
The Role of miR-20 in Health and Disease of the Central Nervous System


Abstract
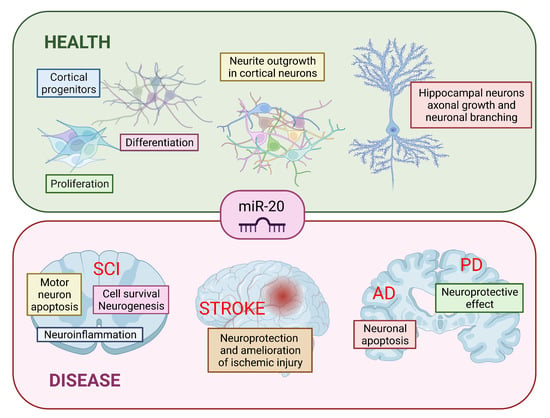
1. Introduction
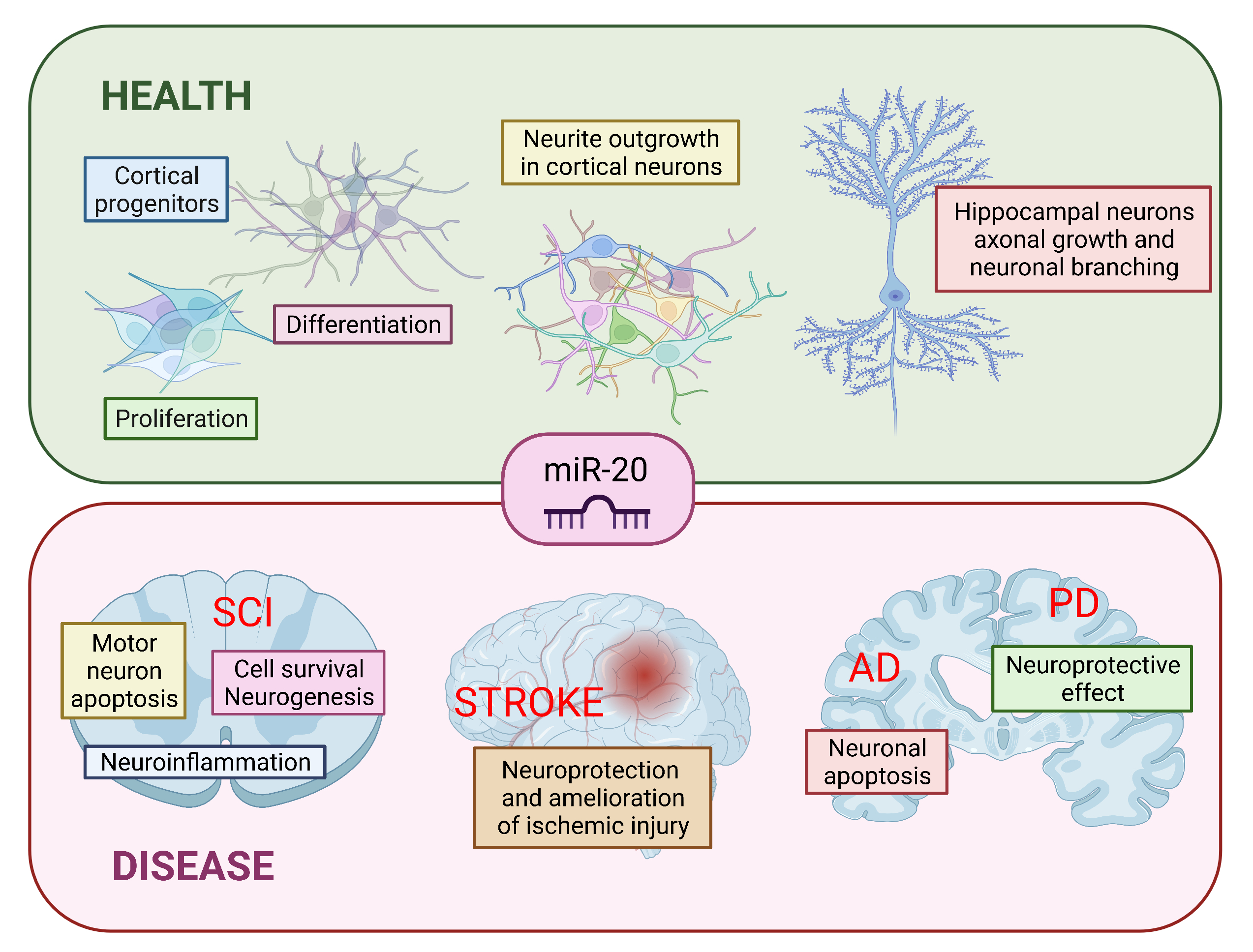
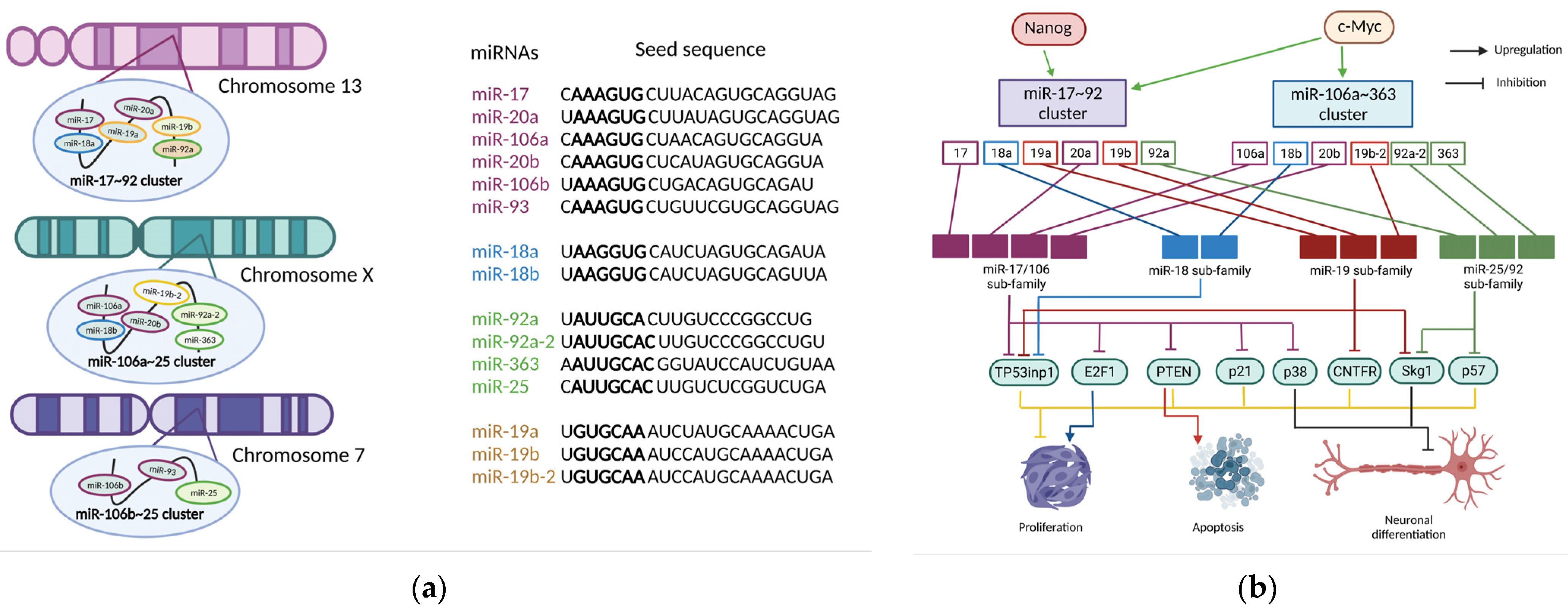
2. miR-17~92 Family
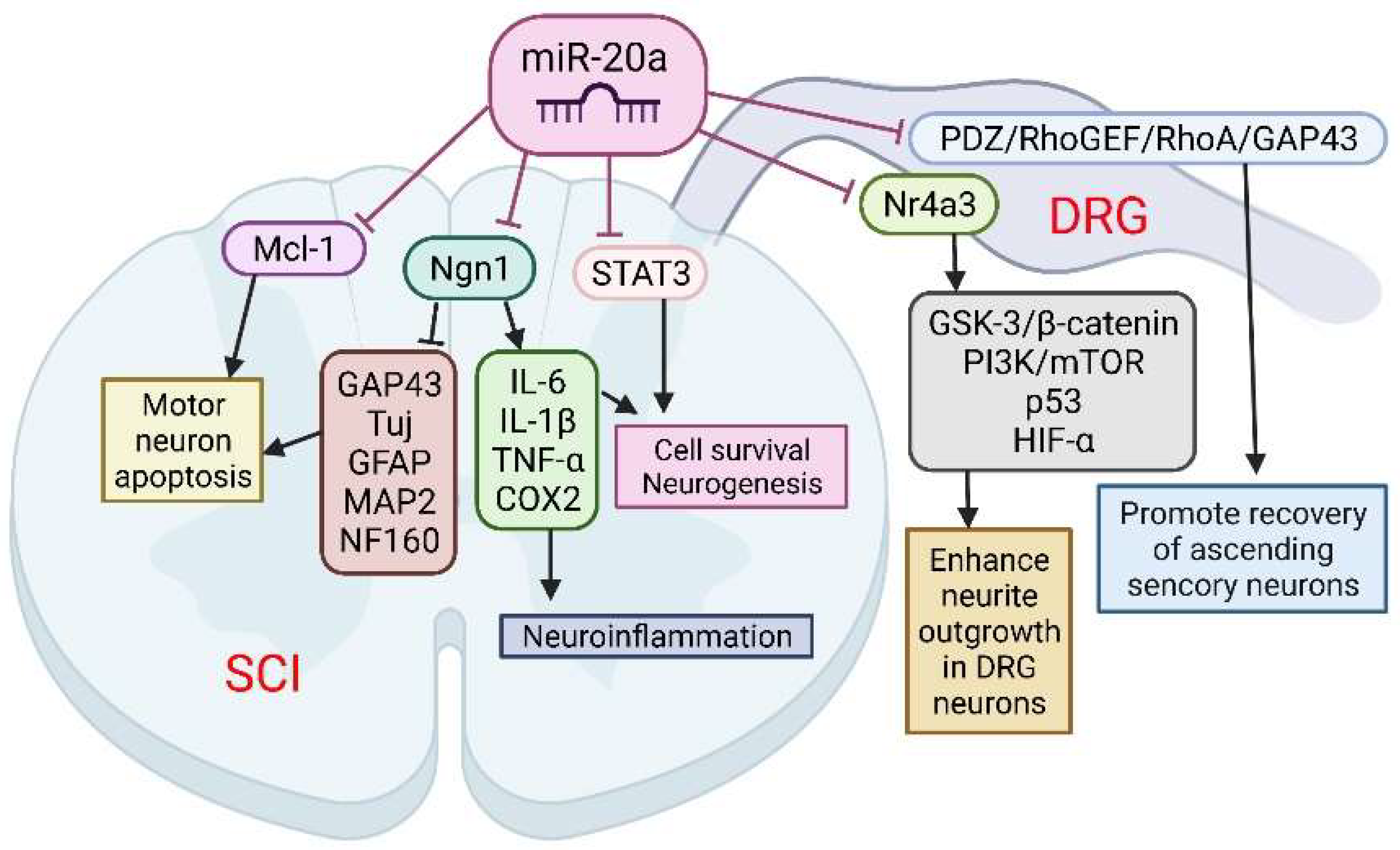
3. Spinal Cord Injury
3.1. Primary Phase
3.2. Secondary Phase
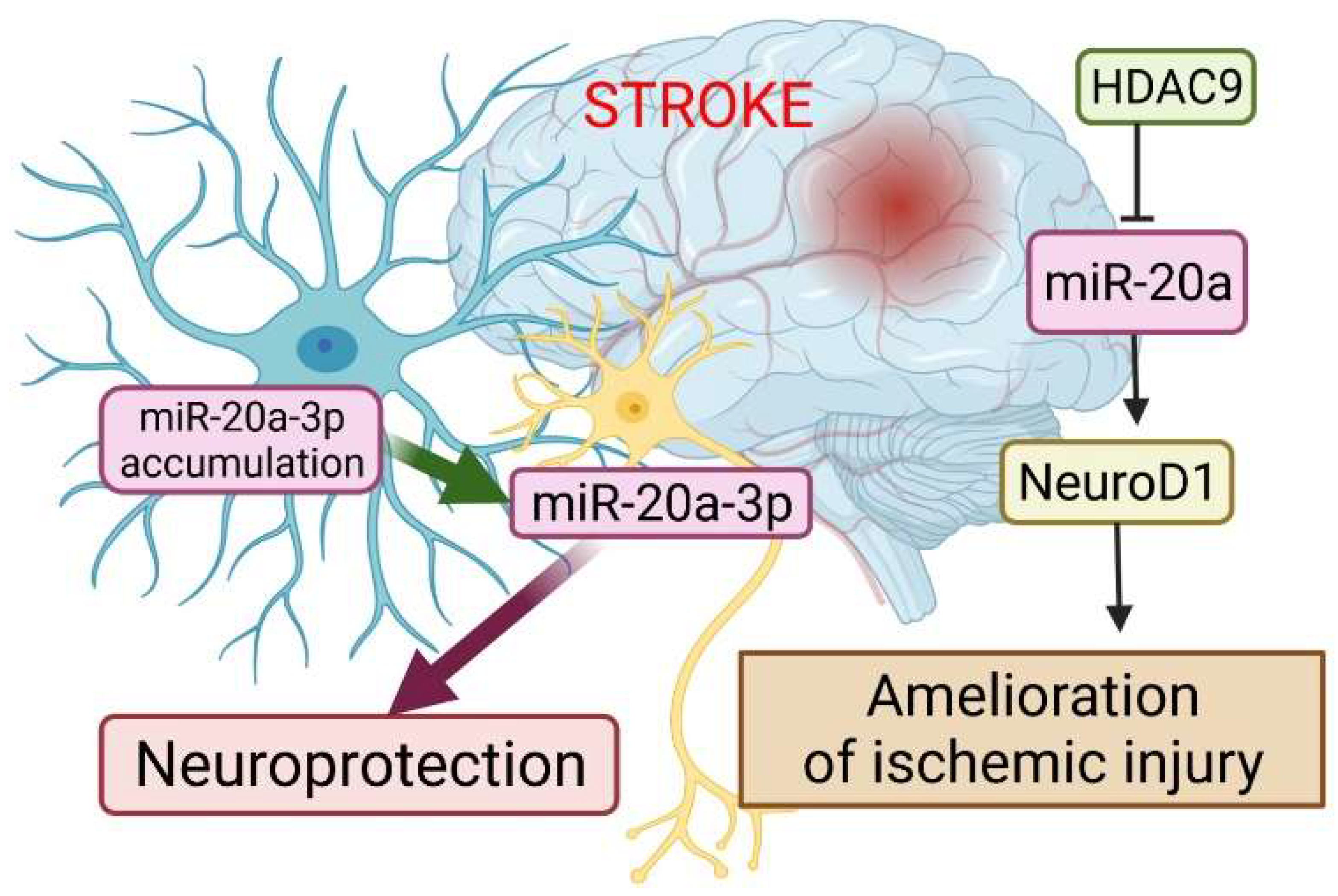
4. Stroke
5. Traumatic Brain Injury
6. Neurodegenerative Diseases
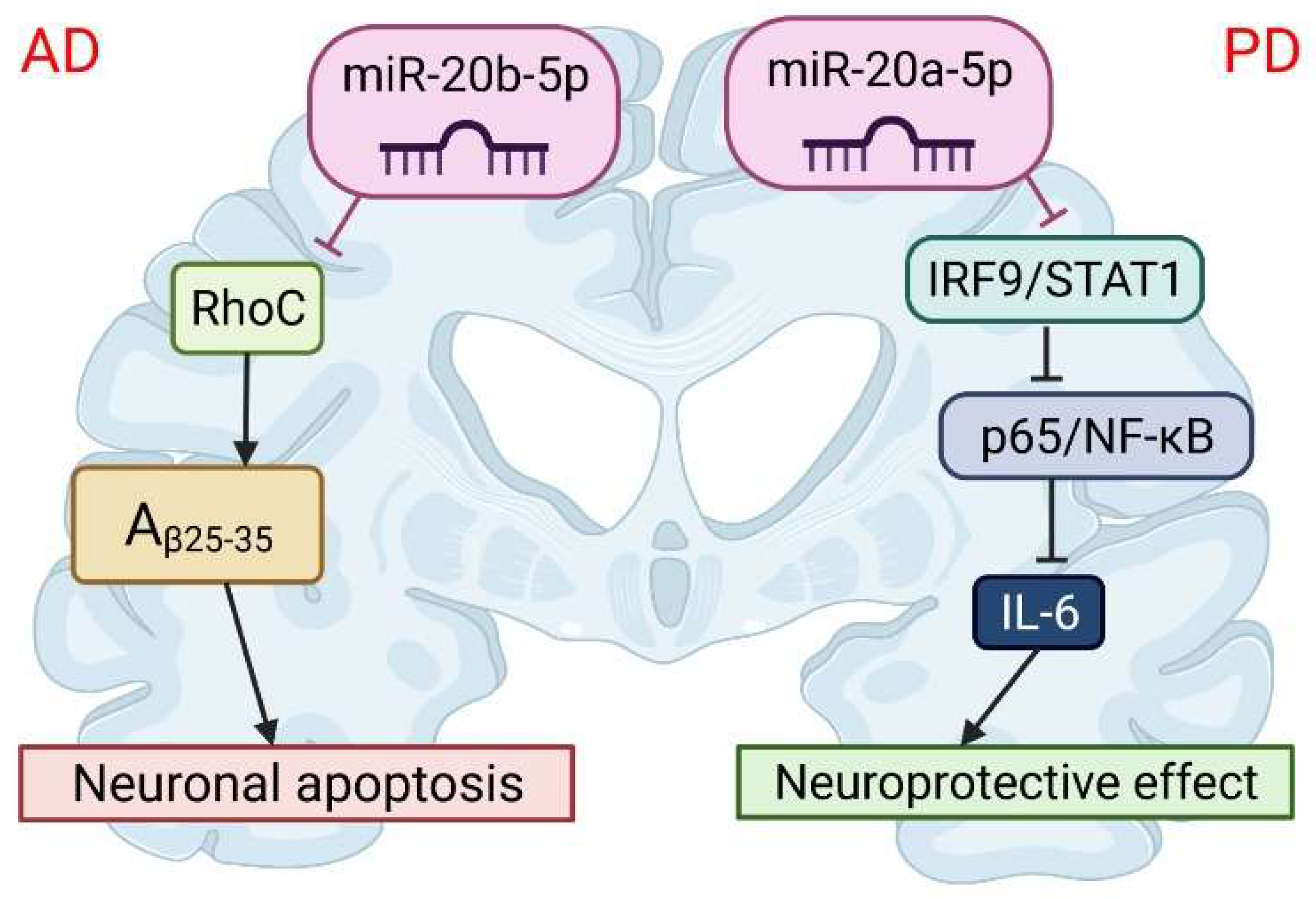
6.1. Alzheimer’s Disease
6.2. Parkinson’s Disease
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bhalala, O.G.; Srikanth, M.; Kessler, J.A. The Emerging Roles of MicroRNAs in CNS Injuries. Nat. Rev. Neurol. 2013, 9, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The Global Prevalence of Dementia: A Systematic Review and Metaanalysis. Alzheimer’s Dement. 2013, 9, 63. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, S.; Curcio, A.; Indolfi, C. Emerging Role of MicroRNAs in Cardiovascular Diseases. Circ. J. 2014, 78, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Dharap, A.; Bowen, K.; Place, R.; Li, L.-C.; Vemuganti, R. Transient Focal Ischemia Induces Extensive Temporal Changes in Rat Cerebral MicroRNAome. J. Cereb. Blood Flow Metab. 2009, 29, 675–687. [Google Scholar] [CrossRef]
- Nieto-Diaz, M.; Esteban, F.J.; Reigada, D.; Munoz-Galdeano, T.; Yunta, M.; Caballero-Lopez, M.; Navarro-Ruiz, R.; Del Aguila, A.; Maza, R.M. MicroRNA Dysregulation in Spinal Cord Injury: Causes, Consequences and Therapeutics. Front. Cell. Neurosci. 2014, 8, 53. [Google Scholar] [CrossRef]
- Ning, B.; Gao, L.; Liu, R.H.; Liu, Y.; Zhang, N.S.; Chen, Z.Y. MicroRNAs in Spinal Cord Injury: Potential Roles and Therapeutic Implications. Int. J. Biol. Sci. 2014, 10, 997–1006. [Google Scholar] [CrossRef]
- Rajgor, D. Macro Roles for MicroRNAs in Neurodegenerative Diseases. Non-Coding RNA Res. 2018, 3, 154–159. [Google Scholar] [CrossRef]
- Quinlan, S.; Kenny, A.; Medina, M.; Engel, T.; Jimenez-Mateos, E.M. MicroRNAs in Neurodegenerative Diseases. Int. Rev. Cell Mol. Biol. 2017, 334, 309–343. [Google Scholar] [CrossRef]
- Iida, A.; Shinoe, T.; Baba, Y.; Mano, H.; Watanabe, S. Dicer Plays Essential Roles for Retinal Development by Regulation of Survival and Differentiation. Investig. Ophthalmol. Vis. Sci. 2011, 52, 3008–3017. [Google Scholar] [CrossRef]
- Yang, P.; Cai, L.; Zhang, G.; Bian, Z.; Han, G. The Role of the MiR-17-92 Cluster in Neurogenesis and Angiogenesis in the Central Nervous System of Adults. J. Neurosci. Res. 2017, 95, 1574–1581. [Google Scholar] [CrossRef]
- Fuziwara, C.S.; Kimura, E.T. Insights into Regulation of the MiR-17-92 Cluster of MiRNAs in Cancer. Front. Med. (Lausanne) 2015, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Hayashita, Y.; Osada, H.; Tatematsu, Y.; Yamada, H.; Yanagisawa, K.; Tomida, S.; Yatabe, Y.; Kawahara, K.; Sekido, Y.; Takahashi, T. A Polycistronic MicroRNA Cluster, MiR-17-92, Is Overexpressed in Human Lung Cancers and Enhances Cell Proliferation. Cancer Res. 2005, 65, 9628–9632. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Liu, Z.; Zhou, L. Roles of MiR-17-92 Cluster in Cardiovascular Development and Common Diseases. Biomed. Res. Int. 2017, 2017, 9102909. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Thomson, J.M.; Hemann, M.T.; Hernando-Monge, E.; Mu, D.; Goodson, S.; Powers, S.; Cordon-Cardo, C.; Lowe, S.W.; Hannon, G.J.; et al. A MicroRNA Polycistron as a Potential Human Oncogene. Nature 2005, 435, 828–833. [Google Scholar] [CrossRef]
- Xia, X.; Wang, Y.; Zheng, J.C. The MicroRNA-17 ~ 92 Family as a Key Regulator of Neurogenesis and Potential Regenerative Therapeutics of Neurological Disorders. Stem Cell Rev. Rep. 2022, 18, 401–411. [Google Scholar] [CrossRef]
- Xia, X.; Lu, H.; Li, C.; Huang, Y.; Wang, Y.; Yang, X.; Zheng, J.C. MiR-106b Regulates the Proliferation and Differentiation of Neural Stem/Progenitor Cells through Tp53inp1-Tp53-Cdkn1a Axis. Stem Cell Res. Ther. 2019, 10, 282. [Google Scholar] [CrossRef]
- Garg, N.; Po, A.; Miele, E.; Campese, A.F.; Begalli, F.; Silvano, M.; Infante, P.; Capalbo, C.; De Smaele, E.; Canettieri, G.; et al. MicroRNA-17-92 Cluster Is a Direct Nanog Target and Controls Neural Stem Cell through Trp53inp1. EMBO J. 2013, 32, 2819–2832. [Google Scholar] [CrossRef]
- Bian, S.; Hong, J.; Li, Q.; Schebelle, L.; Pollock, A.; Knauss, J.L.; Garg, V.; Sun, T. MicroRNA Cluster MiR-17-92 Regulates Neural Stem Cell Expansion and Transition to Intermediate Progenitors in the Developing Mouse Neocortex. Cell Rep. 2013, 3, 1398–1406. [Google Scholar] [CrossRef]
- Toyoshima, M.; Akamatsu, W.; Okada, Y.; Ohnishi, T.; Balan, S.; Hisano, Y.; Iwayama, Y.; Toyota, T.; Matsumoto, T.; Itasaka, N.; et al. Analysis of Induced Pluripotent Stem Cells Carrying 22q11.2 Deletion. Transl. Psychiatry 2016, 6, e934. [Google Scholar] [CrossRef]
- Brett, J.O.; Renault, V.M.; Rafalski, V.A.; Webb, A.E.; Brunet, A. The MicroRNA Cluster MiR-106b~25 Regulates Adult Neural Stem/Progenitor Cell Proliferation and Neuronal Differentiation. Aging 2011, 3, 108–124. [Google Scholar] [CrossRef]
- Naka-Kaneda, H.; Nakamura, S.; Igarashi, M.; Aoi, H.; Kanki, H.; Tsuyama, J.; Tsutsumi, S.; Aburatani, H.; Shimazaki, T.; Okano, H. The MiR-17/106-P38 Axis Is a Key Regulator of the Neurogenic-to-Gliogenic Transition in Developing Neural Stem/Progenitor Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1604–1609. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.L.; Chopp, M.; Fan, B.; Zhang, R.; Wang, X.; Hu, J.; Zhang, X.M.; Zhang, Z.G.; Liu, X.S. Ablation of the MicroRNA-17-92 Cluster in Neural Stem Cells Diminishes Adult Hippocampal Neurogenesis and Cognitive Function. FASEB J. 2019, 33, 5257–5267. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, T.; Aprea, J.; Nardelli, J.; Engel, H.; Selinger, C.; Mombereau, C.; Lemonnier, T.; Moutkine, I.; Schwendimann, L.; Dori, M.; et al. MicroRNAs Establish Robustness and Adaptability of a Critical Gene Network to Regulate Progenitor Fate Decisions during Cortical Neurogenesis. Cell Rep. 2014, 7, 1779–1788. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, T.; Hidaka, R.; Fujimaki, S.; Asashima, M.; Kuwabara, T. MicroRNAs and Epigenetics in Adult Neurogenesis. In Advances in Genetics; Elsevier: Amsterdam, The Netherlands, 2014; Volume 86, pp. 27–44. ISBN 978-0-12-800222-3. [Google Scholar]
- Budde, H.; Schmitt, S.; Fitzner, D.; Opitz, L.; Salinas-Riester, G.; Simons, M. Control of Oligodendroglial Cell Number by the MiR-17-92 Cluster. Development 2010, 137, 2127–2132. [Google Scholar] [CrossRef] [PubMed]
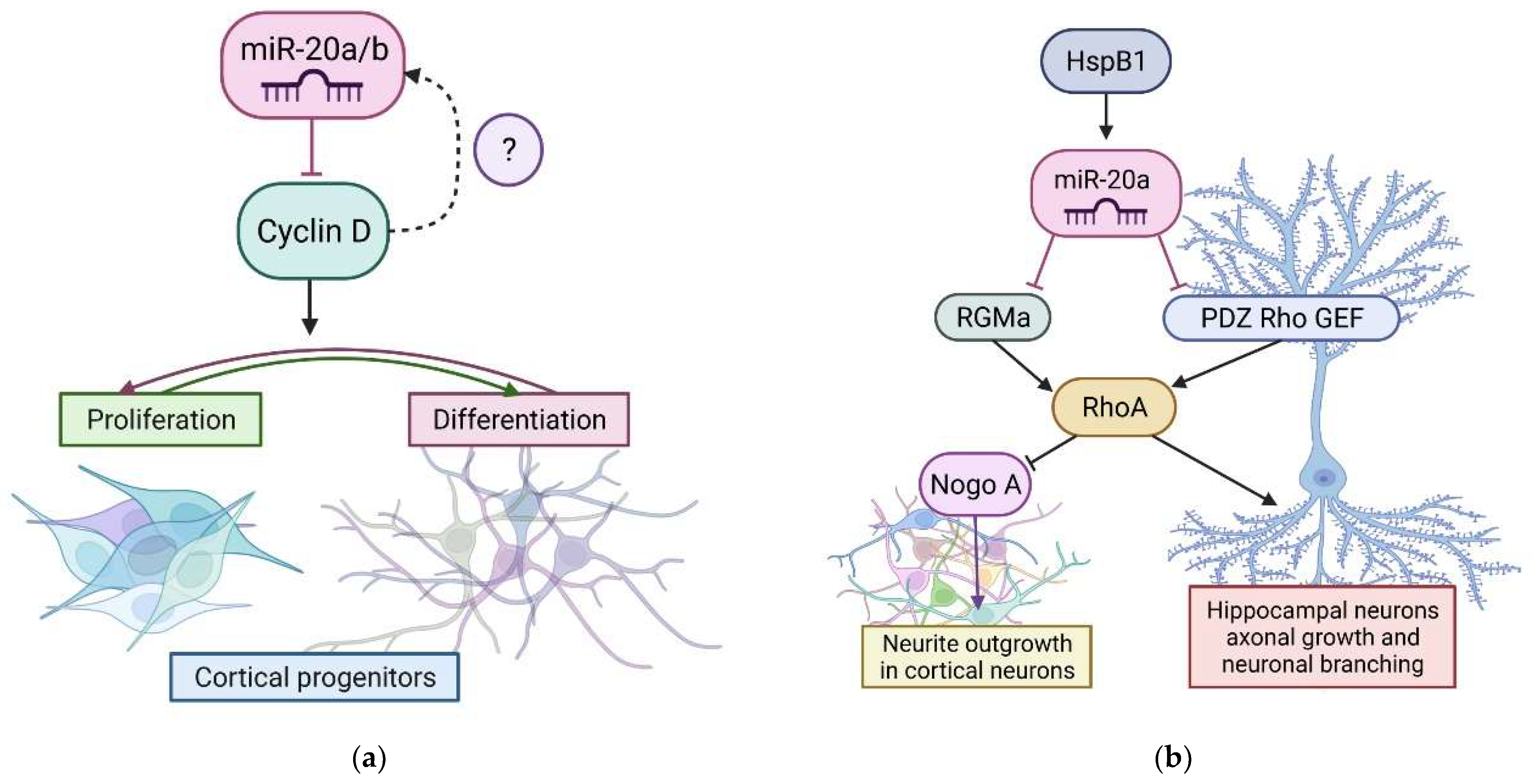
- Sun, X.; Zhou, Z.; Fink, D.J.; Mata, M. HspB1 Silences Translation of PDZ-RhoGEF by Enhancing MiR-20a and MiR-128 Expression to Promote Neurite Extension. Mol. Cell. Neurosci. 2013, 57, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Mymrikov, E.V.; Seit-Nebi, A.S.; Gusev, N.B. Large Potentials of Small Heat Shock Proteins. Physiol. Rev. 2011, 91, 1123–1159. [Google Scholar] [CrossRef]
- Perng, M.D.; Cairns, L.; van den IJssel, P.; Prescott, A.; Hutcheson, A.M.; Quinlan, R.A. Intermediate Filament Interactions Can Be Altered by HSP27 and AlphaB-Crystallin. J. Cell Sci. 1999, 112, 2099–2112. [Google Scholar] [CrossRef]
- Wagstaff, M.J.D.; Collaço-Moraes, Y.; Smith, J.; de Belleroche, J.S.; Coffin, R.S.; Latchman, D.S. Protection of Neuronal Cells from Apoptosis by Hsp27 Delivered with a Herpes Simplex Virus-Based Vector. J. Biol. Chem. 1999, 274, 5061–5069. [Google Scholar] [CrossRef]
- Jin, J.; Kim, S.-N.; Liu, X.; Zhang, H.; Zhang, C.; Seo, J.-S.; Kim, Y.; Sun, T. MiR-17-92 Cluster Regulates Adult Hippocampal Neurogenesis, Anxiety, and Depression. Cell Rep. 2016, 16, 1653–1663. [Google Scholar] [CrossRef]
- Guo, F.; Han, X.; Zhang, J.; Zhao, X.; Lou, J.; Chen, H.; Huang, X. Repetitive Transcranial Magnetic Stimulation Promotes Neural Stem Cell Proliferation via the Regulation of MiR-25 in a Rat Model of Focal Cerebral Ischemia. PLoS ONE 2014, 9, e109267. [Google Scholar] [CrossRef]
- Xin, H.; Katakowski, M.; Wang, F.; Qian, J.-Y.; Liu, X.S.; Ali, M.M.; Buller, B.; Zhang, Z.G.; Chopp, M. MicroRNA-17–92 Cluster in Exosomes Enhance Neuroplasticity and Functional Recovery After Stroke in Rats. Stroke 2017, 48, 747–753. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Yang, S.; Deng, G.; Liu, M.; Zhu, H.; Zhang, W.; Yan, S.; Quan, L.; Bai, J.; Xu, N. Aurora Kinase A Induces MiR-17-92 Cluster through Regulation of E2F1 Transcription Factor. Cell. Mol. Life Sci. 2010, 67, 2069–2076. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Hanse, E.A.; Stedman, K.; Benson, J.M.; Lowman, X.H.; Subramanian, S.; Kelekar, A. Transcription Factor C/EBP-β Induces Tumor-Suppressor Phosphatase PHLPP2 through Repression of the MiR-17-92 Cluster in Differentiating AML Cells. Cell Death Differ. 2016, 23, 1232–1242. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Wang, X.L.; Zhang, L.; Hozeska-Solgot, A.; Tang, T.; Kassis, H.; Zhang, R.L.; Chen, C.; Xu, J.; et al. MicroRNA-17-92 Cluster Mediates the Proliferation and Survival of Neural Progenitor Cells after Stroke. J. Biol. Chem. 2013, 288, 12478–12488. [Google Scholar] [CrossRef]
- Lin, D.; Shi, Y.; Hu, Y.; Du, X.; Tu, G. MiR-329-3p Regulates Neural Stem Cell Proliferation by Targeting E2F1. Mol. Med. Rep. 2019, 19, 4137–4146. [Google Scholar] [CrossRef]
- Cortes-Canteli, M.; Aguilar-Morante, D.; Sanz-Sancristobal, M.; Megias, D.; Santos, A.; Perez-Castillo, A. Role of C/EBPβ Transcription Factor in Adult Hippocampal Neurogenesis. PLoS ONE 2011, 6, e24842. [Google Scholar] [CrossRef]
- Woods, K.; Thomson, J.M.; Hammond, S.M. Direct Regulation of an Oncogenic Micro-RNA Cluster by E2F Transcription Factors. J. Biol. Chem. 2007, 282, 2130–2134. [Google Scholar] [CrossRef]
- Fehlings, M.G.; Tator, C.H. The Relationships among the Severity of Spinal Cord Injury, Residual Neurological Function, Axon Counts, and Counts of Retrogradely Labeled Neurons after Experimental Spinal Cord Injury. Exp. Neurol. 1995, 132, 220–228. [Google Scholar] [CrossRef]
- Arbour, N.; Vanderluit, J.L.; Le Grand, J.N.; Jahani-Asl, A.; Ruzhynsky, V.A.; Cheung, E.C.C.; Kelly, M.A.; MacKenzie, A.E.; Park, D.S.; Opferman, J.T.; et al. Mcl-1 Is a Key Regulator of Apoptosis during CNS Development and after DNA Damage. J. Neurosci. 2008, 28, 6068–6078. [Google Scholar] [CrossRef]
- Dumont, R.J.; Okonkwo, D.O.; Verma, S.; Hurlbert, R.J.; Boulos, P.T.; Ellegala, D.B.; Dumont, A.S. Acute Spinal Cord Injury, Part I: Pathophysiologic Mechanisms. Clin. Neuropharmacol. 2001, 24, 254–264. [Google Scholar] [CrossRef]
- Oyinbo, C.A. Secondary Injury Mechanisms in Traumatic Spinal Cord Injury: A Nugget of This Multiply Cascade. Acta Neurobiol. Exp. (Wars) 2011, 71, 281–299. [Google Scholar] [PubMed]
- Alizadeh, A.; Dyck, S.M.; Karimi-Abdolrezaee, S. Traumatic Spinal Cord Injury: An Overview of Pathophysiology, Models and Acute Injury Mechanisms. Front. Neurol. 2019, 10, 282. [Google Scholar] [CrossRef] [PubMed]
- Mautes, A.E.; Weinzierl, M.R.; Donovan, F.; Noble, L.J. Vascular Events after Spinal Cord Injury: Contribution to Secondary Pathogenesis. Phys. Ther. 2000, 80, 673–687. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.M.; Khazaei, M.; Fehlings, M.G. Translating Mechanisms of Neuroprotection, Regeneration, and Repair to Treatment of Spinal Cord Injury. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2015; Volume 218, pp. 15–54. ISBN 978-0-444-63565-5. [Google Scholar]
- Carmel, J.B.; Galante, A.; Soteropoulos, P.; Tolias, P.; Recce, M.; Young, W.; Hart, R.P. Gene Expression Profiling of Acute Spinal Cord Injury Reveals Spreading Inflammatory Signals and Neuron Loss. Physiol. Genom. 2001, 7, 201–213. [Google Scholar] [CrossRef]
- Aimone, J.; Leasure, J.; Perreau, V.; Thallmair, M. Thechristopherreeveparalysisfounda Spatial and Temporal Gene Expression Profiling of the Contused Rat Spinal Cord. Exp. Neurol. 2004, 189, 204–221. [Google Scholar] [CrossRef]
- Strasser, A.; Puthalakath, H.; Bouillet, P.; Huang, D.C.S.; O’Connor, L.; O’Reilly, L.A.; Cullen, L.; Cory, S.; Adams, J.M. The Role of Bim, a Proapoptotic BH3-Only Member of the Bcl-2 Family, in Cell-Death Control. Ann. N. Y. Acad. Sci. 2006, 917, 541–548. [Google Scholar] [CrossRef]
- Hu, J.-R.; Lv, G.-H.; Yin, B.-L. Altered MicroRNA Expression in the Ischemic-Reperfusion Spinal Cord with Atorvastatin Therapy. J. Pharmacol. Sci. 2013, 121, 343–346. [Google Scholar] [CrossRef]
- Liu, N.-K.; Wang, X.-F.; Lu, Q.-B.; Xu, X.-M. Altered MicroRNA Expression Following Traumatic Spinal Cord Injury. Exp. Neurol. 2009, 219, 424–429. [Google Scholar] [CrossRef]
- Strickland, E.R.; Hook, M.A.; Balaraman, S.; Huie, J.R.; Grau, J.W.; Miranda, R.C. MicroRNA Dysregulation Following Spinal Cord Contusion: Implications for Neural Plasticity and Repair. Neuroscience 2011, 186, 146–160. [Google Scholar] [CrossRef]
- Yunta, M.; Nieto-Díaz, M.; Esteban, F.J.; Caballero-López, M.; Navarro-Ruíz, R.; Reigada, D.; Pita-Thomas, D.W.; del Águila, A.; Muñoz-Galdeano, T.; Maza, R.M. MicroRNA Dysregulation in the Spinal Cord Following Traumatic Injury. PLoS ONE 2012, 7, e34534. [Google Scholar] [CrossRef]
- De Biase, A.; Knoblach, S.M.; Di Giovanni, S.; Fan, C.; Molon, A.; Hoffman, E.P.; Faden, A.I. Gene Expression Profiling of Experimental Traumatic Spinal Cord Injury as a Function of Distance from Impact Site and Injury Severity. Physiol. Genom. 2005, 22, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Buller, B.; Liu, X.; Wang, X.; Zhang, R.L.; Zhang, L.; Hozeska-Solgot, A.; Chopp, M.; Zhang, Z.G. MicroRNA-21 Protects Neurons from Ischemic Death. FEBS J. 2010, 277, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Hafez, M.M.; Hassan, Z.K.; Zekri, A.R.N.; Gaber, A.A.; Al Rejaie, S.S.; Sayed-Ahmed, M.M.; Al Shabanah, O. MicroRNAs and Metastasis-Related Gene Expression in Egyptian Breast Cancer Patients. Asian Pac. J. Cancer Prev. 2012, 13, 591–598. [Google Scholar] [CrossRef]
- Frankel, L.B.; Christoffersen, N.R.; Jacobsen, A.; Lindow, M.; Krogh, A.; Lund, A.H. Programmed Cell Death 4 (PDCD4) Is an Important Functional Target of the MicroRNA MiR-21 in Breast Cancer Cells. J. Biol. Chem. 2008, 283, 1026–1033. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, E.D.; Escobar, Y.; González, G.; Hernández, A.; Galindo, J.M.; García, M.C.; Sánchez, J.A. Posttranscriptional Regulation of the Β2-Subunit of Cardiac L-Type Ca2+ Channels by MicroRNAs during Long-Term Exposure to Isoproterenol in Rats. J. Cardiovasc. Pharmacol. 2011, 58, 470–478. [Google Scholar] [CrossRef]
- Hutchison, E.R.; Kawamoto, E.M.; Taub, D.D.; Lal, A.; Abdelmohsen, K.; Zhang, Y.; Wood, W.H.; Lehrmann, E.; Camandola, S.; Becker, K.G.; et al. Evidence for MiR-181 Involvement in Neuroinflammatory Responses of Astrocytes. Glia 2013, 61, 1018–1028. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of MiR-155 and MiR-125b Levels Following Lipopolysaccharide/TNF-Alpha Stimulation and Their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef]
- Iliopoulos, D.; Jaeger, S.A.; Hirsch, H.A.; Bulyk, M.L.; Struhl, K. STAT3 Activation of MiR-21 and MiR-181b-1 via PTEN and CYLD Are Part of the Epigenetic Switch Linking Inflammation to Cancer. Mol. Cell 2010, 39, 493–506. [Google Scholar] [CrossRef]
- Theis, T.; Yoo, M.; Park, C.S.; Chen, J.; Kügler, S.; Gibbs, K.M.; Schachner, M. Lentiviral Delivery of MiR-133b Improves Functional Recovery After Spinal Cord Injury in Mice. Mol. Neurobiol. 2017, 54, 4659–4671. [Google Scholar] [CrossRef]
- Agostini, M.; Tucci, P.; Steinert, J.R.; Shalom-Feuerstein, R.; Rouleau, M.; Aberdam, D.; Forsythe, I.D.; Young, K.W.; Ventura, A.; Concepcion, C.P.; et al. MicroRNA-34a Regulates Neurite Outgrowth, Spinal Morphology, and Function. Proc. Natl. Acad. Sci. USA 2011, 108, 21099–21104. [Google Scholar] [CrossRef]
- Jee, M.K.; Jung, J.S.; Im, Y.B.; Jung, S.J.; Kang, S.K. Silencing of MiR20a Is Crucial for Ngn1-Mediated Neuroprotection in Injured Spinal Cord. Hum. Gene Ther. 2012, 23, 508–520. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, B.; Yuan, X.; Cui, L.; Wang, Z.; Zhang, Y.; Yu, M.; Xiu, Y.; Zhang, Z.; Li, W.; et al. MiR-20a Plays a Key Regulatory Role in the Repair of Spinal Cord Dorsal Column Lesion via PDZ-RhoGEF/RhoA/GAP43 Axis in Rat. Cell. Mol. Neurobiol. 2019, 39, 87–98. [Google Scholar] [CrossRef]
- Zhao, L.; Gong, L.; Li, P.; Qin, J.; Xu, L.; Wei, Q.; Xie, H.; Mao, S.; Yu, B.; Gu, X.; et al. MiR-20a Promotes the Axon Regeneration of DRG Neurons by Targeting Nr4a3. Neurosci. Bull. 2021, 37, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.-Z.; Tian, Y.; Ander, B.P.; Xu, H.; Stamova, B.S.; Zhan, X.; Turner, R.J.; Jickling, G.; Sharp, F.R. Brain and Blood MicroRNA Expression Profiling of Ischemic Stroke, Intracerebral Hemorrhage, and Kainate Seizures. J. Cereb. Blood Flow Metab. 2010, 30, 92–101. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.S.; Chopp, M.; Zhang, R.L.; Tao, T.; Wang, X.L.; Kassis, H.; Hozeska-Solgot, A.; Zhang, L.; Chen, C.; Zhang, Z.G. MicroRNA Profiling in Subventricular Zone after Stroke: MiR-124a Regulates Proliferation of Neural Progenitor Cells through Notch Signaling Pathway. PLoS ONE 2011, 6, e23461. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Deng, Z.; Huang, H.; Hamblin, M.; Xie, C.; Zhang, J.; Chen, Y.E. MiR-497 Regulates Neuronal Death in Mouse Brain after Transient Focal Cerebral Ischemia. Neurobiol. Dis. 2010, 38, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Yin, K.-J.; Deng, Z.; Hamblin, M.; Xiang, Y.; Huang, H.; Zhang, J.; Jiang, X.; Wang, Y.; Chen, Y.E. Peroxisome Proliferator-Activated Receptor Delta Regulation of MiR-15a in Ischemia-Induced Cerebral Vascular Endothelial Injury. J. Neurosci. 2010, 30, 6398–6408. [Google Scholar] [CrossRef]
- Sepramaniam, S.; Armugam, A.; Lim, K.Y.; Karolina, D.S.; Swaminathan, P.; Tan, J.R.; Jeyaseelan, K. MicroRNA 320a Functions as a Novel Endogenous Modulator of Aquaporins 1 and 4 as Well as a Potential Therapeutic Target in Cerebral Ischemia. J. Biol. Chem. 2010, 285, 29223–29230. [Google Scholar] [CrossRef]
- Zhong, L.; Yan, J.; Li, H.; Meng, L. HDAC9 Silencing Exerts Neuroprotection Against Ischemic Brain Injury via MiR-20a-Dependent Downregulation of NeuroD1. Front. Cell. Neurosci. 2020, 14, 544285. [Google Scholar] [CrossRef]
- Eriksen, J.L.; Mackenzie, I.R.A. Progranulin: Normal Function and Role in Neurodegeneration. J. Neurochem. 2008, 104, 287–297. [Google Scholar] [CrossRef]
- Wang, Y.; Guo, F.; Pan, C.; Lou, Y.; Zhang, P.; Guo, S.; Yin, J.; Deng, Z. Effects of Low Temperatures on Proliferation-Related Signaling Pathways in the Hippocampus after Traumatic Brain Injury. Exp. Biol. Med. (Maywood) 2012, 237, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.-T.; Lei, P.; Wang, H.-C.; Zhang, A.-L.; Han, Z.-L.; Chen, X.; Li, S.-H.; Jiang, R.-C.; Kang, C.-S.; Zhang, J.-N. MiR-21 Improves the Neurological Outcome after Traumatic Brain Injury in Rats. Sci. Rep. 2014, 4, 6718. [Google Scholar] [CrossRef] [PubMed]
- Sabirzhanov, B.; Stoica, B.A.; Zhao, Z.; Loane, D.J.; Wu, J.; Dorsey, S.G.; Faden, A.I. MiR-711 Upregulation Induces Neuronal Cell Death after Traumatic Brain Injury. Cell Death Differ. 2016, 23, 654–668. [Google Scholar] [CrossRef] [PubMed]
- Hébert, S.S.; Horré, K.; Nicolaï, L.; Bergmans, B.; Papadopoulou, A.S.; Delacourte, A.; De Strooper, B. MicroRNA Regulation of Alzheimer’s Amyloid Precursor Protein Expression. Neurobiol. Dis. 2009, 33, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Qin, L.; Tang, B. MicroRNAs in Alzheimer’s Disease. Front. Genet. 2019, 10, 153. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhao, R.; Wu, J.; Wang, Q.; Pang, K.; Shi, Q.; Gao, Q.; Hu, Y.; Dong, X.; Zhang, J.; et al. Melatonin Protects against Aβ-Induced Neurotoxicity in Primary Neurons via MiR-132/PTEN/AKT/FOXO3a Pathway. Biofactors 2018, 44, 609–618. [Google Scholar] [CrossRef]
- Tian, Z.; Dong, Q.; Wu, T.; Guo, J. MicroRNA-20b-5p Aggravates Neuronal Apoptosis Induced by β-Amyloid via down-Regulation of Ras Homolog Family Member C in Alzheimer’s Disease. Neurosci. Lett. 2021, 742, 135542. [Google Scholar] [CrossRef]
- Kanagaraj, N.; Beiping, H.; Dheen, S.T.; Tay, S.S.W. Downregulation of MiR-124 in MPTP-Treated Mouse Model of Parkinson’s Disease and MPP Iodide-Treated MN9D Cells Modulates the Expression of the Calpain/Cdk5 Pathway Proteins. Neuroscience 2014, 272, 167–179. [Google Scholar] [CrossRef]
- Kim, W.; Lee, Y.; McKenna, N.D.; Yi, M.; Simunovic, F.; Wang, Y.; Kong, B.; Rooney, R.J.; Seo, H.; Stephens, R.M.; et al. MiR-126 Contributes to Parkinson’s Disease by Dysregulating the Insulin-like Growth Factor/Phosphoinositide 3-Kinase Signaling. Neurobiol. Aging 2014, 35, 1712–1721. [Google Scholar] [CrossRef]
- Miñones-Moyano, E.; Porta, S.; Escaramís, G.; Rabionet, R.; Iraola, S.; Kagerbauer, B.; Espinosa-Parrilla, Y.; Ferrer, I.; Estivill, X.; Martí, E. MicroRNA Profiling of Parkinson’s Disease Brains Identifies Early Downregulation of MiR-34b/c Which Modulate Mitochondrial Function. Hum. Mol. Genet. 2011, 20, 3067–3078. [Google Scholar] [CrossRef]
- Rezaei, O.; Nateghinia, S.; Estiar, M.A.; Taheri, M.; Ghafouri-Fard, S. Assessment of the Role of Non-Coding RNAs in the Pathophysiology of Parkinson’s Disease. Eur. J. Pharmacol. 2021, 896, 173914. [Google Scholar] [CrossRef] [PubMed]
- Frankel, L.B.; Di Malta, C.; Wen, J.; Eskelinen, E.-L.; Ballabio, A.; Lund, A.H. A Non-Conserved MiRNA Regulates Lysosomal Function and Impacts on a Human Lysosomal Storage Disorder. Nat. Commun. 2014, 5, 5840. [Google Scholar] [CrossRef] [PubMed]
- Rauch, I.; Rosebrock, F.; Hainzl, E.; Heider, S.; Majoros, A.; Wienerroither, S.; Strobl, B.; Stockinger, S.; Kenner, L.; Müller, M.; et al. Noncanonical Effects of IRF9 in Intestinal Inflammation: More than Type I and Type III Interferons. Mol. Cell. Biol. 2015, 35, 2332–2343. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, N.; Castro, D.S.; Guillemot, F. Proneural Genes and the Specification of Neural Cell Types. Nat. Rev. Neurosci. 2002, 3, 517–530. [Google Scholar] [CrossRef] [PubMed]
- Carraro, G.; El-Hashash, A.; Guidolin, D.; Tiozzo, C.; Turcatel, G.; Young, B.M.; De Langhe, S.P.; Bellusci, S.; Shi, W.; Parnigotto, P.P.; et al. MiR-17 Family of MicroRNAs Controls FGF10-Mediated Embryonic Lung Epithelial Branching Morphogenesis through MAPK14 and STAT3 Regulation of E-Cadherin Distribution. Dev. Biol. 2009, 333, 238–250. [Google Scholar] [CrossRef]
- Chen, R.-L.; Balami, J.S.; Esiri, M.M.; Chen, L.-K.; Buchan, A.M. Ischemic Stroke in the Elderly: An Overview of Evidence. Nat. Rev. Neurol. 2010, 6, 256–265. [Google Scholar] [CrossRef]
- Donnan, G.A.; Fisher, M.; Macleod, M.; Davis, S.M. Stroke. Lancet 2008, 371, 1612–1623. [Google Scholar] [CrossRef]
- Roy-O’Reilly, M.; McCullough, L.D. Age and Sex Are Critical Factors in Ischemic Stroke Pathology. Endocrinology 2018, 159, 3120–3131. [Google Scholar] [CrossRef]
- Benjamin, E.J.; Muntner, P.; Alonso, A.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Das, S.R.; et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 2019, 139, e56–e528. [Google Scholar] [CrossRef]
- GBD 2016 Stroke Collaborators Global, Regional, and National Burden of Stroke, 1990-2016: A Systematic Analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 439–458. [CrossRef]
- Puig, B.; Brenna, S.; Magnus, T. Molecular Communication of a Dying Neuron in Stroke. Int. J. Mol. Sci. 2018, 19, 2834. [Google Scholar] [CrossRef] [PubMed]
- Saver, J.L. Time Is Brain—Quantified. Stroke 2006, 37, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Jeyaseelan, K.; Lim, K.Y.; Armugam, A. MicroRNA Expression in the Blood and Brain of Rats Subjected to Transient Focal Ischemia by Middle Cerebral Artery Occlusion. Stroke 2008, 39, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Laterza, O.F.; Lim, L.; Garrett-Engele, P.W.; Vlasakova, K.; Muniappa, N.; Tanaka, W.K.; Johnson, J.M.; Sina, J.F.; Fare, T.L.; Sistare, F.D.; et al. Plasma MicroRNAs as Sensitive and Specific Biomarkers of Tissue Injury. Clin. Chem. 2009, 55, 1977–1983. [Google Scholar] [CrossRef]
- Tan, K.S.; Armugam, A.; Sepramaniam, S.; Lim, K.Y.; Setyowati, K.D.; Wang, C.W.; Jeyaseelan, K. Expression Profile of MicroRNAs in Young Stroke Patients. PLoS ONE 2009, 4, e7689. [Google Scholar] [CrossRef]
- Rink, C.; Khanna, S. MicroRNA in Ischemic Stroke Etiology and Pathology. Physiol Genom. 2011, 43, 521–528. [Google Scholar] [CrossRef]
- Siegel, C.; Li, J.; Liu, F.; Benashski, S.E.; McCullough, L.D. MiR-23a Regulation of X-Linked Inhibitor of Apoptosis (XIAP) Contributes to Sex Differences in the Response to Cerebral Ischemia. Proc. Natl. Acad. Sci. USA 2011, 108, 11662–11667. [Google Scholar] [CrossRef]
- Ouyang, Y.-B.; Lu, Y.; Yue, S.; Xu, L.-J.; Xiong, X.-X.; White, R.E.; Sun, X.; Giffard, R.G. MiR-181 Regulates GRP78 and Influences Outcome from Cerebral Ischemia in Vitro and in Vivo. Neurobiol. Dis. 2012, 45, 555–563. [Google Scholar] [CrossRef]
- Shi, G.; Liu, Y.; Liu, T.; Yan, W.; Liu, X.; Wang, Y.; Shi, J.; Jia, L. Upregulated MiR-29b Promotes Neuronal Cell Death by Inhibiting Bcl2L2 after Ischemic Brain Injury. Exp. Brain Res. 2012, 216, 225–230. [Google Scholar] [CrossRef]
- Selvamani, A.; Sathyan, P.; Miranda, R.C.; Sohrabji, F. An Antagomir to MicroRNA Let7f Promotes Neuroprotection in an Ischemic Stroke Model. PLoS ONE 2012, 7, e32662. [Google Scholar] [CrossRef]
- Branyan, T.E.; Selvamani, A.; Park, M.J.; Korula, K.E.; Kosel, K.F.; Srinivasan, R.; Sohrabji, F. Functional Assessment of Stroke-Induced Regulation of MiR-20a-3p and Its Role as a Neuroprotectant. Transl. Stroke Res. 2021, 13, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Sugo, N.; Yamamoto, N. Visualization of HDAC9 Spatiotemporal Subcellular Localization in Primary Neuron Cultures. Methods Mol. Biol. 2016, 1436, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Lang, B.; Alrahbeni, T.M.A.; Clair, D.S.; Blackwood, D.H.; International Schizophrenia Consortium; McCaig, C.D.; Shen, S. HDAC9 Is Implicated in Schizophrenia and Expressed Specifically in Post-Mitotic Neurons but Not in Adult Neural Stem Cells. Am. J. Stem Cells 2012, 1, 31–41. [Google Scholar] [PubMed]
- Shi, W.; Wei, X.; Wang, Z.; Han, H.; Fu, Y.; Liu, J.; Zhang, Y.; Guo, J.; Dong, C.; Zhou, D.; et al. HDAC9 Exacerbates Endothelial Injury in Cerebral Ischaemia/Reperfusion Injury. J. Cell. Mol. Med. 2016, 20, 1139–1149. [Google Scholar] [CrossRef]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.-S.; Chen, P.-S.; Chuang, D.-M. Histone Deacetylase Inhibitors Exhibit Anti-Inflammatory and Neuroprotective Effects in a Rat Permanent Ischemic Model of Stroke: Multiple Mechanisms of Action. J. Pharmacol. Exp. Ther. 2007, 321, 892–901. [Google Scholar] [CrossRef]
- Management of Concussion/mTBI Working Group. VA/DoD Clinical Practice Guideline for Management of Concussion/Mild Traumatic Brain Injury. J. Rehabil. Res. Dev. 2009, 46, CP1–CP68. [Google Scholar] [CrossRef]
- Finfer, S.R.; Cohen, J. Severe Traumatic Brain Injury. Resuscitation 2001, 48, 77–90. [Google Scholar] [CrossRef]
- Blennow, K.; Hardy, J.; Zetterberg, H. The Neuropathology and Neurobiology of Traumatic Brain Injury. Neuron 2012, 76, 886–899. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lozano, D.; Gonzales-Portillo, G.S.; Acosta, S.; de la Pena, I.; Tajiri, N.; Kaneko, Y.; Borlongan, C.V. Neuroinflammatory Responses to Traumatic Brain Injury: Etiology, Clinical Consequences, and Therapeutic Opportunities. Neuropsychiatr. Dis. Treat. 2015, 11, 97–106. [Google Scholar] [CrossRef]
- Werner, C.; Engelhard, K. Pathophysiology of Traumatic Brain Injury. Br. J. Anaesth. 2007, 99, 4–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.H.; Gustafson, J.; Gangidine, M.; Stepien, D.; Schuster, R.; Pritts, T.A.; Goodman, M.D.; Remick, D.G.; Lentsch, A.B. A Murine Model of Mild Traumatic Brain Injury Exhibiting Cognitive and Motor Deficits. J. Surg. Res. 2013, 184, 981–988. [Google Scholar] [CrossRef] [PubMed]
- Zweckberger, K.; Erös, C.; Zimmermann, R.; Kim, S.-W.; Engel, D.; Plesnila, N. Effect of Early and Delayed Decompressive Craniectomy on Secondary Brain Damage after Controlled Cortical Impact in Mice. J. Neurotrauma 2006, 23, 1083–1093. [Google Scholar] [CrossRef] [PubMed]
- Besenski, N. Traumatic Injuries: Imaging of Head Injuries. Eur. Radiol. 2002, 12, 1237–1252. [Google Scholar] [CrossRef]
- Jalali, R.; Rezaei, M. A Comparison of the Glasgow Coma Scale Score with Full Outline of Unresponsiveness Scale to Predict Patients’ Traumatic Brain Injury Outcomes in Intensive Care Units. Crit. Care Res. Pract. 2014, 2014, 289803. [Google Scholar] [CrossRef][Green Version]
- Redell, J.B.; Moore, A.N.; Ward, N.H.; Hergenroeder, G.W.; Dash, P.K. Human Traumatic Brain Injury Alters Plasma MicroRNA Levels. J. Neurotrauma 2010, 27, 2147–2156. [Google Scholar] [CrossRef]
- Meissner, L.; Gallozzi, M.; Balbi, M.; Schwarzmaier, S.; Tiedt, S.; Terpolilli, N.A.; Plesnila, N. Temporal Profile of MicroRNA Expression in Contused Cortex after Traumatic Brain Injury in Mice. J. Neurotrauma 2016, 33, 713–720. [Google Scholar] [CrossRef]
- Hu, Z.; Yu, D.; Almeida-Suhett, C.; Tu, K.; Marini, A.M.; Eiden, L.; Braga, M.F.; Zhu, J.; Li, Z. Expression of MiRNAs and Their Cooperative Regulation of the Pathophysiology in Traumatic Brain Injury. PLoS ONE 2012, 7, e39357. [Google Scholar] [CrossRef]
- Wang, W.-X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. MiR-107 Regulates Granulin/Progranulin with Implications for Traumatic Brain Injury and Neurodegenerative Disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef]
- Redell, J.B.; Zhao, J.; Dash, P.K. Altered Expression of MiRNA-21 and Its Targets in the Hippocampus after Traumatic Brain Injury. J. Neurosci. Res. 2011, 89, 212–221. [Google Scholar] [CrossRef]
- Bao, T.; Miao, W.; Han, J.; Yin, M.; Yan, Y.; Wang, W.; Zhu, Y. Spontaneous Running Wheel Improves Cognitive Functions of Mouse Associated with MiRNA Expressional Alteration in Hippocampus Following Traumatic Brain Injury. J. Mol. Neurosci. 2014, 54, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Zhou, F.-J.; Chang, Y.-F.; Li, Y.-S.; Liu, G.-C.; Hong, Y.; Chen, H.-L.; Xiyang, Y.-B.; Bao, T. MiR21 Is Associated with the Cognitive Improvement Following Voluntary Running Wheel Exercise in TBI Mice. J. Mol. Neurosci. 2015, 57, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Bhomia, M.; Balakathiresan, N.S.; Wang, K.K.; Papa, L.; Maheshwari, R.K. A Panel of Serum MiRNA Biomarkers for the Diagnosis of Severe to Mild Traumatic Brain Injury in Humans. Sci. Rep. 2016, 6, 28148. [Google Scholar] [CrossRef] [PubMed]
- Di Pietro, V.; Yakoub, K.M.; Scarpa, U.; Di Pietro, C.; Belli, A. MicroRNA Signature of Traumatic Brain Injury: From the Biomarker Discovery to the Point-of-Care. Front. Neurol. 2018, 9, 429. [Google Scholar] [CrossRef]
- Di Pietro, V.; Porto, E.; Ragusa, M.; Barbagallo, C.; Davies, D.; Forcione, M.; Logan, A.; Di Pietro, C.; Purrello, M.; Grey, M.; et al. Salivary MicroRNAs: Diagnostic Markers of Mild Traumatic Brain Injury in Contact-Sport. Front. Mol. Neurosci. 2018, 11, 290. [Google Scholar] [CrossRef]
- Masters, C.L.; Simms, G.; Weinman, N.A.; Multhaup, G.; McDonald, B.L.; Beyreuther, K. Amyloid Plaque Core Protein in Alzheimer Disease and Down Syndrome. Proc. Natl. Acad. Sci. USA 1985, 82, 4245–4249. [Google Scholar] [CrossRef]
- Olsson, B.; Lautner, R.; Andreasson, U.; Öhrfelt, A.; Portelius, E.; Bjerke, M.; Hölttä, M.; Rosén, C.; Olsson, C.; Strobel, G.; et al. CSF and Blood Biomarkers for the Diagnosis of Alzheimer’s Disease: A Systematic Review and Meta-Analysis. Lancet Neurol. 2016, 15, 673–684. [Google Scholar] [CrossRef]
- Rabizadeh, S.; Bitler, C.M.; Butcher, L.L.; Bredesen, D.E. Expression of the Low-Affinity Nerve Growth Factor Receptor Enhances Beta-Amyloid Peptide Toxicity. Proc. Natl. Acad. Sci. USA 1994, 91, 10703–10706. [Google Scholar] [CrossRef]
- Costantini, C.; Rossi, F.; Formaggio, E.; Bernardoni, R.; Cecconi, D.; Della-Bianca, V. Characterization of the Signaling Pathway Downstream P75 Neurotrophin Receptor Involved in Beta-Amyloid Peptide-Dependent Cell Death. J. Mol. Neurosci. 2005, 25, 141–156. [Google Scholar] [CrossRef]
- Shankar, G.M.; Bloodgood, B.L.; Townsend, M.; Walsh, D.M.; Selkoe, D.J.; Sabatini, B.L. Natural Oligomers of the Alzheimer Amyloid-Beta Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway. J. Neurosci. 2007, 27, 2866–2875. [Google Scholar] [CrossRef]
- Baranello, R.J.; Bharani, K.L.; Padmaraju, V.; Chopra, N.; Lahiri, D.K.; Greig, N.H.; Pappolla, M.A.; Sambamurti, K. Amyloid-Beta Protein Clearance and Degradation (ABCD) Pathways and Their Role in Alzheimer’s Disease. Curr. Alzheimer Res. 2015, 12, 32–46. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Kaether, C.; Thinakaran, G.; Sisodia, S. Trafficking and Proteolytic Processing of APP. Cold Spring Harb. Perspect. Med. 2012, 2, a006270. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, D.K.; Farlow, M.R.; Sambamurti, K.; Greig, N.H.; Giacobini, E.; Schneider, L.S. A Critical Analysis of New Molecular Targets and Strategies for Drug Developments in Alzheimer’s Disease. Curr. Drug Targets 2003, 4, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Treatments for Alzheimer’s Disease Emerge. Science 2021, 373, 624–626. [Google Scholar] [CrossRef]
- Zhao, Y.; Zong, Z.; Xu, H. RhoC Expression Level Is Correlated with the Clinicopathological Characteristics of Ovarian Cancer and the Expression Levels of ROCK-I, VEGF, and MMP9. Gynecol. Oncol. 2010, 116, 563–571. [Google Scholar] [CrossRef]
- Zhang, C.; Ge, X.; Lok, K.; Zhao, L.; Yin, M.; Wang, Z.-J. RhoC Involved in the Migration of Neural Stem/Progenitor Cells. Cell. Mol. Neurobiol. 2014, 34, 409–417. [Google Scholar] [CrossRef]
- Narumiya, S.; Thumkeo, D. Rho Signaling Research: History, Current Status and Future Directions. FEBS Lett. 2018, 592, 1763–1776. [Google Scholar] [CrossRef]
- Wang, R.; Chopra, N.; Nho, K.; Maloney, B.; Obukhov, A.G.; Nelson, P.T.; Counts, S.E.; Lahiri, D.K. Human MicroRNA (MiR-20b-5p) Modulates Alzheimer’s Disease Pathways and Neuronal Function, and a Specific Polymorphism Close to the MIR20B Gene Influences Alzheimer’s Biomarkers. Mol. Psychiatry 2022. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Bloem, B.R. The Parkinson Pandemic-A Call to Action. JAMA Neurol. 2018, 75, 9–10. [Google Scholar] [CrossRef]
- Goh, S.Y.; Chao, Y.X.; Dheen, S.T.; Tan, E.-K.; Tay, S.S.-W. Role of MicroRNAs in Parkinson’s Disease. Int. J. Mol. Sci. 2019, 20, 5649. [Google Scholar] [CrossRef]
- Jankovic, J. Parkinson’s Disease: Clinical Features and Diagnosis. J. Neurol. Neurosurg. Psychiatry 2008, 79, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef] [PubMed]
- Hallett, P.J.; Engelender, S.; Isacson, O. Lipid and Immune Abnormalities Causing Age-Dependent Neurodegeneration and Parkinson’s Disease. J. Neuroinflamm. 2019, 16, 153. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.; Takousis, P.; Wohlers, I.; Itua, I.O.G.; Dobricic, V.; Rücker, G.; Binder, H.; Middleton, L.; Ioannidis, J.P.A.; Perneczky, R.; et al. Meta-Analyses Identify Differentially Expressed Micrornas in Parkinson’s Disease. Ann. Neurol. 2019, 85, 835–851. [Google Scholar] [CrossRef]
- Kabaria, S.; Choi, D.C.; Chaudhuri, A.D.; Mouradian, M.M.; Junn, E. Inhibition of MiR-34b and MiR-34c Enhances α-Synuclein Expression in Parkinson’s Disease. FEBS Lett. 2015, 589, 319–325. [Google Scholar] [CrossRef]
- Lehmann, S.M.; Krüger, C.; Park, B.; Derkow, K.; Rosenberger, K.; Baumgart, J.; Trimbuch, T.; Eom, G.; Hinz, M.; Kaul, D.; et al. An Unconventional Role for MiRNA: Let-7 Activates Toll-like Receptor 7 and Causes Neurodegeneration. Nat. Neurosci. 2012, 15, 827–835. [Google Scholar] [CrossRef]
- Wang, Q.; Wang, Y.; Zhou, F.; Li, J.; Lu, G.; Zhao, Y. MiR-20a-5p Regulates MPP+-Induced Oxidative Stress and Neuroinflammation in HT22 Cells by Targeting IRF9/NF-ΚB Axis. Evid. Based Complement. Alternat. Med. 2021, 2021, 6621206. [Google Scholar] [CrossRef]
- Nan, J.; Wang, Y.; Yang, J.; Stark, G.R. IRF9 and Unphosphorylated STAT2 Cooperate with NF-ΚB to Drive IL6 Expression. Proc. Natl. Acad. Sci. USA 2018, 115, 3906–3911. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ND | Dysregulated microRNAs | Validated Targets | References |
|---|---|---|---|
| SCI | miR-21 | Fas-ligand, TPM1, PTEN, PDCD4 | [54,55,56] |
| miR-181, miR-125b | TNF-α | [58,59] | |
| let-7a, miR-181a, miR-30b-5p, miR-30c | IL-6 and IL-1β | [50,52,59,60] | |
| miR-133b | RhoA | [61] | |
| miR-124, miR-34a, miR-219 | Syntaxin-1A, synaptotagmin-1, p53 | [62] | |
| miR-20a | Neurogenin1, IL-6, IL-1β, TNF-α, COX2, caspase-3, STAT3, Mcl-1 | [51,63] | |
| SDLC | miR-20a | GTP-RhoA, Nr4a3 | [64,65] |
| Stroke | miR-124 | JAG-Notch signaling | [66,67] |
| miR-145 | Superoxide dismutase-2 | [4] | |
| miR-497 | Bcl-2, Bcl-w | [68] | |
| miR-15a | Bcl-2 | [69] | |
| miR-320a | Aquaporins | [70] | |
| miR-21 | Fas-ligand | [54] | |
| miR-20a | NeuroD1 | [71] | |
| TBI | miR-107 | Granulin | [72] |
| miR-34a | Notch1 | [73] | |
| miR-144 | Cask, NRF2 | [74] | |
| miR-23a and miR-27a | Bcl-2 | [75] | |
| AD | miR-29a/b-1 | Aβ | [76] |
| miR-29c, miR-107 | BACE1 | [76,77] | |
| miR-132 | PTEN | [77] | |
| miR-124 | PTPN1 | [78] | |
| miR-20a-5p | RhoC | [79] | |
| PD | miR-124 | Calpain/CDK5 | [80] |
| miR-34, miR-126 | IGF-1/PI3K | [81] | |
| miR-34b | PARK2, PARK7 | [82] | |
| miR-95 | α-synuclein, Parkin, SUMF1 | [83,84] | |
| miR-20a-5p | STAT1/IRF9 | [85] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arzhanov, I.; Sintakova, K.; Romanyuk, N. The Role of miR-20 in Health and Disease of the Central Nervous System. Cells 2022, 11, 1525. https://doi.org/10.3390/cells11091525
Arzhanov I, Sintakova K, Romanyuk N. The Role of miR-20 in Health and Disease of the Central Nervous System. Cells. 2022; 11(9):1525. https://doi.org/10.3390/cells11091525
Chicago/Turabian StyleArzhanov, Ivan, Kristyna Sintakova, and Nataliya Romanyuk. 2022. "The Role of miR-20 in Health and Disease of the Central Nervous System" Cells 11, no. 9: 1525. https://doi.org/10.3390/cells11091525
APA StyleArzhanov, I., Sintakova, K., & Romanyuk, N. (2022). The Role of miR-20 in Health and Disease of the Central Nervous System. Cells, 11(9), 1525. https://doi.org/10.3390/cells11091525