
Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Culture and Establishment of Stable Cell Lines
2.3. Determination of Pexophagic Cells
2.4. Confocal Microscopy
2.5. Western Blotting
2.6. ROS Measurement
2.7. ROS Measurement with HyPer Protein
2.8. Statistical Analysis
3. Results
3.1. Inhibition of BRD4 Promotes Pexophagy
3.2. Loss of ATG7 Blocks Molibresib-Induced Peroxisomal Autophagy in HeLa Cells
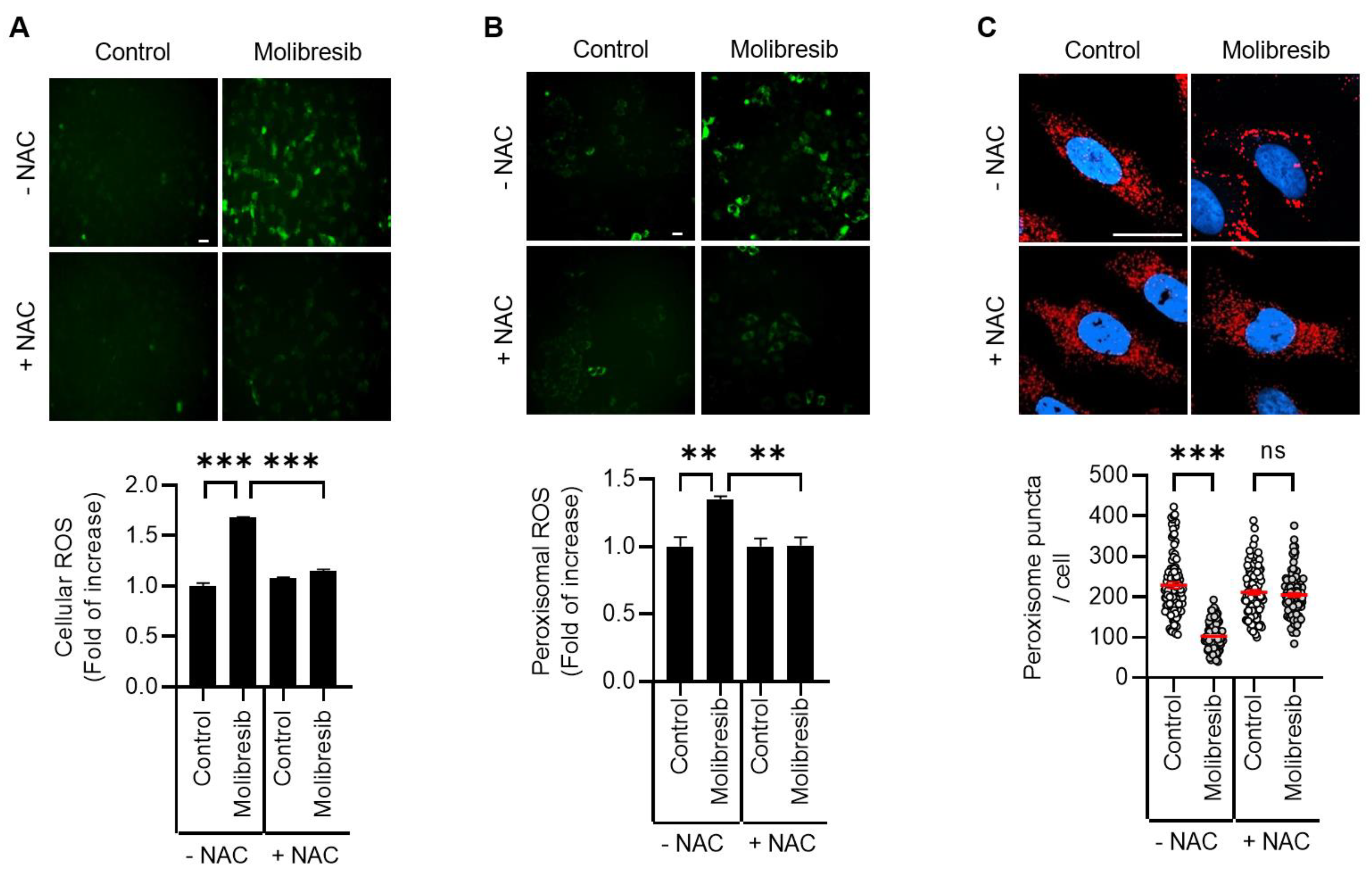
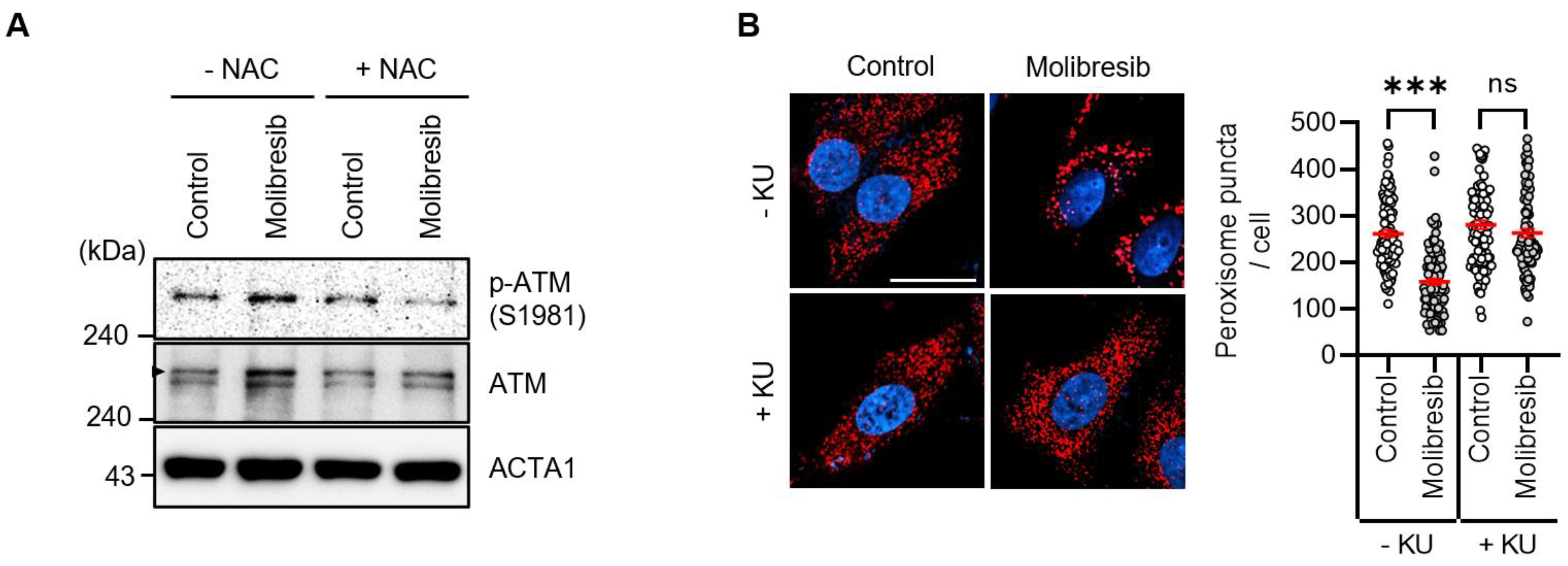
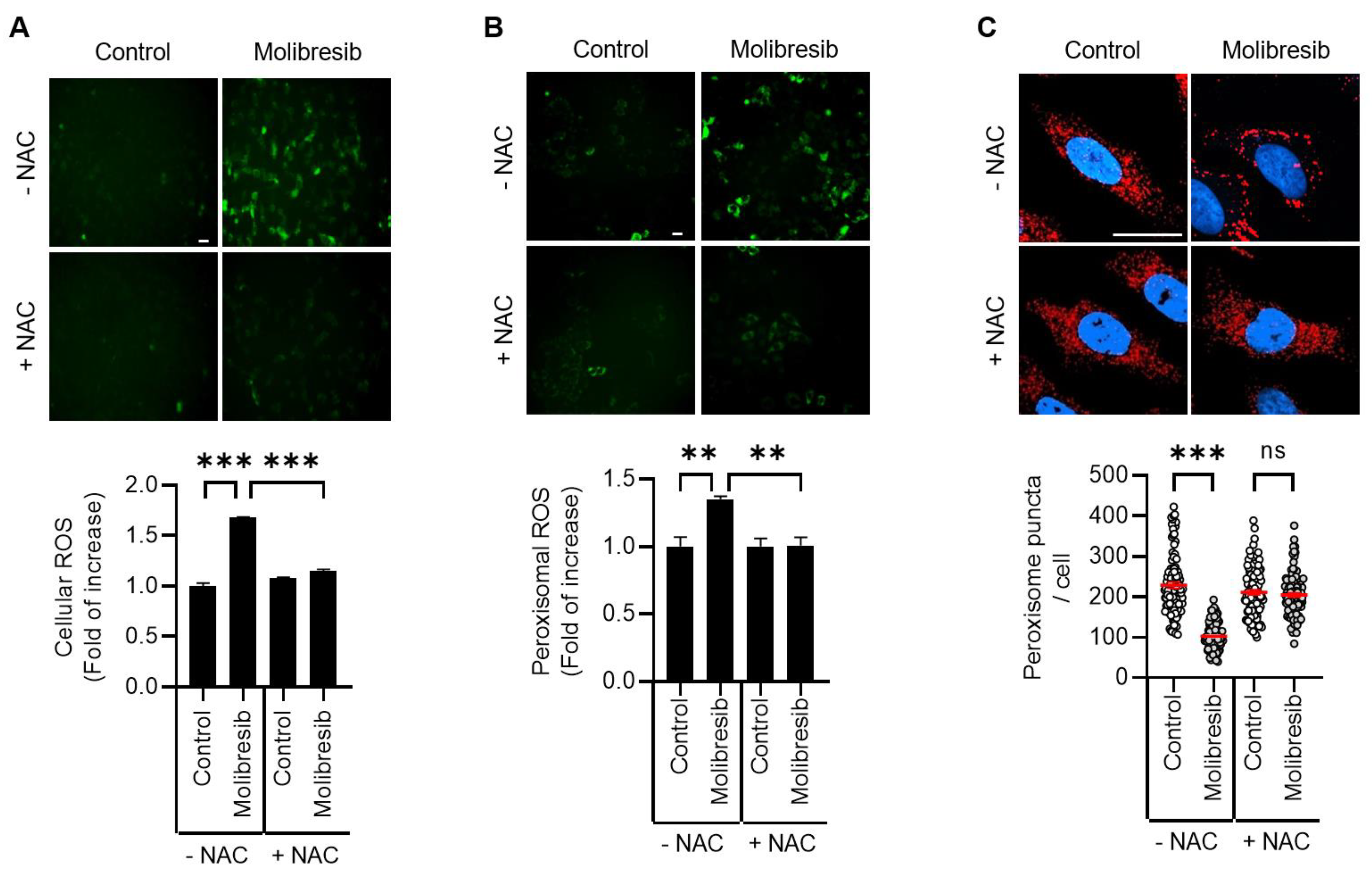
3.3. Molibresib Promotes Pexophagy by Increasing ROS and ATM Activation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Jo, D.S.; Park, N.Y.; Cho, D.-H. Peroxisome quality control and dysregulated lipid metabolism in neurodegenerative diseases. Exp. Mol. Med. 2020, 52, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Uzor, N.-E.; Mccullough, L.; Tsvetkov, A.S. Peroxisomal Dysfunction in Neurological Diseases and Brain Aging. Front. Cell. Neurosci. 2020, 14, 44. [Google Scholar] [CrossRef] [PubMed]
- Ramón, N.M.; Bartel, B. Interdependence of the Peroxisome-targeting Receptors in Arabidopsis thaliana: PEX7 Facilitates PEX5 Accumulation and Import of PTS1 Cargo into Peroxisomes. Mol. Biol. Cell 2010, 21, 1263–1271. [Google Scholar] [CrossRef]
- Kunze, M. Predicting Peroxisomal Targeting Signals to Elucidate the Peroxisomal Proteome of Mammals. Subcell. Biochem. 2018, 89, 157–199. [Google Scholar] [CrossRef]
- Gould, S.J.; Collins, C.S. Opinion: Peroxisomal-protein import: Is it really that complex? Nat. Rev. Mol. Cell Biol. 2002, 3, 382–389. [Google Scholar] [CrossRef]
- Weller, S.; Gould, S.J.; Valle, D. Peroxisome Biogenesis Disorders. Annu. Rev. Genom. Hum. Genet. 2003, 4, 165–211. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, C.; D’Agostino, M.D.; Braverman, N. Peroxisome biogenesis disorders. Transl. Sci. Rare Dis. 2016, 1, 111–144. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (4th edition). Autophagy 2020, 17, 1–382. [Google Scholar] [CrossRef]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Mizushima, N. The ATG conjugation systems in autophagy. Curr. Opin. Cell Biol. 2020, 63, 1–10. [Google Scholar] [CrossRef]
- Sargent, G.; Van Zutphen, T.; Shatseva, T.; Zhang, L.; Di Giovanni, V.; Bandsma, R.; Kim, P.K. PEX2 is the E3 ubiquitin ligase required for pexophagy during starvation. J. Cell Biol. 2016, 214, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Tripathi, D.; Jing, J.; Alexander, A.; Kim, J.; Powell, R.T.; Dere, R.; Tait-Mulder, J.; Lee, J.-H.; Paull, T.T.; et al. ATM functions at the peroxisome to induce pexophagy in response to ROS. Nat. Cell Biol. 2015, 17, 1259–1269. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.-H.; Kim, Y.S.; Jo, D.S.; Choe, S.-K.; Jo, E.-K. Pexophagy: Molecular Mechanisms and Implications for Health and Diseases. Mol. Cells 2018, 41, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Jo, D.S.; Bae, D.-J.; Park, S.J.; Seo, H.M.; Kim, H.B.; Oh, J.S.; Chang, J.W.; Kim, S.-Y.; Shin, J.-W.; Cho, D.-H. Pexophagy is induced by increasing peroxisomal reactive oxygen species in 1′10-phenanthroline-treated cells. Biochem. Biophys. Res. Commun. 2015, 467, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Jin, A.; Lee, J.N.; Kim, M.S.; Kwak, S.; Kim, S.-J.; Song, K.; Choe, S.-K.; Park, R. 2,2′-dipyridyl induces pexophagy. Biochem. Biophys. Res. Commun. 2016, 469, 941–947. [Google Scholar] [CrossRef]
- Lee, J.N.; Dutta, R.K.; Maharjan, Y.; Liu, Z.-Q.; Lim, J.-Y.; Kim, S.-J.; Cho, D.-H.; So, H.-S.; Choe, S.-K.; Park, R. Catalase inhibition induces pexophagy through ROS accumulation. Biochem. Biophys. Res. Commun. 2018, 501, 696–702. [Google Scholar] [CrossRef]
- Hirota, Y.; Yamashita, S.-I.; Kurihara, Y.; Jin, X.; Aihara, M.; Saigusa, T.; Kang, D.; Kanki, T. Mitophagy is primarily due to alternative autophagy and requires the MAPK1 and MAPK14 signaling pathways. Autophagy 2015, 11, 332–343. [Google Scholar] [CrossRef]
- Cheung, K.L.; Kim, C.; Zhou, M.-M. The Functions of BET Proteins in Gene Transcription of Biology and Diseases. Front. Mol. Biosci. 2021, 8, 728777. [Google Scholar] [CrossRef]
- Jo, D.S.; Park, S.J.; Kim, A.-K.; Park, N.Y.; Kim, J.B.; Bae, J.-E.; Park, H.J.; Shin, J.H.; Chang, J.W.; Kim, P.K.; et al. Loss of HSPA9 induces peroxisomal degradation by increasing pexophagy. Autophagy 2020, 16, 1989–2003. [Google Scholar] [CrossRef]
- Cousin, S.; Blay, J.; Garcia, I.B.; de Bono, J.S.; Le Tourneau, C.; Moreno, V.; Trigo, J.; Hann, C.L.; Azad, A.A.; Im, S.A.; et al. Safety, pharmacokinetic, pharmacodynamic and clinical activity of molibresib for the treatment of nuclear protein of the testis carcinoma and other cancers: Results of a Phase I/II open-label, dose escalation study. Int. J. Cancer 2021, 150, 993–1006. [Google Scholar] [CrossRef]
- Sakamaki, J.-I.; Wilkinson, S.; Hahn, M.; Tasdemir, N.; O’Prey, J.; Clark, W.; Hedley, A.; Nixon, C.; Long, J.S.; New, M.; et al. Bromodomain Protein BRD4 Is a Transcriptional Repressor of Autophagy and Lysosomal Function. Mol. Cell 2017, 66, 517–532.e9. [Google Scholar] [CrossRef] [PubMed]
- Wakita, M.; Takahashi, A.; Sano, O.; Loo, T.M.; Imai, Y.; Narukawa, M.; Iwata, H.; Matsudaira, T.; Kawamoto, S.; Ohtani, N.; et al. A BET family protein degrader provokes senolysis by targeting NHEJ and autophagy in senescent cells. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Klionsky, D.J. BRD4 is a newly characterized transcriptional regulator that represses autophagy and lysosomal function. Autophagy 2017, 13, 1801–1803. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, Y.-S.; Kim, D.E.; Lee, J.S.; Song, J.H.; Kim, H.-G.; Cho, D.-H.; Jeong, S.-Y.; Jin, D.-H.; Jang, S.J.; et al. BIX-01294 induces autophagy-associated cell death via EHMT2/G9a dysfunction and intracellular reactive oxygen species production. Autophagy 2013, 9, 2126–2139. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Kim, P.S.; Kim, E.S.; Park, S.J.; Jo, Y.K.; Hwang, J.J.; Park, T.J.; Chang, J.W.; Seo, J.-H.; Cho, D.-H. BIX-01294-induced autophagy regulates elongation of primary cilia. Biochem. Biophys. Res. Commun. 2015, 460, 428–433. [Google Scholar] [CrossRef]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, J.; Lu, X.; Tan, T.-Z.; Chng, W.-J. BET Bromodomain inhibition promotes De-repression of TXNIP and activation of ASK1-MAPK pathway in acute myeloid leukemia. BMC Cancer 2018, 18, 731. [Google Scholar] [CrossRef]
- Chen, Y.; Ning, J.; Cao, W.; Wang, S.; Du, T.; Jiang, J.; Feng, X.; Zhang, B. Research Progress of TXNIP as a Tumor Suppressor Gene Participating in the Metabolic Reprogramming and Oxidative Stress of Cancer Cells in Various Cancers. Front. Oncol. 2020, 10, 568574. [Google Scholar] [CrossRef]
- Coller, H.A. Is Cancer a Metabolic Disease? Am. J. Pathol. 2013, 184, 4–17. [Google Scholar] [CrossRef]
- Dahabieh, M.S.; Di Pietro, E.; Jangal, M.; Goncalves, C.; Witcher, M.; Braverman, N.E.; del Rincón, S.V. Peroxisomes and cancer: The role of a metabolic specialist in a disease of aberrant metabolism. Biochim. Biophys. Acta Rev. Cancer 2018, 1870, 103–121. [Google Scholar] [CrossRef]
- Kim, J.-A. Peroxisome Metabolism in Cancer. Cells 2020, 9, 1692. [Google Scholar] [CrossRef] [PubMed]
- Salcher, S.; Hermann, M.; Kiechl-Kohlendorfer, U.; Ausserlechner, M.J.; Obexer, P. C10ORF10/DEPP-mediated ROS accumulation is a critical modulator of FOXO3-induced autophagy. Mol. Cancer 2017, 16, 95. [Google Scholar] [CrossRef] [PubMed]
- Walter, K.M.; Schönenberger, M.J.; Trötzmüller, M.; Horn, M.; Elsässer, H.-P.; Moser, A.B.; Lucas, M.S.; Schwarz, T.; Gerber, P.A.; Faust, P.L.; et al. Hif-2α Promotes Degradation of Mammalian Peroxisomes by Selective Autophagy. Cell Metab. 2014, 20, 882–897. [Google Scholar] [CrossRef]
- Eberhart, T.; Schönenberger, M.J.; Walter, K.M.; Charles, K.N.; Faust, P.L.; Kovacs, W.J. Peroxisome-Deficiency and HIF-2α Signaling Are Negative Regulators of Ketohexokinase Expression. Front. Cell Dev. Biol. 2020, 8, 566. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Lorenzini, E.; Ciarrocchi, A. BRD4 and Cancer: Going beyond transcriptional regulation. Mol. Cancer 2018, 17, 164. [Google Scholar] [CrossRef]
- Shorstova, T.; Foulkes, W.D.; Witcher, M. Achieving clinical success with BET inhibitors as anti-cancer agents. Br. J. Cancer 2021, 124, 1478–1490. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, Y.H.; Jo, D.S.; Park, N.Y.; Bae, J.-E.; Kim, J.B.; Lee, H.J.; Kim, S.H.; Kim, S.H.; Lee, S.; Son, M.; et al. Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation. Cells 2022, 11, 2839. https://doi.org/10.3390/cells11182839
Kim YH, Jo DS, Park NY, Bae J-E, Kim JB, Lee HJ, Kim SH, Kim SH, Lee S, Son M, et al. Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation. Cells. 2022; 11(18):2839. https://doi.org/10.3390/cells11182839
Chicago/Turabian StyleKim, Yong Hwan, Doo Sin Jo, Na Yeon Park, Ji-Eun Bae, Joon Bum Kim, Ha Jung Lee, So Hyun Kim, Seong Hyun Kim, Sunwoo Lee, Mikyung Son, and et al. 2022. "Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation" Cells 11, no. 18: 2839. https://doi.org/10.3390/cells11182839
APA StyleKim, Y. H., Jo, D. S., Park, N. Y., Bae, J.-E., Kim, J. B., Lee, H. J., Kim, S. H., Kim, S. H., Lee, S., Son, M., Park, K., Jeong, K., Yeom, E., & Cho, D.-H. (2022). Inhibition of BRD4 Promotes Pexophagy by Increasing ROS and ATM Activation. Cells, 11(18), 2839. https://doi.org/10.3390/cells11182839