
Nalfurafine Hydrochloride, a κ-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Reagents and Plasmids
2.2. Cell Culture
2.3. Cell-Based Hormone Library Screening
2.4. Melanin Content Assay
2.5. Autophagy Analysis and Melanophagy Assay with Fluorescent Punctuation
2.6. Western Blotting
2.7. Statistical Analysis
3. Results
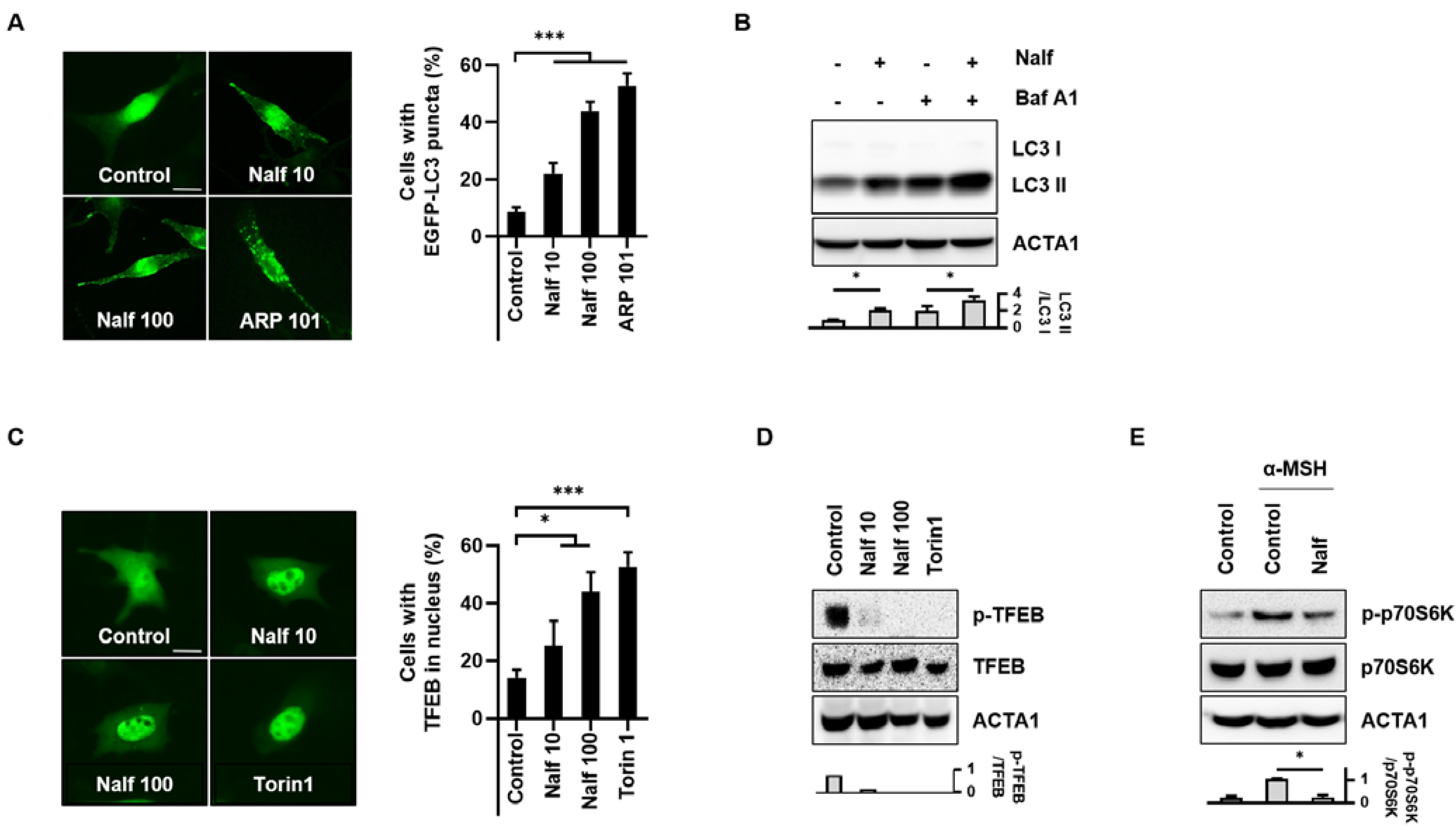
3.1. Nalfurafine Hydrochloride Induces Autophagy Activation in B16F1 Cells
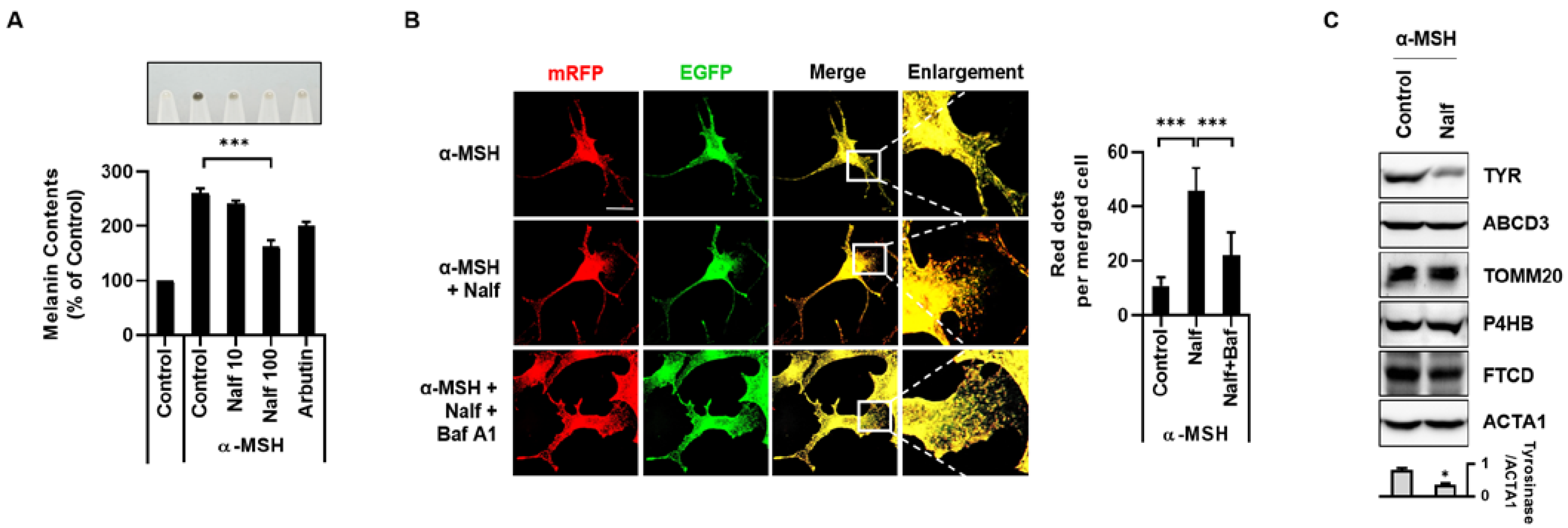
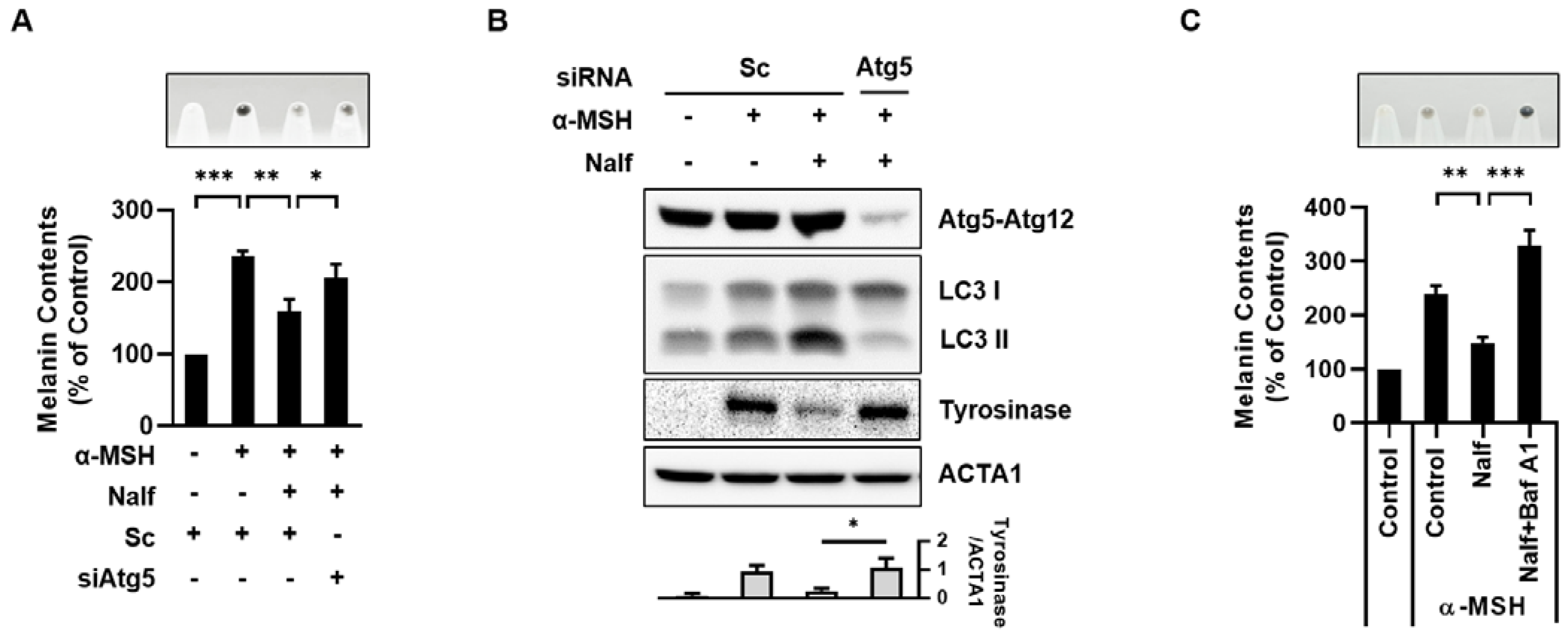
3.2. Nalfurafine Hydrochloride Promotes Melanosomal Degradation by Inducing Melanophagy
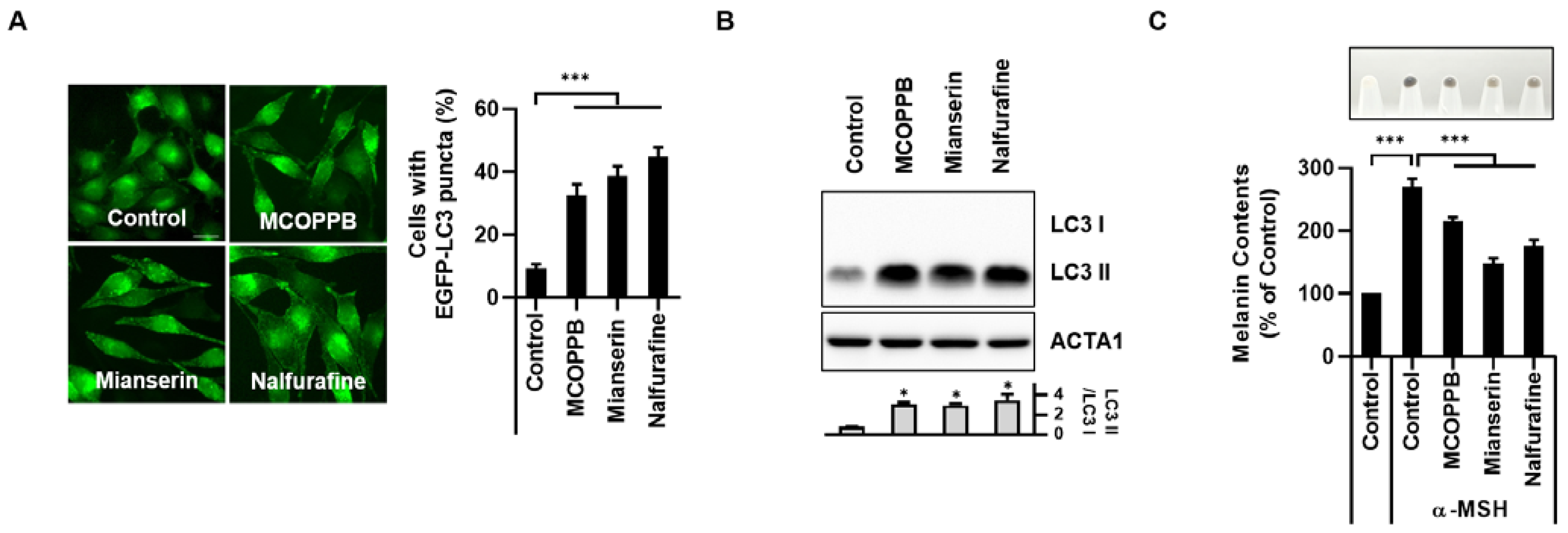
3.3. Activation of the κ-Opioid Receptor Induces Melanophagy in B16F1 Cells
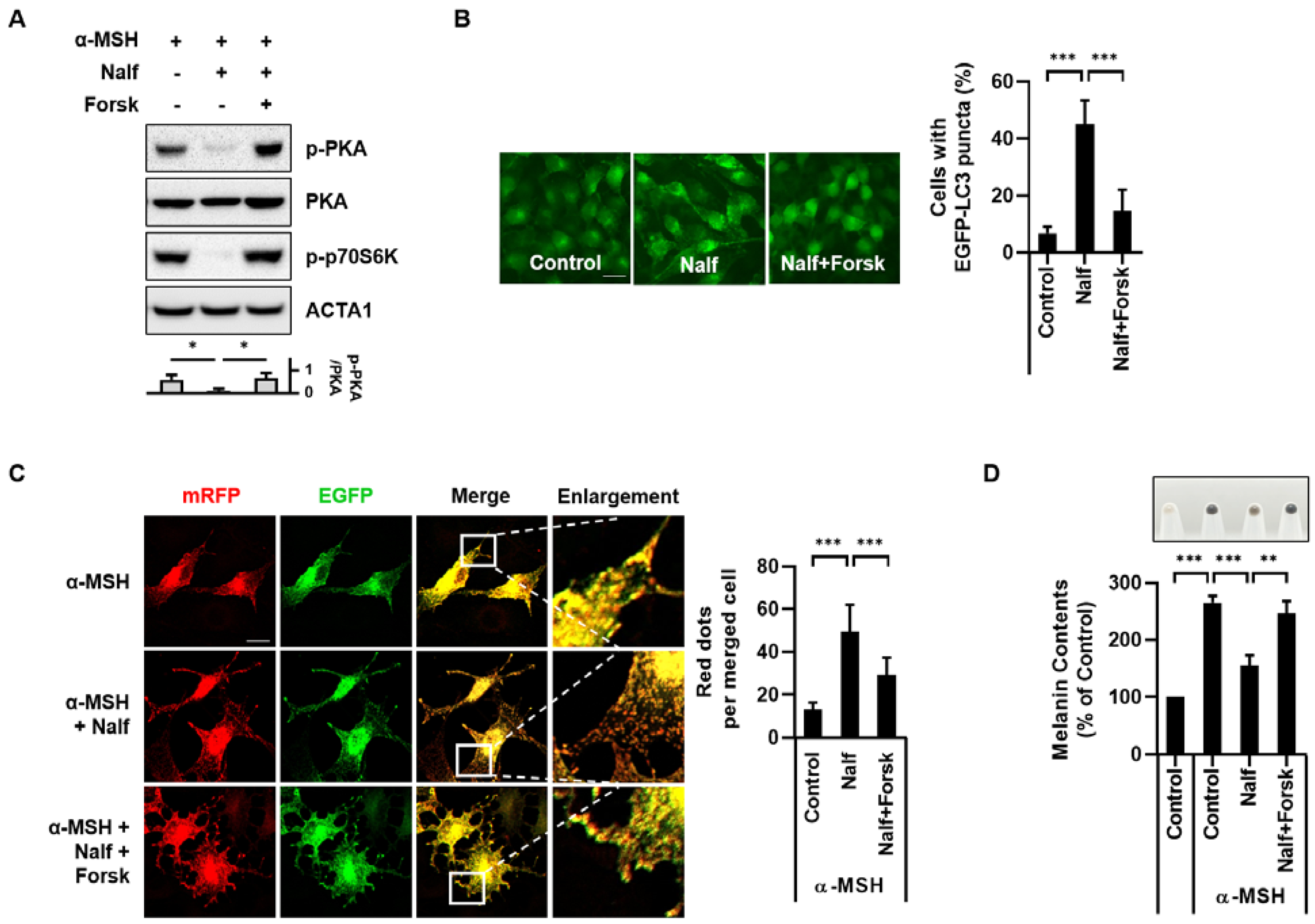
3.4. Inhibition of PKA Mediates Melanophagy in Nalfurafine Hydrochloride-Treated Cells
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition) (1). Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef] [PubMed]
- Glick, D.; Barth, S.; Macleod, K.F. Autophagy: Cellular and molecular mechanisms. J. Pathol. 2010, 221, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell. Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Park, N.Y.; Jo, D.S.; Cho, D.H. Post-Translational Modifications of ATG4B in the Regulation of Autophagy. Cells 2022, 11, 1330. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Kim, Y.S.; Jo, D.S.; Choe, S.K.; Jo, E.K. Pexophagy: Molecular Mechanisms and Implications for Health and Diseases. Mol. Cells 2018, 41, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Kim, J.K.; Jo, E.K. Mitophagy and Innate Immunity in Infection. Mol. Cells 2020, 43, 10–22. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Garcia Arencibia, M.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef]
- Füllgrabe, J.; Ghislat, G.; Cho, D.H.; Rubinsztein, D.C. Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci. 2016, 129, 3059–3066. [Google Scholar] [CrossRef]
- Puertollano, R.; Ferguson, S.M.; Brugarolas, J.; Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 2018, 37, e98804. [Google Scholar] [CrossRef]
- Fore, J. A review of skin and the effects of aging on skin structure and function. Ostomy Wound Manag. 2006, 52, 24–35; quiz 36–27. [Google Scholar]
- D’Alba, L.; Shawkey, M.D. Melanosomes: Biogenesis, Properties, and Evolution of an Ancient Organelle. Physiol. Rev. 2019, 99, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Plensdorf, S.; Livieratos, M.; Dada, N. Pigmentation Disorders: Diagnosis and Management. Am. Fam. Physician 2017, 96, 797–804. [Google Scholar] [PubMed]
- D’Mello, S.A.; Finlay, G.J.; Baguley, B.C.; Askarian-Amiri, M.E. Signaling Pathways in Melanogenesis. Int. J. Mol. Sci. 2016, 17, 1144. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, A.; Fisher, D.E. The master role of microphthalmia-associated transcription factor in melanocyte and melanoma biology. Lab. Investig. 2017, 97, 649–656. [Google Scholar] [CrossRef] [PubMed]
- Park, H.Y.; Wu, C.; Yonemoto, L.; Murphy-Smith, M.; Wu, H.; Stachur, C.M.; Gilchrest, B.A. MITF mediates cAMP-induced protein kinase C-beta expression in human melanocytes. Biochem. J. 2006, 395, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.E.; Barajas, B.; Bussat, R.T.; Yan, K.J.; Neela, P.H.; Flockhart, R.J.; Kovalski, J.; Zehnder, A.; Khavari, P.A. Enhancer-targeted genome editing selectively blocks innate resistance to oncokinase inhibition. Genome Res. 2014, 24, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Ploper, D.; Taelman, V.F.; Robert, L.; Perez, B.S.; Titz, B.; Chen, H.W.; Graeber, T.G.; von Euw, E.; Ribas, A.; De Robertis, E.M. MITF drives endolysosomal biogenesis and potentiates Wnt signaling in melanoma cells. Proc. Natl. Acad. Sci. USA 2015, 112, E420–E429. [Google Scholar] [CrossRef]
- Park, H.J.; Jo, D.S.; Choi, D.S.; Bae, J.E.; Park, N.Y.; Kim, J.B.; Chang, J.H.; Shin, J.J.; Cho, D.H. Ursolic acid inhibits pigmentation by increasing melanosomal autophagy in B16F1 cells. Biochem. Biophys. Res. Commun. 2020, 531, 209–214. [Google Scholar] [CrossRef]
- Zouboulis, C.C. The human skin as a hormone target and an endocrine gland. Hormones (Athens) 2004, 3, 9–26. [Google Scholar] [CrossRef]
- Kamimura, K.; Yokoo, T.; Kamimura, H.; Sakamaki, A.; Abe, S.; Tsuchiya, A.; Takamura, M.; Kawai, H.; Yamagiwa, S.; Terai, S. Long-term efficacy and safety of nalfurafine hydrochloride on pruritus in chronic liver disease patients: Patient-reported outcome based analyses. PLoS ONE 2017, 12, e0178991. [Google Scholar] [CrossRef]
- Jo, Y.K.; Park, S.J.; Shin, J.H.; Kim, Y.; Hwang, J.J.; Cho, D.H.; Kim, J.C. ARP101, a selective MMP-2 inhibitor, induces autophagy-associated cell death in cancer cells. Biochem. Biophys. Res. Commun. 2011, 404, 1039–1043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhai, B.; Gygi, S.P.; Goldberg, A.L. mTOR inhibition activates overall protein degradation by the ubiquitin proteasome system as well as by autophagy. Proc. Natl. Acad. Sci. USA 2015, 112, 15790–15797. [Google Scholar] [CrossRef] [PubMed]
- Park, H.J.; Jo, D.S.; Choi, H.; Bae, J.E.; Park, N.Y.; Kim, J.B.; Choi, J.Y.; Kim, Y.H.; Oh, G.S.; Chang, J.H.; et al. Melasolv induces melanosome autophagy to inhibit pigmentation in B16F1 cells. PLoS ONE 2020, 15, e0239019. [Google Scholar] [CrossRef]
- Boo, Y.C. Arbutin as a Skin Depigmenting Agent with Antimelanogenic and Antioxidant Properties. Antioxidants 2021, 10, 1129. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; He, P.; Huang, Y.; Li, Y.F.; Lu, J.; Li, M.; Kurihara, H.; Luo, Z.; Meng, T.; Onishi, M.; et al. Selective autophagy of intracellular organelles: Recent research advances. Theranostics 2021, 11, 222–256. [Google Scholar] [CrossRef]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef]
- Lopez, A.; Fleming, A.; Rubinsztein, D.C. Seeing is believing: Methods to monitor vertebrate autophagy in vivo. Open Biol. 2018, 8, 180106. [Google Scholar] [CrossRef]
- Ruan, X.; Hall, S.M.; Kaye, A.D. Nalfurafine hydrochloride, a selective κ opioid receptor agonist, has no reinforcing effect on intravenous self-administration in rhesus monkeys. J. Pharmacol. Sci. 2016, 132, 113–114. [Google Scholar] [CrossRef]
- Salemi, S.; Aeschlimann, A.; Reisch, N.; Jüngel, A.; Gay, R.E.; Heppner, F.L.; Michel, B.A.; Gay, S.; Sprott, H. Detection of kappa and delta opioid receptors in skin--outside the nervous system. Biochem. Biophys. Res. Commun. 2005, 338, 1012–1017. [Google Scholar] [CrossRef]
- Zhou, Q.; Zhang, Z.; Long, S.; Li, W.; Wang, B.; Liang, N. Opioids in cancer: The κ-opioid receptor (Review). Mol. Med. Rep. 2022, 25, 44. [Google Scholar] [CrossRef]
- Yamamizu, K.; Furuta, S.; Hamada, Y.; Yamashita, A.; Kuzumaki, N.; Narita, M.; Doi, K.; Katayama, S.; Nagase, H.; Yamashita, J.K.; et al. к Opioids inhibit tumor angiogenesis by suppressing VEGF signaling. Sci. Rep. 2013, 3, 3213. [Google Scholar] [CrossRef] [PubMed]
- Yamamizu, K.; Furuta, S.; Katayama, S.; Narita, M.; Kuzumaki, N.; Imai, S.; Nagase, H.; Suzuki, T.; Narita, M.; Yamashita, J.K. The κ opioid system regulates endothelial cell differentiation and pathfinding in vascular development. Blood 2011, 118, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Alasbahi, R.H.; Melzig, M.F. Forskolin and derivatives as tools for studying the role of cAMP. Pharmazie 2012, 67, 5–13. [Google Scholar] [PubMed]
- Tian, X.; Cui, Z.; Liu, S.; Zhou, J.; Cui, R. Melanosome transport and regulation in development and disease. Pharmacol. Ther. 2021, 219, 107707. [Google Scholar] [CrossRef] [PubMed]
- Edmondson, S.R.; Thumiger, S.P.; Werther, G.A.; Wraight, C.J. Epidermal homeostasis: The role of the growth hormone and insulin-like growth factor systems. Endocr. Rev. 2003, 24, 737–764. [Google Scholar] [CrossRef]
- Lui, A.; New, J.; Ogony, J.; Thomas, S.; Lewis-Wambi, J. Everolimus downregulates estrogen receptor and induces autophagy in aromatase inhibitor-resistant breast cancer cells. BMC Cancer 2016, 16, 487. [Google Scholar] [CrossRef]
- Puigoriol-Illamola, D.; Griñán-Ferré, C.; Vasilopoulou, F.; Leiva, R.; Vázquez, S.; Pallàs, M. 11β-HSD1 Inhibition by RL-118 Promotes Autophagy and Correlates with Reduced Oxidative Stress and Inflammation, Enhancing Cognitive Performance in SAMP8 Mouse Model. Mol. Neurobiol. 2018, 55, 8904–8915. [Google Scholar] [CrossRef]
- Zhou, Y.; Freeman, K.; Setola, V.; Cao, D.; Kaski, S.; Kreek, M.J.; Liu-Chen, L.Y. Preclinical Studies on Nalfurafine (TRK-820), a Clinically Used KOR Agonist. Handb. Exp. Pharmacol. 2022, 271, 137–162. [Google Scholar] [CrossRef]
- Sobczak, M.; Sałaga, M.; Storr, M.A.; Fichna, J. Physiology, signaling, and pharmacology of opioid receptors and their ligands in the gastrointestinal tract: Current concepts and future perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef]
- Gavériaux-Ruff, C.; Peluso, J.; Befort, K.; Simonin, F.; Zilliox, C.; Kieffer, B.L. Detection of opioid receptor mRNA by RT-PCR reveals alternative splicing for the delta- and kappa-opioid receptors. Brain Res. Mol. Brain Res. 1997, 48, 298–304. [Google Scholar] [CrossRef]
- Chen, D.; Chen, Y.; Yan, Y.; Pan, J.; Xing, W.; Li, Q.; Zeng, W. Down-regulation of the tumour suppressor κ-opioid receptor predicts poor prognosis in hepatocellular carcinoma patients. BMC Cancer 2017, 17, 553. [Google Scholar] [CrossRef] [PubMed]
- Finley, M.J.; Happel, C.M.; Kaminsky, D.E.; Rogers, T.J. Opioid and nociceptin receptors regulate cytokine and cytokine receptor expression. Cell. Immunol. 2008, 252, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, Z.; Zeng, C.; Xu, Y.; He, L.; Fang, Q.; Chen, Z. Dynorphin/KOR inhibits neuronal autophagy by activating mTOR signaling pathway to prevent acute seizure epilepsy. Cell Biol. Int. 2022, 46, 1814–1824. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, Y.; Wang, X.; Tian, X.; Zhang, S.; Yang, F.; Guo, H.; Fan, R.; Feng, N.; Jia, M.; et al. The Protective Effects of Κ-Opioid Receptor Stimulation in Hypoxic Pulmonary Hypertension Involve Inhibition of Autophagy Through the AMPK-MTOR Pathway. Cell Physiol. Biochem. 2017, 44, 1965–1979. [Google Scholar] [CrossRef]
- Waldhoer, M.; Bartlett, S.E.; Whistler, J.L. Opioid receptors. Annu. Rev. Biochem. 2004, 73, 953–990. [Google Scholar] [CrossRef]
- Bruchas, M.R.; Chavkin, C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology 2010, 210, 137–147. [Google Scholar] [CrossRef]
- Price, E.R.; Ding, H.F.; Badalian, T.; Bhattacharya, S.; Takemoto, C.; Yao, T.P.; Hemesath, T.J.; Fisher, D.E. Lineage-specific signaling in melanocytes. C-kit stimulation recruits p300/CBP to microphthalmia. J. Biol. Chem. 1998, 273, 17983–17986. [Google Scholar] [CrossRef]
- Passeron, T.; Namiki, T.; Passeron, H.J.; Le Pape, E.; Hearing, V.J. Forskolin protects keratinocytes from UVB-induced apoptosis and increases DNA repair independent of its effects on melanogenesis. J. Investig. Dermatol. 2009, 129, 162–166. [Google Scholar] [CrossRef]
- Djouder, N.; Tuerk, R.D.; Suter, M.; Salvioni, P.; Thali, R.F.; Scholz, R.; Vaahtomeri, K.; Auchli, Y.; Rechsteiner, H.; Brunisholz, R.A.; et al. PKA phosphorylates and inactivates AMPKalpha to promote efficient lipolysis. EMBO J. 2010, 20, 469–481. [Google Scholar] [CrossRef]
- Iannucci, L.F.; Di Benedetto, G.; Lefkimmiatis, K. PRKA/PKA signals and autophagy: Space matters. Autophagy 2021, 17, 1563–1564. [Google Scholar] [CrossRef]
- Grisan, F.; Iannucci, L.F.; Surdo, N.C.; Gerbino, A.; Zanin, S.; Di Benedetto, G.; Pozzan, T.; Lefkimmiatis, K. PKA compartmen talization links cAMP signaling and autophagy. Cell Death Differ. 2021, 28, 2436–2449. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, H.J.; Kim, S.H.; Kim, Y.H.; Kim, S.H.; Oh, G.S.; Bae, J.-E.; Kim, J.B.; Park, N.Y.; Park, K.; Yeom, E.; et al. Nalfurafine Hydrochloride, a κ-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells. Cells 2023, 12, 146. https://doi.org/10.3390/cells12010146
Lee HJ, Kim SH, Kim YH, Kim SH, Oh GS, Bae J-E, Kim JB, Park NY, Park K, Yeom E, et al. Nalfurafine Hydrochloride, a κ-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells. Cells. 2023; 12(1):146. https://doi.org/10.3390/cells12010146
Chicago/Turabian StyleLee, Ha Jung, Seong Hyun Kim, Yong Hwan Kim, So Hyun Kim, Gyeong Seok Oh, Ji-Eun Bae, Joon Bum Kim, Na Yeon Park, Kyuhee Park, Eunbyul Yeom, and et al. 2023. "Nalfurafine Hydrochloride, a κ-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells" Cells 12, no. 1: 146. https://doi.org/10.3390/cells12010146
APA StyleLee, H. J., Kim, S. H., Kim, Y. H., Kim, S. H., Oh, G. S., Bae, J.-E., Kim, J. B., Park, N. Y., Park, K., Yeom, E., Jeong, K., Kim, P., Jo, D. S., & Cho, D.-H. (2023). Nalfurafine Hydrochloride, a κ-Opioid Receptor Agonist, Induces Melanophagy via PKA Inhibition in B16F1 Cells. Cells, 12(1), 146. https://doi.org/10.3390/cells12010146