


NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease



Abstract
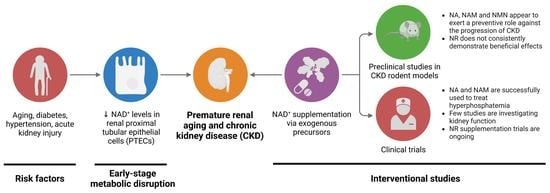
1. Introduction
2. NAD+ Biology in the Kidney
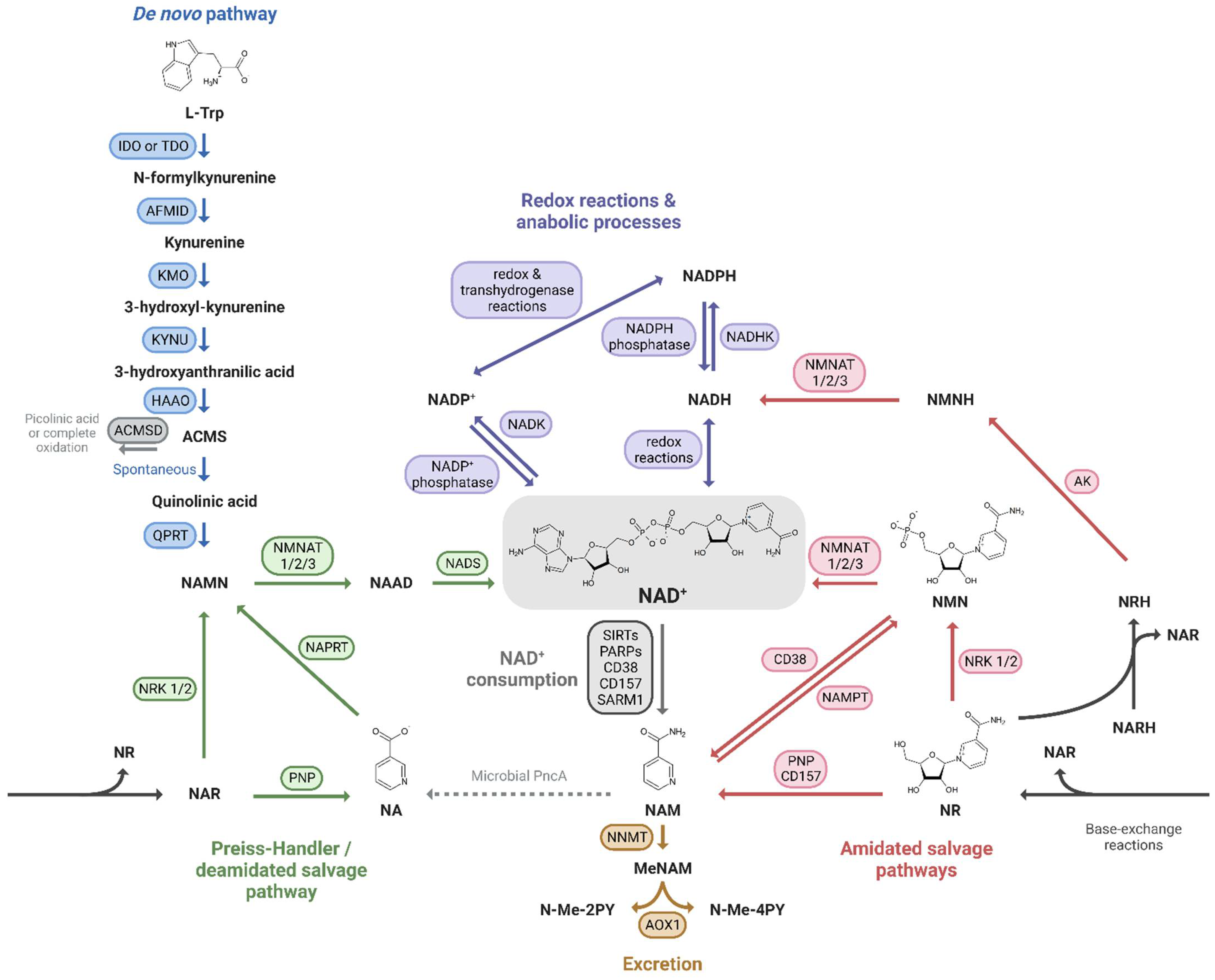
2.1. NAD+ Homeostasis and Precursors
2.1.1. De Novo Pathway
2.1.2. Preiss-Handler Pathway
2.1.3. Amidated Salvage Pathways
2.1.4. Phosphorylation to NADP+
2.1.5. Excretion
2.1.6. Compartmentalization
2.2. NAD+ Cellular Roles
2.2.1. Redox Co-Factor
2.2.2. Enzymatic Substrate
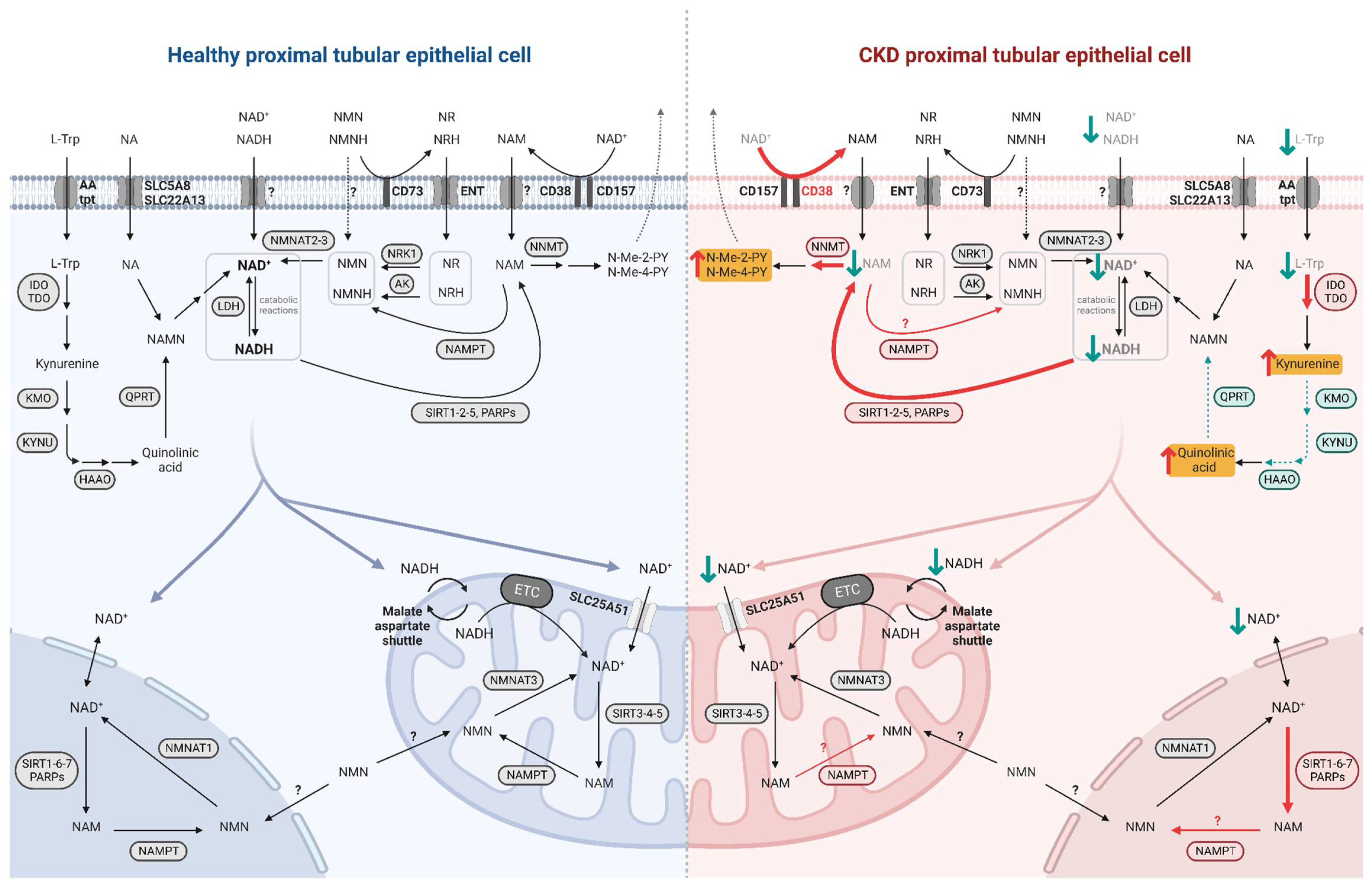
2.3. NAD+ Alterations in the Diseased Kidney
2.3.1. Impaired NAD+ Production
2.3.2. Accelerated NAD+ Consumption
3. NAD+ Boosting Interventions in Premature Renal Aging and CKD
3.1. Preclinical Studies
3.2. Clinical Interventions

{kind=link}
{kind=link}
{kind=link}
| Trial—Year of Completion/Publication | Supplement | Condition | n (NAD+ Precursor Treated Group) | Primary Endpoint | Secondary Endpoints | Main Results | Ref. |
|---|---|---|---|---|---|---|---|
| 2008 | NA 1 g/day, 8 months | CKD on dialysis refractory to treatment of hyperlipidemia and hyperphosphatemia (serum phosphate > 5.5 mg/dL) | 9 | Serum phosphate levels | Lipids profile, MBD and kidney function markers | ↘ Phosphate, TC, TG Unchanged iPTH, LDL-C, albumin, creatinine, BUN | [196] |
| 2010 | NA 1 g/day, 4 weeks then 2 g/day, 20 weeks | Dyslipidemia and eGFR < 60 mL/min/1.73 m2 | 110 (with laropiprant), 67 (without laropiprant) | Serum phosphate levels | N/A | ↘ Phosphate | [210] |
| 2011 | NA 1 g/day, 4 weeks then 2 g/day, 32 weeks | Type 2 diabetes | 446 | Serum phosphate levels | N/A | ↘ Phosphate (No evidence for effect modification by the variable GFR < 60 vs. ≥ 60 mL/min/1.73 m2) | [211] |
| 2012 | NA 400–1000 mg/day, 8 weeks | Hemodialysis | 23 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate Unchanged calcium, iPTH, TC, LDL-C, TG | [212] |
| 2012 | NA 375–1000 mg/day, 12 weeks | Hemodialysis | 14 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate Unchanged calcium, iPTH | [213] |
| NCT00852969—2012 | NA 1 g/day vs. NA 100 mg/day, 14 weeks | CKD stages 2–4 | 30 | Flow mediated dilation by brachial artery reactivity (endothelial function) | HDL-C | No significant differences | - |
| AIM-HIGH NCT00120289—2012 | NA 1.5 or 2 g/day + statin, average of 36 months, max 66 months | >45 years old, prevalent CVD and atherogenic dyslipidaemia, sCr < 2.5 mg/L, eGFRcys+cr < 60 mL/min/1.73 m2 | 352 | Composite End Point of CHD Death | Other CVD endpoints | No CVD benefit from the addition of NA to statin therapy even if: ↗ HDL-C ↘ TG, LDL-C, phosphate Unchanged iPTH, FGF23, calcium | [188,214] |
| NCT00108485—2012 | NA 1.5–2 g/day, 1 year | Diabetic nephropathy (type II diabetes, CKD stage 2–3, eGFR = 30–89 mL/min/1.73 m2, proteinuria < 3.5 g/d, statin treatment) | 5 | Proteinuria | - | Unable to recruit sufficient study subjects | - |
| 2013 | NA 500 mg/day, 6 months | CKD stages 2–4 | 31 | Lipids profile | Serum phosphate levels, GFR | ↗ HDL-C, GFR ↘ TG, phosphate | [199] |
| 2014 | NA 1 g/day, 4 weeks then 2 g/day, 20 weeks | Dyslipidemia and eGFR = 30–74 mL/min/1.73 m2 | 162 (with laropiprant), 97 (without laropiprant) | Serum FGF23 | Other MBD markers | ↘ Phosphate, calcium ↘ FGF23, iPTH (for the NA group without laropiprant) | [215] |
| 2016 | NA 25–100 mg/day, 3 months | Kidney failure and serum phosphate ≥ 5.5 mg/dL | 35 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate Unchanged calcium, PTH, TC, TG, LDL-C | [192] |
| NCT03163576—2022 | NA 750 mg/day, 5 weeks then 1.5 g/day, 6 months | Kidney failure, hemodialysis for > 6 months, serum phosphate > 5.0 mg/dL | 25 | Serum phosphate levels | Lipids profile, MBD markers | ↘ Phosphate, TC, LDL-C, TG, iPTH Unchanged HDL-C, calcium | [216] |
| 1998 | Niceritrol (ester of NA), 750 mg/day, 8 weeks | Hemodialysis | 10 | Serum phosphate levels | Kidney functional markers | ↗ Serum NA, HDL-C ↘ Phosphate Unchanged BUN, creatinine, calcium, TC, TG | [197] |
| 2004 | NAM 500 mg/day, increased by 250 mg/day every 2 weeks until serum phosphate < 6.0 mg/dL (mean dose = 1080 mg/day), 12 weeks total | Hemodialysis and serum phosphate > 6.0 mg/dL | 65 | Serum phosphate levels | Lipids profile, MBD markers | ↗ Serum NAD+, HDL-C ↘ Phosphate, iPTH, LDL-C Unchanged calcium, TG | [217] |
| NCT00508885—2007 | NAM 500–1500 mg/day, 8 weeks | Peritoneal dialysis and serum phosphate > 4.9 mg/dL | 8 | Serum phosphate levels | Lipids profile | ↘ Phosphate Unchanged HDL-C, LDL-C, TG | [218] |
| NCT00316472—2007 | NAM 500–1500 mg/day, 8 weeks | Hemodialysis and serum phosphate ≥ 5.0 mg/dL | 33 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate Unchanged calcium, iPTH, TG, LDL-C, TC | [219] |
| 2008 | NAM 500–1000 mg/day, 8 weeks | Hemodialysis and serum phosphate > 5.0 mg/dL | 24 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate Unchanged calcium, iPTH | [220] |
| 2015 | NAM 200–300 mg/day, 6 months | 6–18 years old, hemodialysis and serum phosphate > 5.0 mg/dL | 30 | Serum phosphate levels | Lipids profile, MBD markers | ↗ HDL-C ↘ Phosphate, iPTH, TG | [221] |
| NIAC-PKD1 NCT02140814—2016 & NIAC-PKD2 NCT02558595—2019 | NAM 30 mg/kg/day, 12 months | Autosomal dominant polycystic kidney disease | 10 & 18 | Sirtuin deacetylase activity (acetylated/total p53 ratio in PBMCs) | eGFR, kidney volume, biomarker levels, pain | No sustained inhibition of sirtuin activity Unchanged secondary outcomes | [198] |
| NICOREN NCT01011699—2017 | NAM 0.5–2 g/day, 24 weeks | CKD and hemodialysis | 49 | Serum phosphate levels | Safety, MBD markers | ↘ Phosphate, α-klotho ↗ Calcium, HDL-C, N-Me-2PY Unchanged iPTH, FGF23 | [194] |
| COMBINE NCT02258074—2019 | NAM 1.5 g/day, 1 year | CKD stages 3–4 (eGFR = 20–45 mL/min/1.73 m2; serum phosphate ≥ 2.8 mg/dL) | 51 | FGF23, serum phosphate levels | MBD and kidney function markers | ↗ Calcium, ACR Unchanged FGF23, phosphate, iPTH, eGFR | [191] |
| NOPHOS—2020 | NAM 250–1500 mg/day, 12 weeks | Hemodialysis and serum phosphate > 4.5 mg/dL | 239 | Serum phosphate levels | MBD and kidney function markers | ↘ Phosphate, iPTH | [193] |
| CoNR NCT03579693—2021 | NR 1.2 g/day, 6 weeks | CKD (eGFR < 50 mL/min/1.73 m2) without dialysis or kidney transplantation | 25 (n total) | Aerobic capacity | PBMCs mitochondrial energetics, fatigue, physical function, heart failure symptoms, oxidative stress, inflammation | Not yet disclosed | - |
| NCT04040959—Recruiting | NR 1 g/day, 3 months | 35–80 years, CKD stages 3–4 (eGFR = 20–60 mL/min/1.73 m2), blood pressure < 140/90 mmHg | 118 (n total) | Carotid-femoral pulse wave velocity | Systolic blood pressure, safety, NAD+ metabolome in PBMCs | Active recruitment | - |
| NCT04893733—Recruiting | Intravenous vitamin B complex, 5 days | AKI | 180 (n total) | Serum creatinine | Progression to CKD | Active recruitment | - |
4. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- O’Sullivan, E.D.; Hughes, J.; Ferenbach, D.A. Renal Aging: Causes and Consequences. J. Am. Soc. Nephrol. 2017, 28, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Li, S.; Wang, C.; Huang, C.; Becker, B.N.; Bomback, A.S.; Brown, W.W.; Burrows, N.R.; Jurkovitz, C.T.; McFarlane, S.I.; et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: Results from the Kidney Early Evaluation Program (KEEP). Am. J. Kidney Dis. 2010, 55 (Suppl. S2), S23–S33. [Google Scholar] [CrossRef] [PubMed]
- Kovesdy, C.P. Epidemiology of chronic kidney disease: An update 2022. Kidney Int. Suppl. 2022, 12, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Romagnani, P.; Remuzzi, G.; Glassock, R.; Levin, A.; Jager, K.J.; Tonelli, M.; Massy, Z.; Wanner, C.; Anders, H.J. Chronic kidney disease. Nat. Rev. Dis. Primers 2017, 3, 17088. [Google Scholar] [CrossRef]
- Zhong, J.; Yang, H.C.; Fogo, A.B. A perspective on chronic kidney disease progression. Am. J. Physiol. Renal Physiol. 2017, 312, F375–F384. [Google Scholar] [CrossRef] [PubMed]
- Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 1–150. [Google Scholar]
- Nangaku, M. Chronic hypoxia and tubulointerstitial injury: A final common pathway to end-stage renal failure. J. Am. Soc. Nephrol. 2006, 17, 17–25. [Google Scholar] [CrossRef]
- Yamanouchi, M.; Hoshino, J.; Ubara, Y.; Takaichi, K.; Kinowaki, K.; Fujii, T.; Ohashi, K.; Mise, K.; Toyama, T.; Hara, A.; et al. Clinicopathological predictors for progression of chronic kidney disease in nephrosclerosis: A biopsy-based cohort study. Nephrol. Dial. Transplant. 2019, 34, 1182–1188. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, Y.; Shen, L.; Li, X.; Jiao, Q.; Li, Z.; Jia, J.; He, L.; Zhang, Q.; Wang, N.; et al. Clinical and Histological Predictors of Renal Survival in Patients with Biopsy-Proven Diabetic Nephropathy. Kidney Dis. 2022, 8, 93–101. [Google Scholar] [CrossRef]
- Ruiz-Ortega, M.; Rayego-Mateos, S.; Lamas, S.; Ortiz, A.; Rodrigues-Diez, R.R. Targeting the progression of chronic kidney disease. Nat. Rev. Nephrol. 2020, 16, 269–288. [Google Scholar] [CrossRef]
- Cano, N.; Fiaccadori, E.; Tesinsky, P.; Toigo, G.; Druml, W.; DGEM; Kuhlmann, M.; Mann, H.; Horl, W.H.; ESPEN. ESPEN Guidelines on Enteral Nutrition: Adult renal failure. Clin. Nutr. 2006, 25, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Burrowes, J.D.; Byham-Gray, L.D.; Campbell, K.L.; Carrero, J.J.; Chan, W.; Fouque, D.; Friedman, A.N.; Ghaddar, S.; Goldstein-Fuchs, D.J.; et al. KDOQI Clinical Practice Guideline for Nutrition in CKD: 2020 Update. Am. J. Kidney Dis. 2020, 76 (Suppl. S1), S1–S107. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Borges, N.A.; Lindholm, B.; Shiels, P.G.; Evenepoel, P.; Stenvinkel, P. Food as medicine: Targeting the uraemic phenotype in chronic kidney disease. Nat. Rev. Nephrol. 2021, 17, 153–171. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L. The proximal tubule is the primary target of injury and progression of kidney disease: Role of the glomerulotubular junction. Am. J. Physiol. Renal Physiol. 2016, 311, F145–F161. [Google Scholar] [CrossRef]
- Fassett, R.G.; Venuthurupalli, S.K.; Gobe, G.C.; Coombes, J.S.; Cooper, M.A.; Hoy, W.E. Biomarkers in chronic kidney disease: A review. Kidney Int. 2011, 80, 806–821. [Google Scholar] [CrossRef]
- Vaidya, V.S.; Ferguson, M.A.; Bonventre, J.V. Biomarkers of acute kidney injury. Annu. Rev. Pharmacol. Toxicol. 2008, 48, 463–493. [Google Scholar] [CrossRef]
- Cohen, J.J. Relationship between energy requirements for Na+ reabsorption and other renal functions. Kidney Int. 1986, 29, 32–40. [Google Scholar] [CrossRef]
- van der Rijt, S.; Leemans, J.C.; Florquin, S.; Houtkooper, R.H.; Tammaro, A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 2022, 18, 588–603. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Gai, Z.; Wang, T.; Visentin, M.; Kullak-Ublick, G.A.; Fu, X.; Wang, Z. Lipid Accumulation and Chronic Kidney Disease. Nutrients 2019, 11, 722. [Google Scholar] [CrossRef]
- Li, H.; Dixon, E.E.; Wu, H.; Humphreys, B.D. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022, 34, 1977–1998.e9. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, R.; Kanda, T.; Komatsu, M.; Itoh, T.; Minakuchi, H.; Urai, H.; Kuroita, T.; Shigaki, S.; Tsukamoto, T.; Higuchi, N.; et al. The significance of NAD + metabolites and nicotinamide N-methyltransferase in chronic kidney disease. Sci. Rep. 2022, 12, 6398. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; Wang, S.R.; Huang, X.Z.; Xie, Q.H.; Xu, Y.Y.; Shang, D.; Hao, C.M. Nicotinamide Mononucleotide, an NAD(+) Precursor, Rescues Age-Associated Susceptibility to AKI in a Sirtuin 1-Dependent Manner. J. Am. Soc. Nephrol. 2017, 28, 2337–2352. [Google Scholar] [CrossRef] [PubMed]
- Katsyuba, E.; Romani, M.; Hofer, D.; Auwerx, J. NAD(+) homeostasis in health and disease. Nat. Metab. 2020, 2, 9–31. [Google Scholar] [CrossRef] [PubMed]
- Fang, E.F.; Lautrup, S.; Hou, Y.; Demarest, T.G.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. NAD+ in Aging: Molecular Mechanisms and Translational Implications. Trends Mol. Med. 2017, 23, 899–916. [Google Scholar] [CrossRef]
- Ciarlo, E.; Joffraud, M.; Hayat, F.; Giner, M.P.; Giroud-Gerbetant, J.; Sanchez-Garcia, J.L.; Rumpler, M.; Moco, S.; Migaud, M.E.; Canto, C. Nicotinamide Riboside and Dihydronicotinic Acid Riboside Synergistically Increase Intracellular NAD(+) by Generating Dihydronicotinamide Riboside. Nutrients 2022, 14, 2752. [Google Scholar] [CrossRef]
- Giroud-Gerbetant, J.; Joffraud, M.; Giner, M.P.; Cercillieux, A.; Bartova, S.; Makarov, M.V.; Zapata-Perez, R.; Sanchez-Garcia, J.L.; Houtkooper, R.H.; Migaud, M.E.; et al. A reduced form of nicotinamide riboside defines a new path for NAD(+) biosynthesis and acts as an orally bioavailable NAD(+) precursor. Mol. Metab. 2019, 30, 192–202. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J., 3rd; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018, 27, 1067–1080.e5. [Google Scholar] [CrossRef]
- Faivre, A.; Katsyuba, E.; Verissimo, T.; Lindenmeyer, M.; Rajaram, R.D.; Naesens, M.; Heckenmeyer, C.; Mottis, A.; Feraille, E.; Cippa, P.; et al. Differential role of nicotinamide adenine dinucleotide deficiency in acute and chronic kidney disease. Nephrol. Dial. Transplant. 2021, 36, 60–68. [Google Scholar] [CrossRef]
- Kempson, S.A.; Colon-Otero, G.; Ou, S.Y.; Turner, S.T.; Dousa, T.P. Possible role of nicotinamide adenine dinucleotide as an intracellular regulator of renal transport of phosphate in the rat. J. Clin. Investig. 1981, 67, 1347–1360. [Google Scholar] [CrossRef]
- Gopal, E.; Fei, Y.J.; Miyauchi, S.; Zhuang, L.; Prasad, P.D.; Ganapathy, V. Sodium-coupled and electrogenic transport of B-complex vitamin nicotinic acid by slc5a8, a member of the Na/glucose co-transporter gene family. Biochem. J. 2005, 388, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, A.; Dolle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011, 286, 21767–21778. [Google Scholar] [CrossRef]
- Kandasamy, P.; Gyimesi, G.; Kanai, Y.; Hediger, M.A. Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci. 2018, 43, 752–789. [Google Scholar] [CrossRef]
- Ikeda, M.; Tsuji, H.; Nakamura, S.; Ichiyama, A.; Nishizuka, Y.; Hayaishi, O. Studies on the Biosynthesis of Nicotinamide Adenine Dinucleotide. Ii. A Role of Picolinic Carboxylase in the Biosynthesis of Nicotinamide Adenine Dinucleotide from Tryptophan in Mammals. J. Biol. Chem. 1965, 240, 1395–1401. [Google Scholar] [CrossRef]
- Shi, H.; Enriquez, A.; Rapadas, M.; Martin, E.; Wang, R.; Moreau, J.; Lim, C.K.; Szot, J.O.; Ip, E.; Hughes, J.N.; et al. NAD Deficiency, Congenital Malformations, and Niacin Supplementation. N. Engl. J. Med. 2017, 377, 544–552. [Google Scholar] [CrossRef]
- Preiss, J.; Handler, P. Biosynthesis of diphosphopyridine nucleotide. I. Identification of intermediates. J. Biol. Chem. 1958, 233, 488–492. [Google Scholar] [CrossRef]
- Hara, N.; Yamada, K.; Terashima, M.; Osago, H.; Shimoyama, M.; Tsuchiya, M. Molecular identification of human glutamine- and ammonia-dependent NAD synthetases. Carbon-nitrogen hydrolase domain confers glutamine dependency. J. Biol. Chem. 2003, 278, 10914–10921. [Google Scholar] [CrossRef]
- Collins, P.B.; Chaykin, S. The management of nicotinamide and nicotinic acid in the mouse. J. Biol. Chem. 1972, 247, 778–783. [Google Scholar] [CrossRef]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014, 9, e113939. [Google Scholar] [CrossRef]
- Canto, C.; Menzies, K.J.; Auwerx, J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015, 22, 31–53. [Google Scholar] [CrossRef]
- Rongvaux, A.; Shea, R.J.; Mulks, M.H.; Gigot, D.; Urbain, J.; Leo, O.; Andris, F. Pre-B-cell colony-enhancing factor, whose expression is up-regulated in activated lymphocytes, is a nicotinamide phosphoribosyltransferase, a cytosolic enzyme involved in NAD biosynthesis. Eur. J. Immunol. 2002, 32, 3225–3234. [Google Scholar] [CrossRef] [PubMed]
- Bieganowski, P.; Brenner, C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell 2004, 117, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef]
- Nakahata, Y.; Sahar, S.; Astarita, G.; Kaluzova, M.; Sassone-Corsi, P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009, 324, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, K.M.; Yoshino, J.; Brace, C.S.; Abrassart, D.; Kobayashi, Y.; Marcheva, B.; Hong, H.K.; Chong, J.L.; Buhr, E.D.; Lee, C.; et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009, 324, 651–654. [Google Scholar] [CrossRef]
- Revollo, J.R.; Korner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin regulates insulin secretion in beta cells as a systemic NAD biosynthetic enzyme. Cell Metab. 2007, 6, 363–375. [Google Scholar] [CrossRef]
- Dall, M.; Trammell, S.A.J.; Asping, M.; Hassing, A.S.; Agerholm, M.; Vienberg, S.G.; Gillum, M.P.; Larsen, S.; Treebak, J.T. Mitochondrial function in liver cells is resistant to perturbations in NAD(+) salvage capacity. J. Biol. Chem. 2019, 294, 13304–13326. [Google Scholar] [CrossRef]
- Ratajczak, J.; Joffraud, M.; Trammell, S.A.; Ras, R.; Canela, N.; Boutant, M.; Kulkarni, S.S.; Rodrigues, M.; Redpath, P.; Migaud, M.E.; et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat. Commun. 2016, 7, 13103. [Google Scholar] [CrossRef]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef]
- Grozio, A.; Mills, K.F.; Yoshino, J.; Bruzzone, S.; Sociali, G.; Tokizane, K.; Lei, H.C.; Cunningham, R.; Sasaki, Y.; Migaud, M.E.; et al. Slc12a8 is a nicotinamide mononucleotide transporter. Nat. Metab. 2019, 1, 47–57. [Google Scholar] [CrossRef]
- Tempel, W.; Rabeh, W.M.; Bogan, K.L.; Belenky, P.; Wojcik, M.; Seidle, H.F.; Nedyalkova, L.; Yang, T.; Sauve, A.A.; Park, H.W.; et al. Nicotinamide riboside kinase structures reveal new pathways to NAD+. PLoS Biol. 2007, 5, e263. [Google Scholar] [CrossRef] [PubMed]
- Belenky, P.; Christensen, K.C.; Gazzaniga, F.; Pletnev, A.A.; Brenner, C. Nicotinamide riboside and nicotinic acid riboside salvage in fungi and mammals. Quantitative basis for Urh1 and purine nucleoside phosphorylase function in NAD+ metabolism. J. Biol. Chem. 2009, 284, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Mohammed, F.S.; Zhang, N.; Sauve, A.A. Dihydronicotinamide riboside is a potent NAD(+) concentration enhancer in vitro and in vivo. J. Biol. Chem. 2019, 294, 9295–9307. [Google Scholar] [CrossRef] [PubMed]
- Zapata-Perez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Canto, C.; Lopez-Leonardo, C.; McIntyre, R.L.; et al. Reduced nicotinamide mononucleotide is a new and potent NAD(+) precursor in mammalian cells and mice. FASEB J. 2021, 35, e21456. [Google Scholar] [CrossRef]
- Lerner, F.; Niere, M.; Ludwig, A.; Ziegler, M. Structural and functional characterization of human NAD kinase. Biochem. Biophys. Res. Commun. 2001, 288, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Kruger, N.J.; von Schaewen, A. The oxidative pentose phosphate pathway: Structure and organisation. Curr. Opin. Plant Biol. 2003, 6, 236–246. [Google Scholar] [CrossRef]
- Hanukoglu, I.; Rapoport, R. Routes and regulation of NADPH production in steroidogenic mitochondria. Endocr. Res. 1995, 21, 231–241. [Google Scholar] [CrossRef]
- Kawai, S.; Murata, K. Structure and function of NAD kinase and NADP phosphatase: Key enzymes that regulate the intracellular balance of NAD(H) and NADP(H). Biosci. Biotechnol. Biochem. 2008, 72, 919–930. [Google Scholar] [CrossRef]
- Aksoy, S.; Szumlanski, C.L.; Weinshilboum, R.M. Human liver nicotinamide N-methyltransferase. cDNA cloning, expression, and biochemical characterization. J. Biol. Chem. 1994, 269, 14835–14840. [Google Scholar] [CrossRef]
- Felsted, R.L.; Chaykin, S. N1-methylnicotinamide oxidation in a number of mammals. J. Biol. Chem. 1967, 242, 1274–1279. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Kale, A.; Perrone, R.; Lopez-Dominguez, J.A.; Pisco, A.O.; Kasler, H.G.; Schmidt, M.S.; Heckenbach, I.; Kwok, R.; Wiley, C.D.; et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat. Metab. 2020, 2, 1265–1283. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.C.; Mofarrahi, M.; Vassilakopoulos, T.; Maltais, F.; Sigala, I.; Debigare, R.; Bellenis, I.; Hussain, S.N. Expression and functional significance of nicotinamide N-methyl transferase in skeletal muscles of patients with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 181, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Gokarn, R.; Solon-Biet, S.M.; Cogger, V.C.; Cooney, G.J.; Wahl, D.; McMahon, A.C.; Mitchell, J.R.; Mitchell, S.J.; Hine, C.; de Cabo, R.; et al. Long-term Dietary Macronutrients and Hepatic Gene Expression in Aging Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 1618–1625. [Google Scholar] [CrossRef]
- Schmeisser, K.; Mansfeld, J.; Kuhlow, D.; Weimer, S.; Priebe, S.; Heiland, I.; Birringer, M.; Groth, M.; Segref, A.; Kanfi, Y.; et al. Role of sirtuins in lifespan regulation is linked to methylation of nicotinamide. Nat. Chem. Biol. 2013, 9, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Luongo, T.S.; Eller, J.M.; Lu, M.J.; Niere, M.; Raith, F.; Perry, C.; Bornstein, M.R.; Oliphint, P.; Wang, L.; McReynolds, M.R.; et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020, 588, 174–179. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005, 280, 36334–36341. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007, 130, 1095–1107. [Google Scholar] [CrossRef]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD(+). Science 2016, 352, 1474–1477. [Google Scholar] [CrossRef]
- Di Lisa, F.; Ziegler, M. Pathophysiological relevance of mitochondria in NAD(+) metabolism. FEBS Lett. 2001, 492, 4–8. [Google Scholar] [CrossRef]
- Tischler, M.E.; Friedrichs, D.; Coll, K.; Williamson, J.R. Pyridine nucleotide distributions and enzyme mass action ratios in hepatocytes from fed and starved rats. Arch. Biochem. Biophys. 1977, 184, 222–236. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Canto, C.; Wanders, R.J.; Auwerx, J. The secret life of NAD+: An old metabolite controlling new metabolic signaling pathways. Endocr. Rev. 2010, 31, 194–223. [Google Scholar] [CrossRef] [PubMed]
- Harden, A.; Young, W. The alcoholic ferment of yeast-juice. Proc. R. Soc. Lond. 1906, 77, 405–420. [Google Scholar]
- Warburg, O.; Christian, W. Pyridine, the hydrogen transfusing component of fermentative enzymes. Helv. Chim. Acta 1936, 19, 79–88. [Google Scholar] [CrossRef]
- Krebs, H.A.; Veech, R.L. Equilibrium relations between pyridine nucleotides and adenine nucleotides and their roles in the regulation of metabolic processes. Adv. Enzym. Regul. 1969, 7, 397–413. [Google Scholar] [CrossRef]
- Ying, W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: Regulation and biological consequences. Antioxid. Redox Signal. 2008, 10, 179–206. [Google Scholar] [CrossRef]
- Ratliff, B.B.; Abdulmahdi, W.; Pawar, R.; Wolin, M.S. Oxidant Mechanisms in Renal Injury and Disease. Antioxid. Redox. Signal. 2016, 25, 119–146. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Kaeberlein, M.; McVey, M.; Guarente, L. The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev. 1999, 13, 2570–2580. [Google Scholar] [CrossRef]
- Guarente, L. Calorie restriction and sirtuins revisited. Genes Dev. 2013, 27, 2072–2085. [Google Scholar] [CrossRef]
- Sinclair, D.A. Toward a unified theory of caloric restriction and longevity regulation. Mech. Ageing Dev. 2005, 126, 987–1002. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Pirinen, E.; Auwerx, J. Sirtuins as regulators of metabolism and healthspan. Nat. Rev. Mol. Cell Biol. 2012, 13, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Khan, S.; Wang, Y.; Charron, G.; He, B.; Sebastian, C.; Du, J.; Kim, R.; Ge, E.; Mostoslavsky, R.; et al. SIRT6 regulates TNF-alpha secretion through hydrolysis of long-chain fatty acyl lysine. Nature 2013, 496, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Anderson, R.M.; Bitterman, K.J.; Wood, J.G.; Medvedik, O.; Sinclair, D.A. Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003, 423, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Bitterman, K.J.; Anderson, R.M.; Cohen, H.Y.; Latorre-Esteves, M.; Sinclair, D.A. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J. Biol. Chem. 2002, 277, 45099–45107. [Google Scholar] [CrossRef]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef]
- Kong, L.; Wu, H.; Zhou, W.; Luo, M.; Tan, Y.; Miao, L.; Cai, L. Sirtuin 1: A Target for Kidney Diseases. Mol. Med. 2015, 21, 87–97. [Google Scholar] [CrossRef]
- Cheng, H.L.; Mostoslavsky, R.; Saito, S.; Manis, J.P.; Gu, Y.; Patel, P.; Bronson, R.; Appella, E.; Alt, F.W.; Chua, K.F. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc. Natl. Acad. Sci. USA 2003, 100, 10794–10799. [Google Scholar] [CrossRef]
- He, W.; Wang, Y.; Zhang, M.Z.; You, L.; Davis, L.S.; Fan, H.; Yang, H.C.; Fogo, A.B.; Zent, R.; Harris, R.C.; et al. Sirt1 activation protects the mouse renal medulla from oxidative injury. J. Clin. Investig. 2010, 120, 1056–1068. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Simic, P.; Sakamaki, Y.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; et al. Renal tubular Sirt1 attenuates diabetic albuminuria by epigenetically suppressing Claudin-1 overexpression in podocytes. Nat. Med. 2013, 19, 1496–1504. [Google Scholar] [CrossRef]
- Hirschey, M.D.; Shimazu, T.; Goetzman, E.; Jing, E.; Schwer, B.; Lombard, D.B.; Grueter, C.A.; Harris, C.; Biddinger, S.; Ilkayeva, O.R.; et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010, 464, 121–125. [Google Scholar] [CrossRef]
- Koyama, T.; Kume, S.; Koya, D.; Araki, S.; Isshiki, K.; Chin-Kanasaki, M.; Sugimoto, T.; Haneda, M.; Sugaya, T.; Kashiwagi, A.; et al. SIRT3 attenuates palmitate-induced ROS production and inflammation in proximal tubular cells. Free Radic. Biol. Med. 2011, 51, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Wen, P.; Luo, J.; Ding, H.; Cao, H.; He, W.; Zen, K.; Zhou, Y.; Yang, J.; Jiang, L. Sirtuin 3 regulates mitochondrial protein acetylation and metabolism in tubular epithelial cells during renal fibrosis. Cell Death Dis. 2021, 12, 847. [Google Scholar] [CrossRef] [PubMed]
- Cai, J.; Liu, Z.; Huang, X.; Shu, S.; Hu, X.; Zheng, M.; Tang, C.; Liu, Y.; Chen, G.; Sun, L.; et al. The deacetylase sirtuin 6 protects against kidney fibrosis by epigenetically blocking beta-catenin target gene expression. Kidney Int. 2020, 97, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Muraoka, H.; Hasegawa, K.; Sakamaki, Y.; Minakuchi, H.; Kawaguchi, T.; Yasuda, I.; Kanda, T.; Tokuyama, H.; Wakino, S.; Itoh, H. Role of Nampt-Sirt6 Axis in Renal Proximal Tubules in Extracellular Matrix Deposition in Diabetic Nephropathy. Cell Rep. 2019, 27, 199–212.e195. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Huang, X.Z.; Wen, D.; Zhang, M.; Xie, Q.; Ma, L.; Guan, Y.; Ren, Y.; Chen, J.; Hao, C.M. Sirt1 activation ameliorates renal fibrosis by inhibiting the TGF-β/Smad3 pathway. J. Cell Biochem. 2014, 115, 996–1005. [Google Scholar] [CrossRef]
- Li, J.; Qu, X.; Ricardo, S.D.; Bertram, J.F.; Nikolic-Paterson, D.J. Resveratrol Inhibits Renal Fibrosis in the Obstructed Kidney: Potential Role in Deacetylation of Smad3. Am. J. Pathol. 2010, 177, 1065–1071. [Google Scholar] [CrossRef]
- Xiao, Z.; Chen, C.; Meng, T.; Zhang, W.; Zhou, Q. Resveratrol attenuates renal injury and fibrosis by inhibiting transforming growth factor-β pathway on matrix metalloproteinase 7. Exp. Biol. Med. 2016, 241, 140–146. [Google Scholar] [CrossRef]
- Quan, Y.; Park, W.; Jin, J.; Kim, W.; Park, S.K.; Kang, K.P. Sirtuin 3 Activation by Honokiol Decreases Unilateral Ureteral Obstruction-Induced Renal Inflammation and Fibrosis via Regulation of Mitochondrial Dynamics and the Renal NF-κBTGF-β1/Smad Signaling Pathway. Int. J. Mol. Sci. 2020, 21, 402. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Chaudhuri, A.R.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C. The role of PARP-1 and PARP-2 enzymes in metabolic regulation and disease. Cell Metab. 2012, 16, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Mouchiroud, L.; Houtkooper, R.H.; Moullan, N.; Katsyuba, E.; Ryu, D.; Canto, C.; Mottis, A.; Jo, Y.S.; Viswanathan, M.; Schoonjans, K.; et al. The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 2013, 154, 430–441. [Google Scholar] [CrossRef] [PubMed]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Zheng, J.; Devalaraja-Narashimha, K.; Singaravelu, K.; Padanilam, B.J. Poly(ADP-ribose) polymerase-1 gene ablation protects mice from ischemic renal injury. Am. J. Physiol. Renal Physiol. 2005, 288, F387–F398. [Google Scholar] [CrossRef]
- Kim, J.; Padanilam, B.J. Loss of poly(ADP-ribose) polymerase 1 attenuates renal fibrosis and inflammation during unilateral ureteral obstruction. Am. J. Physiol. Renal Physiol. 2011, 301, F450–F459. [Google Scholar] [CrossRef]
- Shevalye, H.; Maksimchyk, Y.; Watcho, P.; Obrosova, I.G. Poly(ADP-ribose) polymerase-1 (PARP-1) gene deficiency alleviates diabetic kidney disease. Biochim. Biophys. Acta 2010, 1802, 1020–1027. [Google Scholar] [CrossRef]
- Yaku, K.; Palikhe, S.; Izumi, H.; Yoshida, T.; Hikosaka, K.; Hayat, F.; Karim, M.; Iqbal, T.; Nitta, Y.; Sato, A.; et al. BST1 regulates nicotinamide riboside metabolism via its glycohydrolase and base-exchange activities. Nat. Commun. 2021, 12, 6767. [Google Scholar] [CrossRef]
- Quarona, V.; Zaccarello, G.; Chillemi, A.; Brunetti, E.; Singh, V.K.; Ferrero, E.; Funaro, A.; Horenstein, A.L.; Malavasi, F. CD38 and CD157: A long journey from activation markers to multifunctional molecules. Cytom. Part B Clin. Cytom. 2013, 84, 207–217. [Google Scholar] [CrossRef]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef]
- Ernst, I.M.; Fliegert, R.; Guse, A.H. Adenine Dinucleotide Second Messengers and T-lymphocyte Calcium Signaling. Front. Immunol. 2013, 4, 259. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef] [PubMed]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarrago, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef] [PubMed]
- Preugschat, F.; Carter, L.H.; Boros, E.E.; Porter, D.J.; Stewart, E.L.; Shewchuk, L.M. A pre-steady state and steady state kinetic analysis of the N-ribosyl hydrolase activity of hCD157. Arch. Biochem. Biophys. 2014, 564, 156–163. [Google Scholar] [CrossRef]
- Tarrago, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018, 27, 1081–1095. [Google Scholar] [CrossRef]
- Angeletti, C.; Amici, A.; Gilley, J.; Loreto, A.; Trapanotto, A.G.; Antoniou, C.; Merlini, E.; Coleman, M.P.; Orsomando, G. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 2022, 25, 103812. [Google Scholar] [CrossRef]
- Essuman, K.; Summers, D.W.; Sasaki, Y.; Mao, X.; DiAntonio, A.; Milbrandt, J. The SARM1 Toll/Interleukin-1 Receptor Domain Possesses Intrinsic NAD(+) Cleavage Activity that Promotes Pathological Axonal Degeneration. Neuron 2017, 93, 1334–1343.e1335. [Google Scholar] [CrossRef]
- Gerdts, J.; Brace, E.J.; Sasaki, Y.; DiAntonio, A.; Milbrandt, J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science 2015, 348, 453–457. [Google Scholar] [CrossRef]
- Mink, M.; Fogelgren, B.; Olszewski, K.; Maroy, P.; Csiszar, K. A novel human gene (SARM) at chromosome 17q11 encodes a protein with a SAM motif and structural similarity to Armadillo/beta-catenin that is conserved in mouse, Drosophila, and Caenorhabditis elegans. Genomics 2001, 74, 234–244. [Google Scholar] [CrossRef]
- Elvehjem, C.A.; Madden, R.J.; Strong, F.M.; Woolley, D.W. RELATION OF NICOTINIC ACID AND NICOTINIC ACID AMIDE TO CANINE BLACK TONGUE. J. Am. Chem. Soc. 1937, 59, 1767–1768. [Google Scholar] [CrossRef]
- Covarrubias, A.J.; Perrone, R.; Grozio, A.; Verdin, E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat. Rev. Mol. Cell Biol. 2021, 22, 119–141. [Google Scholar] [CrossRef] [PubMed]
- Cercillieux, A.; Ratajczak, J.; Joffraud, M.; Sanchez-Garcia, J.L.; Jacot, G.; Zollinger, A.; Métairon, S.; Giroud-Gerbetant, J.; Rumpler, M.; Ciarlo, E.; et al. Nicotinamide riboside kinase 1 protects against diet and age-induced pancreatic β-cell failure. Mol. Metab. 2022, 66, 101605. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Tarrago, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Poyan Mehr, A.; Tran, M.T.; Ralto, K.M.; Leaf, D.E.; Washco, V.; Messmer, J.; Lerner, A.; Kher, A.; Kim, S.H.; Khoury, C.C.; et al. De novo NAD(+) biosynthetic impairment in acute kidney injury in humans. Nat. Med. 2018, 24, 1351–1359. [Google Scholar] [CrossRef] [PubMed]
- Bignon, Y.; Rinaldi, A.; Nadour, Z.; Poindessous, V.; Nemazanyy, I.; Lenoir, O.; Fohlen, B.; Weill-Raynal, P.; Hertig, A.; Karras, A.; et al. Cell stress response impairs de novo NAD+ biosynthesis in the kidney. JCI Insight 2022, 7, e153019. [Google Scholar] [CrossRef]
- Zhang, W.; Korstanje, R.; Thaisz, J.; Staedtler, F.; Harttman, N.; Xu, L.; Feng, M.; Yanas, L.; Yang, H.; Valdar, W.; et al. Genome-wide association mapping of quantitative traits in outbred mice. G3 Genes Genomes Genet. 2012, 2, 167–174. [Google Scholar] [CrossRef][Green Version]
- Korstanje, R.; Deutsch, K.; Bolanos-Palmieri, P.; Hanke, N.; Schroder, P.; Staggs, L.; Brasen, J.H.; Roberts, I.S.; Sheehan, S.; Savage, H.; et al. Loss of Kynurenine 3-Mono-oxygenase Causes Proteinuria. J. Am. Soc. Nephrol. 2016, 27, 3271–3277. [Google Scholar] [CrossRef]
- Liu, X.; Luo, D.; Huang, S.; Liu, S.; Zhang, B.; Wang, F.; Lu, J.; Chen, J.; Li, S. Impaired Nicotinamide Adenine Dinucleotide Biosynthesis in the Kidney of Chronic Kidney Disease. Front. Physiol. 2021, 12, 723690. [Google Scholar] [CrossRef]
- Schefold, J.C.; Zeden, J.P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef]
- Konan, K.V.; Taylor, M.W. Importance of the two interferon-stimulated response element (ISRE) sequences in the regulation of the human indoleamine 2,3-dioxygenase gene. J. Biol. Chem. 1996, 271, 19140–19145. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Clish, C.B.; Ghorbani, A.; Larson, M.G.; Elmariah, S.; McCabe, E.; Yang, Q.; Cheng, S.; Pierce, K.; Deik, A.; et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 2013, 24, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Goek, O.N.; Prehn, C.; Sekula, P.; Romisch-Margl, W.; Doring, A.; Gieger, C.; Heier, M.; Koenig, W.; Wang-Sattler, R.; Illig, T.; et al. Metabolites associate with kidney function decline and incident chronic kidney disease in the general population. Nephrol. Dial. Transplant. 2013, 28, 2131–2138. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Jang, H.B.; Yoo, M.G.; Park, S.I.; Lee, H.J. Amino Acid Metabolites Associated with Chronic Kidney Disease: An Eight-Year Follow-Up Korean Epidemiology Study. Biomedicines 2020, 8, 222. [Google Scholar] [CrossRef]
- Debnath, S.; Velagapudi, C.; Redus, L.; Thameem, F.; Kasinath, B.; Hura, C.E.; Lorenzo, C.; Abboud, H.E.; O’Connor, J.C. Tryptophan Metabolism in Patients With Chronic Kidney Disease Secondary to Type 2 Diabetes: Relationship to Inflammatory Markers. Int. J. Tryptophan Res. 2017, 10, 1178646917694600. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argiles, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Brandacher, G.; Cakar, F.; Winkler, C.; Schneeberger, S.; Obrist, P.; Bösmüller, C.; Werner-Felmayer, G.; Werner, E.R.; Bonatti, H.; Margreiter, R.; et al. Non-invasive monitoring of kidney allograft rejection through IDO metabolism evaluation. Kidney Int. 2007, 71, 60–67. [Google Scholar] [CrossRef]
- Tran, M.T.; Zsengeller, Z.K.; Berg, A.H.; Khankin, E.V.; Bhasin, M.K.; Kim, W.; Clish, C.B.; Stillman, I.E.; Karumanchi, S.A.; Rhee, E.P.; et al. PGC1alpha drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 2016, 531, 528–532. [Google Scholar] [CrossRef]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef]
- Yoshida, M.; Satoh, A.; Lin, J.B.; Mills, K.F.; Sasaki, Y.; Rensing, N.; Wong, M.; Apte, R.S.; Imai, S.I. Extracellular Vesicle-Contained eNAMPT Delays Aging and Extends Lifespan in Mice. Cell Metab. 2019, 30, 329–342.e325. [Google Scholar] [CrossRef]
- Ugur, S.; Ulu, R.; Dogukan, A.; Gurel, A.; Yigit, I.P.; Gozel, N.; Aygen, B.; Ilhan, N. The renoprotective effect of curcumin in cisplatin-induced nephrotoxicity. Ren. Fail. 2015, 37, 332–336. [Google Scholar] [CrossRef]
- Morigi, M.; Perico, L.; Rota, C.; Longaretti, L.; Conti, S.; Rottoli, D.; Novelli, R.; Remuzzi, G.; Benigni, A. Sirtuin 3-dependent mitochondrial dynamic improvements protect against acute kidney injury. J. Clin. Investig. 2015, 125, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Imai, S.; Yoshino, J. The importance of NAMPT/NAD/SIRT1 in the systemic regulation of metabolism and ageing. Diabetes Obes. Metab. 2013, 15 (Suppl. S3), 26–33. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Song, H.K.; Lee, M.H.; Ko, G.J.; Han, J.Y.; Han, S.Y.; Han, K.H.; Kim, H.K.; Cha, D.R. Visfatin is upregulated in type-2 diabetic rats and targets renal cells. Kidney Int. 2010, 78, 170–181. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liang, Y.; Hu, T.; Wei, R.; Cai, C.; Wang, P.; Wang, L.; Qiao, W.; Feng, L. Endogenous Nampt upregulation is associated with diabetic nephropathy inflammatory-fibrosis through the NF-κB p65 and Sirt1 pathway; NMN alleviates diabetic nephropathy inflammatory-fibrosis by inhibiting endogenous Nampt. Exp. Ther. Med. 2017, 14, 4181–4193. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Benito-Martin, A.; Ucero, A.C.; Izquierdo, M.C.; Santamaria, B.; Picatoste, B.; Carrasco, S.; Lorenzo, O.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A. Endogenous NAMPT dampens chemokine expression and apoptotic responses in stressed tubular cells. Biochim. Biophys. Acta 2014, 1842, 293–303. [Google Scholar] [CrossRef]
- Song, H.K.; Lee, M.H.; Kim, B.K.; Park, Y.G.; Ko, G.J.; Kang, Y.S.; Han, J.Y.; Han, S.Y.; Han, K.H.; Kim, H.K.; et al. Visfatin: A new player in mesangial cell physiology and diabetic nephropathy. Am. J. Physiol. Renal Physiol. 2008, 295, F1485–F1494. [Google Scholar] [CrossRef]
- Rutkowski, B.; Slominska, E.; Szolkiewicz, M.; Smolenski, R.T.; Striley, C.; Rutkowski, P.; Swierczynski, J. N-methyl-2-pyridone-5-carboxamide: A novel uremic toxin? Kidney Int. 2003, 63, S19–S21. [Google Scholar] [CrossRef]
- Altmeyer, M.; Hottiger, M.O. Poly(ADP-ribose) polymerase 1 at the crossroad of metabolic stress and inflammation in aging. Aging 2009, 1, 458–469. [Google Scholar] [CrossRef]
- Martin, D.R.; Lewington, A.J.; Hammerman, M.R.; Padanilam, B.J. Inhibition of poly(ADP-ribose) polymerase attenuates ischemic renal injury in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2000, 279, R1834–R1840. [Google Scholar] [CrossRef]
- O’Valle, F.; Del Moral, R.G.; Benitez Mdel, C.; Martin-Oliva, D.; Gomez-Morales, M.; Aguilar, D.; Aneiros-Fernandez, J.; Hernandez-Cortes, P.; Osuna, A.; Moreso, F.; et al. Poly[ADP-ribose] polymerase-1 expression is related to cold ischemia, acute tubular necrosis, and delayed renal function in kidney transplantation. PLoS ONE 2009, 4, e7138. [Google Scholar] [CrossRef] [PubMed]
- O’Valle, F.; Gomez-Morales, M.; Del Moral, R.M.; Seron, D.; Moreso, F.; Osuna, A.; Oliver, F.J.; Del Moral, R.G. Poly(ADP-ribose) polymerase expression in kidney transplantation: From alfa (alpha) to Omega (Omega). Transplant. Proc. 2007, 39, 2099–2101. [Google Scholar] [CrossRef]
- Ogura, Y.; Kitada, M.; Monno, I.; Kanasaki, K.; Watanabe, A.; Koya, D. Renal mitochondrial oxidative stress is enhanced by the reduction of Sirt3 activity, in Zucker diabetic fatty rats. Redox. Rep. 2018, 23, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ogura, Y.; Kitada, M.; Xu, J.; Monno, I.; Koya, D. CD38 inhibition by apigenin ameliorates mitochondrial oxidative stress through restoration of the intracellular NAD(+)/NADH ratio and Sirt3 activity in renal tubular cells in diabetic rats. Aging 2020, 12, 11325–11336. [Google Scholar] [CrossRef] [PubMed]
- Shu, B.; Feng, Y.; Gui, Y.; Lu, Q.; Wei, W.; Xue, X.; Sun, X.; He, W.; Yang, J.; Dai, C. Blockade of CD38 diminishes lipopolysaccharide-induced macrophage classical activation and acute kidney injury involving NF-kappaB signaling suppression. Cell Signal. 2018, 42, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Baur, J.A.; Imai, S.I. NAD(+) Intermediates: The Biology and Therapeutic Potential of NMN and NR. Cell Metab. 2018, 27, 513–528. [Google Scholar] [CrossRef]
- Ralto, K.M.; Rhee, E.P.; Parikh, S.M. NAD(+) homeostasis in renal health and disease. Nat. Rev. Nephrol. 2020, 16, 99–111. [Google Scholar] [CrossRef]
- Wee, H.N.; Liu, J.J.; Ching, J.; Kovalik, J.P.; Lim, S.C. The Kynurenine Pathway in Acute Kidney Injury and Chronic Kidney Disease. Am. J. Nephrol. 2021, 52, 771–787. [Google Scholar] [CrossRef]
- Fukuwatari, T.; Shibata, K. Nutritional aspect of tryptophan metabolism. Int. J. Tryptophan Res. 2013, 6 (Suppl. S1), 3–8. [Google Scholar] [CrossRef]
- Gazzaniga, F.; Stebbins, R.; Chang, S.Z.; McPeek, M.A.; Brenner, C. Microbial NAD metabolism: Lessons from comparative genomics. Microbiol. Mol. Biol. Rev. 2009, 73, 529–541. [Google Scholar] [CrossRef]
- Shats, I.; Williams, J.G.; Liu, J.; Makarov, M.V.; Wu, X.; Lih, F.B.; Deterding, L.J.; Lim, C.; Xu, X.; Randall, T.A.; et al. Bacteria Boost Mammalian Host NAD Metabolism by Engaging the Deamidated Biosynthesis Pathway. Cell Metab. 2020, 31, 564–579.e7. [Google Scholar] [CrossRef] [PubMed]
- Hamzaoui, M.; Djerada, Z.; Brunel, V.; Mulder, P.; Richard, V.; Bellien, J.; Guerrot, D. 5/6 nephrectomy induces different renal, cardiac and vascular consequences in 129/Sv and C57BL/6JRJ mice. Sci. Rep. 2020, 10, 1524. [Google Scholar] [CrossRef] [PubMed]
- Cho, K.H.; Kim, H.J.; Rodriguez-Iturbe, B.; Vaziri, N.D. Niacin ameliorates oxidative stress, inflammation, proteinuria, and hypertension in rats with chronic renal failure. Am. J. Physiol. Renal Physiol. 2009, 297, F106–F113. [Google Scholar] [CrossRef]
- Cho, K.H.; Kim, H.J.; Kamanna, V.S.; Vaziri, N.D. Niacin improves renal lipid metabolism and slows progression in chronic kidney disease. Biochim. Biophys. Acta 2010, 1800, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Chevalier, R.L.; Forbes, M.S.; Thornhill, B.A. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009, 75, 1145–1152. [Google Scholar] [CrossRef]
- Zheng, M.; Cai, J.; Liu, Z.; Shu, S.; Wang, Y.; Tang, C.; Dong, Z. Nicotinamide reduces renal interstitial fibrosis by suppressing tubular injury and inflammation. J. Cell Mol. Med. 2019, 23, 3995–4004. [Google Scholar] [CrossRef]
- Zhen, X.; Zhang, S.; Xie, F.; Zhou, M.; Hu, Z.; Zhu, F.; Nie, J. Nicotinamide Supplementation Attenuates Renal Interstitial Fibrosis via Boosting the Activity of Sirtuins. Kidney Dis. 2021, 7, 186–199. [Google Scholar] [CrossRef]
- Basile, D.P.; Bonventre, J.V.; Mehta, R.; Nangaku, M.; Unwin, R.; Rosner, M.H.; Kellum, J.A.; Ronco, C.; Group, A.X.W. Progression after AKI: Understanding Maladaptive Repair Processes to Predict and Identify Therapeutic Treatments. J. Am. Soc. Nephrol. 2016, 27, 687–697. [Google Scholar] [CrossRef]
- Jia, Y.; Kang, X.; Tan, L.; Ren, Y.; Qu, L.; Tang, J.; Liu, G.; Wang, S.; Xiong, Z.; Yang, L. Nicotinamide Mononucleotide Attenuates Renal Interstitial Fibrosis After AKI by Suppressing Tubular DNA Damage and Senescence. Front. Physiol. 2021, 12, 649547. [Google Scholar] [CrossRef]
- Morevati, M.; Egstrand, S.; Nordholm, A.; Mace, M.L.; Andersen, C.B.; Salmani, R.; Olgaard, K.; Lewin, E. Effect of NAD+ boosting on kidney ischemia-reperfusion injury. PLoS ONE 2021, 16, e0252554. [Google Scholar] [CrossRef]
- Baudoux, T.; Jadot, I.; Decleves, A.E.; Antoine, M.H.; Colet, J.M.; Botton, O.; De Prez, E.; Pozdzik, A.; Husson, C.; Caron, N.; et al. Experimental Aristolochic Acid Nephropathy: A Relevant Model to Study AKI-to-CKD Transition. Front. Med. 2022, 9, 822870. [Google Scholar] [CrossRef] [PubMed]
- Diwan, V.; Brown, L.; Gobe, G.C. Adenine-induced chronic kidney disease in rats. Nephrology 2018, 23, 5–11. [Google Scholar] [CrossRef]
- Kumakura, S.; Sato, E.; Sekimoto, A.; Hashizume, Y.; Yamakage, S.; Miyazaki, M.; Ito, S.; Harigae, H.; Takahashi, N. Nicotinamide Attenuates the Progression of Renal Failure in a Mouse Model of Adenine-Induced Chronic Kidney Disease. Toxins 2021, 13, 50. [Google Scholar] [CrossRef]
- Eto, N.; Miyata, Y.; Ohno, H.; Yamashita, T. Nicotinamide prevents the development of hyperphosphataemia by suppressing intestinal sodium-dependent phosphate transporter in rats with adenine-induced renal failure. Nephrol. Dial. Transplant. 2005, 20, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, J.M.; Wang, Z.V.; Park, A.S.; Zhang, J.; Zhang, D.; Hu, M.C.; Moe, O.W.; Susztak, K.; Scherer, P.E. Adiponectin promotes functional recovery after podocyte ablation. J. Am. Soc. Nephrol. 2013, 24, 268–282. [Google Scholar] [CrossRef] [PubMed]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef]
- Yasuda, I.; Hasegawa, K.; Sakamaki, Y.; Muraoka, H.; Kawaguchi, T.; Kusahana, E.; Ono, T.; Kanda, T.; Tokuyama, H.; Wakino, S.; et al. Pre-emptive Short-term Nicotinamide Mononucleotide Treatment in a Mouse Model of Diabetic Nephropathy. J. Am. Soc. Nephrol. 2021, 32, 1355–1370. [Google Scholar] [CrossRef]
- Myakala, K.; Wang, X.X.; Jones, B.A.; Hirschey, M.D.; Yang, X.; Rosenberg, A.Z.; Ginley, B.; Sarder, P.; Brodsky, L.; Jang, Y.; et al. NAD metabolism modulates mitochondrial function and inflammation and prevents progression of diabetic kidney disease. bioRxiv 2021. [Google Scholar] [CrossRef]
- Katai, K.; Tanaka, H.; Tatsumi, S.; Fukunaga, Y.; Genjida, K.; Morita, K.; Kuboyama, N.; Suzuki, T.; Akiba, T.; Miyamoto, K.; et al. Nicotinamide inhibits sodium-dependent phosphate cotransport activity in rat small intestine. Nephrol. Dial. Transplant. 1999, 14, 1195–1201. [Google Scholar] [CrossRef]
- Wu, K.I.; Bacon, R.A.; Al-Mahrouq, H.A.; Kempson, S.A. Nicotinamide as a rapid-acting inhibitor of renal brush-border phosphate transport. Am. J. Physiol. 1988, 255, F15–F21. [Google Scholar] [CrossRef]
- Papadimitriou, A.; Silva, K.C.; Peixoto, E.B.; Borges, C.M.; Lopes de Faria, J.M.; Lopes de Faria, J.B. Theobromine increases NAD(+)/Sirt-1 activity and protects the kidney under diabetic conditions. Am. J. Physiol. Renal Physiol. 2015, 308, F209–F225. [Google Scholar] [CrossRef] [PubMed]
- Birjmohun, R.S.; Hutten, B.A.; Kastelein, J.J.; Stroes, E.S. Efficacy and safety of high-density lipoprotein cholesterol-increasing compounds: A meta-analysis of randomized controlled trials. J. Am. Coll. Cardiol. 2005, 45, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; Sidhu, M.S.; Toth, P.P. The therapeutic role of niacin in dyslipidemia management. J. Cardiovasc. Pharmacol. Ther. 2014, 19, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Poynten, A.M.; Gan, S.K.; Kriketos, A.D.; O’Sullivan, A.; Kelly, J.J.; Ellis, B.A.; Chisholm, D.J.; Campbell, L.V. Nicotinic acid-induced insulin resistance is related to increased circulating fatty acids and fat oxidation but not muscle lipid content. Metabolism 2003, 52, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Capuzzi, D.M.; Guyton, J.R.; Morgan, J.M.; Goldberg, A.C.; Kreisberg, R.A.; Brusco, O.A.; Brody, J. Efficacy and safety of an extended-release niacin (Niaspan): A long-term study. Am. J. Cardiol. 1998, 82, 74U–81U; discussion 85U–86U. [Google Scholar] [CrossRef]
- Wise, A.; Foord, S.M.; Fraser, N.J.; Barnes, A.A.; Elshourbagy, N.; Eilert, M.; Ignar, D.M.; Murdock, P.R.; Steplewski, K.; Green, A.; et al. Molecular identification of high and low affinity receptors for nicotinic acid. J. Biol. Chem. 2003, 278, 9869–9874. [Google Scholar] [CrossRef]
- Benyo, Z.; Gille, A.; Bennett, C.L.; Clausen, B.E.; Offermanns, S. Nicotinic acid-induced flushing is mediated by activation of epidermal langerhans cells. Mol. Pharmacol. 2006, 70, 1844–1849. [Google Scholar] [CrossRef]
- Investigators, A.-H.; Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Raikou, V.D. Serum phosphate and chronic kidney and cardiovascular disease: Phosphorus potential implications in general population. World J. Nephrol. 2021, 10, 76–87. [Google Scholar] [CrossRef]
- Sabbagh, Y.; O’Brien, S.P.; Song, W.; Boulanger, J.H.; Stockmann, A.; Arbeeny, C.; Schiavi, S.C. Intestinal npt2b plays a major role in phosphate absorption and homeostasis. J. Am. Soc. Nephrol. 2009, 20, 2348–2358. [Google Scholar] [CrossRef] [PubMed]
- Ix, J.H.; Isakova, T.; Larive, B.; Raphael, K.L.; Raj, D.S.; Cheung, A.K.; Sprague, S.M.; Fried, L.F.; Gassman, J.J.; Middleton, J.P.; et al. Effects of Nicotinamide and Lanthanum Carbonate on Serum Phosphate and Fibroblast Growth Factor-23 in CKD: The COMBINE Trial. J. Am. Soc. Nephrol. 2019, 30, 1096–1108. [Google Scholar] [CrossRef] [PubMed]
- Zahed, N.S.; Zamanifar, N.; Nikbakht, H. Effect of low dose nicotinic acid on hyperphosphatemia in patients with end stage renal disease. Indian J. Nephrol. 2016, 26, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Ketteler, M.; Wiecek, A.; Rosenkranz, A.R.; Pasch, A.; Rekowski, J.; Hellmann, B.; Karus, M.; Ammer, R. Efficacy and Safety of a Novel Nicotinamide Modified-Release Formulation in the Treatment of Refractory Hyperphosphatemia in Patients Receiving Hemodialysis-A Randomized Clinical Trial. Kidney Int. Rep. 2021, 6, 594–604. [Google Scholar] [CrossRef]
- Lenglet, A.; Liabeuf, S.; Esper, N.E.; Brisset, S.; Mansour, J.; Lemaire-Hurtel, A.S.; Mary, A.; Brazier, M.; Kamel, S.; Mentaverri, R.; et al. Efficacy and safety of nicotinamide in haemodialysis patients: The NICOREN study. Nephrol. Dial. Transplant. 2017, 32, 1597. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Ma, T.; Zhang, P. Efficacy and safety of nicotinamide on phosphorus metabolism in hemodialysis patients: A systematic review and meta-analysis. Medicine 2018, 97, e12731. [Google Scholar] [CrossRef]
- Restrepo Valencia, C.A.; Cruz, J. Safety and effectiveness of nicotinic acid in the management of patients with chronic renal disease and hyperlipidemia associated to hyperphosphatemia. Nefrologia 2008, 28, 61–66. [Google Scholar]
- Shimoda, K.; Akiba, T.; Matsushima, T.; Rai, T.; Abe, K.; Hoshino, M. Niceritrol decreases serum phosphate levels in chronic hemodialysis patients. Nihon Jinzo Gakkai Shi 1998, 40, 1–7. [Google Scholar]
- El Ters, M.; Zhou, X.; Lepping, R.J.; Lu, P.; Karcher, R.T.; Mahnken, J.D.; Brooks, W.M.; Winklhofer, F.T.; Li, X.; Yu, A.S.L. Biological Efficacy and Safety of Niacinamide in Patients With ADPKD. Kidney Int. Rep. 2020, 5, 1271–1279. [Google Scholar] [CrossRef]
- Jin Kang, H.; Kim, D.K.; Mi Lee, S.; Han Kim, K.; Hee Han, S.; Hyun Kim, K.; Eun Kim, S.; Ki Son, Y.; An, W.S. Effects of low-dose niacin on dyslipidemia and serum phosphorus in patients with chronic kidney disease. Kidney Res. Clin. Pract. 2013, 32, 21–26. [Google Scholar] [CrossRef]
- Zhou, X.; Fan, L.X.; Sweeney, W.E., Jr.; Denu, J.M.; Avner, E.D.; Li, X. Sirtuin 1 inhibition delays cyst formation in autosomal-dominant polycystic kidney disease. J. Clin. Investig. 2013, 123, 3084–3098. [Google Scholar] [CrossRef]
- Canto, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012, 15, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Airhart, S.E.; Shireman, L.M.; Risler, L.J.; Anderson, G.D.; Nagana Gowda, G.A.; Raftery, D.; Tian, R.; Shen, D.D.; O’Brien, K.D. An open-label, non-randomized study of the pharmacokinetics of the nutritional supplement nicotinamide riboside (NR) and its effects on blood NAD+ levels in healthy volunteers. PLoS ONE 2017, 12, e0186459. [Google Scholar] [CrossRef] [PubMed]
- Conze, D.; Brenner, C.; Kruger, C.L. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci. Rep. 2019, 9, 9772. [Google Scholar] [CrossRef] [PubMed]
- Dellinger, R.W.; Santos, S.R.; Morris, M.; Evans, M.; Alminana, D.; Guarente, L.; Marcotulli, E. Repeat dose NRPT (nicotinamide riboside and pterostilbene) increases NAD(+) levels in humans safely and sustainably: A randomized, double-blind, placebo-controlled study. NPJ Aging Mech. Dis. 2017, 3, 17. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic nicotinamide riboside supplementation is well-tolerated and elevates NAD(+) in healthy middle-aged and older adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Trammell, S.A.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 12948. [Google Scholar] [CrossRef]
- Brakedal, B.; Dolle, C.; Riemer, F.; Ma, Y.; Nido, G.S.; Skeie, G.O.; Craven, A.R.; Schwarzlmuller, T.; Brekke, N.; Diab, J.; et al. The NADPARK study: A randomized phase I trial of nicotinamide riboside supplementation in Parkinson’s disease. Cell Metab. 2022, 34, 396–407.e396. [Google Scholar] [CrossRef]
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.J.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.I.; et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 2021, 372, 1224–1229. [Google Scholar] [CrossRef]
- Pirinen, E.; Auranen, M.; Khan, N.A.; Brilhante, V.; Urho, N.; Pessia, A.; Hakkarainen, A.; Ulla Heinonen, J.K.; Schmidt, M.S.; Haimilahti, K.; et al. Niacin Cures Systemic NAD(+) Deficiency and Improves Muscle Performance in Adult-Onset Mitochondrial Myopathy. Cell Metab. 2020, 32, 144. [Google Scholar] [CrossRef]
- Maccubbin, D.; Tipping, D.; Kuznetsova, O.; Hanlon, W.A.; Bostom, A.G. Hypophosphatemic effect of niacin in patients without renal failure: A randomized trial. Clin. J. Am. Soc. Nephrol. 2010, 5, 582–589. [Google Scholar] [CrossRef]
- Bostom, A.G.; Maclean, A.A.; Maccubbin, D.; Tipping, D.; Giezek, H.; Hanlon, W.A. Extended-release niacin/laropiprant lowers serum phosphorus concentrations in patients with type 2 diabetes. J. Clin. Lipidol. 2011, 5, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Edalat-Nejad, M.; Zameni, F.; Talaiei, A. The effect of niacin on serum phosphorus levels in dialysis patients. Indian J. Nephrol. 2012, 22, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Aramwit, P.; Srisawadwong, R.; Supasyndh, O. Effectiveness and safety of extended-release nicotinic acid for reducing serum phosphorus in hemodialysis patients. J. Nephrol. 2012, 25, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, R.; Katz, R.; Hoofnagle, A.; Bostom, A.; Rifkin, D.E.; McBride, R.; Probstfield, J.; Block, G.; Ix, J.H. The Effect of Extended Release Niacin on Markers of Mineral Metabolism in CKD. Clin. J. Am. Soc. Nephrol. 2018, 13, 36–44. [Google Scholar] [CrossRef]
- Rao, M.; Steffes, M.; Bostom, A.; Ix, J.H. Effect of niacin on FGF23 concentration in chronic kidney disease. Am. J. Nephrol. 2014, 39, 484–490. [Google Scholar] [CrossRef]
- Ahmed, H.M.; Yossif, E.; Abd-Elkader, A.S.; Aziz, E.M.A. The efficacy and safety of niacin on hyperphosphatemia in ESRD patients undergoing hemodialysis: Randomized controlled trial. Egypt. J. Intern. Med. 2022, 34, 1–11. [Google Scholar] [CrossRef]
- Takahashi, Y.; Tanaka, A.; Nakamura, T.; Fukuwatari, T.; Shibata, K.; Shimada, N.; Ebihara, I.; Koide, H. Nicotinamide suppresses hyperphosphatemia in hemodialysis patients. Kidney Int. 2004, 65, 1099–1104. [Google Scholar] [CrossRef]
- Young, D.O.; Cheng, S.C.; Delmez, J.A.; Coyne, D.W. The effect of oral niacinamide on plasma phosphorus levels in peritoneal dialysis patients. Perit. Dial. Int. 2009, 29, 562–567. [Google Scholar] [CrossRef]
- Cheng, S.C.; Young, D.O.; Huang, Y.; Delmez, J.A.; Coyne, D.W. A randomized, double-blind, placebo-controlled trial of niacinamide for reduction of phosphorus in hemodialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 1131–1138. [Google Scholar] [CrossRef]
- Shahbazian, H.; Zafar Mohtashami, A.; Ghorbani, A.; Abbaspour, M.R.; Belladi Musavi, S.S.; Hayati, F.; Lashkarara, G.R. Oral nicotinamide reduces serum phosphorus, increases HDL, and induces thrombocytopenia in hemodialysis patients: A double-blind randomized clinical trial. Nefrologia 2011, 31, 58–65. [Google Scholar] [CrossRef]
- El Borolossy, R.; El Wakeel, L.M.; El Hakim, I.; Sabri, N. Efficacy and safety of nicotinamide in the management of hyperphosphatemia in pediatric patients on regular hemodialysis. Pediatr. Nephrol. 2016, 31, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Boulware, L.E.; Dember, L.M.; Freedman, B.I.; Furth, S.L.; Holzman, L.B.; Ketchum, C.J.; Little, M.H.; Mehrotra, R.; Moe, S.M.; et al. The kidney research national dialogue: Gearing up to move forward. Clin. J. Am. Soc. Nephrol. 2014, 9, 1806–1811. [Google Scholar] [CrossRef]
- Neelakantan, H.; Vance, V.; Wetzel, M.D.; Wang, H.L.; McHardy, S.F.; Finnerty, C.C.; Hommel, J.D.; Watowich, S.J. Selective and membrane-permeable small molecule inhibitors of nicotinamide N-methyltransferase reverse high fat diet-induced obesity in mice. Biochem. Pharmacol. 2018, 147, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Gardell, S.J.; Hopf, M.; Khan, A.; Dispagna, M.; Hampton Sessions, E.; Falter, R.; Kapoor, N.; Brooks, J.; Culver, J.; Petucci, C.; et al. Boosting NAD(+) with a small molecule that activates NAMPT. Nat. Commun. 2019, 10, 3241. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Han, T.; Nijhawan, D.; Theodoropoulos, P.; Naidoo, J.; Yadavalli, S.; Mirzaei, H.; Pieper, A.A.; Ready, J.M.; McKnight, S.L. P7C3 neuroprotective chemicals function by activating the rate-limiting enzyme in NAD salvage. Cell 2014, 158, 1324–1334. [Google Scholar] [CrossRef]
- Katsyuba, E.; Mottis, A.; Zietak, M.; De Franco, F.; van der Velpen, V.; Gariani, K.; Ryu, D.; Cialabrini, L.; Matilainen, O.; Liscio, P.; et al. De novo NAD(+) synthesis enhances mitochondrial function and improves health. Nature 2018, 563, 354–359. [Google Scholar] [CrossRef]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Renal Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef]
- Lynch, M.R.; Tran, M.T.; Ralto, K.M.; Zsengeller, Z.K.; Raman, V.; Bhasin, S.S.; Sun, N.; Chen, X.; Brown, D.; Rovira, I.I.; et al. TFEB-driven lysosomal biogenesis is pivotal for PGC1alpha-dependent renal stress resistance. JCI Insight 2019, 5. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Lautrup, S.; Jensen, M.B.; Yang, B.; SenGupta, T.; Caponio, D.; Khezri, R.; Demarest, T.G.; Aman, Y.; et al. NAD(+) augmentation restores mitophagy and limits accelerated aging in Werner syndrome. Nat. Commun. 2019, 10, 5284. [Google Scholar] [CrossRef]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-beta proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Romani, M.; Sorrentino, V.; Oh, C.M.; Li, H.; de Lima, T.I.; Zhang, H.; Shong, M.; Auwerx, J. NAD(+) boosting reduces age-associated amyloidosis and restores mitochondrial homeostasis in muscle. Cell Rep. 2021, 34, 108660. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Dan, X.; Hou, Y.; Lee, J.H.; Wechter, N.; Krishnamurthy, S.; Kimura, R.; Babbar, M.; Demarest, T.; McDevitt, R.; et al. NAD(+) supplementation prevents STING-induced senescence in ataxia telangiectasia by improving mitophagy. Aging Cell 2021, 20, e13329. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, K.J.; O’Sullivan, E.D.; Humphries, D.; Baird, D.P.; Docherty, M.H.; Neely, S.A.; Krimpenfort, P.J.; Melk, A.; Schmitt, R.; Ferreira-Gonzalez, S.; et al. Cellular senescence inhibits renal regeneration after injury in mice, with senolytic treatment promoting repair. Sci. Transl. Med. 2021, 13. [Google Scholar] [CrossRef] [PubMed]


| Preclinical Model | Description | Phenotype | Interventions & Outcomes | |||
|---|---|---|---|---|---|---|
| NA | NAM | NMN | NR | |||
| 5/6 nephrectomy (5/6 Nx) | 2/3 of a kidney is excised and the second kidney is removed | Model of progressive renal failure after loss of renal mass: persistent decrease in GFR, proteinuria, glomerular sclerosis and hypertension [162] | ↘ Hypertension, proteinuria, tubular injury, inflammation markers and lipid accumulation [163,164] | - | - | - |
| Unilateral Ureter Obstruction (UUO) | Complete ureteral obstruction | Tubulointerstitial fibrosis: interstitial inflammation, tubular dilation, tubular atrophy, macrophage infiltration and extracellular matrix deposition [165] | - | ↘ Fibrosis, tubular atrophy, apoptosis, immune cell infiltration, tubular injury and inflammation markers [166,167] ↘ Acetylated levels of Smad 3 and p53 [167] | - | No effect on interstitial fibrosis [29] |
| Ischemia-Reperfusion Injury (IRI) | Clamping of one or both renal artery and vein for a variable length of time | After 2–9 weeks: AKI-to-CKD transition: renal hypoxia and maladaptive repair leading to interstitial fibrosis [168] | - | - | ↘ Fibrosis, PTECs senescence, DNA damage and cytokine expression [169] | ↗ Kidney NADH No effect on plasma creatinine, plasma phosphate, tubular damage, profibrotic genes expression [170] |
| Aristolochic acid (AA) nephropathy | i.p. injection of AA for a variable number of days | After 5–20 days: AKI-to-CKD transition: progressive tubular atrophy and interstitial fibrosis [171] | - | ↘ Tubular injury, NGAL levels and cytokine expression (Il-1β and Il-6) [167] | - | - |
| Adenine-induced CKD | Adenine supplementation (0.25–0.75%) in the diet for 3–6 weeks | Progressive kidney damage and cardiovascular disease: tubular and glomerular damage, tubulointerstitial fibrosis, inflammation, oxidative stress, loss of renal function, hyperphosphatemia, crystal formation and vessel calcification [172] | - | Prophylactic administration: ↗ Kidney NAD+, plasma NAM ↘ Progression of plasma creatinine, BUN and indoxyl sulfate, tubular injury, fibrosis and inflammation markers, accumulation of glycolysis and Krebs cycle metabolites [173] ↘ Serum phosphate, NaPi-2b expression, elevations in BUN and serum creatinine [174] | - | - |
| POD-ATTAC | Induction of apoptosis in podocytes upon injection of a construct-specific agent | Proteinuria, foot process effacement, mesangial expansion, glomerulosclerosis and tubulointerstitial damage [175] | - | - | - | ↗ Kidney NAD+ No effect on GFR, plasma BUN and creatinine, and interstitial fibrosis [29] |
| db/db | Deletion mutation in the leptin receptor (LepRdb/db) | Type 2 diabetes model: obesity, hyperlipidemia, hyperinsulinemia and insulin resistance Albuminuria, glomerular basement thickening, podocyte loss, oxidative stress and inflammation [176] | - | No effect on albuminuria and HbA1c levels [177] | ↗ Kidney NAD+, SIRT1, synaptopodin, NAMPT and NMNAT1 expression ↘ Albuminuria, fibrosis, foot process effacement, Claudin-1 expression [177] | ↗ Kidney NAD+, SIRT3 ↘ Albuminuria, fibrosis, tubular injury, inflammation and oxidative stress markers, and cGAS-STING activation [178] |
| Streptotozin (STZ)-induced diabetes | i.p. injection of STZ for a variable number of days | Type 1 diabetes model: absolute insulin deficiency Albuminuria, glomerular hypertrophy, glomerular basement thickening, oxidative stress, inflammation and tubulointerstitial fibrosis [176] | - | - | ↗ Sirt1 ↘ Nampt, NF-KB p65, Vimentin [145] | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chanvillard, L.; Tammaro, A.; Sorrentino, V. NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease. Cells 2023, 12, 21. https://doi.org/10.3390/cells12010021
Chanvillard L, Tammaro A, Sorrentino V. NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease. Cells. 2023; 12(1):21. https://doi.org/10.3390/cells12010021
Chicago/Turabian StyleChanvillard, Lucie, Alessandra Tammaro, and Vincenzo Sorrentino. 2023. "NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease" Cells 12, no. 1: 21. https://doi.org/10.3390/cells12010021
APA StyleChanvillard, L., Tammaro, A., & Sorrentino, V. (2023). NAD+ Metabolism and Interventions in Premature Renal Aging and Chronic Kidney Disease. Cells, 12(1), 21. https://doi.org/10.3390/cells12010021