manner and is maximally active in G2-phase cells, with significantly reduced levels in G1 [15,16].
Alternative end-joining (alt-EJ) still remains incompletely characterized, and it is widely accepted that it functions by default when KU- and DNA-dependent serine/threonine protein kinase catalytic subunits (DNA-PKcs), Ligase 4 (LIG4) or other c-NHEJ factors are defective [17,18,19,20]. Alt-EJ may make greater use of microhomology (MH) compared to c-NHEJ [21,22,23]. However, it remains questionable whether MH-dependency is a universal requirement for all alt-EJ events at all conditions. Indeed, by studying the utilization of homologies in c-NHEJ-proficient and -deficient cells, Mansour demonstrated that alt-EJ was MH-independent [24]. In conclusion, alt-EJ was defined as any c-NHEJ-independent end-joining process, and dependence on MH has been well characterized, but it may not always be required. Based on these findings, two kinds of systems of detection and characterization of alt-EJ were established: residual repair of IR-induced DSBs after c-NHEJ factor deactivation or the presence of MH at junctions generated in model DSBs—typically enzymatically induced.
Adopting the concept of MH detection, appropriately designed reporter assays monitoring GFP-expression after processing of I-Sce I-induced DSBs by a specific DSB repair pathway are frequently used to characterize alt-EJ [10,25,26]. Such studies have reported that MRE11 is required for the resection of as few as 20 bp during alt-EJ [10], and similar conclusions have been drawn for CtIP and NBS1 [10,19,27,28,29]. However, in these reporter assays, microhomology is built-in, which may result in an overestimation of the contributions of resection to alt-EJ.
Measuring DSBs directly, using pulsed-field gel electrophoresis (PFGE), allows the study of DSB induction and repair kinetics, and in these experiments residual repair activity after c-NHEJ inactivation is taken to reflect the contribution of alt-EJ. Using PFGE, we have recently shown that CtIP depletion suppresses alt-EJ in both exponentially growing and in G2-phase-enriched cells [30]. One key characteristic of alt-EJ uncovered using PFGE is its crucial dependence on the state of growth. Thus, alt-EJ activity is low during G1-phase and steadily increases as cells move to G2-phase. Notably, alt-EJ is almost completely abrogated in some cell lines in G0-phase [31,32,33]. Epidermal growth factor receptor (EGFR) inhibition in proliferating cells suppresses alt-EJ only marginally, and the addition of EGF in G0-phase cells increases alt-EJ marginally, which suggests that the inhibition of alt-EJ in G0 cells is not a direct consequence of suppressed growth factor signaling [34]. While the mechanism underlying the suppression of alt-EJ in G0-phase cells still remains to be elucidated, a tenable hypothesis is that suppression of alt-EJ derives from a further reduction in resection in G0, which immediately raises the question as to the underpinning mechanism.
The cell-cycle-dependent regulation of resection is achieved through CDKs [35]. CtIP phosphorylation by CDKs on Thr847 and Ser327 critically regulates resection in vertebrates [36,37,38]. Interestingly, CDK2-dependent phosphorylation of Ser276 and Thr315 also promotes CtIP binding to PIN1 to dampen resection [39]. CDKs also promote resection by phosphorylating EXO1 [40], NBS1 [41,42,43] and DNA2 [44]. Finally, CDKs regulate resection by suppressing resection blocks raised by 53BP1 and DNA helicase B (HELB) [45,46,47].
The oscillating activity of CDKs throughout the cell cycle is regulated by the periodic degradation, via the ubiquitin–proteasome system, of cyclins and CDK inhibitors (CKIs) [48]. Central in this process is a pair of RING-type E3 ubiquitin ligases: SCF (for SKP1/Cullin/F-box protein) and the anaphase-promoting complex/cyclosome (APC/C) [49]. SCF remains active from late G1- to late G2-phase and selectively degrades proteins through priming for degradation. S-phase kinase-associated protein 2 (SKP2) is the main substrate recognition factor of SCF [49]. CDC20 homolog-1 (CDH1) is the main substrate recognition factor of the APC/C complex, which is activated in late M- to early/mid-G1-phase and retains activity until the inception of DNA replication [49,50].
Notably, in addition to CDKs, APCCDH1 also regulates resection by regulating cyclins (and, thus, indirectly CDK activity) and CtIP abundance. IR activates APCCDH1 and mediates CtIP degradation to limit resection in G2-phase cells [51]. In addition, APCCDH1 competes with SCF, and indeed, SKP2 depletion causes the hyper-activation of APCCDH1, and thus, the full degradation of CtIP that compromises alt-EJ in G2-phase-enriched cells [30]. These results suggest that the regulation of alt-EJ is achieved through CDK-dependent activation of key components of the resection apparatus and the SCF-APC/C-dependent regulation of their levels by protein degradation. The regulation of alt-EJ by CtIP levels and its CDK-dependent activation remains to be elucidated in G1- and, particularly, in G0-phase cells.
In the present study, we continue with the elucidation of the mechanistic regulation of alt-EJ throughout the cell cycle by focusing on cells in G1- and G0-phase. While notable exceptions have been reported [52,53], HR is generally thought to be suppressed in this phase of the cell cycle. The same holds true for SSA, which is known to require longer resection than HR. These characteristics of repair pathway activity in G1/G0-phase cells make it possible to, rather specifically, investigate the role of resection in alt-EJ [10,54,55,56] by simply inhibiting c-NHEJ—the only DSB repair pathway that retains detectable activity in these cells.
We report that CtIP is required for alt-EJ activity in G1-phase and that its degradation or reduced phosphorylation abrogates resection and alt-EJ. Interestingly, G0-phase cells are more radioresistant to killing than G1-phase cells, which suggests that the suppression of alt-EJ can be beneficial for the cell. Since almost all terminally differentiated, post-mitotic cells in the human body are non-cycling, and because unstimulated stem cells frequently rest in G0-phase, it is possible that the suppression of error-prone alt-EJ is a natural mechanism safeguarding genomic stability and protecting against tumorigenesis.
2. Materials and Methods
2.1. Cell Culture and Irradiation
Normal, immortalized human fibroblasts, 82-6 hTert (provided by Drs. M. Lobrich and P. Jeggo) [16]; and human lung carcinoma cells, A549 (ATCC, CCL-185) and its derivatives, CDH1−/− [30] and POLQ−/−; human osteosarcoma cells, U2OS and its POLQ mutant (provided by Dr. J. Stark) [57] were grown and were irradiated as reported earlier [15,30]. POLQ−/− A549 cells were generated using CRISPR/Cas9Nickase. Two guide RNAs (gRNAs, Table S1) targeting exon 2 were cloned into all-in-one plasmids (PX460) and delivered to the cells by nucleofection using the Nucleofector-2B device.
2.2. Cell Survival Determination
Cell survival was measured using the colony formation assay, as described previously [30].
2.3. RNA Interference
Knockdown experiments were carried out using specific siRNAs against the proteins of interest. The siRNAs (Table S1) were delivered to the cells by nucleofection, as described previously [15,30].
2.4. Treatment of Cells with Inhibitors
All inhibitors (Table S2) were added 1 h before irradiation unless indicated otherwise and were kept until the time of analysis.
2.5. Flow Cytometry (FC) Analysis of Resection
Resection analysis by FC through RPA70 detection was measured as described earlier [15,30]. A similar protocol was adopted for the analysis of resection through BrdU detection. In this case, BrdU at a concentration of 2 µM was added 24 h before irradiation in exponentially growing (Expo) cells or before serum deprivation (SD) in G0-phase cells. Antibodies used for this purpose are listed in Table S3. Experiments were replicated three times and representative histograms from one experiment are shown.
2.6. FC Analysis of γH2AX Intensity
γH2AX intensity is measured by FC, as described earlier [58]. Experiments were replicated three times and representative histograms from one experiment are shown. Antibodies used for this purpose are listed in Table S3.
2.7. Polyacrylamide Gel Electrophoresis (SDS-PAGE) and Western Blotting
SDS-PAGE and immunoblotting were employed, as previously described [15,30]. Antibodies used are listed in Table S3.
2.8. Real-Time Polymerase Chain Reaction (PCR)
mRNA levels of CtIP were determined by real-time PCR. We used commercially available kits to extract total RNAs (Roche, 11828665001) and synthesize the first cDNA strands (Thermo fisher scientific, K1631). The PCR primers and conditions used are listed in Tables S4 and S5, respectively. Data were analyzed using Light-Cycler software Version 4.1 (Roche).
2.9. Pulsed-Field Gel Electrophoresis (PFGE)
To analyze repair of DSBs, in general, and specifically alt-EJ, PFGE was used, as described earlier [30]. The equivalent dose (Deq), rather than the fraction of DNA released (FDR), calculated using the corresponding dose–response curve, was adopted here as a parameter because it corrects the repair kinetics analysis for fluctuations in the dose–response curves. Six determinations from two independent experiments were used to calculate means and standard errors (SEs). Repair kinetics were fitted assuming two exponential components (fast and slow) of rejoining according to the equation, Deq = Ae−bt + Ce−dt [59], using a nonlinear regression analysis tool (Sigma-plot 14; Systat Software GmbH). The parameters A and C provide information on the fraction of DSBs processed by fast versus slow kinetics. The parameters b and d allow the calculation of the repair half times for the fast and the slow components of repair.
2.10. G1-Phase Cell Synchronization
A thymidine block combined with a nocodazole block was used to synchronize Expo cells in G1-phase. For details, see the legend of Figure S1.
2.11. Statistical Analyses
Results were expressed as mean ± standard error (SE) calculated from three or more repeats. Statistical significance between experimental groups was determined by using t-test. The significance of differences between individual measurements was indicated by using symbols: * p < 0.05, ** p < 0.01.
3. Results
3.1. Alt-EJ Is Suppressed as G1-Phase Cells Enter G0-Phase
We previously demonstrated using PFGE that alt-EJ is suppressed in mouse and hamster c-NHEJ mutants when they grow into a plateau- and enter G0-phase [31,60,61]. These results were later also confirmed using appropriately designed reporter assays [62]. Here, we extend these observations to human cells and begin with the elucidation of the underpinning mechanisms. Figure 1a shows that, under the conditions employed, 82-6 hTert cells grow logarithmically for 3 days and enter later, if not refed, a plateau phase. In the plateau phase, the majority of cells accumulate with a G1-phase-equivalent DNA content (Figure 1b, left panels). Figure 1b (right panel) shows that levels of Ki67, a widely used marker for discrimination between cells of G1- and G0 phases, decrease pronouncedly when cells enter plateau phase. These results are confirmed using Pyronin Y, an alternative marker of G0 cells (Figure S1c). This shows that plateau-phase cultures mainly comprise G0-phase cells.
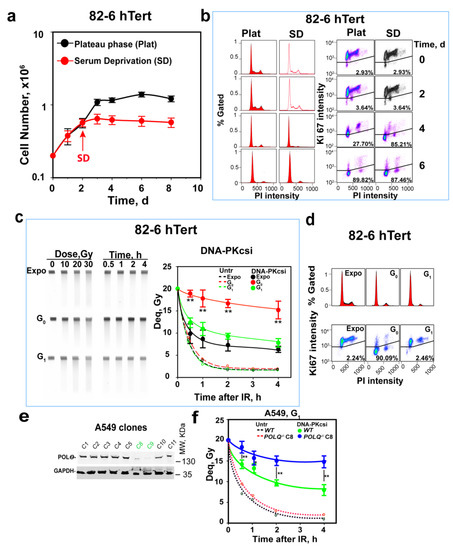
Figure 1.
Suppression of alt-EJ in G0-phase cells. (a) Proliferation of 82-6 hTert cells under normal growth conditions, as well as after transfer to SD-medium two days later. Plot shows mean ± SE from 3 independent experiments. (b) Cell cycle distribution and cell growth status determination by PI combined with Ki67 staining at the indicated times of growth. All experiments are repeated 3 times, and a representative one is shown. (c) Kinetics of DSB repair measured by PFGE after exposure of cells to 20 Gy in the presence or absence of DNA-PKcsi, NU7441, at a concentration of 2.5 µM. The image on the left shows a typical PFGE gel with the dose–response and the repair kinetics analysis in the presence of DNA-PKcsi for exponentially growing as well as G0 and G1-enriched cells. The plot on the right shows results obtained by densitometry analysis of six similar gels in two experiments. Broken lines show results obtained with untreated cells and are shown without error bars to avoid congestion in the figure. The solid lines reflect the repair kinetics measured in the presence of DNA-PKcsi and are generated by fitting to the sum of two exponentials, as described under Materials and Methods. (d) PI and Ki67 staining of cells used in (c) generated to confirm the growth status of the cells used. (e) Western blot validation of Polθ depletion in selected A549 clones. Clones C8 and C9 show clear depletion of the protein. (f) Kinetics of DSB repair after exposure to 20 Gy in the presence or absence of DNA-PKcsi in POLQ wild-type and mutant (clone 8) G1-phase A549 cells. Plot shows mean ± SE of 6 determinations from 2 independent experiments. Untr, Expo, DNA-Pkcsi, WT and Deq represent untreated, exponentially growing, DNA-PKcs-inhibitor-treated, wild-type and dose equivalent, respectively; Symbols ** in (c,f) indicate p < 0.01 in statistical significance calculation, symbols * indicate p < 0.05.
To reduce irreproducibility associated with unfed plateau-phase cultures [15], we generated G0-phase 82-6 hTert cell populations using serum deprivation (SD) starting at day 2 of growth (Figure 1a). Figure 1a,b show good stability and enrichment in G0-phase in SD cultures, similar to that of plateau-phase cultures, at day 3. SD-cultures are, therefore, exclusively used in the following experiments. To compare the responses of G0- to those of G1-phase cells, we also employed the protocol outlined in Figure S1a to obtain enriched populations of G1-phase cells. Figure S1b shows that this protocol generates highly enriched G1-phase cells, 4 h after release from nocodazole block.
Analysis of DSB repair using PFGE after exposure to 20 Gy (Figure 1c, broken lines) shows indistinguishable repair potential in Expo, as well as in enriched G0- and G1-phase 82-6 hTert cells. Figure 1d summarizes the cell cycle distribution and growth state through PI and Ki67 staining. This repair activity mainly reflects the function of c-NHEJ [5,63], which removes over 90% of DSBs within 2 h, independently of the cell growth stage and cell cycle phase. The treatment of cells with the DNA-PKcs inhibitor NU7441 (2.5 µM, DNA-PKcsi) inhibits c-NHEJ and allows alt-EJ to come to the fore, since as mentioned above, HR (and SSA) is largely inactive in G0 and G1 cells [6]. This is also confirmed by the absence of RAD51 foci in these cells, despite efficient formation in G2-phase cells (Figure S1d). In contrast, the formation of γH2AX foci increases linearly as a function of IR dose and shows the expected increase in G2-phase cells owing to their higher DNA content.
Exponentially growing cells treated with DNA-PKcsi show a profound reduction in DSB repair activity, but alt-EJ and, possibly, also HR and SSA repair over 50% of DSBs within 2 h; G1-phase cells show only slightly reduced DSB repair activity in the presence of DNA-PKcsi, which can mainly be attributed to alt-EJ, as HR and SSA are inactive. Additionally, the small difference in the kinetics between exponentially growing and enriched G1-phase cells that fails to reach statistical significance suggests that the contribution of HR and SSA in the former cells is small under the conditions employed here [5].
Strikingly, G0-phase cells treated with DNA-PKcsi are profoundly deficient in DSB repair, particularly at early stages after IR and repair less than 20% of induced DSBs in 4 h (Figure 1c). This is evidence for strong alt-EJ suppression, as cells transit from G1- to G0-phase of the cell cycle. Fitting of these curves to the sum of two exponentials, as described earlier [59], shows that in G0, the half time of repair (t50) of both the fast as well as the slow components of DSB rejoining increases (Figure S1e) and that the contribution of the fast component is dramatically reduced. We conclude that the fast component of alt-EJ becomes strongly compromised as cells enter G0-phase.
Lung adenocarcinoma A549 cells can also be maintained under analogous growth conditions to generate highly enriched cultures of G1- and G0-phase cells (Figure S1f,g). When DSB repair is analyzed in the presence of DNA-PKcsi, a similar, strong suppression of alt-EJ is observed in G0-phase cells (Figure S1h,i). We conclude that suppression of alt-EJ in G0-phase cells is a general phenotype, detectable in several cell lines from different species [31,32,33], and focus below on the elucidation of the underpinning mechanisms.
To solidify our postulate that DSB rejoining in G1-phase cells after the inhibition of DNA-PKcs reflects mainly alt-EJ, we tested a mutant of A549 cells defective in the POLQ gene that encodes a key component of alt-EJ, Polθ [64]. The approach used to generate this mutant is described under “Materials and Methods”, and Figure 1e shows representative clones examined for a reduction in Polθ levels. We selected clone 8 for further experiments, and Figure 1f shows the DSB repair kinetics in POLQ-deficient and wild-type G1-A549 cells. In the absence of DNA-PKcsi, when c-NHEJ is fully active, deficiency in POLQ reduces repair only slightly (broken lines, less than a 5% reduction in any time point), with no statistical significance reached at any of the time points examined. Strikingly, in the presence of DNA-PKcsi, residual repair activity is significantly compromised in POLQ−/−A549 cells: 25% repair within 30 min and 50% repair within 2 h in wild-type A549 cells, but less than 20% repair in POLQ−/−A549 cells at 4 h after IR. Since Polθ is a component of alt-EJ, we surmise that residual repair activity after the inhibition of DNA-PKcs reflects the function of this pathway.
To confirm this postulate with a different cell line, we tested, in a similar experiment, a pair of U2OS cell lines proficient and deficient in Polθ [57]. The results obtained are summarized in Figure S1j. Here, again, Polθ deficiency strongly compromises alt-EJ brought to the fore by the inhibition of DNA-PKcs. We also demonstrated earlier the dependence of this residual repair activity on LIG1/3 and PARP 1 [65,66], further confirming that, after c-NHEJ inhibition, repair in G1-phase mainly reflects the function of alt-EJ.
The results presented here and the majority of those reported earlier [31,60,61], analyze DSB repair at high IR doses. We recently reported profound adaptations in the DSB repair pathway choice with increasing IR doses up to 20 Gy [5,67,68,69]. We, therefore, examined the above-described suppression of alt-EJ in human G0-phase cells after exposure to 2 Gy, using analysis of γH2AX signal intensity by FC (Figure S2). We employed a modified method similar to that described previously [5,67,68,69] measuring the γH2AX signal in a cell-cycle-specific manner, as outlined in Figure S2a,b.
A γH2AX signal reduction of nearly 50% within 6 h in untreated cells was observed (Figure S2c), showing, as expected, that DSB repair can also be followed with this assay but that the kinetics measured are markedly slower than in the PFGE experiments discussed above. However, even with this assay, the repair kinetics of G1-phase and G0-phase cells are similar, with only a slight trend for slower repair kinetics in G0-phase cells (Figure S2c). In the presence of DNA-PKcsi, the repair is not detectable in G0-phase cells using γH2AX analysis and becomes detectable in G1-phase cells only at 6 h post-IR (Figure S2d).
We are currently investigating whether the slower repair kinetics and the reduced growth-state effect on alt-EJ at low IR doses reflects the earlier discussed [70] divergence between DSB repair analysis with assays measuring the physical DNA integrity versus the decay of the DDR protein γH2AX. We also explore whether larger differences become evident after longer periods of follow-up post-IR and whether they reflect the reported dose dependency of alt-EJ [71,72]. Therefore, in the experiments presented below, we focus on high-IR-dose effects.
3.2. DNA End-Resection Is Undetectable in G0-Phase Cells
As resection is believed to facilitate alt-EJ, we investigated whether reduced resection underpins its suppression in G0- versus G1-phase cells. Using methods similar to γH2AX detection, we measured RPA signals in cells in G0- and G1-phase.
Figure 2a,b show, as already reported [16], low-level resection in G1-phase cells of exponentially growing cultures after exposure to 10 Gy. Figure 2c confirms this low-level resection in G1 using BrdU signal analysis as an alternative to RPA for ssDNA detection and, thus, resection analysis, in irradiated cells [68]. In accordance with recent results in G2-phase [68,69], DNA-PKcsi fails to modify resection in G1-phase (Figure S3a). Therefore, subsequent resection experiments were carried out in the absence of DNA-PKcsi. Similar patterns of resection were also observed for A549 cells, with low but detectable resection in G1-phase cells of exponentially growing cultures after exposure to 10 Gy (Figure S3b), which remain unaffected by DNA-PKcsi (Figure S3c).
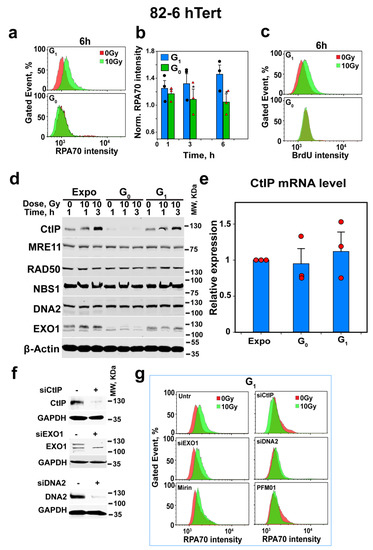
Figure 2.
Depletion of CtIP as cells enter G0-phase suppresses resection. (a) Representative analysis of resection at DSBs, specifically in G1-phase cells discriminated by using PI- and EdU-combined staining after exposure to 10 Gy using RPA signal intensity as parameter and quantification by FC in 82-6 hTert cells; for details, see under “Material and Methods”. (b) Combined results from three independent experiments similar to those shown in (a) as a bar plot showing the normalized RPA70 signal intensity, as well as the associated SE and the significance of the observed differences. Black circles and red triangles show the results of individual experiments used in the analysis. Symbol * indicates p < 0.05 in statistical significance calculation. (c) As in (a), but for BrdU signal intensity to label single-stranded DNA and analyze resection using an alternative method to RPA staining. Experiments were repeated 3 times, and a representative one is shown. (d) WB analysis showing the levels of CtIP, MRE11, RAD50, DNA2 and EXO1 in 82-6 hTert cells in different stages of growth and phases of the cell cycle, measured at different times after exposure to 0 Gy or 10 Gy of IR. Experiments were repeated 3 times and a representative one is shown. (e) mRNA levels of CtIP in 82-6 hTert cells at different stages of growth and phases of the cell cycle. Red circles show the results of individual experiments used in the analysis Plot shows mean ± SE from 3 independent experiments. (f) Western blot analysis of CtIP, EXO1 and DNA2 after 48 h protein knockdown using specific siRNAs, which was conducted to confirm the efficient depletion of targeted protein for the experiment shown in (g). (g) Resection analysis as in (a) after knockdown or inhibition of CtIP, MRE11, EXO1 or DNA2 in G1-phase 82-6 hTert cells.
Notably, similar analysis in G0-phase in both cell lines failed to detect resection (Figure 2a–c and Figure S3b). These observations point to a direct link between reduced alt-EJ activity in G0- versus G1-phase cells and the associated reduction in resection activity. Because resection in G1-phase cells was relatively small (Figure 2a–c), we sought additional experimental validation of the putative connection between resection and alt-EJ activity by examining, under similar conditions, the changes in the enzymatic machinery of resection.
3.3. CtIP Levels Are Undetectable in G0-Phase Cells
The undetectable resection in G0-phase cells may derive either from a reduction in the abundance of components of the resection machinery or from a down-regulation of their activities. We, therefore, measured the levels of key proteins of the resection apparatus in exponentially growing, as well as in G0- and G1-phase 82-6 hTert cells, before and 1 or 3 h after exposure to 10 Gy. Figure 2d shows that while MRE11, RAD50, NBS1 and DNA2 show only minor fluctuations, CtIP and EXO1 are markedly reduced in G0-phase cells, as compared to G1-phase or exponentially growing cells. Irradiation profoundly increases the levels of CtIP in exponentially growing and G1-phase cells, but CtIP, as well as its radiation-induced increase, are undetectable in G0-phase cells. Because CtIP is a central regulatory component of the resection apparatus, its depletion in G0-phase cells explains the absence of resection.
The reduction in CtIP-protein levels in G0-phase cells may reflect the normally occurring reduction in gene expression during quiescence. We, therefore, measured the mRNA levels of CtIP in exponentially growing as well as in G1- and G0-phase cells. Figure 2e shows similar mRNA levels in the different conditions analyzed, thus excluding transcriptional regulation as a candidate mechanism.
We, therefore, turned our attention to CtIP stability. Figure 2f shows the successful depletion of CtIP, EXO1 and DNA2 in exponentially growing 82-6 hTert cells, and Figure 2g shows that CtIP and DNA2 depletion suppresses resection in G1-phase, whereas EXO1 depletion is ineffective. Additionally, the inhibition of MRE11 endonuclease activity with PFM01 inhibits resection, whereas Mirin, an inhibitor of MRE11 exonuclease activity [12], is ineffective (Figure 2g). Figure S4a summarizes the results from several similar experiments, confirms the conclusions and shows their statistical power.
To directly connect resection with the function of alt-EJ in DSB repair, we analyzed G1-phase cells after depletion of CtIP, EXO1 or DNA2, or after the inhibition of MRE11 endonuclease or exonuclease activity. The results in Figure 3a show that CtIP knockdown not only suppresses resection profoundly, but also inhibits alt-EJ, whereas EXO1 knockdown, which has no effect on resection, leaves alt-EJ unaffected. In this experiment, DNA2 knockdown has no detectable effect on alt-EJ, which we interpret as evidence that short-range resection in G1-phase cells is sufficient for alt-EJ. Indeed, PFM01, which inhibits the endonuclease activity of MRE11 and is, thus, expected to specifically inhibit short-range resection, strongly suppresses alt-EJ, whereas Mirin fails to inhibit resection or alt-EJ.
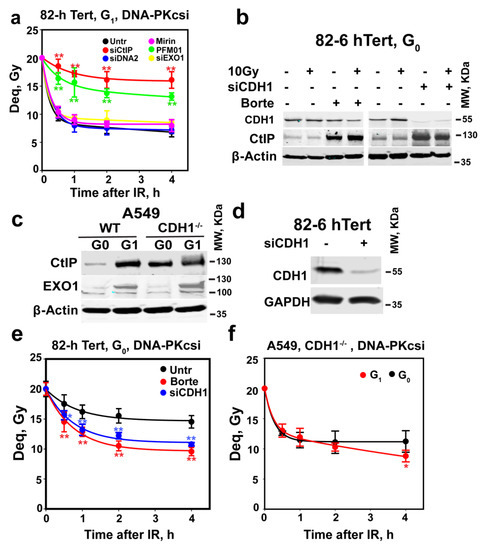
Figure 3.
Depletion of CtIP suppresses alt-EJ in G0-phase cells. (a) As in Figure 1c for 82-6 hTert cells exposed to 20 Gy and maintained in the presence of DNA-PKcsi after knockdown or inhibition of CtIP, MRE11, EXO1 and DNA2. Plot shows mean ± SE from 6 determinations from 2 independent experiments. (b) WB analysis of CtIP levels in cells treated with 2 µM bortezomib, or depleted of CDH1 using specific siRNAs, in G0-phase 82-6 hTert cells. Bortezomib was added 2 h before IR and kept for 3 h after IR. (c) WB analysis of CtIP and EXO1 in CDH1 wild-type and mutant G0- or G1-phase A549 cells; all experiments were repeated 3 times in (b,c), and a representative one is shown. (d) WB analysis of CDH1 knockdown in cells used in (e). (e) Rescue of alt-EJ in G0-phase 82-6 hTert cells after treatment with bortezomib for 2 h, or 48 h CDH1 knockdown, as shown in (d). (f) Alt-EJ in CDH1−/− A549 cells in G0- and G1-phase of the cell cycle. Borte in (b) represents bortezomib. Plots in (e,f) show mean ± SE of 6 determinations from 2 independent experiments. Symbol ** in (a,e) indicates p < 0.01 in statistical significance calculation, symbols * indicate p < 0.05.
We conclude that, in G1-phase cells, alt-EJ benefits from resection initiated by the MRN/CtIP and that the depletion of CtIP as cells enter G0-phase is causative to the suppression of alt-EJ observed. Combined with the results in Figure 1c, our observations further suggest that resection is required for fast DSB rejoining by alt-EJ. These observations, of course, immediately raise the question as to the mechanisms underpinning CtIP depletion in G0-phase cells.
3.4. APCCDH1 Depletes CtIP in G0-Phase Cells
APCCDH1 ubiquitinates CtIP and mediates its degradation by the proteasome in G2-phase [39,51,73]. We, therefore, examined the effect of the proteasome inhibitor bortezomib on CtIP levels in G0 cells. The treatment of G0-phase 82-6 hTert cells with 2 µM bortezomib for 2 h markedly increases CtIP levels (Figure 3b). Moreover, CDH1 knockdown similarly upregulates the expression of CtIP (Figure 3b). Finally, in A549 CDH1−/− cells [30], CtIP is clearly detectable in G0-phase and increases further somewhat in G1-phase cells. Interestingly, EXO1 levels remain unaffected by CDH1 status and show the above-documented decrease in G0-phase (Figure 3c). These results convincingly show that increased APC/CCDH1 activity in G0-phase cells causes the degradation of CtIP.
We next investigated whether suppression of CtIP degradation using the above treatments restores alt-EJ and resection in G0-phase cells. Figure 3d,e show that CDH1 depletion, or treatment with bortezomib, markedly restores alt-EJ in G0-phase cells (Figure 3e), without affecting their cell cycle distribution (Figure S4b). However, both CDH1 depletion and Bortezomib failed to clearly elevate resection in G0-phase cells (Figure S4c,d). We think this might be due to the relatively low sensitivity of the resection analysis method and the ability of alt-EJ to also function after only short-range resection of the DNA ends, as discussed above (see below for further discussion on the topic). Moreover, alt-EJ remains at G1-phase-levels when A549 CDH1−/− cells enter G0-phase (Figure 3f), as expected from the associated absence of CtIP degradation (Figure 3c). Collectively, these results identify APC/C as a regulator of alt-EJ activity operating by adjusting CtIP levels.
3.5. Low CDK Activity in G0-Phase Cells Keeps CtIP Inactive and alt-EJ Suppressed
CtIP activity is also regulated by CDK-dependent phosphorylations [35,36,37,38,39]. High CDK activity is a feature of proliferating cells and is low in G0-phase cells [74,75]. When mitotic cells divide and enter G1-phase, or when G0-phase cells are stimulated to proliferate by growth factors, CDK4/6 is activated by D-type cyclins, the expression of which is growth-factor-inducible [76]. In late G1-phase, Cyclin D-CDK4/6 complexes phosphorylate pocket proteins (RB1, p107 and p130) to release E2F transcription factors to induce the transcription of G1/S target genes, including the one encoding for cyclin E, thus activating CDK2/Cyclin E [77].
We considered the possibility that active CDK4/6 in G1-phase cells phosphorylates CtIP, and that this phosphorylation is important for CtIP activation and, thus, for resection and alt-EJ. We tested the effects of CDK inhibition on resection and alt-EJ in enriched G1-phase 82-6 hTert cells. Figure 4a shows that CDK4/6 inhibition, confirmed by the suppression of retinoblastoma protein (RB1) phosphorylation in serine 807 and 811 (Figure 4b), suppresses to G0 levels alt-EJ in G1-phase without affecting cell cycle distribution in a detectable manner (Figure 4c). On the other hand, the inhibition of CDK2 or CDK1 has no effect on DSB repair activity under these conditions. This can be explained by CDK4/6 being the sole CDK activity at this stage of G1-phase in non-transformed 82-6 hTert cells. Figure 4d shows that CDK4/6 inhibition also suppresses resection in G1-phase cells, whereas the effect of CDK2 or CDK1 inhibition fails to reach statistical significance. Figure 4e,f show that the elevation of CtIP levels by IR is suppressed by CDK4/6 inhibition, which suggests that the upregulation of CtIP in G1-phase cells induced by IR is CDK4/6-dependent. In addition, CDK4/6 inhibition also suppresses IR-induced CtIP phosphorylation at threonine 847 (CtIP-T847). These results suggest that CDK4/6-mediated stabilization, as well as CDK-mediated phosphorylation, contribute to the regulation of CtIP and resection in G1-phase cells.
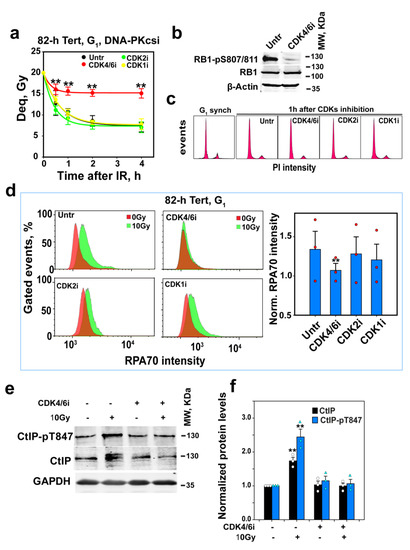
Figure 4.
CDK4/6 inhibition suppresses alt-EJ and resection in G1-phase cells. (a) As in Figure 3a after inhibition of CDK4/6, CDK2 or CDK1 in G1-phase 82-6 hTert cells; CDK4/6i, CDK2i and CDK1i were added 1 h before IR at a concentration of 500 nM, 5 µM and 10 µM, respectively. Plot shows mean ± SE from 6 determinations in 2 independent experiments. (b) WB analysis of RB1-pS807/811 in G1-phase 82-6 hTert cells to confirm the efficacy of CDK4/6 inhibition by PD032991. Cells were treated with PD032991 1h before collection. Experiments were repeated 3 times, and a representative one is shown. (c) Cell cycle distribution of cells before and after CDK inhibition used in (a). (d) Resection analysis, as in Figure 2a, after CDK4/6, CDK2 or CDK1 inhibition in 82-6 hTert cells in G1-phase. Red circles show the results of individual experiments used in the analysis. Plot on the right shows mean ± SE from 3 independent experiments. (e) WB analysis of the effect of CDK4/6 inhibition on CtIP and CtIP-pT847 3h after exposure to 0 Gy or 10 Gy in G1-phase 82-6 hTert cells. Experiments were repeated 3 times, and a representative one is shown. (f) Measurements of band density and statistical analysis of results shown in (e). White circles and turquoise triangles show the results of individual experiments used in the analysis Plot shows mean ± SE from 3 independent experiments. Symbol ** in (a,d,f) indicates p < 0.01 in statistical significance calculation.
It has been reported, when using confluent (practically G0-phase) 82-6 hTert cultures, that PLK3 phosphorylates and activates CtIP for resection to support a special form of resection-dependent c-NHEJ [78]. We, therefore, investigated the effects of PLK3 inhibition on the regulation of G1-phase alt-EJ and resection. PLK3 is known to be required for cell entry into S-phase by promoting the expression of cyclin E1 [79]. We confirmed PLK3 inhibition by analyzing cyclin E1 levels after the release of cells from G0 via transfer to a fresh growth medium in the presence or absence of the PLK1/3 inhibitor, GW843682X (2 µM). Figure S5a shows that the levels of cyclin E1 strongly increase after 12 h in these cells and that GW843682X abrogates this increase. Notably, PLK3 inhibition fails to suppress alt-EJ (Figure S5b) or resection (Figure S5c,d) in G1-phase 82-6 hTert cells. Thus, PLK3 may be specifically utilized in G0-phase cells to regulate resection-dependent c-NHEJ, when CDK4/6 activity is low and alt-EJ is suppressed.
We investigated how the inhibition of CDKs affects alt-EJ in WT and CDH1−/− A549 cells, tested in G1- and G0-phase (Figure 5a). Notably, similar to 82-6 hTert cells, alt-EJ only depends on CDK4/6 in WT-enriched G1-phase A549 cells. In contrast, in CDH1−/− A549 cells, the single inhibition of CDK4/6, CDK1 or CDK2 exerts only small inhibitory effects on alt-EJ. However, a cocktail including all inhibitors causes a strong suppression of alt-EJ, both in enriched G1- as well as G0-phase cells (Figure 5a). As mentioned above, DSB induction upregulates CtIP levels in a CDK4/6-dependent manner in G1-phase-synchronized 82-6 hTert cells. Figure S6a shows the same CtIP regulatory pattern in G1-phase A549 cells, which suggests that the upregulation of CtIP by IR in G1-phase cells is not specific for 82-6 hTert cells. However, the IR-dependent upregulation of CtIP is absent in CDH1−/− A549 G1 or G0-phase cells, and CDKsi also fails to affect CtIP levels detectably (Figure S6b). Taken together, these results suggest that IR suppresses the activity of APC/CCDH1 in wild-type G1-phase cells in a CDK-dependent manner and, thus, upregulates CtIP.
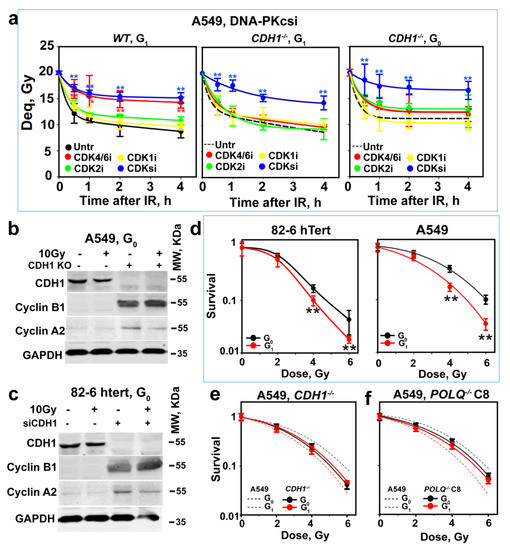
Figure 5.
CDH1 depletion rescues alt-EJ after CtIP depletion but also alters the CDK dependence of CtIP activation. (a) As in Figure 3a after inhibition of CDK4/6, CDK2 or CDK1, alone or combined, in G1- or G0-phase wild-type or CDH1−/− A549 cells. Plot shows mean ± SE from 6 determinations in 2 independent experiments. (b) WB analysis showing the effects of CDH1 depletion on cyclins A2 and B1 in G0-phase A549 cells. Experiments were repeated 3 times, and a representative one is shown. (c) As in (b) for 82-6 hTert cells. (d) Radiosensitivity of G0- and G1-phase 82-6 hTert and A549 cells determined using colony formation, as described in Material and Methods. (e) As in (d) for CDH1−/−A549 cells and their WT counterparts (broken lines). (f) As in (e) for POLQ−/− A549 cells, clone 8, and their WT counterparts (broken lines). Plots in (d–f) show mean ± SE from 3 independent experiments. Symbol * and ** in (a,d) indicate p < 0.05 and p < 0.01, respectively, in statistical significance calculation.
The shift in the absolute dependence of alt-EJ on a specific set of CDKs likely reflects the general stabilization of cyclins in G1-phase following APC/CCDH1 inactivation in the above experiments, which leaves several CDKs active to interchangeably activate CtIP. Indeed, Figure 5b shows that in CDH1−/− A549 cells, cyclins A2 and B1 are stabilized, while the levels of cyclin D1 and cyclin E1 remain unaffected (Figure S6c). The same trends are also observed in 82-6 hTert cells after CDH1 knockdown (Figure 5c and Figure S6d). Figure S6c,d should be changed.
Thus, CDH1 depletion shifts CDK activity in early G1-phase from specific CDK4/6 activation to general CDK4/6, CDK1 and CDK2 activation, which explains the results obtained. Specifically, the degradation characteristics by APC/C of cyclins and CtIP in G1-phase allow CDK-dependent CtIP activation in the G1-phase of the cell cycle and, thus, the resection and alt-EJ activities observed. Notably, the results obtained with CDH1−/− cells also show that when CtIP is present in cells, its activity strictly requires CDK-dependent phosphorylation.
3.6. Suppression of Alt-EJ in G0-Phase Cells Enhances Radioresistance to Killing
The above results uncover the well-designed, programmed suppression of alt-EJ in G0-phase cells, which is part of the more general cell cycle regulation of CtIP levels and activity, which, in turn, regulate resection and alt-EJ. However, are there benefits for irradiated cells from such regulatory adaptations of the activities of specific DSB repair pathways?
It is relevant that alt-EJ is error-prone and, therefore, a putative major source of genomic instability. To address this question, we exposed cells to IR, either in G0- or G1-phase, and plated them at a lower density to allow for colony formation 6 h later. Figure 5d shows that, in both cell lines, G0-phase cells are significantly more radioresistant than G1-phase cells. In addition, in CDH1−/− A549 cells, G0-phase cells that retain resection and, therefore, alt-EJ are equally radiosensitive to G1-phase cells (Figure 5e). Notably, the suppression of alt-EJ by the inactivation of POLQ also compromises the differences in radiosensitivity between cells in G1- and G0-phase (Figure 5f). In addition, CDK4/6 inhibition renders A549 cells irradiated in G1-phase as radioresistant (Figure S6e), providing further support to the concept and to the uncovered regulatory circuitry. We surmise that in G1-phase, cell survival benefits from the suppression of alt-EJ.
4. Discussion
4.1. CtIP Levels Regulate Resection and Alt-EJ throughout the Cell Cycle
We show above that CtIP cooperates with MRE11 to regulate resection in G1-phase cells. Indeed, CtIP is involved in the regulation of alt-EJ throughout the cell cycle in two distinct ways: First, CtIP levels fluctuate throughout the cell cycle and are, as we show here, undetectable in G0-phase phase owing to the activity of APCCDH1, which mediates nearly complete CtIP degradation. The inactivation of APC/CCDH1, as cells progress from G0- to G1- and then to S-phase [51,80], allows CtIP levels to increase, reaching a maximum at the end of G2-phase. Notably, our past and present work show that alt-EJ is almost fully suppressed in G0-phase and maximally active in G2-phase [31,32,33]. It remains to be investigated whether residual alt-EJ activity in G0-phase cells reflects residual CtIP activity or resection-independent alt-EJ proceeding with slow kinetics.
Second, CtIP activity is regulated by CDKs [36,37,38,39]. It is relevant, in this regard, that CDK activity is at its lowest level in quiescent cells, increases in G1-phase cells, and is again at maximum levels in G2-phase cells [81]. APC/CCDH1 activity marks cyclins for degradation, and, thus, suppresses CDK activity, which is required for CtIP activation [82]. This parallel evolution of ubiquitination and phosphorylation activities ensures that when CtIP is present, there will be CDK activity to prime it. It also explains why CDH1 deficiency or treatment with bortezomib has effects on both of these endpoints.
4.2. CtIP-Stability Regulation during the Cell Cycle and in Response to DNA Damage
HR is conceptually designed to faithfully restore the genome and to, thus, maintain genomic stability. CtIP-mediated resection exposes a 3′ single-stranded DNA that invades the sister chromatid during HR. In G2-phase cells, APC/CCDH1 mediates the clearance of CtIP from DSBs through ubiquitin-mediated degradation, thus limiting resection and adjusting it to the HR requirements [51]. We show here that CtIP enhances alt-EJ in G1-phase cells, thus promoting genomic instability (Figure 5d). CtIP is stabilized after IR in G1-phase cells (Figure 2d), which is opposite to the destabilization induced in G2-phase cells. This suggests benefits from CtIP stabilization, possibly at the expense of fidelity, at least when c-NHEJ is inactive. Indeed, CtIP-mutated chicken B cells (DT40) are sensitive to IR in G1-phase [83]. On the other hand, CtIP depletion rescues the DSB repair defect of Artemis mutants in G1-phase human fibroblasts but exerts only small effects on wild-type cells [16]. This observation suggests that CtIP depletion causes a switch from resection-dependent to resection-independent DSB processing in G1-phase cells [16]. Taken together, these data suggest that CtIP is involved in the repair of DSBs throughout the cell cycle, and that these specific functions and their regulation have a strong cell cycle component.
4.3. Relevance of Alt-EJ Suppression to Radiotherapy and Carcinogenesis
Processing of DSBs by alt-EJ generates chromosomal translocations that cause genomic instability and cancer [9]. DSBs naturally occur in a cell in the order of 10 to 50 DSBs per cell per day, depending on the cell cycle phase and tissue studied [84]. Most recent estimates put the number of cells in an average human body at around 37 trillion, most of which are non-cycling. These numbers indicate that a very high number of DSBs are induced every day in the human body. Therefore, protection from carcinogenesis may be achieved by suppressing the mutagenetic repair of spontaneous DSBs in the majority of cells.
On the other hand, tumor recurrence after radiation treatment is thought to be caused by radioresistant cells, which may derive from the pool of G0-phase cancer cells, which, as we show, are radioresistant. Here, the suppression of alt-EJ we show in quiescent cells might contribute to tumor recurrence by increasing the survival of tumor cells. Collectively, we, therefore, conclude that the default regulation of alt-EJ by CtIP suppresses carcinogenesis but may contribute to tumor recurrence. It will be important to develop means to harness this regulation in an effort to suppress carcinogenesis in normal cells or to decrease radioresistance in tumor cells.
4.4. Alt-EJ in G1-Phase Benefits from Resection When c-NHEJ Is Suppressed
The initiation of 5’-3’ resection at DNA ends is a critical determinant of the repair pathway choice, as it commits DSBs to HR while suppressing c-NHEJ [54,56,85]. Until recently, it was thought that resection only occurs in S- and G2-phase cells, and that it was inhibited in G1-phase by the Ku70/80 heterodimer and other proteins [54]. However, RPA loading at DSBs has, indeed, been observed in irradiated G1-phase cells, suggesting limited resection [56,86,87]. It has been suggested that in G1-phase, resection is dependent on the DSB number, with three or fewer endonuclease-induced DSBs per cell being insufficient and four or more being sufficient to activate resection [88]. Notably, resection has been reported in G1-phase human cells, particularly for complex DSBs induced by high LET particles or at high doses of X-rays [78,89]. Interestingly, 2 Gy of α-particles induced G1-resection-dependent DSB repair, which is independent of PARP1 and LIG1/3 but dependent on Artemis and DNA-PKcs. Because DNA-PKcs inhibition leaves the repair kinetics of Artemis deficient cells unchanged, it was concluded that this form of resection-dependent DSB repair reflects special forms of c-NHEJ rather than alt-EJ [16]. More work is needed to generate a mechanistic framework accommodating both sets of observations.
We previously demonstrated the suppression of alt-EJ when cells enter G0-phase [31,60]. Here, we extend these studies to human cells, uncover the role of resection in the regulation of alt-EJ and correlate the suppression of alt-EJ with compromised resection in G0-cells. Together, the above-discussed reports and our present results show that, on the one hand, resection can be induced in G1-phase, even in G0-phase cells by complex DSBs, to facilitate special forms of c-NHEJ [16]. On the other hand, simple DSBs of a c-NHEJ-deficient background activate resection specifically in G1-phase, which supports alt-EJ.
4.5. Collaboration between APCCDH1 and CDKs in the Regulation of DSB Repair in G1-Phase
CDK1 and CDK2 phosphorylate CtIP, NBS1, EXO1 and DNA2 to regulate resection [90,91,92] in G2-phase cells. Moreover, the necessity of CDK2-dependent CtIP phosphorylation on Thr847 is known for resection in G1-phase cells. However, here, using appropriately synchronized cells, we provide evidence for the contribution of CDK4/6 to CtIP activation and resection in G1-phase. Thus, it seems plausible that all cell cycle CDKs activate CtIP.
In addition to the phosphorylation-dependent regulation of CtIP activity, protein levels are also directly regulated and modulate resection in DSBs. APC/CCDH1 targets CtIP for proteasomal degradation after mitotic exit to generate the low resection environment that persists during G1-phase [51]. Moreover, it is well known that the oscillating activity of CDKs throughout the cell cycle is regulated by the periodic degradation of cyclins and CDK inhibitors (CKIs) by the ubiquitin–proteasome system [48], in which the APC/CCDH1 complex plays a central role, as it targets cyclin B1 and cyclin A2 for degradation. On the other hand, the affinity of CDH1 toward APC/C is suppressed by CDK phosphorylation [49,50]. These feedback mechanisms define the crosstalk between APCCDH1 and CDKs in the regulation of the cell cycle with extensions to DSB repair. These findings place the ubiquitination pathway and, particularly APCCDH1, in an intermediate node in the DSB repair pathway choice network.
Supplementary Materials
The following supporting information can be downloaded from: https://www.mdpi.com/article/10.3390/cells12111530/s1, Table S1. Sequence of siRNAs and gRNAs; Table S2. Inhibitors; Table S3. Antibodies; Table S4. Primers for real time-PCR; Table S5. Conditions for real time-PCR; Figure S1. Approaches for cell cycle dependent analysis; Figure S2. DSB repair analysis after 2 Gy of IR in G1 phase 82-6 hTert cells, using γH2AX as endpoint; Figure S3. Resection analysis in G0- and G1-phase cells exposed to IR; Figure S4. Effects of inhibition or knockdown of components of the resection apparatus on DNA end resection in G1-phase 82-6 hTert cells; Figure S5. Effects of PLK3 inhibition on alt-EJ and resection in G1-phase 82-6 hTert cells; Figure S6. Effects of CDKs on CtIP level in CDH1 wild type and CDH1 deficient cells.
Author Contributions
Conceptualization, F.L. and G.I.; methodology, F.L., E.M., Y.S., A.S. and G.I.; formal analysis, F.L., E.M., Y.S. and G.I.; investigation, F.L. and G.I.; resources, M.S., B.T. and G.I.; data curation, F.L., E.M., Y.S., A.S. and G.I.; writing—original draft preparation, F.L. and G.I.; writing—review and editing, F.L. and G.I.; visualization, F.L., E.M., Y.S. and G.I.; supervision, M.S., B.T. and G.I.; project administration, M.S., B.T. and G.I.; funding acquisition, M.S., B.T. and G.I. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the German Federal Ministry of Education and Research (BMBF-02NUK037B, BMBF-02NUK043B and BMBF-02NUK054B), the German Research Foundation (IL51.10, IL51.11, GRK1739) and the German Federal Ministry for Economic Affairs (BMWi-50WB1836), as well as by DAAD (project number: 57515880).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
As requested.
Acknowledgments
The authors are indebted to M. Löbrich, P. Jeggo, A. Sartori and J. Stark for providing cell lines and reagents.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Schipler, A.; Iliakis, G. DNA double-strand-break complexity levels and their possible contributions to the probability for error-prone processing and repair pathway choice. Nucleic Acids Res. 2013, 41, 7589–7605. [Google Scholar] [CrossRef] [PubMed]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Le Guen, T.; Ragu, S.; Guirouilh-Barbat, J.; Lopez, B.S. Role of the double-strand break repair pathway in the maintenance of genomic stability. Mol. Cell. Oncol. 2015, 2, e968020. [Google Scholar] [CrossRef] [PubMed]
- Murmann-Konda, T.; Soni, A.; Stuschke, M.; Iliakis, G. Analysis of chromatid-break-repair detects a homologous recombination to non-homologous end-joining switch with increasing load of DNA double-strand breaks. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2021, 867, 503372. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Staudt, C.; Soni, A.; Murmann-Konda, T.; Siemann-Loekes, M.; Iliakis, G. Strong suppression of gene conversion with increasing DNA double-strand break load delimited by 53BP1 and RAD52. Nucleic Acids Res. 2020, 48, 1905–1924. [Google Scholar] [CrossRef]
- Iliakis, G.; Murmann, T.; Soni, A. Alternative end-joining repair pathways are the ultimate backup for abrogated classical non-homologous end-joining and homologous recombination repair: Implications for the formation of chromosome translocations. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2015, 793, 166–175. [Google Scholar] [CrossRef]
- Ahrabi, S.; Sarkar, S.; Pfister, S.X.; Pirovano, G.; Higgins, G.S.; Porter, A.C.; Humphrey, T.C. A role for human homologous recombination factors in suppressing microhomology-mediated end joining. Nucleic Acids Res. 2016, 44, 5743–5757. [Google Scholar] [CrossRef]
- Lieber, M.R. NHEJ and its backup pathways in chromosomal translocations. Nat. Struct. Mol. Biol. 2010, 17, 393–395. [Google Scholar] [CrossRef]
- Feng, W.; Simpson, D.A.; Carvajal-Garcia, J.; Price, B.A.; Kumar, R.J.; Mose, L.E.; Wood, R.D.; Rashid, N.; Purvis, J.E.; Parker, J.S.; et al. Genetic determinants of cellular addiction to DNA polymerase theta. Nat. Commun. 2019, 10, 4286. [Google Scholar] [CrossRef]
- Truong, L.N.; Li, Y.; Shi, L.Z.; Hwang, P.Y.; He, J.; Wang, H.; Razavian, N.; Berns, M.W.; Wu, X. Microhomology-mediated End Joining and Homologous Recombination share the initial end resection step to repair DNA double-strand breaks in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 7720–7725. [Google Scholar] [CrossRef]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509–514. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.-M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA Double-Strand Break Repair Pathway Choice Is Directed by Distinct MRE11 Nuclease Activities. Mol. Cell. 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Garcia, V.; Phelps, S.E.; Gray, S.; Neale, M.J. Bidirectional resection of DNA double-strand breaks by Mre11 and Exo1. Nature 2011, 479, 241–244. [Google Scholar] [CrossRef]
- Nimonkar, A.V.; Genschel, J.; Kinoshita, E.; Polaczek, P.; Campbell, J.L.; Wyman, C.; Modrich, P.; Kowalczykowski, S.C. BLM-DNA2-RPA-MRN and EXO1-BLM-RPA-MRN constitute two DNA end resection machineries for human DNA break repair. Genes. Dev. 2011, 25, 350–362. [Google Scholar] [CrossRef]
- Li, F.; Mladenov, E.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Shift in G1-Checkpoint from ATM-Alone to a Cooperative ATM Plus ATR Regulation with Increasing Dose of Radiation. Cells 2022, 11, 63. [Google Scholar] [CrossRef]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhasz, S.; Kunzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Lobrich, M. DNA Double-Strand Break Resection Occurs during Non-homologous End Joining in G1 but Is Distinct from Resection during Homologous Recombination. Mol. Cell. 2017, 65, 671–684 e675. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, E.; Schmiemann, V.; Goedecke, W.; Reichenberger, S.; Pfeiffer, P. DNA double-strand break repair in cell-free extracts from Ku80-deficient cells: Implications for Ku serving as an alignment factor in non-homologous DNA end joining. Nucleic Acids Res. 2000, 28, 2585–2596. [Google Scholar] [CrossRef]
- Verkaik, N.S.; Esveldt-van Lange, R.E.E.; van Heemst, D.; Brüggenwirth, H.T.; Hoeijmakers, J.H.J.; Zdzienicka, M.Z.; van Gent, D.C. Different types of V(D)J recombination and end-joining defects in DNA double-strand break repair mutant mammalian cells. Eur. J. Immunol. 2002, 32, 701–709. [Google Scholar] [CrossRef]
- Della-Maria, J.; Zhou, Y.; Tsai, M.-S.; Kuhnlein, J.; Carney, J.P.; Paull, T.T.; Tomkinson, A.E. Human Mre11/Human Rad50/Nbs1 and DNA Ligase IIIα/XRCC1 Protein Complexes Act Together in an Alternative Nonhomologous End Joining Pathway. J. Biol. Chem. 2011, 286, 33845–33853. [Google Scholar] [CrossRef]
- Wang, H.; Perrault, A.R.; Takeda, Y.; Qin, W.; Wang, H.; Iliakis, G. Biochemical evidence for Ku-independent backup pathways of NHEJ. Nucleic Acids Res. 2020, 48, 5200. [Google Scholar] [CrossRef]
- Lieber, M.R. The Mechanism of Double-Strand DNA Break Repair by the Nonhomologous DNA End-Joining Pathway. Annu. Rev. Biochem. 2010, 79, 181–211. [Google Scholar] [CrossRef]
- Decottignies, A. Alternative end-joining mechanisms: A historical perspective. Front. Genet. 2013, 4, 48. [Google Scholar] [CrossRef] [PubMed]
- Boboila, C.; Alt, F.W.; Schwer, B. Chapter One—Classical and Alternative End-Joining Pathways for Repair of Lymphocyte-Specific and General DNA Double-Strand Breaks. In Advances in Immunology; Frederick, W.A., Ed.; Academic Press: Cambridge, MA, USA, 2012; Volume 116, pp. 1–49. [Google Scholar]
- Mansour, W.Y.; Rhein, T.; Dahm-Daphi, J. The alternative end-joining pathway for repair of DNA double-strand breaks requires PARP1 but is not dependent upon microhomologies. Nucleic Acids Res. 2010, 38, 6065–6077. [Google Scholar] [CrossRef] [PubMed]
- Bennardo, N.; Cheng, A.; Huang, N.; Stark, J.M. Alternative-NHEJ Is a Mechanistically Distinct Pathway of Mammalian Chromosome Break Repair. PLoS Genet. 2008, 4, e1000110. [Google Scholar] [CrossRef] [PubMed]
- Frit, P.; Barboule, N.; Yuan, Y.; Gomez, D.; Calsou, P. Alternative end-joining pathway(s): Bricolage at DNA breaks. DNA Repair. 2014, 17, 81–97. [Google Scholar] [CrossRef]
- Cheng, Q.; Barboule, N.; Frit, P.; Gomez, D.; Bombarde, O.; Couderc, B.; Ren, G.-S.; Salles, B.; Calsou, P. Ku counteracts mobilization of PARP1 and MRN in chromatin damaged with DNA double-strand breaks. Nucleic Acids Res. 2011, 39, 9605–9619. [Google Scholar] [CrossRef]
- Lee-Theilen, M.; Matthews, A.J.; Kelly, D.; Zheng, S.; Chaudhuri, J. CtIP promotes microhomology-mediated alternative end joining during class-switch recombination. Nat. Struct. Mol. Biol. 2011, 18, 75–79. [Google Scholar] [CrossRef]
- Zhang, Y.; Jasin, M. An essential role for CtIP in chromosomal translocation formation through an alternative end-joining pathway. Nat. Struct. Mol. Biol. 2011, 18, 80–84. [Google Scholar] [CrossRef]
- Li, F.; Mladenov, E.; Mortoga, S.; Iliakis, G. SCF(SKP2) regulates APC/C(CDH1)-mediated degradation of CTIP to adjust DNA-end resection in G2-phase. Cell. Death Dis. 2020, 11, 548. [Google Scholar] [CrossRef]
- Windhofer, F.; Wu, W.; Wang, M.; Singh, S.K.; Saha, J.; Rosidi, B.; Iliakis, G. Marked dependence on growth state of backup pathways of NHEJ. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 1462–1470. [Google Scholar] [CrossRef]
- Wu, W.; Wang, M.; Wu, W.; Singh, S.K.; Mussfeldt, T.; Iliakis, G. Repair of radiation induced DNA double strand breaks by backup NHEJ is enhanced in G2. DNA Repair. 2008, 7, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Wang, M.; Mussfeldt, T.; Iliakis, G. Enhanced Use of Backup Pathways of NHEJ in G2 in Chinese Hamster Mutant Cells with Defects in the Classical Pathway of NHEJ. Radiat. Res. 2008, 170, 512–520. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Bednar, T.; Zhang, L.; Wu, W.; Mladenov, E.; Iliakis, G. Inhibition of B-NHEJ in Plateau-Phase Cells Is Not a Direct Consequence of Suppressed Growth Factor Signaling. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, e237–e243. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. End Resection at Double-Strand Breaks: Mechanism and Regulation. Cold Spring Harb. Perspect. Biol. 2014, 6, a016436. [Google Scholar] [CrossRef]
- Huertas, P.; Jackson, S.P. Human CtIP Mediates Cell Cycle Control of DNA End Resection and Double Strand Break Repair. J. Biol. Chem. 2009, 284, 9558–9565. [Google Scholar] [CrossRef]
- Yu, X.; Wu, L.C.; Bowcock, A.M.; Aronheim, A.; Baer, R. The C-terminal (BRCT) domains of BRCA1 interact in vivo with CtIP, a protein implicated in the CtBP pathway of transcriptional repression. J. Biol. Chem. 1998, 273, 25388–25392. [Google Scholar] [CrossRef]
- Yu, X.; Chen, J. DNA Damage-Induced Cell Cycle Checkpoint Control Requires CtIP, a Phosphorylation-Dependent Binding Partner of BRCA1 C-Terminal Domains. Mol. Cell. Biol. 2004, 24, 9478–9486. [Google Scholar] [CrossRef]
- Steger, M.; Murina, O.; Hühn, D.; Ferretti, L.P.; Walser, R.; Hänggi, K.; Lafranchi, L.; Neugebauer, C.; Paliwal, S.; Janscak, P.; et al. Prolyl Isomerase PIN1 Regulates DNA Double-Strand Break Repair by Counteracting DNA End Resection. Mol. Cell. 2013, 50, 333–343. [Google Scholar] [CrossRef]
- Tomimatsu, N.; Mukherjee, B.; Catherine Hardebeck, M.; Ilcheva, M.; Vanessa Camacho, C.; Louise Harris, J.; Porteus, M.; Llorente, B.; Khanna, K.K.; Burma, S. Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat. Commun. 2014, 5, 3561. [Google Scholar] [CrossRef]
- Wohlbold, L.; Merrick, K.A.; De, S.; Amat, R.; Kim, J.H.; Larochelle, S.; Allen, J.J.; Zhang, C.; Shokat, K.M.; Petrini, J.H.J.; et al. Chemical Genetics Reveals a Specific Requirement for Cdk2 Activity in the DNA Damage Response and Identifies Nbs1 as a Cdk2 Substrate in Human Cells. PLoS Genet. 2012, 8, e1002935. [Google Scholar] [CrossRef]
- Falck, J.; Forment, J.V.; Coates, J.; Mistrik, M.; Lukas, J.; Bartek, J.; Jackson, S.P. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012, 13, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, L.P.; Lafranchi, L.; Sartori, A.A. Controlling DNA-end resection: A new task for CDKs. Front. Genet. 2013, 4, 99. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Niu, H.; Chung, W.H.; Zhu, Z.; Papusha, A.; Shim, E.Y.; Lee, S.E.; Sung, P.; Ira, G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat. Struct. Mol. Biol. 2011, 18, 1015–1019. [Google Scholar] [CrossRef]
- Tkáč, J.; Xu, G.; Adhikary, H.; Young, J.T.F.; Gallo, D.; Escribano-Díaz, C.; Krietsch, J.; Orthwein, A.; Munro, M.; Sol, W.; et al. HELB Is a Feedback Inhibitor of DNA End Resection. Mol. Cell. 2016, 61, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Guler, G.D.; Liu, H.; Vaithiyalingam, S.; Arnett, D.R.; Kremmer, E.; Chazin, W.J.; Fanning, E. Human DNA helicase B (HDHB) binds to replication protein A and facilitates cellular recovery from replication stress. J. Biol. Chem. 2012, 287, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Xia, X.; Yan, P.; Liu, H.; Podust, V.N.; Reynolds, A.B.; Fanning, E. Cell cycle-dependent regulation of a human DNA helicase that localizes in DNA damage foci. Mol. Biol. Cell. 2004, 15, 3320–3332. [Google Scholar] [CrossRef]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef]
- Nakayama, K.I.; Nakayama, K. Ubiquitin ligases: Cell-cycle control and cancer. Nat. Rev. Cancer 2006, 6, 369–381. [Google Scholar] [CrossRef]
- Wei, W.; Ayad, N.G.; Wan, Y.; Zhang, G.-J.; Kirschner, M.W.; Kaelin, J.W.G. Degradation of the SCF component Skp2 in cell-cycle phase G1 by the anaphase-promoting complex. Nature 2004, 428, 194–198. [Google Scholar] [CrossRef]
- Lafranchi, L.; de Boer, H.R.; de Vries, E.G.; Ong, S.E.; Sartori, A.A.; van Vugt, M.A. APC/CCdh1 controls CtIP stability during the cell cycle and in response to DNA damage. EMBO J. 2014, 33, 2860–2879. [Google Scholar] [CrossRef]
- Yilmaz, D.; Furst, A.; Meaburn, K.; Lezaja, A.; Wen, Y.; Altmeyer, M.; Reina-San-Martin, B.; Soutoglou, E. Activation of homologous recombination in G1 preserves centromeric integrity. Nature 2021, 600, 748–753. [Google Scholar] [CrossRef] [PubMed]
- Orthwein, A.; Noordermeer, S.M.; Wilson, M.D.; Landry, S.; Enchev, R.I.; Sherker, A.; Munro, M.; Pinder, J.; Salsman, J.; Dellaire, G.; et al. A mechanism for the suppression of homologous recombination in G1 cells. Nature 2015, 528, 422–426. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Huang, J. DNA End Resection: Facts and Mechanisms. Genom. Proteom. Bioinform. 2016, 14, 126–130. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S. Mechanism and regulation of DNA end resection in eukaryotes. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 195–212. [Google Scholar] [CrossRef]
- Huertas, P. DNA resection in eukaryotes: Deciding how to fix the break. Nat. Struct. Mol. Biol. 2010, 17, 11–16. [Google Scholar] [CrossRef]
- Kelso, A.A.; Lopezcolorado, F.W.; Bhargava, R.; Stark, J.M. Distinct roles of RAD52 and POLQ in chromosomal break repair and replication stress response. PLoS Genet. 2019, 15, e1008319. [Google Scholar] [CrossRef]
- Cheng, Y.; Li, F.; Mladenov, E.; Iliakis, G. The yield of DNA double strand breaks determined after exclusion of those forming from heat-labile lesions predicts tumor cell radiosensitivity to killing. Radiother. Oncol. 2015, 116, 366–373. [Google Scholar] [CrossRef]
- DiBiase, S.J.; Zeng, Z.-C.; Chen, R.; Hyslop, T.; Curran, W.J., Jr.; Iliakis, G. DNA-dependent protein kinase stimulates an independently active, nonhomologous, end-joining apparatus. Cancer Res. 2000, 60, 1245–1253. [Google Scholar]
- Singh, S.K.; Wu, W.; Zhang, L.; Klammer, H.; Wang, M.; Iliakis, G. Widespread Dependence of Backup NHEJ on Growth State: Ramifications for the Use of DNA-PK Inhibitors. Int. J. Radiat. Oncol. Biol. Phys. 2011, 79, 540–548. [Google Scholar] [CrossRef]
- Iliakis, G. Backup pathways of NHEJ in cells of higher eukaryotes: Cell cycle dependence. Radiother. Oncol. 2009, 92, 310–315. [Google Scholar] [CrossRef]
- Bindra, R.S.; Goglia, A.G.; Jasin, M.; Powell, S.N. Development of an assay to measure mutagenic non-homologous end-joining repair activity in mammalian cells. Nucleic Acids Res. 2013, 41, e115. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zeng, Z.-C.; Bui, T.-A.; Sonoda, E.; Takata, M.; Takeda, S.; Iliakis, G. Efficient rejoining of radiation-induced DNA double-strand breaks in vertebrate cells deficient in genes of the RAD52 epistasis group. Oncogene 2001, 20, 2212–2224. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.; Chandramouly, G.; McDevitt, S.M.; Ozdemir, A.Y.; Pomerantz, R.T. Mechanism of microhomology-mediated end-joining promoted by human DNA polymerase theta. Nat. Struct. Mol. Biol. 2015, 22, 230–237. [Google Scholar] [CrossRef]
- Paul, K.; Wang, M.; Mladenov, E.; Bencsik-Theilen, A.A.; Bednar, T.; Wu, W.; Arakawa, H.; Iliakis, G. DNA ligases I and III cooperate in alternative non-homologous end-joining in vertebrates. PLoS ONE 2013, 8, e59505. [Google Scholar] [CrossRef]
- Soni, A.; Li, F.; Wang, Y.; Grabos, M.; Krieger, L.M.; Chaudhary, S.; Hasan, M.S.M.; Ahmed, M.; Coleman, C.N.; Teicher, B.A.; et al. Inhibition of Parp1 by BMN673 Effectively Sensitizes Cells to Radiotherapy by Upsetting the Balance of Repair Pathways Processing DNA Double-Strand Breaks. Mol. Cancer Ther. 2018, 17, 2206–2216. [Google Scholar] [CrossRef] [PubMed]
- Mladenov, E.; Fan, X.; Paul-Konietzko, K.; Soni, A.; Iliakis, G. DNA-PKcs and ATM epistatically suppress DNA end resection and hyperactivation of ATR-dependent G2-checkpoint in S-phase irradiated cells. Sci. Rep. 2019, 9, 14597. [Google Scholar] [CrossRef]
- Mladenov, E.; Fan, X.; Dueva, R.; Soni, A.; Iliakis, G. Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase. Sci. Rep. 2019, 9, 8255. [Google Scholar] [CrossRef]
- Xiao, H.; Li, F.; Mladenov, E.; Soni, A.; Mladenova, V.; Pan, B.; Dueva, R.; Stuschke, M.; Timmermann, B.; Iliakis, G. Increased Resection at DSBs in G2-Phase Is a Unique Phenotype Associated with DNA-PKcs Defects That Is Not Shared by Other Factors of c-NHEJ. Cells 2022, 11, 2099. [Google Scholar] [CrossRef]
- Kinner, A.; Wu, W.Q.; Staudt, C.; Iliakis, G. gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef]
- Iliakis, G.; Mehta, R.; Jackson, M. Level of DNA double-strand break rejoining in Chinese hamster xrs-5 cells is dose-dependent: Implications for the mechanism of radiosensitivity. Int. J. Radiat. Biol. 1992, 61, 315–321. [Google Scholar] [CrossRef]
- Chang, C.; Biedermann, K.A.; Mezzina, M.; Brown, J.M. Characterization of the DNA double strand break repair defect in scid mice. Cancer Res. 1993, 53, 1244–1248. [Google Scholar] [PubMed]
- Ferretti, L.P.; Himmels, S.F.; Trenner, A.; Walker, C.; von Aesch, C.; Eggenschwiler, A.; Murina, O.; Enchev, R.I.; Peter, M.; Freire, R.; et al. Cullin3-KLHL15 ubiquitin ligase mediates CtIP protein turnover to fine-tune DNA-end resection. Nat. Commun. 2016, 7, 12628. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Kaldis, P. Cdks, cyclins and CKIs: Roles beyond cell cycle regulation. Development 2013, 140, 3079–3093. [Google Scholar] [CrossRef]
- Suryadinata, R.; Sadowski, M.; Sarcevic, B. Control of cell cycle progression by phosphorylation of cyclin-dependent kinase (CDK) substrates. Biosci. Rep. 2010, 30, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Matsushime, H.; Roussel, M.F.; Ashmun, R.A.; Sherr, C.J. Colony-stimulating factor 1 regulates novel cyclins during the G1 phase of the cell cycle. Cell 1991, 65, 701–713. [Google Scholar] [CrossRef]
- Donjerkovic, D.; Scott, D.W. Regulation of the G1 phase of the mammalian cell cycle. Cell. Res. 2000, 10, 1–16. [Google Scholar] [CrossRef]
- Barton, O.; Naumann, S.C.; Diemer-Biehs, R.; Künzel, J.; Steinlage, M.; Conrad, S.; Makharashvili, N.; Wang, J.; Feng, L.; Lopez, B.S.; et al. Polo-like kinase 3 regulates CtIP during DNA double-strand break repair in G1. J. Cell. Biol. 2014, 206, 877–894. [Google Scholar] [CrossRef]
- Zimmerman, W.C.; Erikson, R.L. Polo-like kinase 3 is required for entry into S phase. Proc. Natl. Acad. Sci. USA 2007, 104, 1847–1852. [Google Scholar] [CrossRef]
- Cappell, S.D.; Chung, M.; Jaimovich, A.; Spencer, S.L.; Meyer, T. Irreversible APC(Cdh1) Inactivation Underlies the Point of No Return for Cell-Cycle Entry. Cell 2016, 166, 167–180. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef]
- Skaar, J.R.; Pagano, M. Cdh1: A master G0/G1 regulator. Nat. Cell. Biol. 2008, 10, 755–757. [Google Scholar] [CrossRef] [PubMed]
- Yun, M.H.; Hiom, K. CtIP-BRCA1 modulates the choice of DNA double-strand-break repair pathway throughout the cell cycle. Nature 2009, 459, 460–463. [Google Scholar] [CrossRef] [PubMed]
- Vilenchik, M.M.; Knudson, A.G. Endogenous DNA double-strand breaks: Production, fidelity of repair, and induction of cancer. Proc. Natl. Acad. Sci. USA 2003, 100, 12871–12876. [Google Scholar] [CrossRef] [PubMed]
- Symington, L.S.; Gautier, J. Double-Strand Break End Resection and Repair Pathway Choice. Annu. Rev. Genet. 2011, 45, 247–271. [Google Scholar] [CrossRef] [PubMed]
- Lisby, M.; Barlow, J.H.; Burgess, R.C.; Rothstein, R. Choreography of the DNA Damage Response: Spatiotemporal Relationships among Checkpoint and Repair Proteins. Cell 2004, 118, 699–713. [Google Scholar] [CrossRef]
- Barlow, J.H.; Lisby, M.; Rothstein, R. Differential Regulation of the Cellular Response to DNA Double-Strand Breaks in G1. Mol. Cell. 2008, 30, 73–85. [Google Scholar] [CrossRef]
- Zierhut, C.; Diffley, J.F.X. Break dosage, cell cycle stage and DNA replication influence DNA double strand break response. EMBO J. 2008, 27, 1875–1885. [Google Scholar] [CrossRef]
- Averbeck, N.B.; Ringel, O.; Herrlitz, M.; Jakob, B.; Durante, M.; Taucher-Scholz, G. DNA end resection is needed for the repair of complex lesions in G1-phase human cells. Cell. Cycle 2014, 13, 2509–2516. [Google Scholar] [CrossRef]
- Aylon, Y.; Liefshitz, B.; Kupiec, M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J. 2004, 23, 4868–4875. [Google Scholar] [CrossRef]
- Ira, G.; Pellicioli, A.; Balijja, A.; Wang, X.; Fiorani, S.; Carotenuto, W.; Liberi, G.; Bressan, D.; Wan, L.; Hollingsworth, N.M.; et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004, 431, 1011–1017. [Google Scholar] [CrossRef]
- Deans, A.J.; Khanna, K.K.; McNees, C.J.; Mercurio, C.; Heierhorst, J.; McArthur, G.A. Cyclin-Dependent Kinase 2 Functions in Normal DNA Repair and Is a Therapeutic Target in BRCA1-Deficient Cancers. Cancer Res. 2006, 66, 8219–8226. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).