
Role of Liver CD38 in the Regulation of Metabolic Pathways during Cold-Induced Thermogenesis in Mice


 ,
,  , , and
, , and 
Abstract
1. Introduction
2. Materials and Methods
2.1. In Vivo Experiments
2.2. Measurement of NAD(P)(H) Content
2.3. Enzymatic Assays
2.3.1. NAD+-Ase
2.3.2. NAD+ Synthesis
2.3.3. NMNAT Activity
2.3.4. NAD+ Kinase Activity
2.3.5. LDH Activity
2.3.6. Glucose-6 Phosphate Dehydrogenase Activity
2.4. Glucose-6 Phosphate Content
2.5. Western Blot Analysis
2.6. Glycogen
2.7. Bile Acids, Cholesterol, Triglyceride and Glycerol Measurements
2.8. qPCR Analyses
2.9. Statistical Analyses
3. Results
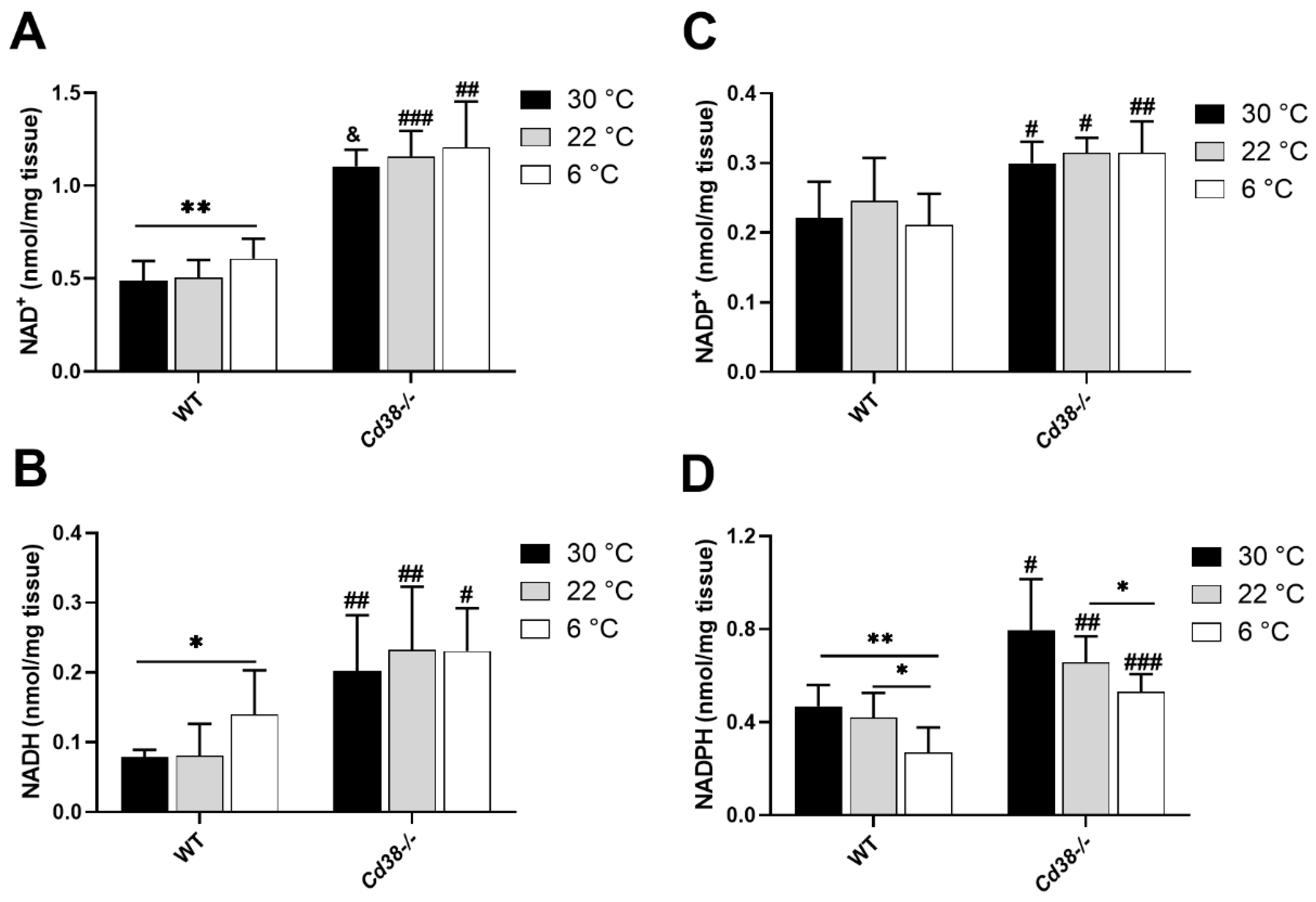
3.1. NAD(H) and NADP(H) Levels Are Modified in Liver during Cold Exposure
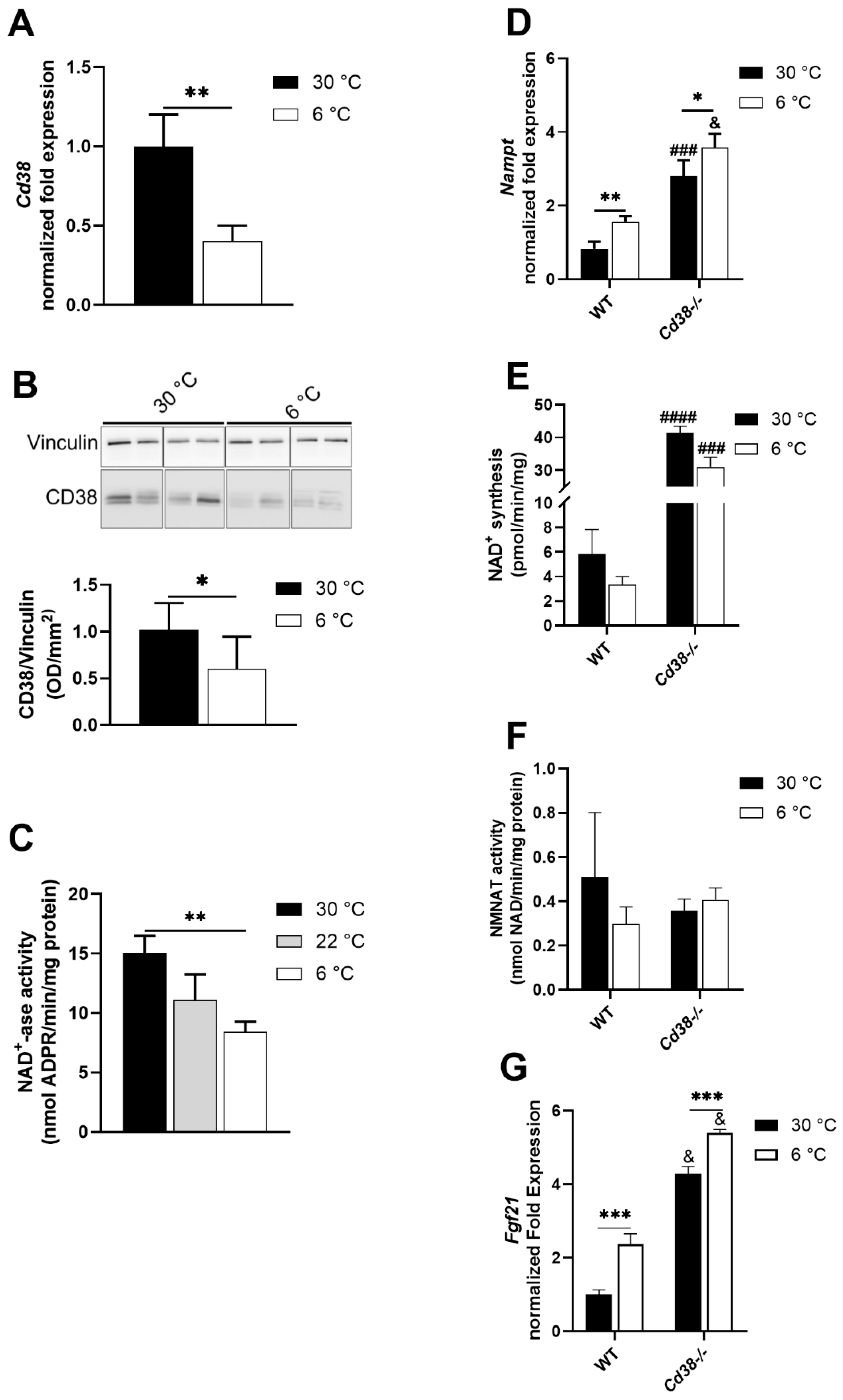
3.2. CD38 Expression Is Downregulated in Liver upon Cold Exposure
3.3. NAMPT and FGF21 Are Upregulated in Liver upon Cold Exposure
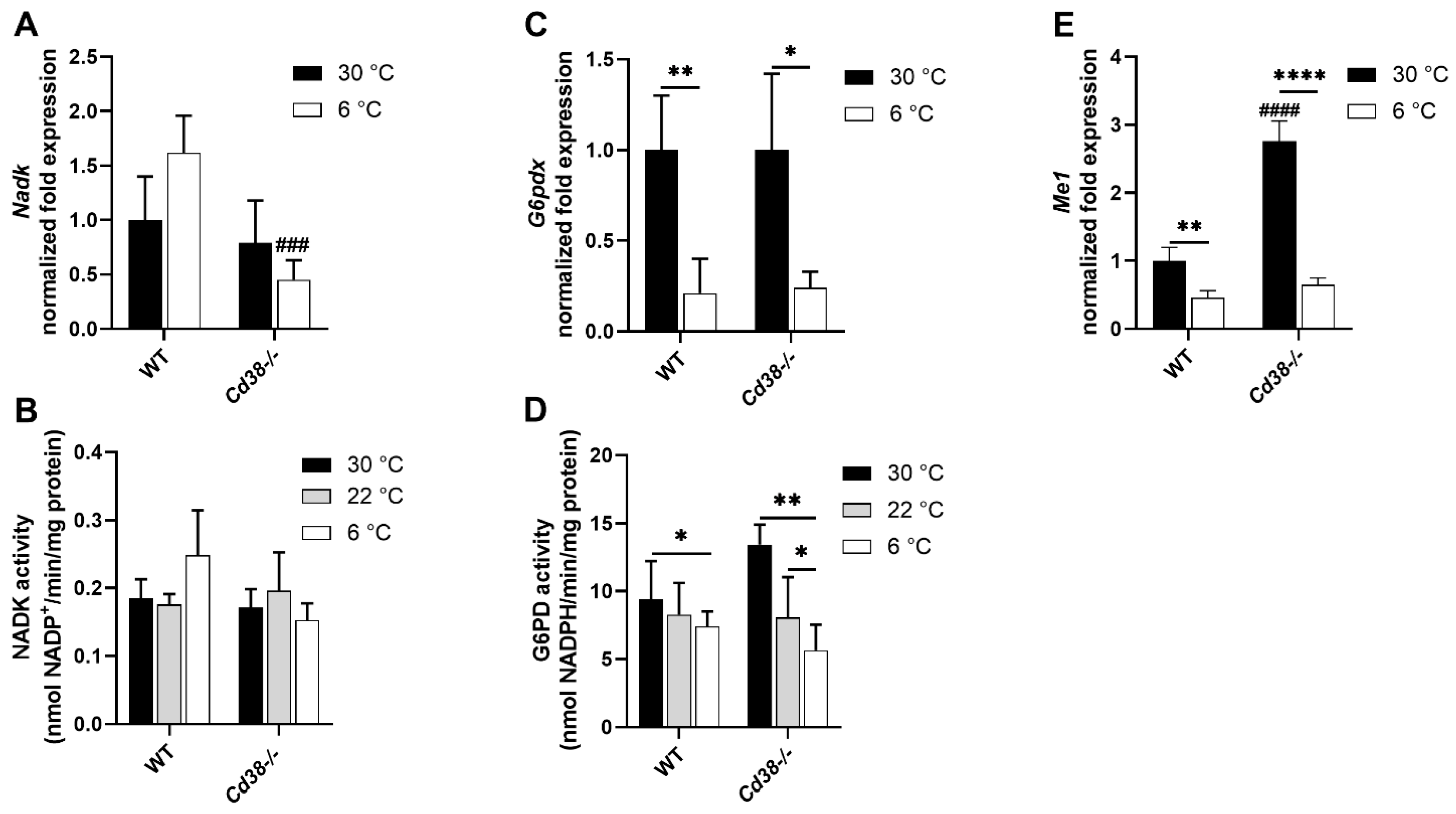
3.4. Downregulated Expression of G6PD and Malic Enzyme Are Responsible for the Decrease in NADPH in Liver of WT and Cd38−/− Mice upon Cold Stimulation
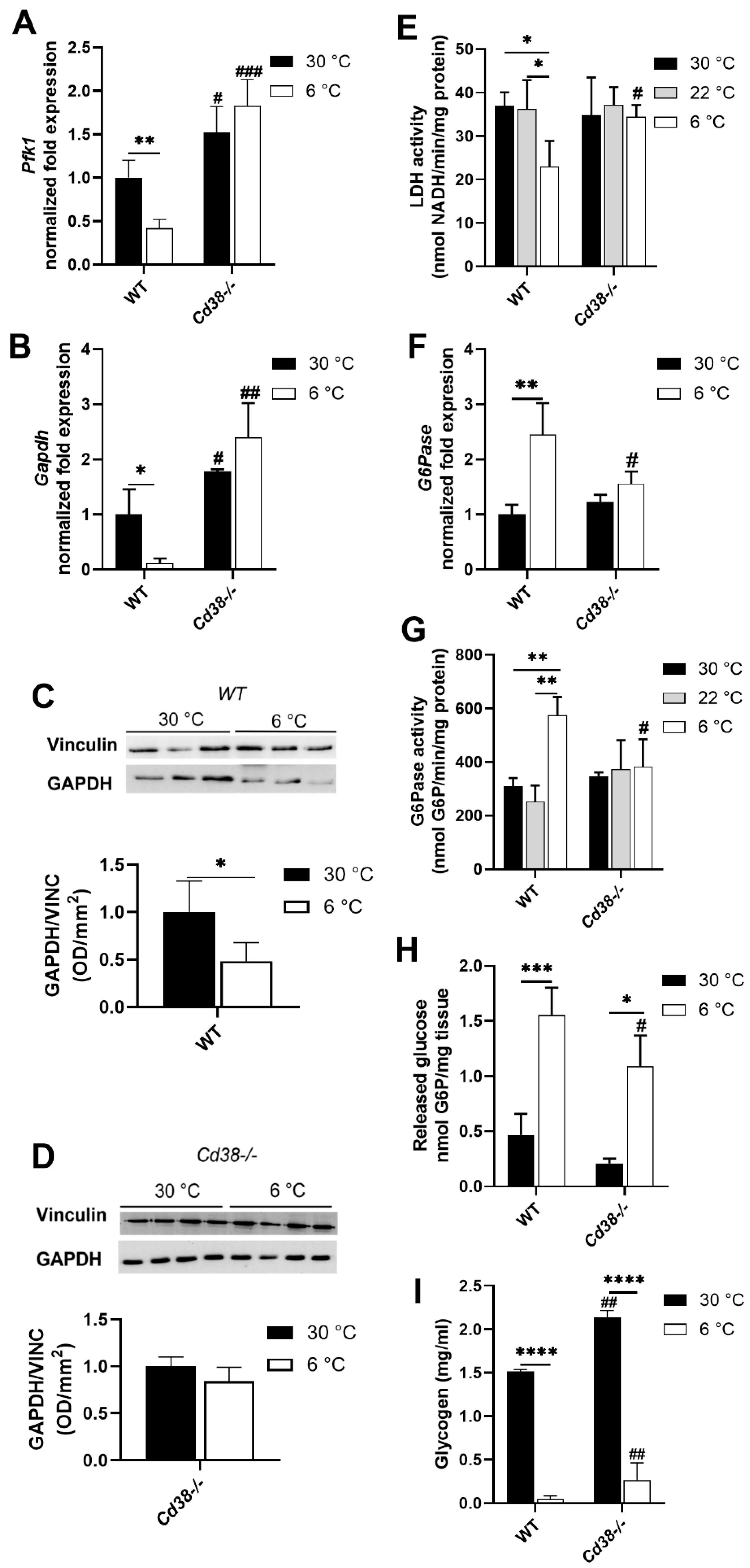
3.5. Glycolytic and Gluconeogenic Pathways Are Not Affected by Cold Exposure in Cd38−/− Mice
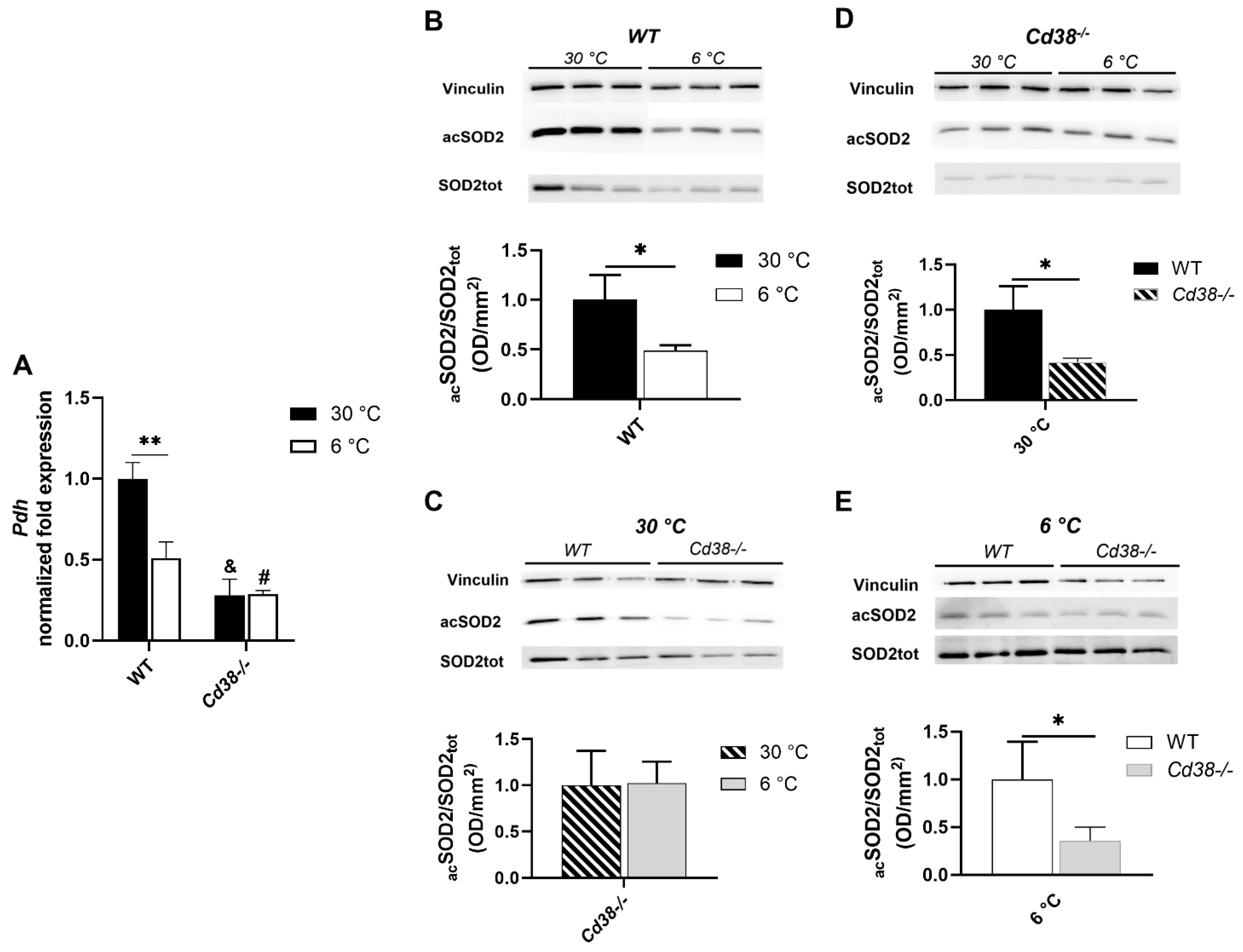
3.6. CD38 Deletion Affects Pdh Expression and SIRT3 Enzymatic Activity in Liver
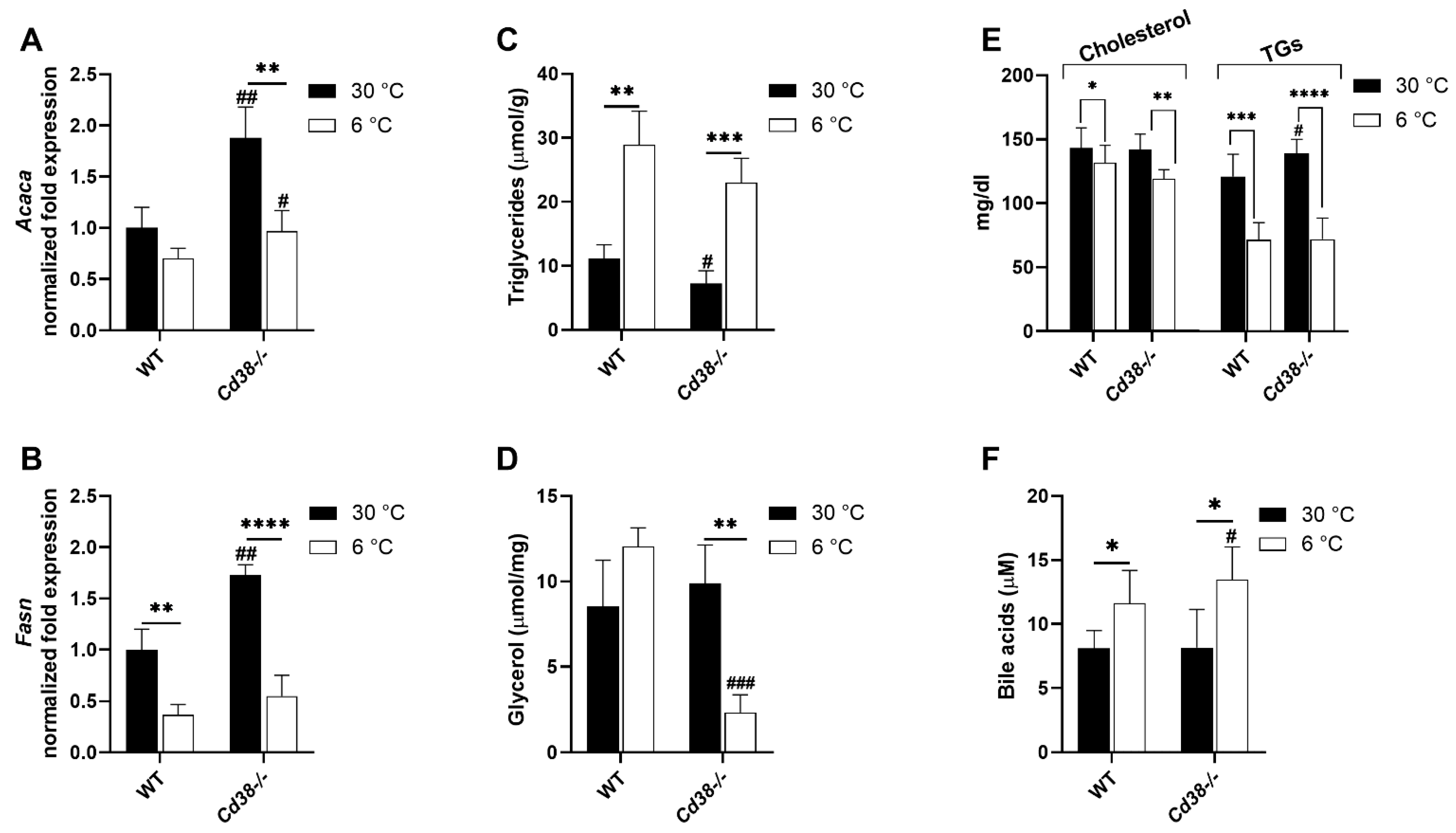
3.7. Cd38 Deletion Promotes Hepatic Lipid Synthesis and Release at Warm Temperature and BAs Release at Cold Temperature
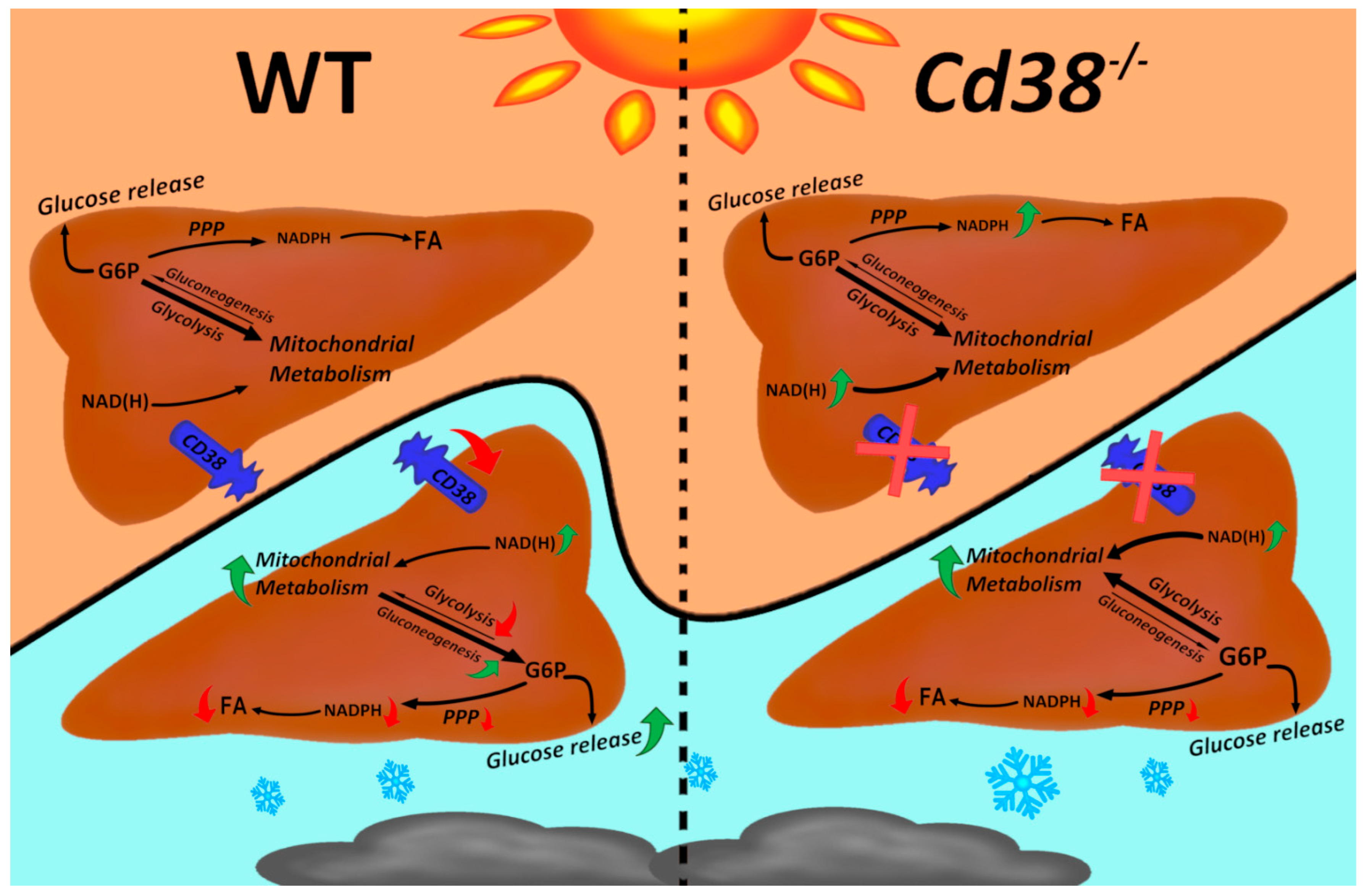
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Rui, L. Energy metabolism in the liver. Compr. Physiol. 2014, 4, 177–197. [Google Scholar] [PubMed]
- Li, Y.; Adeniji, N.T.; Fan, W.; Kunimoto, K.; Török, N.J. Non-alcoholic Fatty Liver Disease and Liver Fibrosis during Aging. Aging Dis. 2022, 13, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Abumrad, N.A. The Liver as a Hub in Thermogenesis. Cell Metab. 2017, 26, 454–455. [Google Scholar] [CrossRef] [PubMed]
- Simcox, J.; Geoghegan, G.; Maschek, J.A.; Bensard, C.L.; Pasquali, M.; Miao, R.; Lee, S.; Jiang, L.; Huck, I.; Kershaw, E.E.; et al. Global Analysis of Plasma Lipids Identifies Liver-Derived Acylcarnitines as a Fuel Source for Brown Fat Thermogenesis. Cell Metab. 2017, 26, 509–522. [Google Scholar] [CrossRef]
- Chang, S.H.; Song, N.J.; Choi, J.H.; Yun, U.J.; Park, K.W. Mechanisms underlying UCP1 dependent and independent adipocyte thermogenesis. Obes. Rev. 2019, 20, 241–251. [Google Scholar]
- Worthmann, A.; John, C.; Rühlemann, M.C.; Baguhl, M.; Heinsen, F.A.; Schaltenberg, N.; Heine, M.; Schlein, C.; Evangelakos, I.; Mineo, C.; et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017, 23, 839–849. [Google Scholar] [CrossRef]
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718. [Google Scholar] [CrossRef]
- Xiao, W.; Wang, R.S.; Handy, D.E.; Loscalzo, J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid. Redox Signal. 2018, 28, 251–272. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018, 39, 424–436. [Google Scholar] [CrossRef]
- Strømland, Ø.; Diab, J.; Ferrario, E.; Sverkeli, L.J.; Ziegler, M. The balance between NAD+ biosynthesis and consumption in ageing. Mech. Ageing Dev. 2021, 199, 111569. [Google Scholar] [CrossRef]
- Navas, L.E.; Carnero, A. NAD+ metabolism, stemness, the immune response, and cancer. Signal Transduct. Target Ther. 2021, 6, 2. [Google Scholar] [PubMed]
- Lee, H.C.; Graeff, R.M.; Walseth, T.F. ADP-ribosyl cyclase and CD38. Multi-functional enzymes in Ca2+ signaling. Adv. Exp. Med. Biol. 1997, 419, 411–419. [Google Scholar] [PubMed]
- Fliegert, R.; Bauche, A.; Wolf Pérez, A.M.; Watt, J.M.; Rozewitz, M.D.; Winzer, R.; Janus, M.; Gu, F.; Rosche, A.; Harneit, A.; et al. 2′-Deoxyadenosine 5′-diphosphoribose is an endogenous TRPM2 superagonist. Nat. Chem. Biol. 2017, 13, 1036–1044. [Google Scholar] [CrossRef]
- Van de Donk, N.W.C.J.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Morandi, F.; Airoldi, I.; Marimpietri, D.; Bracci, C.; Faini, A.C.; Gramignoli, R. CD38, a Receptor with Multifunctional Activities: From Modulatory Functions on Regulatory Cell Subsets and Extracellular Vesicles to a Target for Therapeutic Strategies. Cells 2019, 8, 1527. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, D.A.; Guedes, A.G.P.; Lund, F.E.; Subramanian, S.; Walseth, T.F.; Kannan, M.S. CD38 in the pathogenesis of allergic airway disease: Potential therapeutic targets. Pharmacol. Ther. 2017, 172, 116–126. [Google Scholar] [CrossRef] [PubMed]
- Zuo, W.; Liu, N.; Zeng, Y.; Liu, Y.; Li, B.; Wu, K.; Xiao, Y.; Liu, Q. CD38: A Potential Therapeutic Target in Cardiovascular Disease. Cardiovasc. Drugs Ther. 2020, 35, 815–828. [Google Scholar]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharmacol. 2011, 61, 301–332. [Google Scholar] [PubMed]
- Barbosa, M.T.; Soares, S.M.; Novak, C.M.; Sinclair, D.; Levine, J.A.; Aksoy, P.; Chini, E.N. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007, 21, 3629–3639. [Google Scholar] [CrossRef]
- Escande, C.; Nin, V.; Price, N.L.; Capellini, V.; Gomes, A.P.; Barbosa, M.T.; O’Neil, L.; White, T.A.; Sinclair, D.A.; Chini, E.N. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: Implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes 2013, 62, 1084–1093. [Google Scholar] [CrossRef]
- De Zélicourt, A.; Fayssoil, A.; Dakouane-Giudicelli, M.; De Jesus, I.; Karoui, A.; Zarrouki, F.; Lefebvre, F.; Mansart, A.; Launay, J.M.; Piquereau, J.; et al. CD38-NADase is a new major contributor to Duchenne muscular dystrophic phenotype. EMBO Mol. Med. 2022, 14, e12860. [Google Scholar] [CrossRef]
- Tarragó, M.G.; Chini, C.C.S.; Kanamori, K.S.; Warner, G.M.; Caride, A.; de Oliveira, G.C.; Rud, M.; Samani, A.; Hein, K.Z.; Huang, R.; et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD+ Decline. Cell Metab. 2018, 27, 1081–1095. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, J.; Mills, K.F.; Yoon, M.J.; Imai, S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011, 14, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, A.A.; Choudhury, M.; Rahman, S.M.; McCurdy, C.E.; Friederich, M.; Van Hove, J.L.; Watson, P.A.; Birdsey, N.; Bao, J.; Gius, D.; et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem. J. 2011, 433, 505–514. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Guillemin, G.J.; Mansour, H.; Chan-Ling, T.; Poljak, A.; Grant, R. Age related changes in NAD+ metabolism oxidative stress and Sirt1 activity in wistar rats. PLoS ONE 2011, 6, e19194. [Google Scholar] [CrossRef]
- Xie, L.; Wen, K.; Li, Q.; Huang, C.C.; Zhao, J.L.; Zhao, Q.H.; Xiao, Y.F.; Guan, X.H.; Qian, Y.S.; Gan, L.; et al. CD38 Deficiency Protects Mice from High Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Activating NAD(+)/Sirtuins Signaling Pathways-Mediated Inhibition of Lipid Accumulation and Oxidative Stress in Hepatocytes. Int. J. Biol. Sci. 2021, 17, 4305–4315. [Google Scholar] [CrossRef]
- Chini, C.C.S.; Peclat, T.R.; Warner, G.M.; Kashyap, S.; Espindola-Netto, J.M.; de Oliveira, G.C.; Gomez, L.S.; Hogan, K.A.; Tarragó, M.G.; Puranik, A.S.; et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat. Metab. 2020, 2, 1284–1304. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Cho, B.H.; Kim, U.H. CD38-mediated Ca2+ signaling contributes to angiotensin II-induced activation of hepatic stellate cells: Attenuation of hepatic fibrosis by CD38 ablation. J. Biol. Chem. 2010, 285, 576–582. [Google Scholar] [CrossRef]
- Benzi, A.; Sturla, L.; Heine, M.; Fischer, A.W.; Spinelli, S.; Magnone, M.; Sociali, G.; Parodi, A.; Fenoglio, D.; Emionite, L.; et al. CD38 downregulation modulates NAD+ and NADP(H) levels in thermogenic adipose tissues. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2021, 1866, 158819. [Google Scholar] [CrossRef]
- Benzi, A.; Grozio, A.; Spinelli, S.; Sturla, L.; Guse, A.H.; De Flora, A.; Zocchi, E.; Heeren, J.; Bruzzone, S. Role of CD38 in Adipose Tissue: Tuning Coenzyme Availability? Nutrients 2021, 13, 3734. [Google Scholar] [CrossRef]
- Sociali, G.; Grozio, A.; Caffa, I.; Schuster, S.; Becherini, P.; Damonte, P.; Sturla, L.; Fresia, C.; Passalacqua, M.; Mazzola, F.; et al. SIRT6 deacetylase activity regulates NAMPT activity and NAD(P)(H) pools in cancer cells. FASEB J. 2019, 33, 3704–3717. [Google Scholar] [CrossRef] [PubMed]
- Grozio, A.; Sociali, G.; Sturla, L.; Caffa, I.; Soncini, D.; Salis, A.; Raffaelli, N.; De Flora, A.; Nencioni, A.; Bruzzone, S. CD73 protein as a source of extracellular precursors for sustained NAD+ biosynthesis in FK866-treated tumor cells. J. Biol. Chem. 2013, 288, 25938–25949. [Google Scholar] [CrossRef] [PubMed]
- Sturla, L.; Fresia, C.; Guida, L.; Bruzzone, S.; Scarfi, S.; Usai, C.; Fruscione, F.; Magnone, M.; Millo, E.; Basile, G.; et al. LANCL2 is necessary for abscisic acid binding and signaling in human granulocytes and in rat insulinoma cells. J. Biol. Chem. 2009, 284, 28045–28057. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using realtime quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Higgins, C.B.; Mayer, A.L.; Zhang, Y.; Franczyk, M.; Ballentine, S.; Yoshino, J.; DeBosch, B.J. SIRT1 selectively exerts the metabolic protective effects of hepatocyte nicotinamide phosphoribosyltransferase. Nat. Commun. 2022, 13, 1074. [Google Scholar] [CrossRef] [PubMed]
- Ameka, M.; Markan, K.R.; Morgan, D.A.; BonDurant, L.D.; Idiga, S.O.; Naber, M.C.; Zhu, Z.; Zingman, L.V.; Grobe, J.L.; Rahmouni, K.; et al. Liver Derived FGF21 Maintains Core Body Temperature During Acute Cold Exposure. Sci. Rep. 2019, 9, 630. [Google Scholar] [CrossRef]
- Rah, S.Y.; Kim, U.H. CD38-mediated Ca(2+) signaling contributes to glucagon-induced hepatic gluconeogenesis. Sci. Rep. 2015, 5, 10741. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef]
- Wei, X.; Jia, R.; Yang, Z.; Jiang, J.; Huang, J.; Yan, J.; Luo, X. NAD+/sirtuin metabolism is enhanced in response to cold-induced changes in lipid metabolism in mouse liver. FEBS Lett. 2020, 594, 1711–1725. [Google Scholar] [CrossRef]
- Rah, S.Y.; Lee, Y.H.; Kim, U.H. NAADP-mediated Ca2+ signaling promotes autophagy and protects against LPS-induced liver injury. FASEB J. 2017, 31, 3126–3137. [Google Scholar] [CrossRef] [PubMed]
- Cosker, F.; Cheviron, N.; Yamasaki, M.; Menteyne, A.; Lund, F.E.; Moutin, M.J.; Galione, A.; Cancela, J.M. The ecto-enzyme CD38 is a nicotinic acid adenine dinucleotide phosphate (NAADP) synthase that couples receptor activation to Ca2+ mobilization from lysosomes in pancreatic acinar cells. J. Biol. Chem. 2010, 285, 38251–38259. [Google Scholar] [CrossRef] [PubMed]
- Toews, C.J.; Lowy, C.; Ruderman, N.B. The regulation of gluconeogenesis. The effect of pent-4-enoic acid on gluconeogenesis and on the gluconeogenic metabolite concentrations of isolated perfused rat liver. J. Biol. Chem. 1970, 245, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Sistare, F.D.; Haynes, R.C., Jr. The interaction between the cytosolic pyridine nucleotide redox potential and gluconeogenesis from lactate/pyruvate in isolated rat hepatocytes. Implications for investigations of hormone action. J. Biol. Chem. 1985, 260, 12748–12753. [Google Scholar] [CrossRef]
- Grefhorst, A.; van den Beukel, J.C.; Dijk, W.; Steenbergen, J.; Voortman, G.J.; Leeuwenburgh, S.; Visser, T.J.; Kersten, S.; Friesema, E.C.H.; Themmen, A.P.N.; et al. Multiple effects of cold exposure on livers of male mice. J. Endocrinol. 2018, 238, 91–106. [Google Scholar] [CrossRef]
- Heine, M.; Fischer, A.W.; Schlein, C.; Jung, C.; Straub, L.G.; Gottschling, K.; Mangels, N.; Yuan, Y.; Nilsson, S.K.; Liebscher, G.; et al. Lipolysis Triggers a Systemic Insulin Response Essential for Efficient Energy Replenishment of Activated Brown Adipose Tissue in Mice. Cell Metab. 2018, 28, 644–655. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018, 27, 529–547. [Google Scholar] [PubMed]
- Yang, Y.; Zhang, N.; Zhang, G.; Sauve, A.A. NRH salvage and conversion to NAD(+) requires NRH kinase activity by adenosine kinase. Nat. Metab. 2020, 2, 364–379. [Google Scholar] [CrossRef]
- Pham, T.X.; Bae, M.; Kim, M.B.; Lee, Y.; Hu, S.; Kang, H.; Park, Y.K.; Lee, J.Y. Nicotinamide riboside, an NAD+ precursor, attenuates the development of liver fibrosis in a diet-induced mouse model of liver fibrosis. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 2451–2463. [Google Scholar] [CrossRef]
- Zapata-Pérez, R.; Tammaro, A.; Schomakers, B.V.; Scantlebery, A.M.L.; Denis, S.; Elfrink, H.L.; Giroud-Gerbetant, J.; Cantó, C.; López-Leonardo, C.; McIntyre, R.L.; et al. Reduced nicotinamide mononucleotide is a new and potent NAD+ precursor in mammalian cells and mice. FASEB J. 2021, 35, e21456. [Google Scholar] [CrossRef]
- Radenkovic, D.; Verdin, E. Clinical Evidence for Targeting NAD Therapeutically. Pharmaceuticals 2020, 13, 247. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, M.; Yoshino, J.; Kayser, B.D.; Patti, G.J.; Franczyk, M.P.; Mills, K.F.; Sindelar, M.; Pietka, T.; Patterson, B.W.; Imai, S.I.; et al. Nicotinamide mononucleotide increases muscle insulin sensitivity in prediabetic women. Science 2021, 372, 1224–1229. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Mouse Gene | Accession Number | Sequence, 5′-3′ |
|---|---|---|
| Nadk | NM_001159637 | Forward 5′-CCAAGTCTCGGAGCCTGTC-3′ Reverse 5′-AAATGTTGTCACTGGGCACG-3′ |
| Nampt | NM_021524 | Forward 5′-AATGTCTCCTTCGGTTCTGGTG-3′ Reverse 5′-CCCGCTGGTGTCCTATGTAAAG-3′ |
| Cd38 | NM_007646 | Forward 5′-GGTCCTGATCGCCTTGGTAGTAG-3′ Reverse 5′-ATCTCCTGGCAGTTCTGATCTCTC-3′ |
| Acaca | NM_133360 | Forward 5′-CACTGTGGCTTCTCCAGCA-3′ Reverse 5′-CACCGACGGATAGATCGCAT-3′ |
| Fasn | NM_007988 | Forward 5′-ATGGGTGTGGAAGTTCGTCAG-3′ Reverse 5′-AGTGTGCTCAGGTTCAGTTGG-3′ |
| Me1 | NM_008615 | Forward 5′-GGACCCGCATCTCAACAAGG-3′ Reverse 5′-AGGGCGGCAACAATCCATGA-3′ |
| G6pdx | NM_008062 | Forward 5′-TGATCGAGAAAAGCCCCAGC-3′ Reverse 5′-GTGAGGGTTCACCCACTTGT-3′ |
| Gapdh | GU214026 | Forward 5′-CGTGCCGCCTGGAGAAACCTG-3′ Reverse 5′-TGGAAGAGTGGGAGTTGCTGTTGAAG-3′ |
| Pfk1 | NM_001163487 | Forward 5′-AGTTGGTATCTTCACGGGCG-3′ Reverse 5′-CATAGACACGCTCTCCCACG-3′ |
| Pdha1 | NM_008810 | Forward 5′-GATGGAGCTAAAGGCGGATCA-3′ Reverse 5′-TCCGTAGGGTTTATGCCAGC-3′ |
| G6pase | NM_008061 | Forward 5′-AGCCAAGAGATGGTGTGAGC-3′ Reverse 5′-TACATGCTGGAGTTGAGGGC-3′ |
| β-2 Microglobulin | NM_009735 | Forward 5′-CGGTCGCTTCAGTCGTCAG-3′ Reverse 5′-CAGTTCAGTATGTTCGGCTTCC-3′ |
| Ubiquitin | NM_019639 | Forward 5′-GACAGGCAAGACCATCAC-3′ Reverse 5′-TCTGAGGCGAAGGACTAAG-3′ |
| Tbp | NM_013684 | Forward 5′-GAAGCTGCGGTACAATTCCAG-3′ Reverse 5′-CCCCTTGTACCCTTCACCAAT-3′ |
| β-actin | NM_007393 | Forward 5′-GCGAGAAGATGACCCAGATC-3′ Reverse 5′-GGATAGCACAGCCTGGATAG-3′ |
| Fgf21 | NM_020013 | Forward 5′-CACACCGCAGTCCAGAAAGT-3′ Reverse 5′-CCTAGAGGCTTTGACACCCA-3′ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benzi, A.; Spinelli, S.; Sturla, L.; Heine, M.; Fischer, A.W.; Koch-Nolte, F.; Mittrücker, H.-W.; Guse, A.H.; De Flora, A.; Heeren, J.; et al. Role of Liver CD38 in the Regulation of Metabolic Pathways during Cold-Induced Thermogenesis in Mice. Cells 2022, 11, 3812. https://doi.org/10.3390/cells11233812
Benzi A, Spinelli S, Sturla L, Heine M, Fischer AW, Koch-Nolte F, Mittrücker H-W, Guse AH, De Flora A, Heeren J, et al. Role of Liver CD38 in the Regulation of Metabolic Pathways during Cold-Induced Thermogenesis in Mice. Cells. 2022; 11(23):3812. https://doi.org/10.3390/cells11233812
Chicago/Turabian StyleBenzi, Andrea, Sonia Spinelli, Laura Sturla, Markus Heine, Alexander W. Fischer, Friedrich Koch-Nolte, Hans-Willi Mittrücker, Andreas H. Guse, Antonio De Flora, Joerg Heeren, and et al. 2022. "Role of Liver CD38 in the Regulation of Metabolic Pathways during Cold-Induced Thermogenesis in Mice" Cells 11, no. 23: 3812. https://doi.org/10.3390/cells11233812
APA StyleBenzi, A., Spinelli, S., Sturla, L., Heine, M., Fischer, A. W., Koch-Nolte, F., Mittrücker, H.-W., Guse, A. H., De Flora, A., Heeren, J., & Bruzzone, S. (2022). Role of Liver CD38 in the Regulation of Metabolic Pathways during Cold-Induced Thermogenesis in Mice. Cells, 11(23), 3812. https://doi.org/10.3390/cells11233812