
An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Antibodies and Flow Cytometry
2.2. Enzyme-Linked Immunosorbent Cytokine Assays (ELISA)
2.3. Lentiviral Vectors
2.4. Human Monocyte-Derived Macrophage (MDM) Culture and Polarization
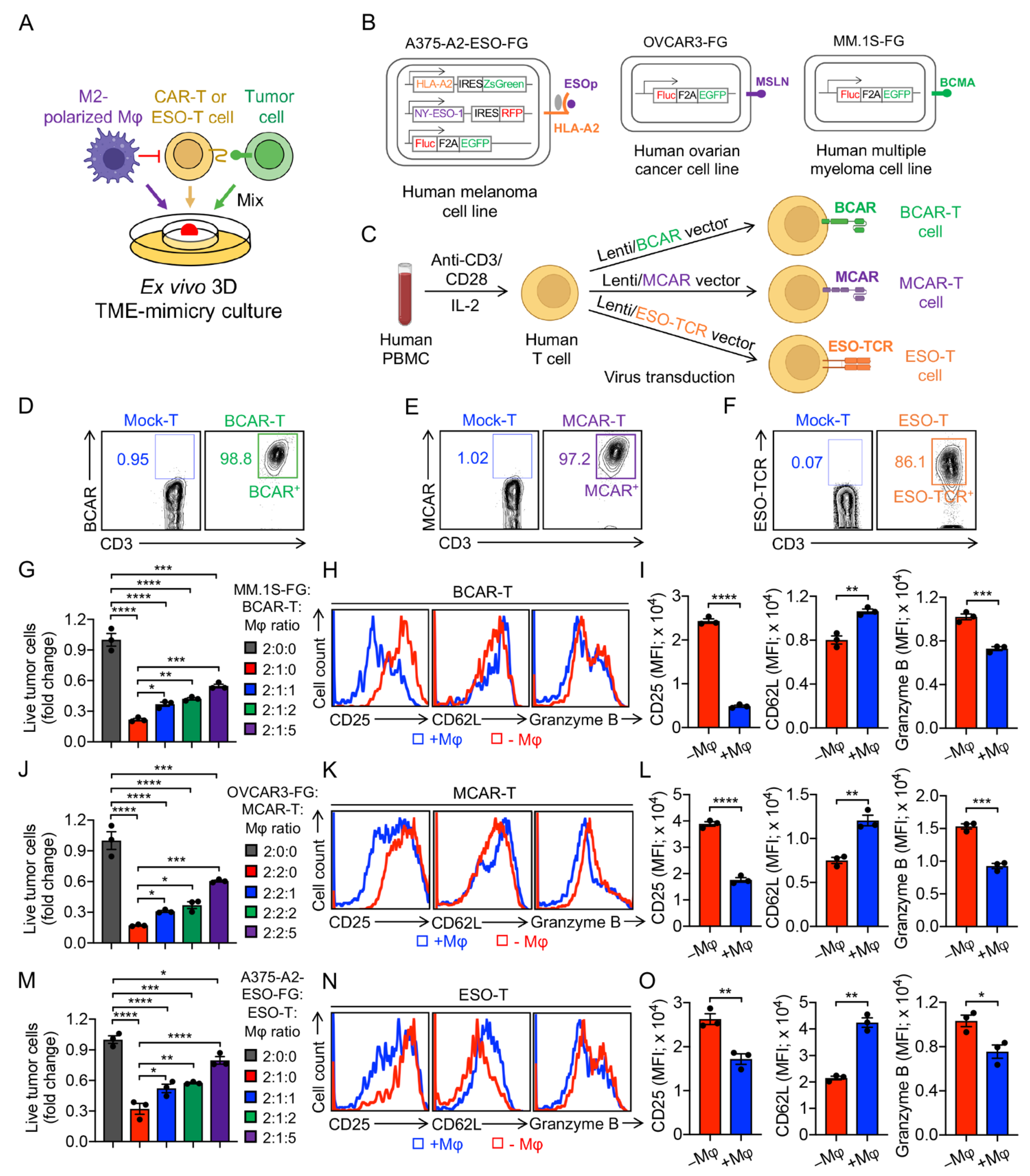
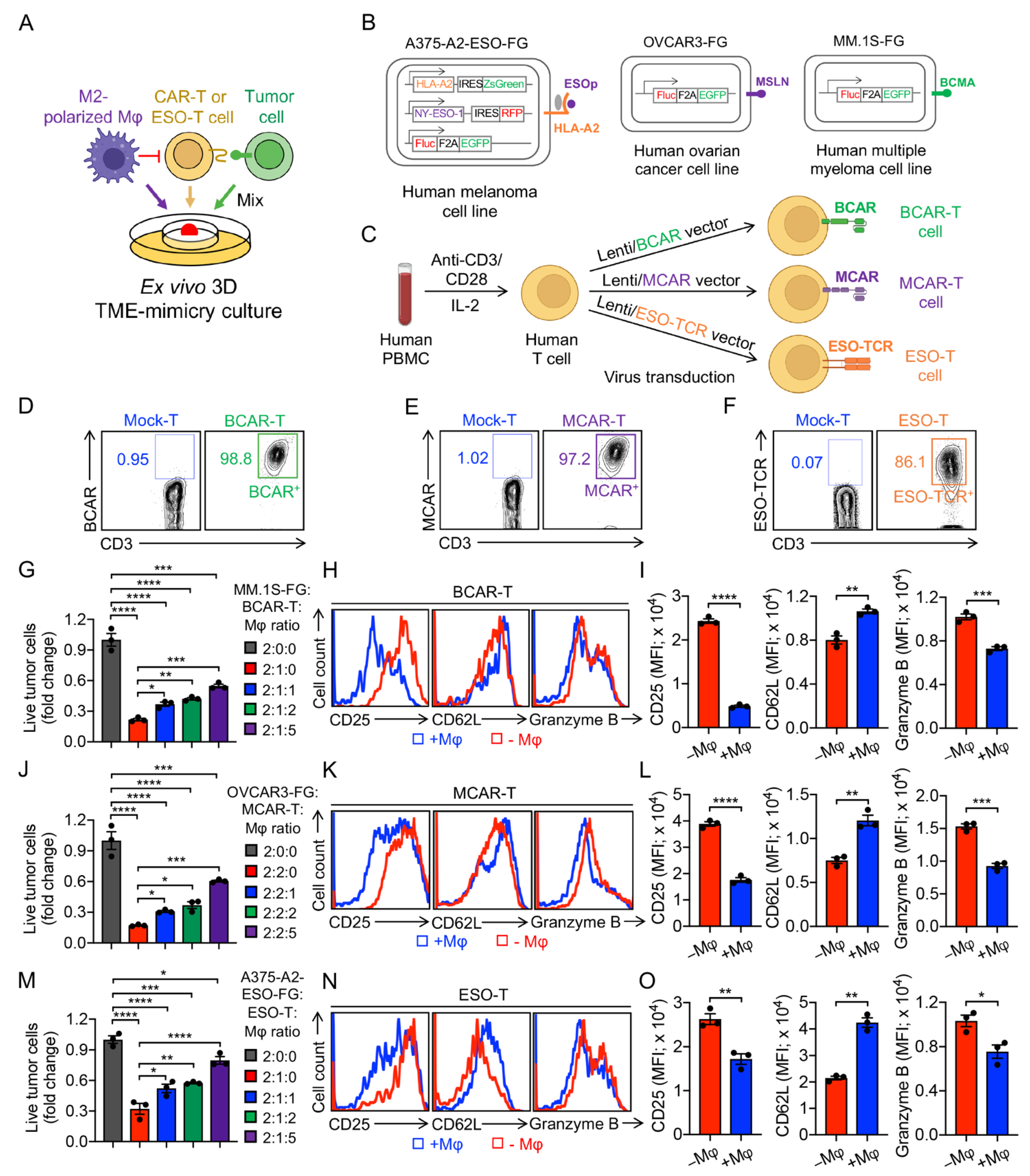
2.5. Generation of Human BCMA CAR-Engineered T (BCAR-T) Cells, Mesothelin CAR-Engineered T (MCAR-T) Cells, and NY-ESO-1-Specific TCR-Engineered T (ESO-T) Cells
2.6. In Vitro Mixed Mφ/T-Cell Reaction Assay
2.7. In Vitro Tumor Cell Killing Assay
2.8. Ex Vivo 3D TME-Mimicry Culture
3. Results
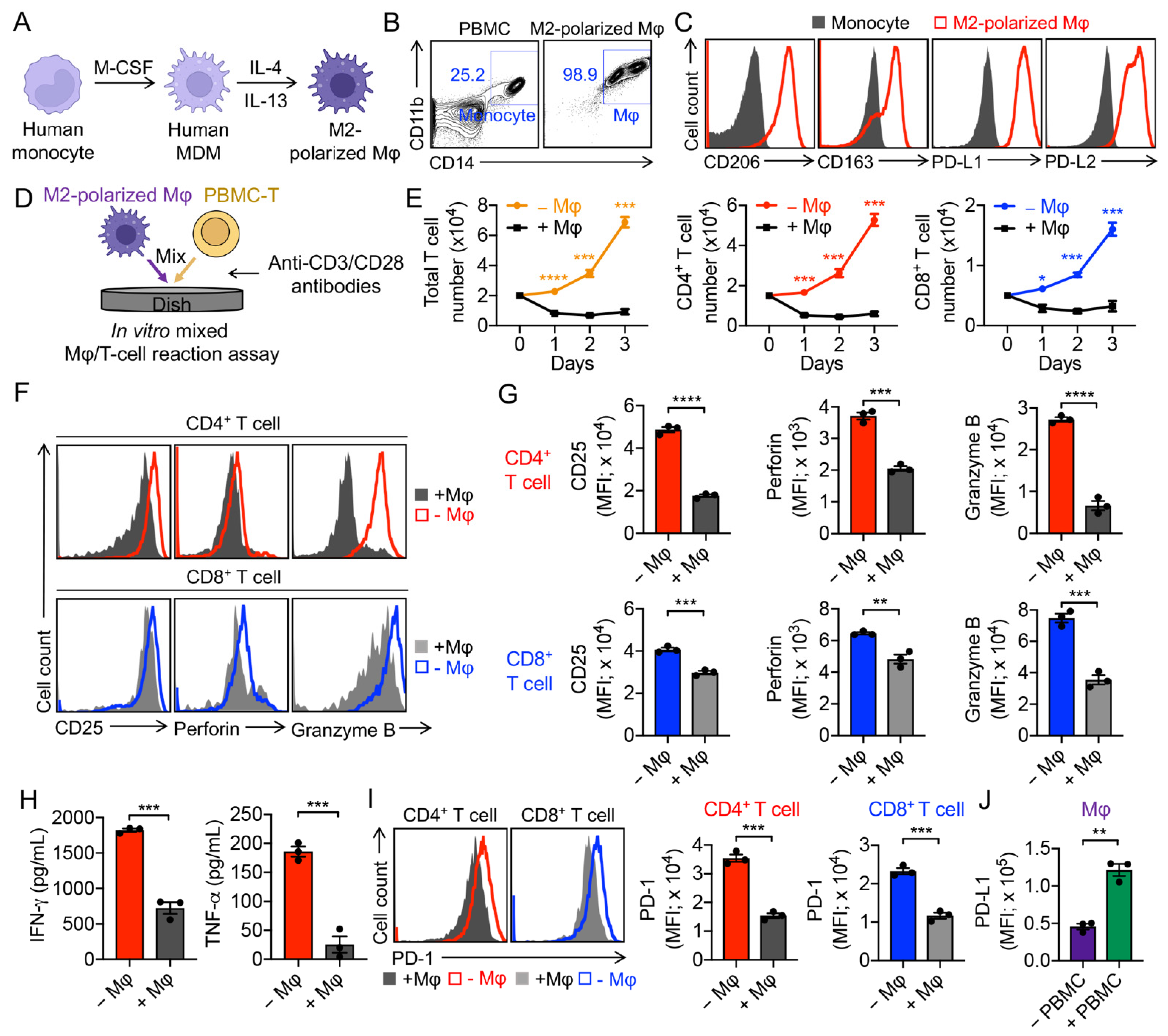
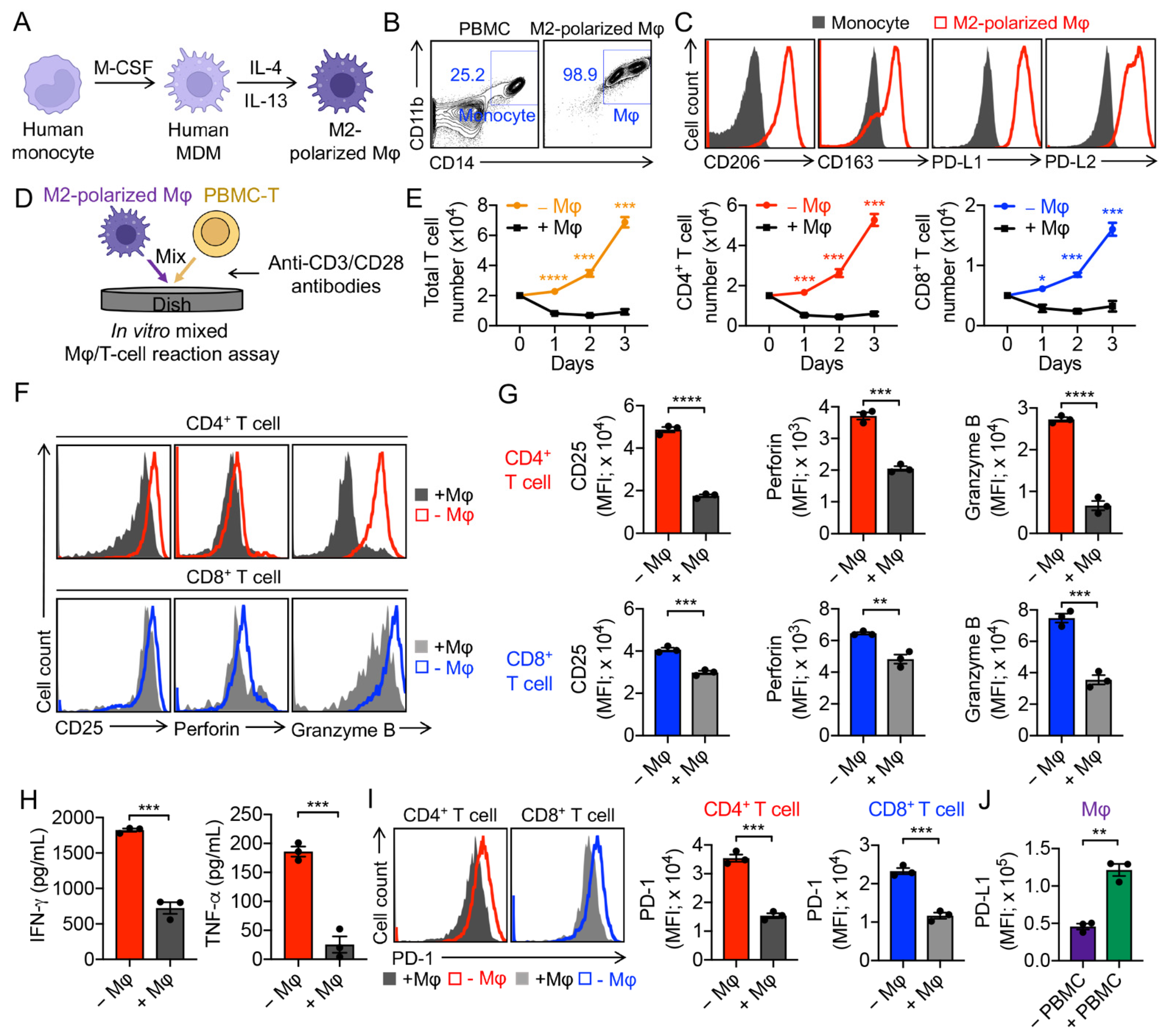
3.1. Generation of Human Monocyte-Derived M2-Polarized Macrophages
3.2. Validation of M2-Polarized Macrophage Immunosuppressive Function
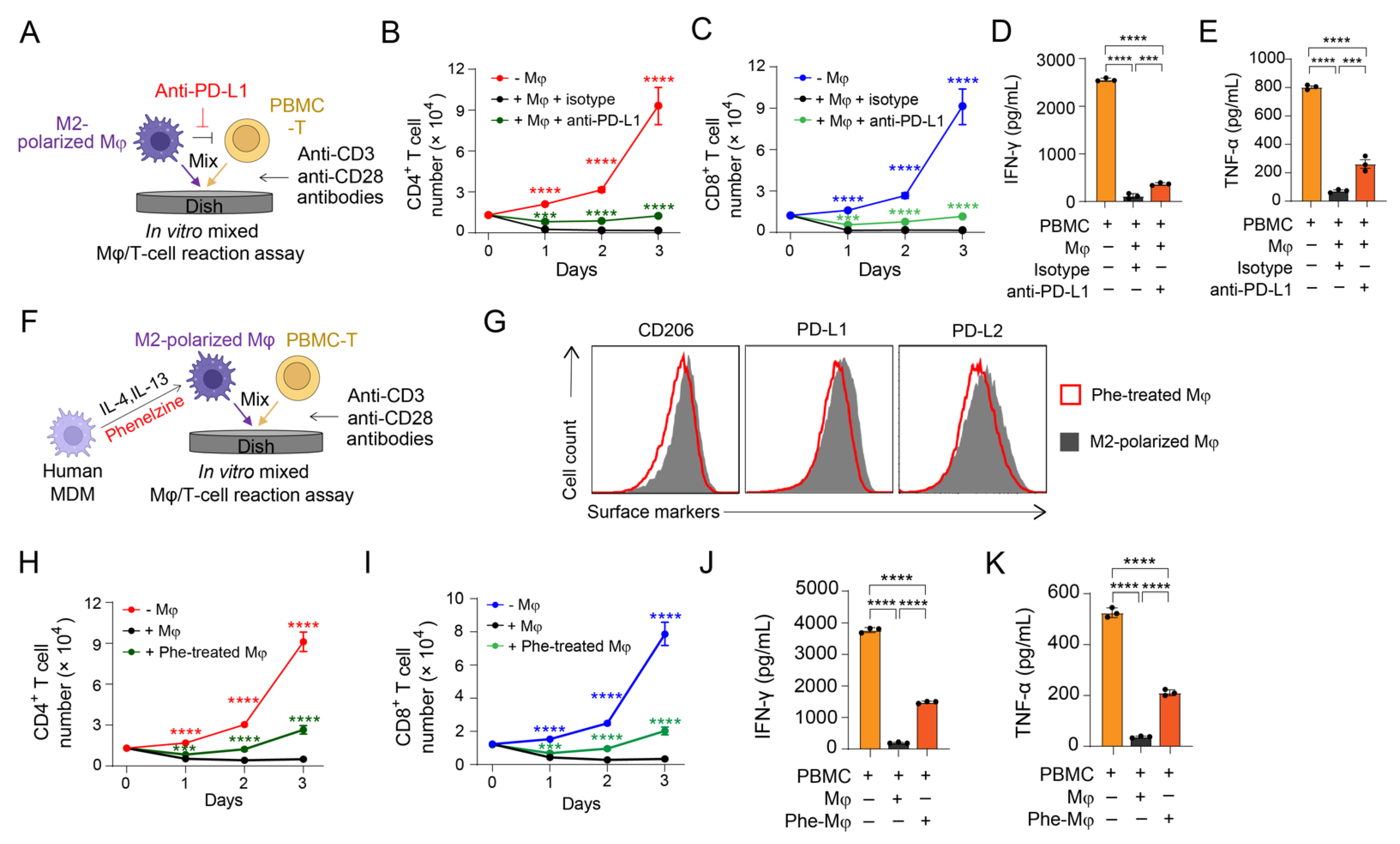
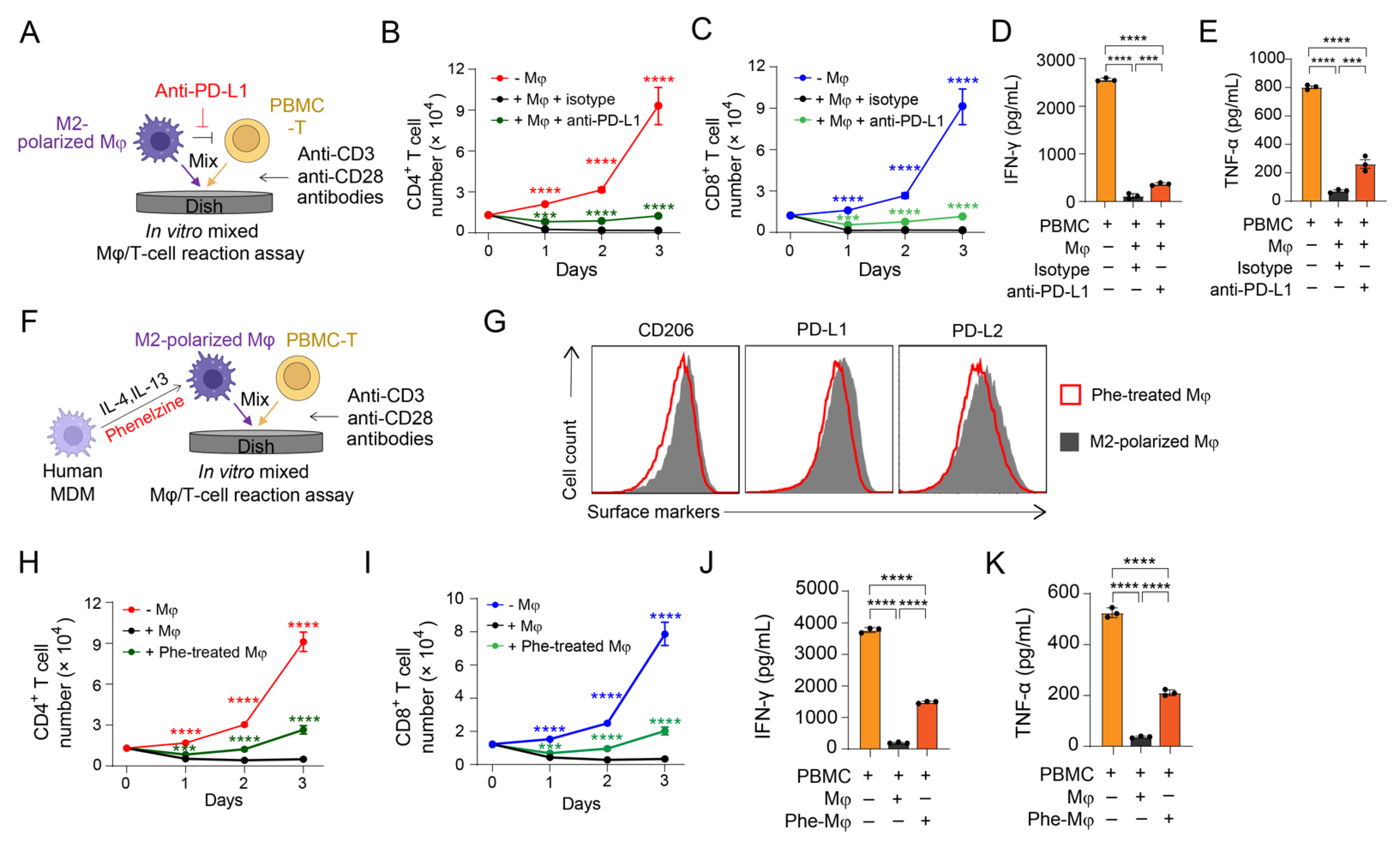
3.3. Validation of PD-1/PD-L1 Blockade on Antagonizing M2 Macrophage-Mediated Immunosuppression
3.4. Validation of MAO-A Blockade on Antagonizing M2 Macrophage-Mediated Immunosuppression
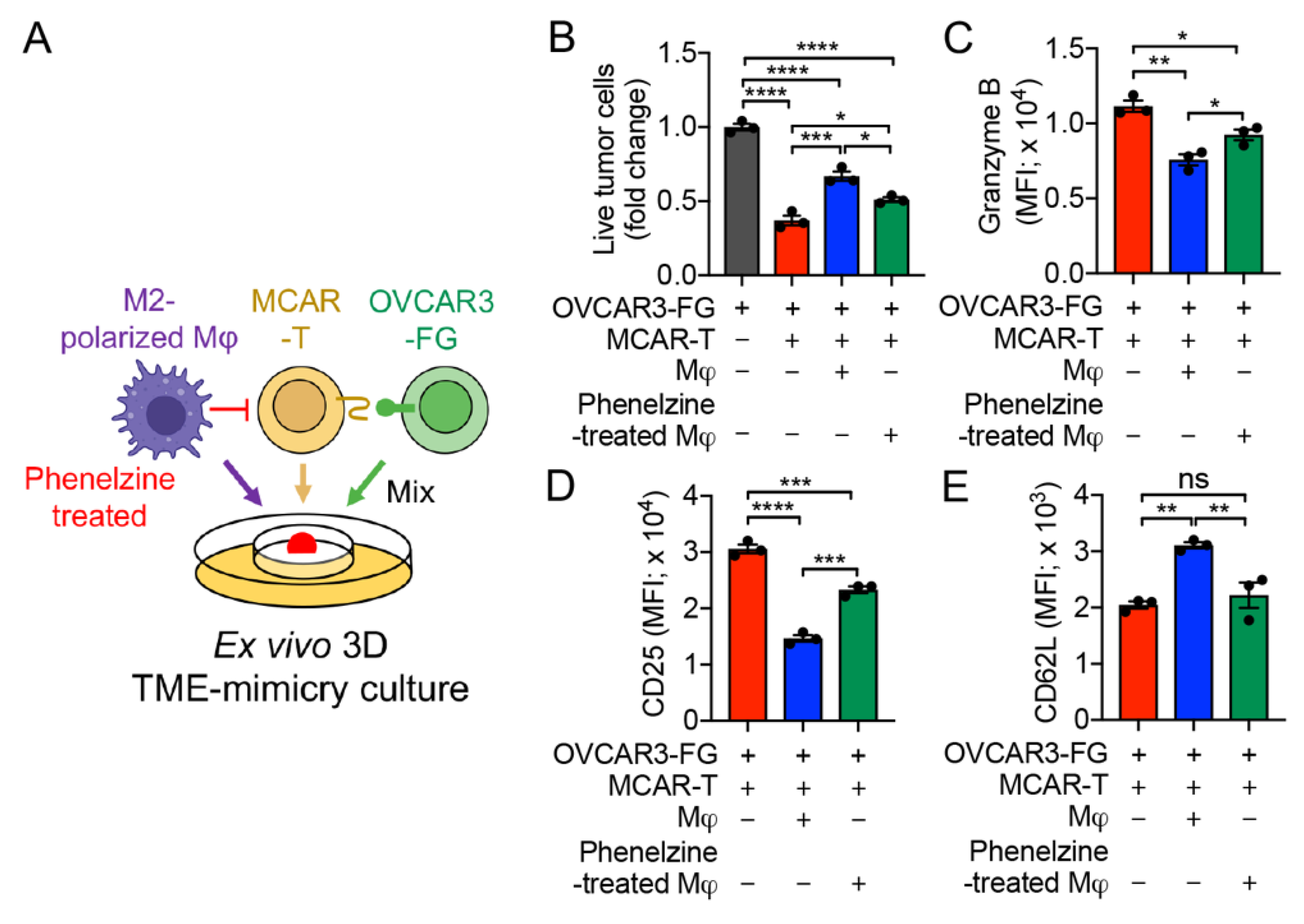
3.5. Development of an Ex Vivo 3D TME-Mimicry Culture to Study TAM Modulation of T-Cell Antitumor Reactivity
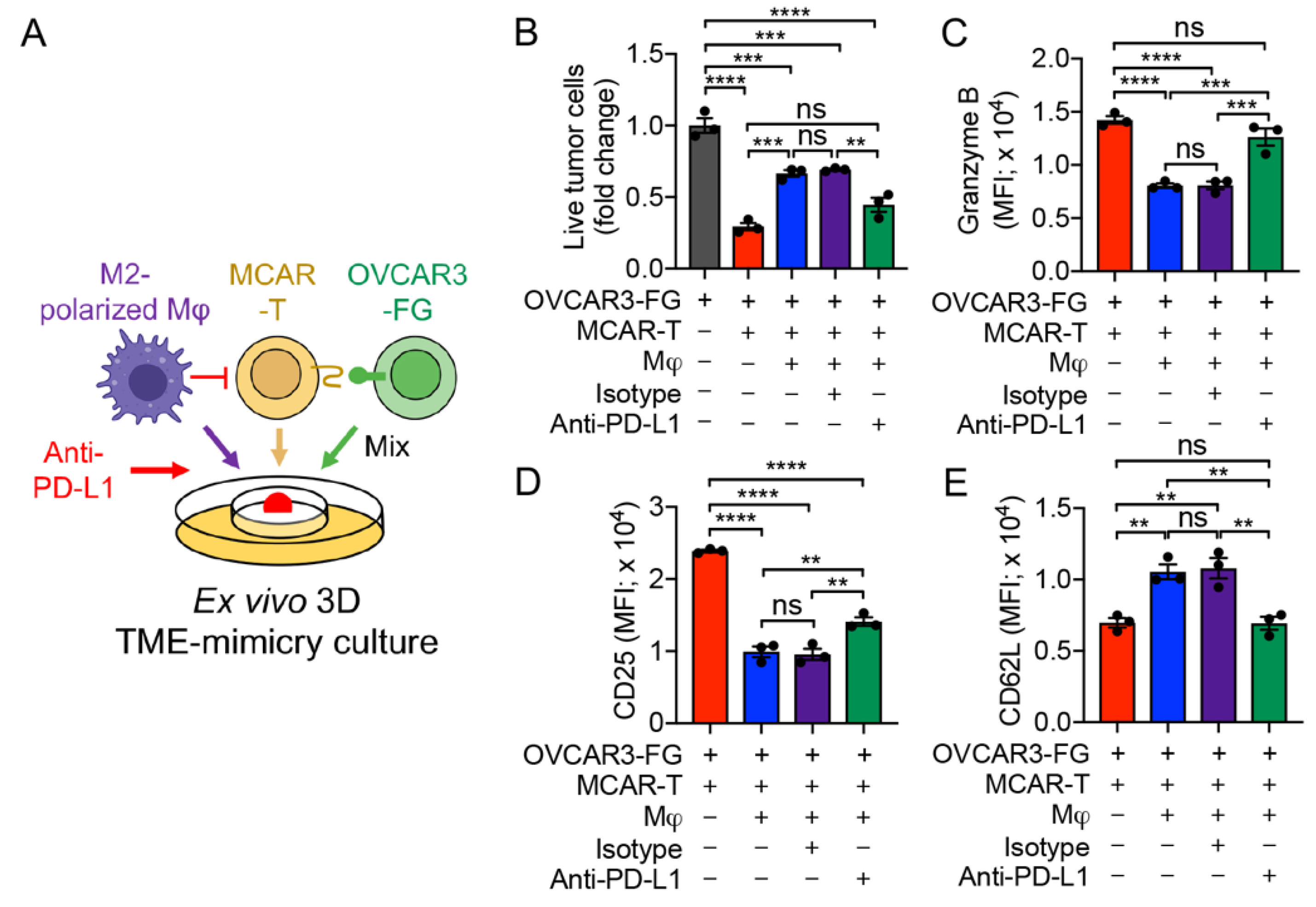
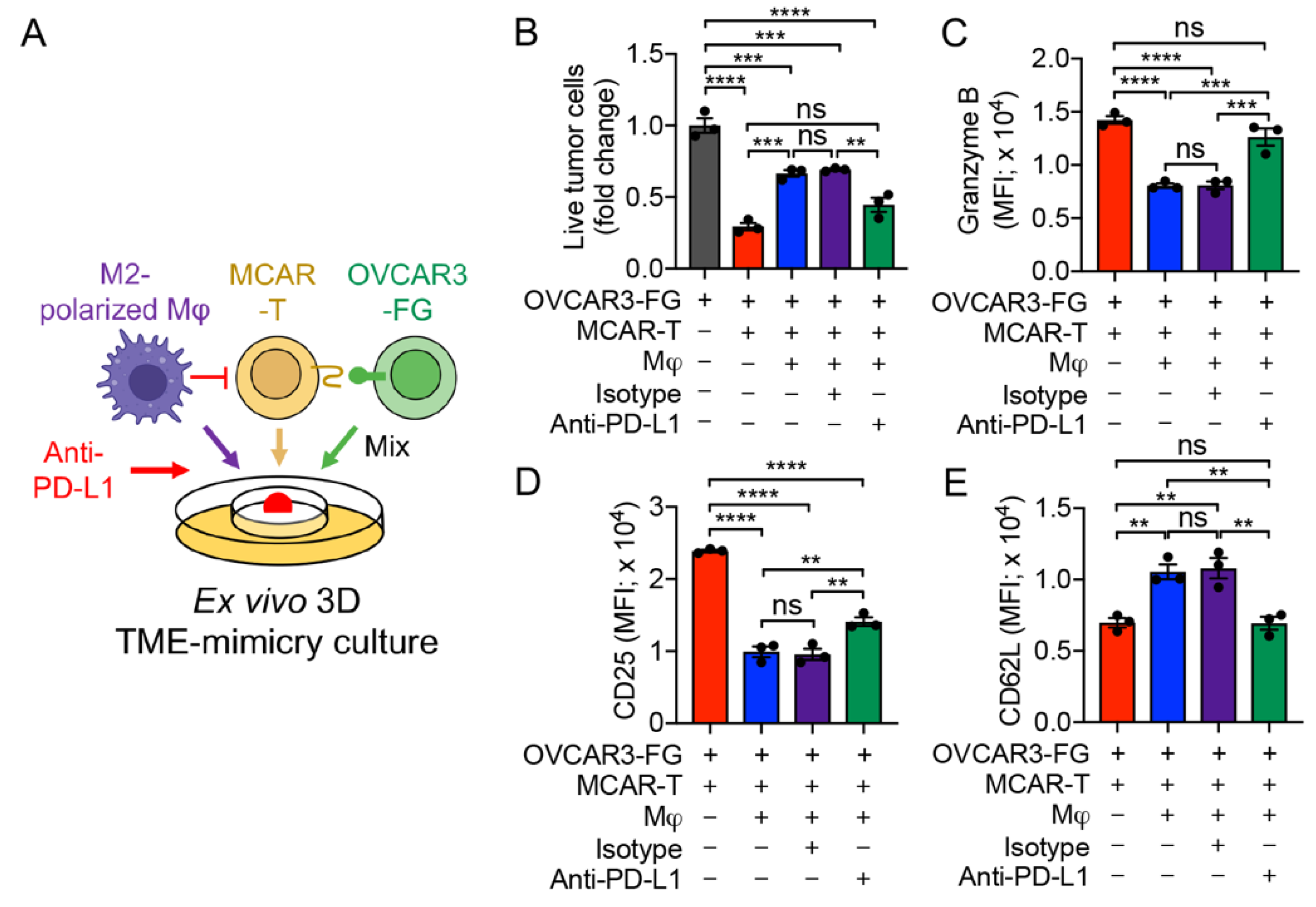
3.6. PD-1/PD-L1 Blockade Antagonizes TAM-Suppression of T-Cell Antitumor Reactivity
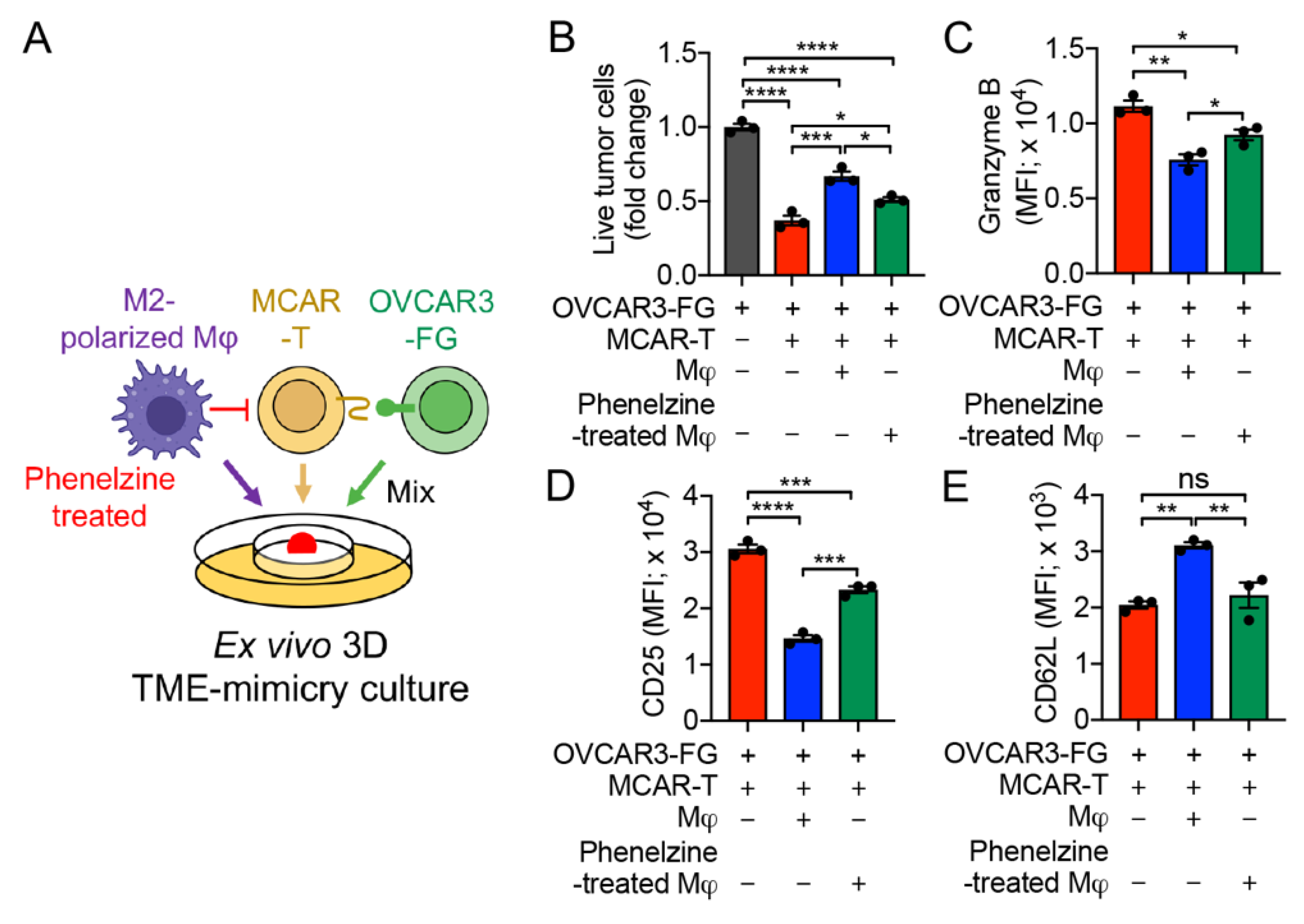
3.7. MAO-A Blockade Antagonizes TAM-Suppression of T-Cell Antitumor Reactivity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chen, Y.; Song, Y.; Du, W.; Gong, L.; Chang, H.; Zou, Z. Tumor-associated macrophages: An accomplice in solid tumor progression. J. Biomed. Sci. 2019, 26, 78. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Lin, K.; Li, X.; Yuan, X.; Xu, P.; Ni, P.; Xu, D. Redefining Tumor-Associated Macrophage Subpopulations and Functions in the Tumor Microenvironment. Front. Immunol. 2020, 11, 1731. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Yrigoyen, M.; Cassetta, L.; Pollard, J.W. Macrophage targeting in cancer. Ann. N. Y. Acad. Sci. 2021, 1499, 18–41. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Gyamfi, J.; Jang, H.; Koo, J.S. The role of tumor-associated macrophage in breast cancer biology. Histol. Histopathol. 2018, 33, 133–145. [Google Scholar] [CrossRef] [PubMed]
- Yahaya, M.A.F.; Lila, M.A.M.; Ismail, S.; Zainol, M.; Afizan, N.A.R.N.M. Tumour-Associated Macrophages (TAMs) in Colon Cancer and How to Reeducate Them. J. Immunol. Res. 2019, 2019, 2368249. [Google Scholar] [CrossRef] [PubMed]
- Ngambenjawong, C.; Gustafson, H.H.; Pun, S.H. Progress in tumor-associated macrophage (TAM)-targeted therapeutics. Adv. Drug Deliv. Rev. 2017, 114, 206–221. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Cheng, T.; Zhan, J.; Peng, X.; Zhang, Y.; Wen, J.; Chen, X.; Ying, M. Targeting tumor-associated macrophages in the tumor microenvironment. Oncol. Lett. 2020, 20, 234. [Google Scholar] [CrossRef]
- Lin, Y.; Xu, J.; Lan, H. Tumor-associated macrophages in tumor metastasis: Biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019, 12, 76. [Google Scholar] [CrossRef]
- Wu, Q.; Zhou, W.; Yin, S.; Zhou, Y.; Chen, T.; Qian, J.; Su, R.; Hong, L.; Lu, H.; Zhang, F.; et al. Blocking Triggering Receptor Expressed on Myeloid Cells-1-Positive Tumor-Associated Macrophages Induced by Hypoxia Reverses Immunosuppression and Anti-Programmed Cell Death Ligand 1 Resistance in Liver Cancer. Hepatology 2019, 70, 198–214. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774.e7. [Google Scholar] [CrossRef] [Green Version]
- Giannone, G.; Ghisoni, E.; Genta, S.; Scotto, G.; Tuninetti, V.; Turinetto, M.; Valabrega, G. Immuno-Metabolism and Microenvironment in Cancer: Key Players for Immunotherapy. Int. J. Mol. Sci. 2020, 21, 4414. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gao, A.; Zhang, F.; Yang, Z.; Wang, S.; Fang, Y.; Li, J.; Wang, J.; Shi, W.; Wang, L.; et al. ILT4 inhibition prevents TAM- and dysfunctional T cell-mediated immunosuppression and enhances the efficacy of anti-PD-L1 therapy in NSCLC with EGFR activation. Theranostics 2021, 11, 3392–3416. [Google Scholar] [CrossRef] [PubMed]
- Petty, A.J.; Owen, D.H.; Yang, Y.; Huang, X. Targeting Tumor-Associated Macrophages in Cancer Immunotherapy. Cancers 2021, 13, 5318. [Google Scholar] [CrossRef] [PubMed]
- Timmers, M.; Roex, G.; Wang, Y.; Campillo-Davo, D.; Van Tendeloo, V.F.I.; Chu, Y.; Berneman, Z.N.; Luo, F.; Van Acker, H.H.; Anguille, S. Chimeric antigen receptor-modified T cell therapy in multiple myeloma: Beyond B cell maturation antigen. Front. Immunol. 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Aftab, B.T.; Sasu, B.; Krishnamurthy, J.; Gschweng, E.; Alcazer, V.; Depil, S. Toward “off-the-shelf” allogeneic CAR T cells. Adv. Cell Gene Ther. 2020, 3, 1–11. [Google Scholar] [CrossRef]
- Cohen, A.D. CAR T Cells and Other Cellular Therapies for Multiple Myeloma: 2018 Update. Am. Soc. Clin. Oncol. Educ. B. 2018, 38, e6–e15. [Google Scholar] [CrossRef]
- Jensen, M.C.; Riddell, S.R. Designing chimeric antigen receptors to effectively and safely target tumors. Curr. Opin. Immunol. 2015, 33, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell Jr, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Petrova, P.S.; Viller, N.N.; Wong, M.; Pang, X.; Lin, G.H.Y.; Dodge, K.; Chai, V.; Chen, H.; Lee, V.; House, V.; et al. TTI-621 (SIRPαFc): A CD47-Blocking Innate Immune Checkpoint Inhibitor with Broad Antitumor Activity and Minimal Erythrocyte Binding. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 1068–1079. [Google Scholar] [CrossRef] [Green Version]
- Weiskopf, K.; Jahchan, N.S.; Schnorr, P.J.; Cristea, S.; Ring, A.M.; Maute, R.L.; Volkmer, A.K.; Volkmer, J.-P.; Liu, J.; Lim, J.S.; et al. CD47-blocking immunotherapies stimulate macrophage-mediated destruction of small-cell lung cancer. J. Clin. Invest. 2016, 126, 2610–2620. [Google Scholar] [CrossRef]
- Sanford, D.E.; Belt, B.A.; Panni, R.Z.; Mayer, A.; Deshpande, A.D.; Carpenter, D.; Mitchem, J.B.; Plambeck-Suess, S.M.; Worley, L.A.; Goetz, B.D.; et al. Inflammatory monocyte mobilization decreases patient survival in pancreatic cancer: A role for targeting the CCL2/CCR2 axis. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2013, 19, 3404–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Knolhoff, B.L.; Meyer, M.A.; Nywening, T.M.; West, B.L.; Luo, J.; Wang-Gillam, A.; Goedegebuure, S.P.; Linehan, D.C.; DeNardo, D.G. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res. 2014, 74, 5057–5069. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoves, S.; Ooi, C.-H.; Wolter, C.; Sade, H.; Bissinger, S.; Schmittnaegel, M.; Ast, O.; Giusti, A.M.; Wartha, K.; Runza, V.; et al. Rapid activation of tumor-associated macrophages boosts preexisting tumor immunity. J. Exp. Med. 2018, 215, 859–876. [Google Scholar] [CrossRef]
- Xiang, X.; Wang, J.; Lu, D.; Xu, X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal. Transduct. Target. Ther. 2021, 6, 75. [Google Scholar] [CrossRef] [PubMed]
- Barré-Sinoussi, F.; Montagutelli, X. Animal models are essential to biological research: Issues and perspectives. Futur. Sci. OA 2015, 1, FSO63. [Google Scholar] [CrossRef] [Green Version]
- Pinto, C.; Estrada, M.F.; Brito, C. In Vitro and Ex Vivo Models—The Tumor Microenvironment in a Flask. Adv. Exp. Med. Biol. 2020, 1219, 431–443. [Google Scholar] [CrossRef]
- Li, Y.-R.; Zhou, Y.; Kim, Y.J.; Zhu, Y.; Ma, F.; Yu, J.; Wang, Y.-C.; Chen, X.; Li, Z.; Zeng, S.; et al. Development of allogeneic HSC-engineered iNKT cells for off-the-shelf cancer immunotherapy. Cell Rep. Med. 2021, 2, 100449. [Google Scholar] [CrossRef]
- Zhu, Y.; Smith, D.J.; Zhou, Y.; Li, Y.R.; Yu, J.; Lee, D.; Wang, Y.C.; Di Biase, S.; Wang, X.; Hardoy, C.; et al. Development of Hematopoietic Stem Cell-Engineered Invariant Natural Killer T Cell Therapy for Cancer. Cell Stem Cell 2019, 25, 542–557.e9. [Google Scholar] [CrossRef]
- Jin, X.; Kruth, H.S. Culture of Macrophage Colony-stimulating Factor Differentiated Human Monocyte-derived Macrophages. J. Vis. Exp. 2016, 112, 54244. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8. [Google Scholar] [CrossRef]
- Wang, Y.-C.; Wang, X.; Yu, J.; Ma, F.; Li, Z.; Zhou, Y.; Zeng, S.; Ma, X.; Li, Y.-R.; Neal, A.; et al. Targeting monoamine oxidase A-regulated tumor-associated macrophage polarization for cancer immunotherapy. Nat. Commun. 2021, 12, 3530. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Tummala, S.; Kebriaei, P.; Wierda, W.; Gutierrez, C.; Locke, F.L.; Komanduri, K.V.; Lin, Y.; Jain, N.; Daver, N.; et al. Chimeric antigen receptor T-cell therapy—Assessment and management of toxicities. Nat. Rev. Clin. Oncol. 2018, 15, 47–62. [Google Scholar] [CrossRef]
- Sadelain, M.; Brentjens, R.; Rivière, I. The basic principles of chimeric antigen receptor design. Cancer Discov. 2013, 3, 388–398. [Google Scholar] [CrossRef] [Green Version]
- Perez, C.; Gruber, I.; Arber, C. Off-the-Shelf Allogeneic T Cell Therapies for Cancer: Opportunities and Challenges Using Naturally Occurring “Universal” Donor T Cells. Front. Immunol. 2020, 11, 583716. [Google Scholar] [CrossRef] [PubMed]
- Ghorashian, S.; Kramer, A.M.; Onuoha, S.; Wright, G.; Bartram, J.; Richardson, R.; Albon, S.J.; Casanovas-Company, J.; Castro, F.; Popova, B.; et al. Enhanced CAR T cell expansion and prolonged persistence in pediatric patients with ALL treated with a low-affinity CD19 CAR. Nat. Med. 2019, 25, 1408–1414. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-R.; Zhou, Y.; Kramer, A.; Yang, L. Engineering stem cells for cancer immunotherapy. Trends Cancer 2021, 7, 1059–1073. [Google Scholar] [CrossRef]
- Yu, B.; Jiang, T.; Liu, D. BCMA-targeted immunotherapy for multiple myeloma. J. Hematol. Oncol. 2020, 13, 125. [Google Scholar] [CrossRef]
- Klampatsa, A.; Dimou, V.; Albelda, S.M. Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert Opin. Biol. Ther. 2021, 21, 473–486. [Google Scholar] [CrossRef]
- Thomas, R.; Al-Khadairi, G.; Roelands, J.; Hendrickx, W.; Dermime, S.; Bedognetti, D.; Decock, J. NY-ESO-1 Based Immunotherapy of Cancer: Current Perspectives. Front. Immunol. 2018, 9, 947. [Google Scholar] [CrossRef]
- Wang, X.; Li, B.; Kim, Y.J.; Wang, Y.-C.; Li, Z.; Yu, J.; Zeng, S.; Ma, X.; Choi, I.Y.; Di Biase, S.; et al. Targeting monoamine oxidase A for T cell-based cancer immunotherapy. Sci. Immunol. 2021, 6, 2383. [Google Scholar] [CrossRef] [PubMed]
- Breslin, S.; O’Driscoll, L. The relevance of using 3D cell cultures, in addition to 2D monolayer cultures, when evaluating breast cancer drug sensitivity and resistance. Oncotarget 2016, 7, 45745–45756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, A.; Schlederer, M.; Pudelko, K.; Stadler, M.; Walter, S.; Unterleuthner, D.; Unger, C.; Kramer, N.; Hengstschläger, M.; Kenner, L.; et al. Comparison of cancer cells in 2D vs. 3D culture reveals differences in AKT-mTOR-S6K signaling and drug responses. J. Cell Sci. 2017, 130, 203–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melissaridou, S.; Wiechec, E.; Magan, M.; Jain, M.V.; Chung, M.K.; Farnebo, L.; Roberg, K. The effect of 2D and 3D cell cultures on treatment response, EMT profile and stem cell features in head and neck cancer. Cancer Cell Int. 2019, 19, 16. [Google Scholar] [CrossRef] [Green Version]
- Däster, S.; Amatruda, N.; Calabrese, D.; Ivanek, R.; Turrini, E.; Droeser, R.A.; Zajac, P.; Fimognari, C.; Spagnoli, G.C.; Iezzi, G.; et al. Induction of hypoxia and necrosis in multicellular tumor spheroids is associated with resistance to chemotherapy treatment. Oncotarget 2017, 8, 1725–1736. [Google Scholar] [CrossRef] [Green Version]
- Diao, W.; Tong, X.; Yang, C.; Zhang, F.; Bao, C.; Chen, H.; Liu, L.; Li, M.; Ye, F.; Fan, Q.; et al. Behaviors of Glioblastoma Cells in in Vitro Microenvironments. Sci. Rep. 2019, 9, 85. [Google Scholar] [CrossRef] [Green Version]
- Berthiaume, F.; Moghe, P.V.; Toner, M.; Yarmush, M.L. Effect of extracellular matrix topology on cell structure, function, and physiological responsiveness: Hepatocytes cultured in a sandwich configuration. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1996, 10, 1471–1484. [Google Scholar] [CrossRef]
- Wen, Z.; Liao, Q.; Hu, Y.; You, L.; Zhou, L.; Zhao, Y. A spheroid-based 3-D culture model for pancreatic cancer drug testing, using the acid phosphatase assay. Braz. J. Med. Biol. Res. Rev. Bras. Pesqui. Med. E Biol. 2013, 46, 634–642. [Google Scholar] [CrossRef] [Green Version]
- de la Puente, P.; Muz, B.; Gilson, R.C.; Azab, F.; Luderer, M.; King, J.; Achilefu, S.; Vij, R.; Azab, A.K. 3D tissue-engineered bone marrow as a novel model to study pathophysiology and drug resistance in multiple myeloma. Biomaterials 2015, 73, 70–84. [Google Scholar] [CrossRef] [Green Version]
- Dillard, P.; Köksal, H.; Inderberg, E.-M.; Wälchli, S. A Spheroid Killing Assay by CAR T Cells. J. Vis. Exp. 2018, 142, e58785. [Google Scholar] [CrossRef] [Green Version]
- Sureban, S.M.; Berahovich, R.; Zhou, H.; Xu, S.; Wu, L.; Ding, K.; May, R.; Qu, D.; Bannerman-Menson, E.; Golubovskaya, V.; et al. DCLK1 Monoclonal Antibody-Based CAR-T Cells as a Novel Treatment Strategy against Human Colorectal Cancers. Cancers 2019, 12, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dangles-Marie, V.; Richon, S.; El-Behi, M.; Echchakir, H.; Dorothée, G.; Thiery, J.; Validire, P.; Vergnon, I.; Menez, J.; Ladjimi, M.; et al. A three-dimensional tumor cell defect in activating autologous CTLs is associated with inefficient antigen presentation correlated with heat shock protein-70 down-regulation. Cancer Res. 2003, 63, 3682–3687. [Google Scholar] [PubMed]
- Giannattasio, A.; Weil, S.; Kloess, S.; Ansari, N.; Stelzer, E.H.K.; Cerwenka, A.; Steinle, A.; Koehl, U.; Koch, J. Cytotoxicity and infiltration of human NK cells in in vivo-like tumor spheroids. BMC Cancer 2015, 15, 351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fei, L.; Ren, X.; Yu, H.; Zhan, Y. Targeting the CCL2/CCR2 Axis in Cancer Immunotherapy: One Stone, Three Birds? Front. Immunol. 2021, 12, 771210. [Google Scholar] [CrossRef]
- Beatty, G.L.; Chiorean, E.G.; Fishman, M.P.; Saboury, B.; Teitelbaum, U.R.; Sun, W.; Huhn, R.D.; Song, W.; Li, D.; Sharp, L.L.; et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science 2011, 331, 1612–1616. [Google Scholar] [CrossRef] [Green Version]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef] [PubMed]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Han, C.; Wang, S.; Fang, P.; Ma, Z.; Xu, L.; Yin, R. Cancer-associated fibroblasts: An emerging target of anti-cancer immunotherapy. J. Hematol. Oncol. 2019, 12, 86. [Google Scholar] [CrossRef]
- Lv, M.; Wang, K.; Huang, X.-J. Myeloid-derived suppressor cells in hematological malignancies: Friends or foes. J. Hematol. Oncol. 2019, 12, 105. [Google Scholar] [CrossRef] [Green Version]
- Scott, E.N.; Gocher, A.M.; Workman, C.J.; Vignali, D.A.A. Regulatory T Cells: Barriers of Immune Infiltration Into the Tumor Microenvironment. Front. Immunol. 2021, 12, 702726. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.-R.; Yu, Y.; Kramer, A.; Hon, R.; Wilson, M.; Brown, J.; Yang, L. An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells 2022, 11, 1583. https://doi.org/10.3390/cells11091583
Li Y-R, Yu Y, Kramer A, Hon R, Wilson M, Brown J, Yang L. An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells. 2022; 11(9):1583. https://doi.org/10.3390/cells11091583
Chicago/Turabian StyleLi, Yan-Ruide, Yanqi Yu, Adam Kramer, Ryan Hon, Matthew Wilson, James Brown, and Lili Yang. 2022. "An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy" Cells 11, no. 9: 1583. https://doi.org/10.3390/cells11091583
APA StyleLi, Y.-R., Yu, Y., Kramer, A., Hon, R., Wilson, M., Brown, J., & Yang, L. (2022). An Ex Vivo 3D Tumor Microenvironment-Mimicry Culture to Study TAM Modulation of Cancer Immunotherapy. Cells, 11(9), 1583. https://doi.org/10.3390/cells11091583