
Comparison of Whole Blood Cryopreservation Methods for Extensive Flow Cytometry Immunophenotyping

 , , ,
, , ,
Abstract
:1. Introduction
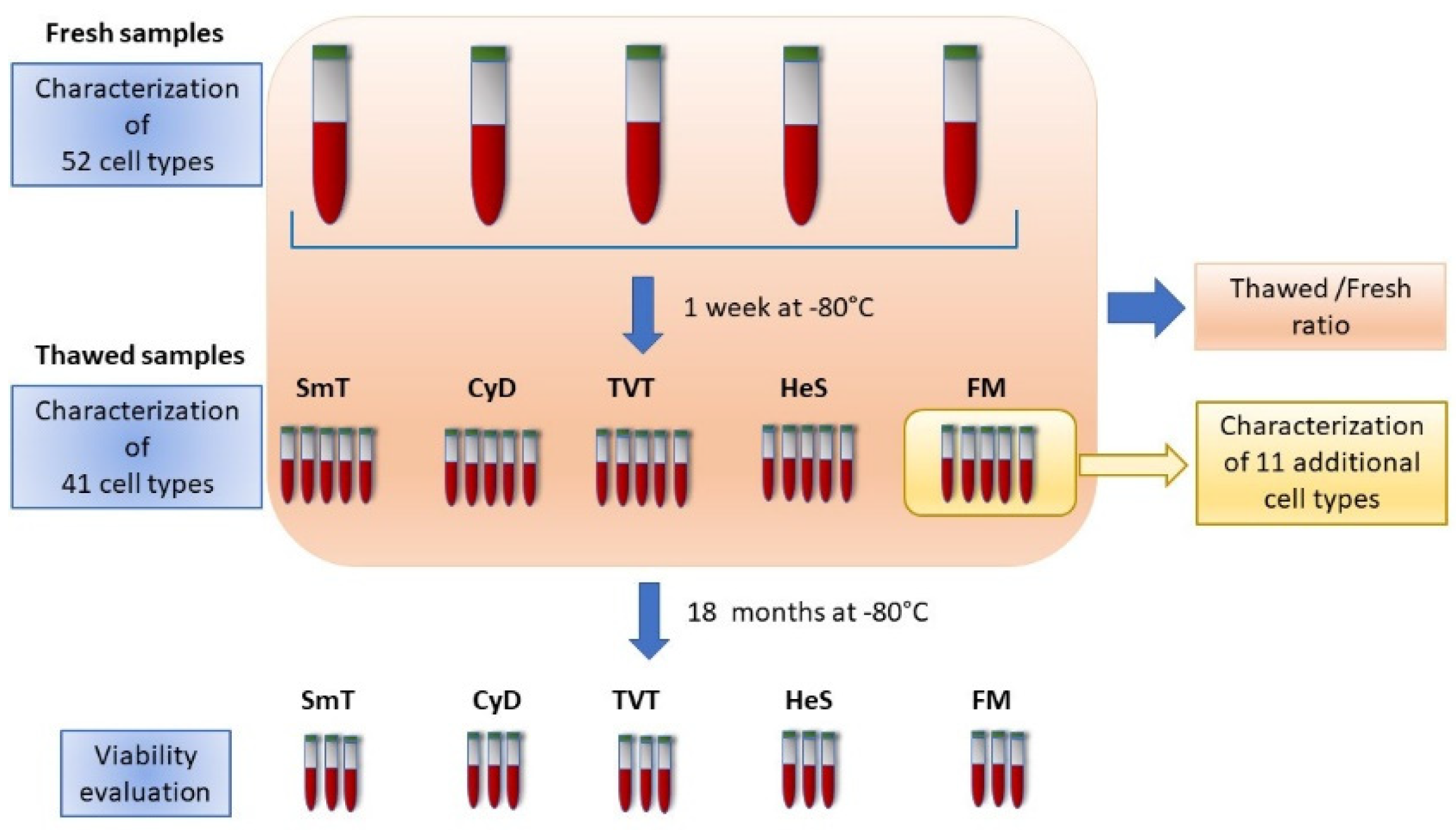
2. Materials and Methods
2.1. The SardiNIA Dataset
2.2. WB Cryopreservation and Thawing
2.2.1. Method A: Proteomic Stabilizer from Smart Tube (SmT) (# MTS1P 100/CS, San Carlos, CA, USA)
2.2.2. Method B: CytoDelics Whole Blood Cell Stabilizer from CytoDelics (CyD, #hC001-1000)
2.2.3. Method C: Freezing Mix (FM) Solution of 10% DMSO (Sigma-Aldrich, St. Luis, MO, USA, #D2650) Diluted in RPMI 1640 Medium (Lonza,#BE12-167F)
2.2.4. Method D: Red Blood Cell Depletion Kit, HetaSep (HeS) from Stemcells (#07906)
2.2.5. Method E: TransFix/EDTA Vacuum Blood Collection Tubes from Cytomark (TVTs, #TVT-03-50)
2.3. Staining Protocols
2.3.1. B Cell Panel
- CD24 vs. CD38 allowed the discrimination of transitional (CD24+ CD38hi), memory (CD24+ CD38−/dim) and naïve-mature (CD24− CD38−/dim) subsets (Supplementary Figure S1D);
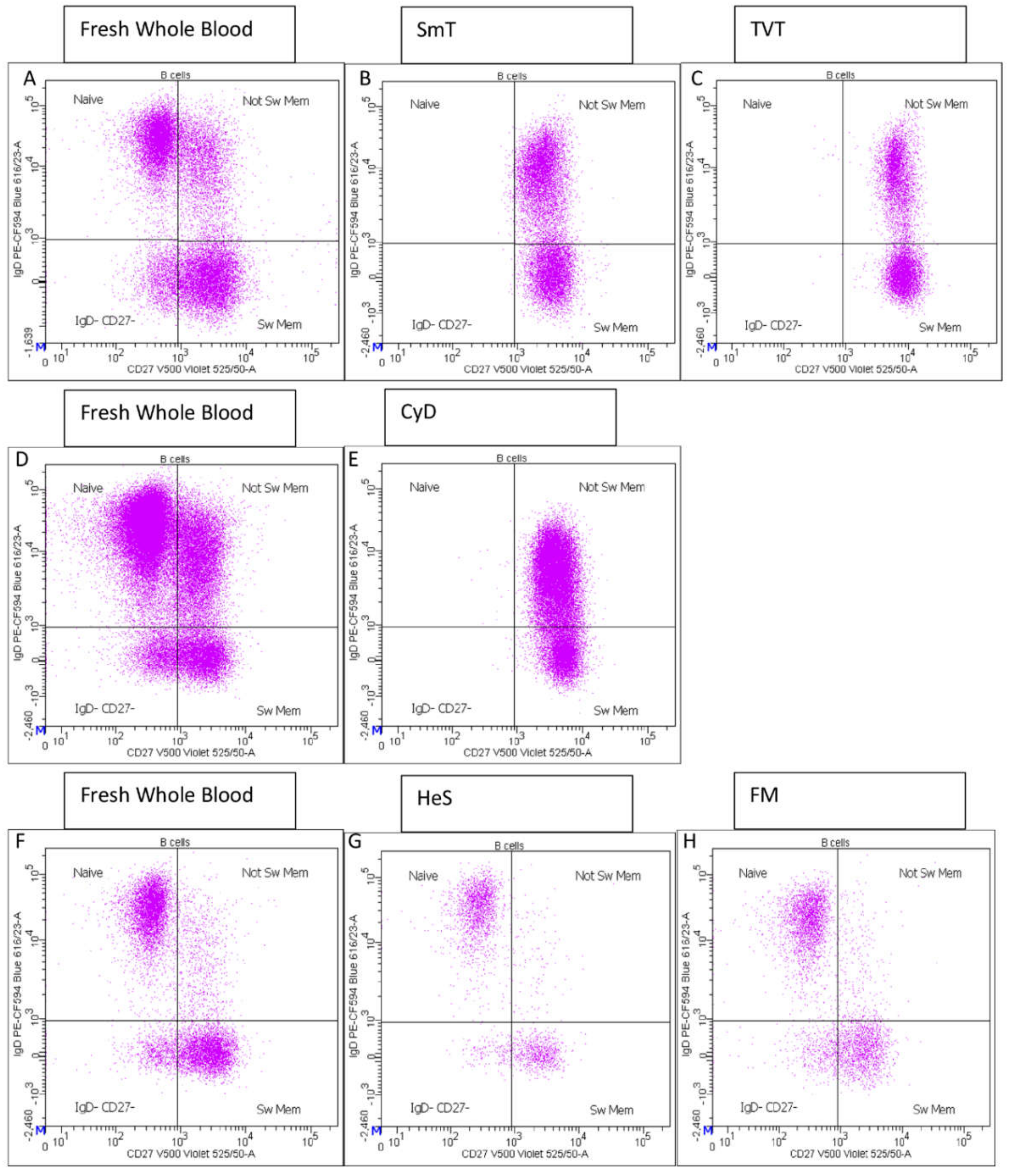
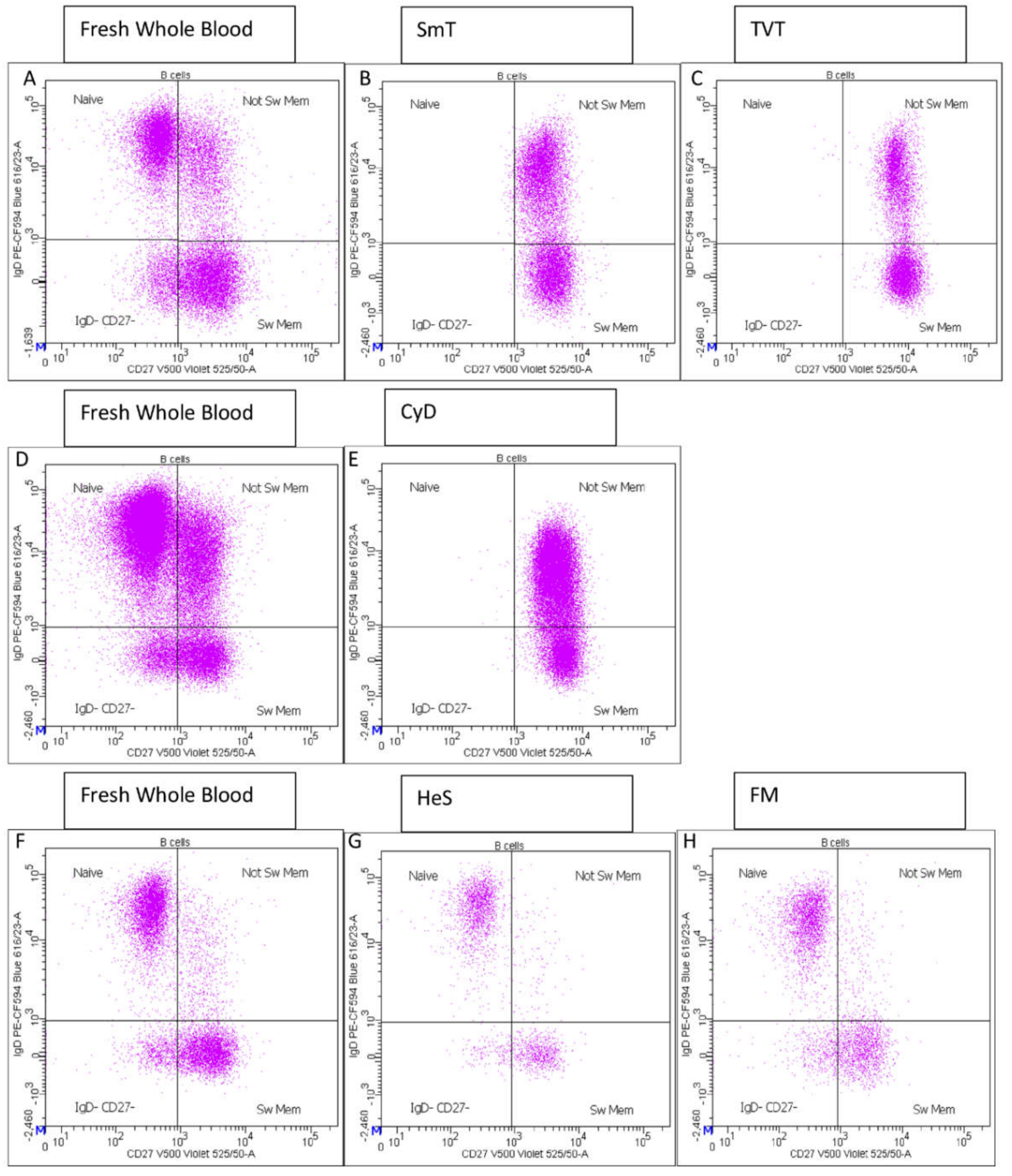
- CD27 vs. IgD identified switched memory (CD27+ IgD−), un-switched memory (CD27+ IgD+), naïve (CD27− IgD+) and CD27− IgD− B cells (Supplementary Figure S1E);
- IgD vs. CD38, also known as a Bm1-Bm5 classification [14], distinguished six B cell subsets: Bm1 (IgD+ CD38−) mainly virgin-naïve and un-switched memory cells; Bm2 (IgD+ CD38dim) activated-naïve cells; Bm2′ (IgD+ CD38br) pre-germinal center (GC) cells; Bm3-Bm4 (IgD− CD38br) centroblasts and centrocytes present in GC but very low/absent in blood; early Bm5 (IgD− CD38dim) and late Bm5 (IgD− CD38−) memory cells (Supplementary Figure S1F);
- CD24 vs. CD27 identified CD24+ CD27+ memory cells (Supplementary Figure S1G);
- CD20 vs. CD38 discriminated plasma blasts/plasma cells (as CD20− CD38hi) (Supplementary Figure S1H);
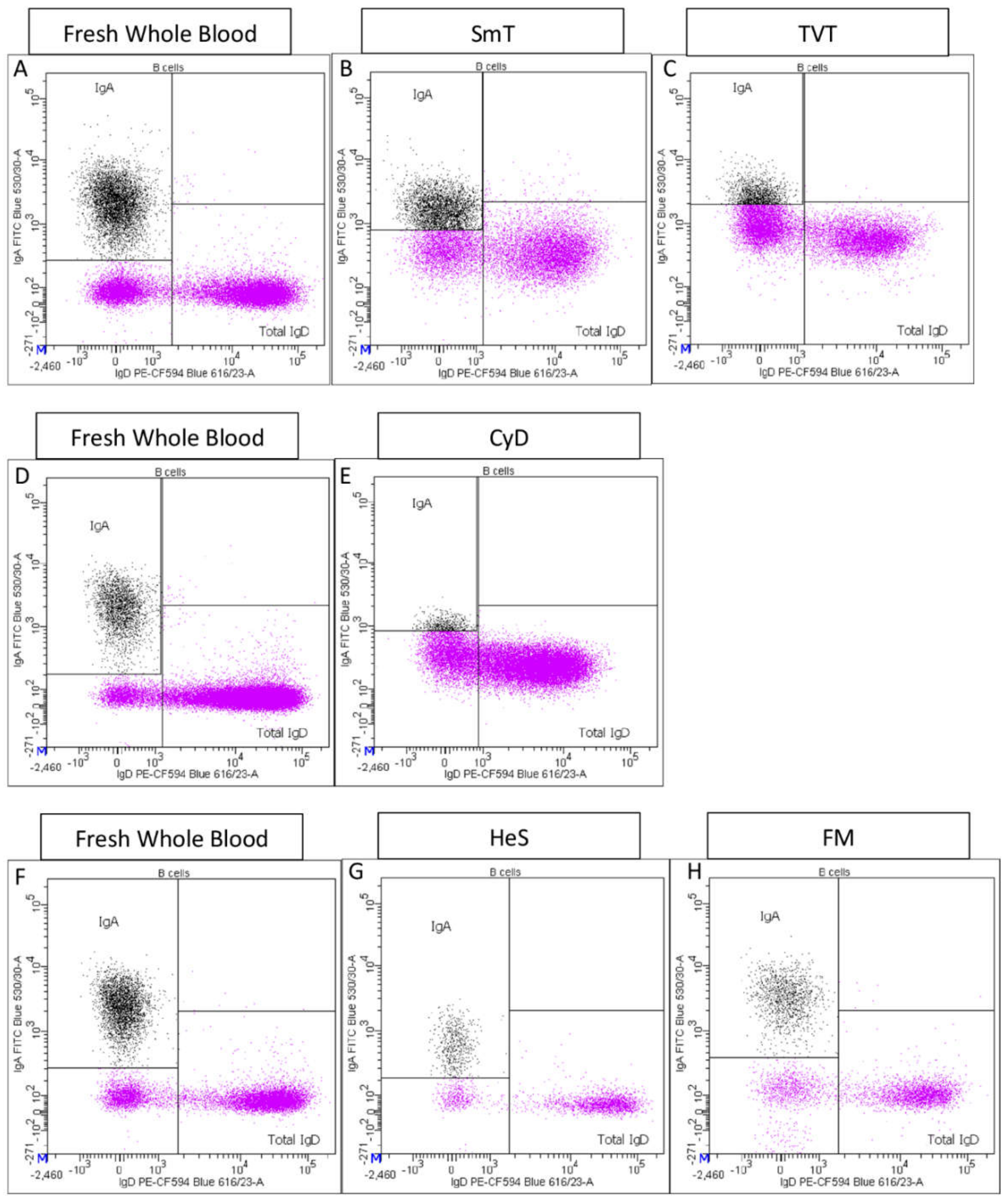
- Total IgA+ B cells and total IgD+ B cells were also identified (Supplementary Figure S1I).
2.3.2. CD4 T Cell Panel
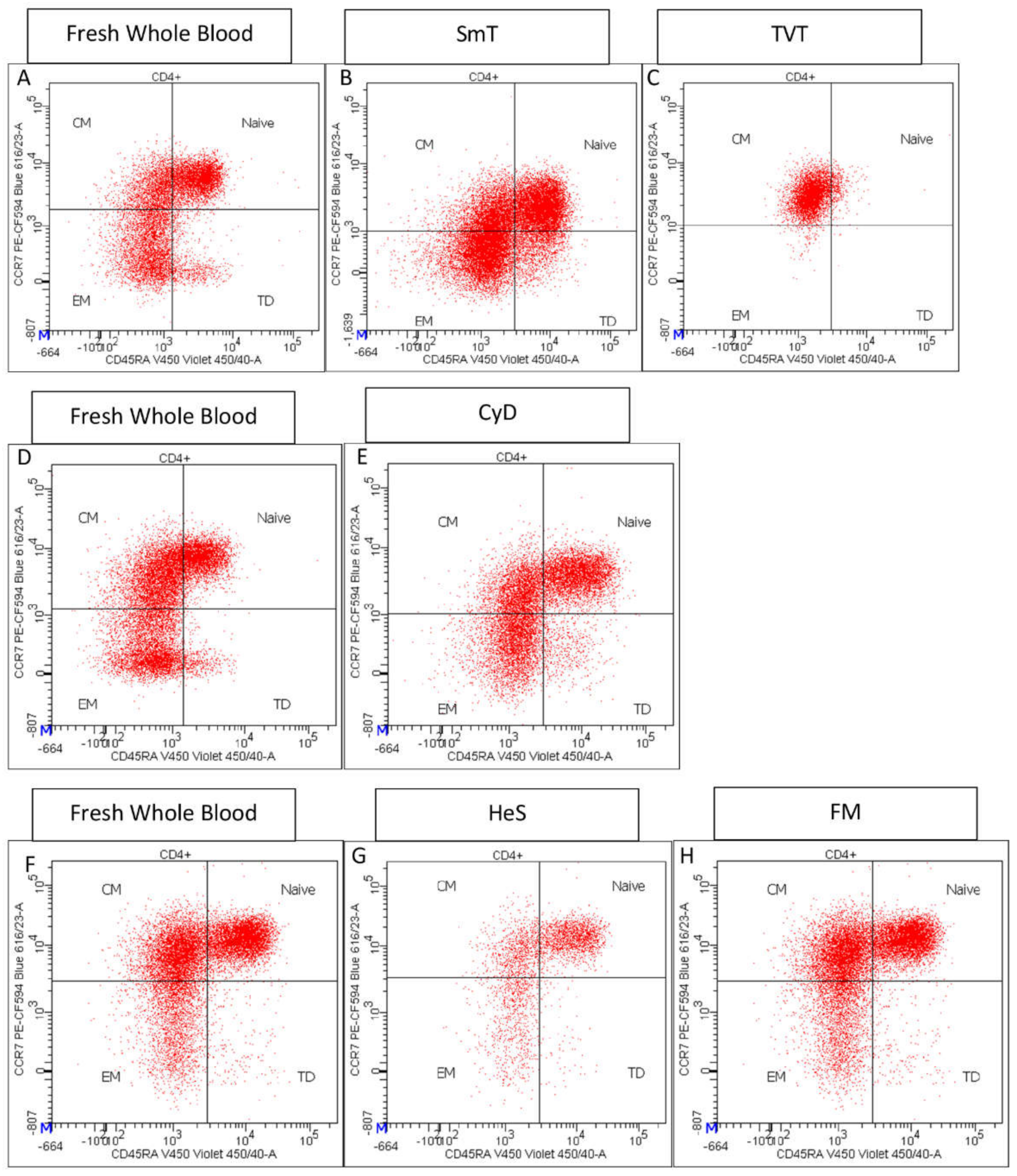
- CD45RA and CCR7 expression identified CD4+ T cell maturation stages: Naïve (CD45RA+ CCR7+), Central Memory (CM) (CD45RA−- CCR7+), Effector Memory (EM) (CCR7− CD45RA−), and Terminally Differentiated (TD) CD45RA+ CCR7+) (Supplementary Figure S2E) [15];
- CXCR3 and CCR6 expression discriminated Th1-Th17 (CXCR3+ CCR6+), Th17 (CXCR3− CCR6+) Th1 (CXCR3+ CCR6−), and Th2 (CXCR3− CCR6−) (Supplementary Figure S2F);
- High expression of CD25 and low expression of CD127 characterized regulatory CD4+ T cells (Tregs) (CD127− CD25 high) (Supplementary Figure S2G). We note that a rigorous Treg identification would require the use of an antibody against the transcription factor FoxP3. However, to avoid fixation and permeabilization steps required to detect FoxP3, we used the surface marker CD127 and CD25, the combination of which provides a reliable inference of Tregs, as identified by FoxP3. Tregs were further subdivided into resting (CD45RA+ CD25+), secreting (CD45RA− CD25+), and activated (CD45RA− CD25++) cells (Supplementary Figure S2H) [16];
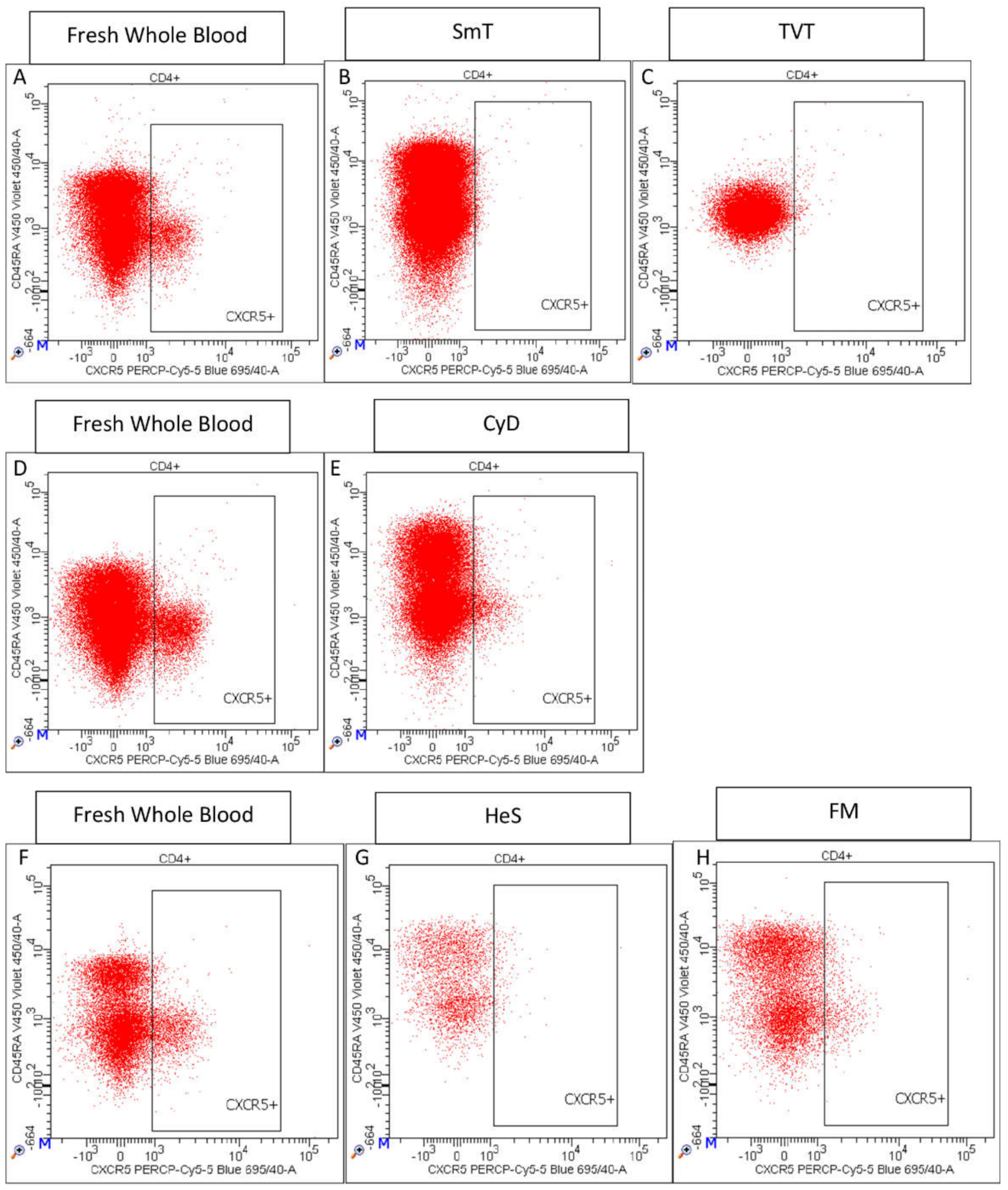
- Among CD4 positive T cells, PD1+ (or CD279+) (Supplementary Figure S2I), CXCR5+ (Supplementary Figure S2J), CCR4+ (Supplementary Figure S3K), ICOS+ (or CD278+) (Supplementary Figure S2L), and CD161+ T cells (Supplementary Figure S2M) were also assessed.
2.3.3. Viability Panel
2.3.4. M8NK Panel
2.4. Cytometer Settings and Sample Analysis
2.5. Data Analysis
3. Results
3.1. B Cell Characterization in Thawed Samples
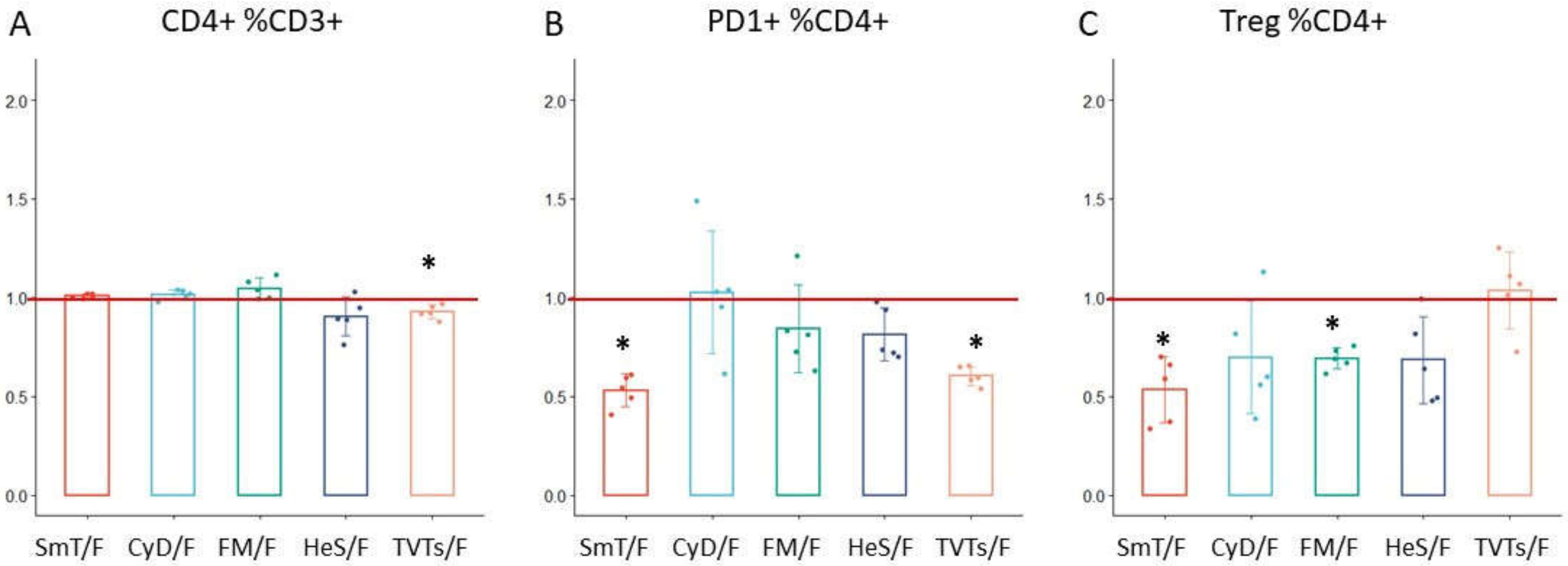
3.2. CD4 T Cell Characterization in Thawed Samples
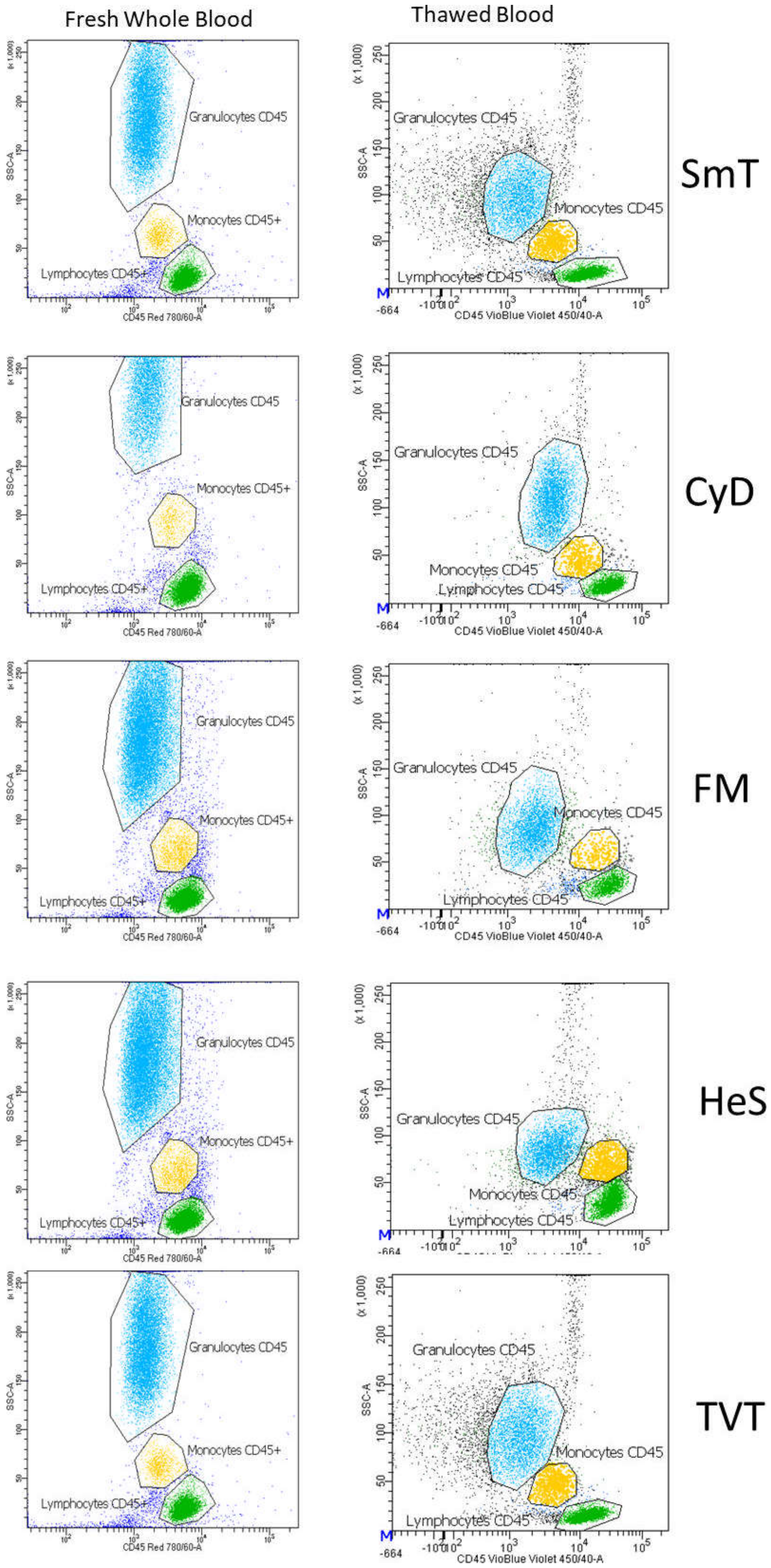
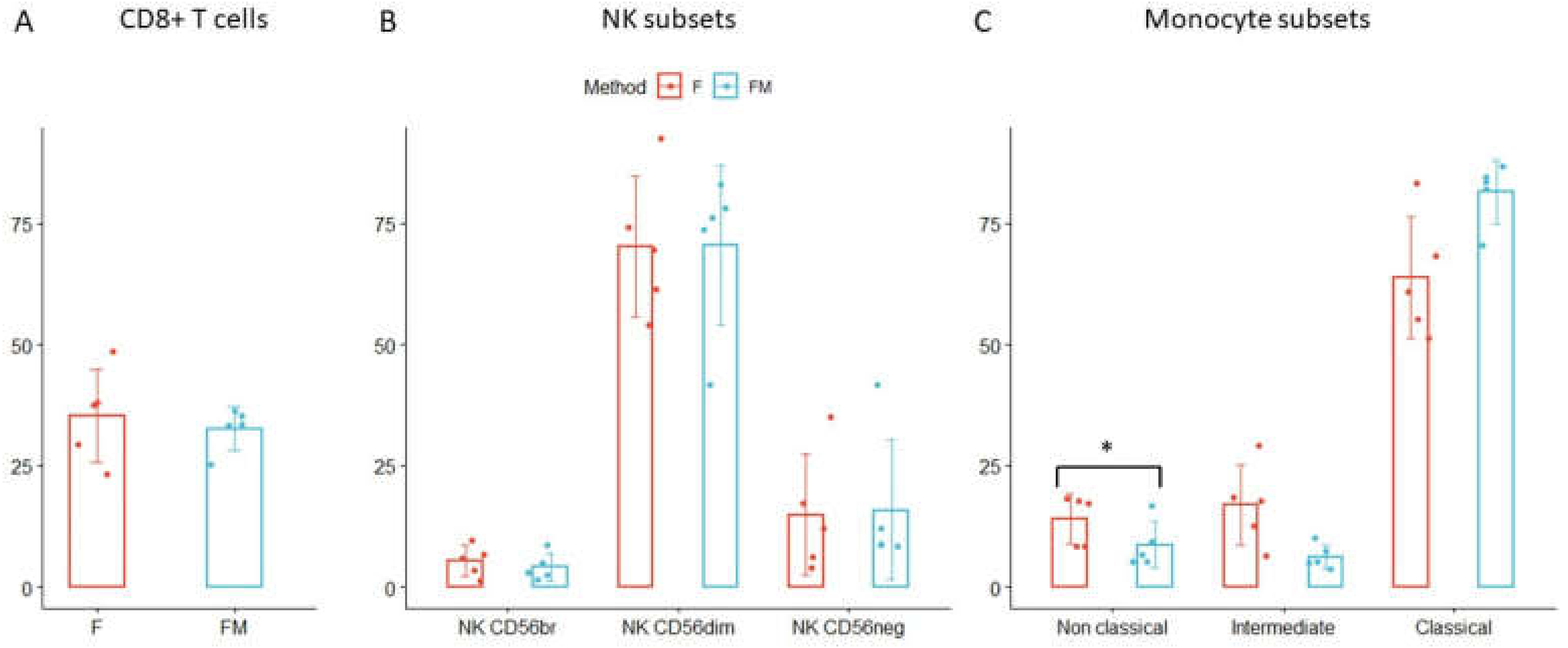
3.3. Monocytes and Granulocytes Detection in Thawed Samples
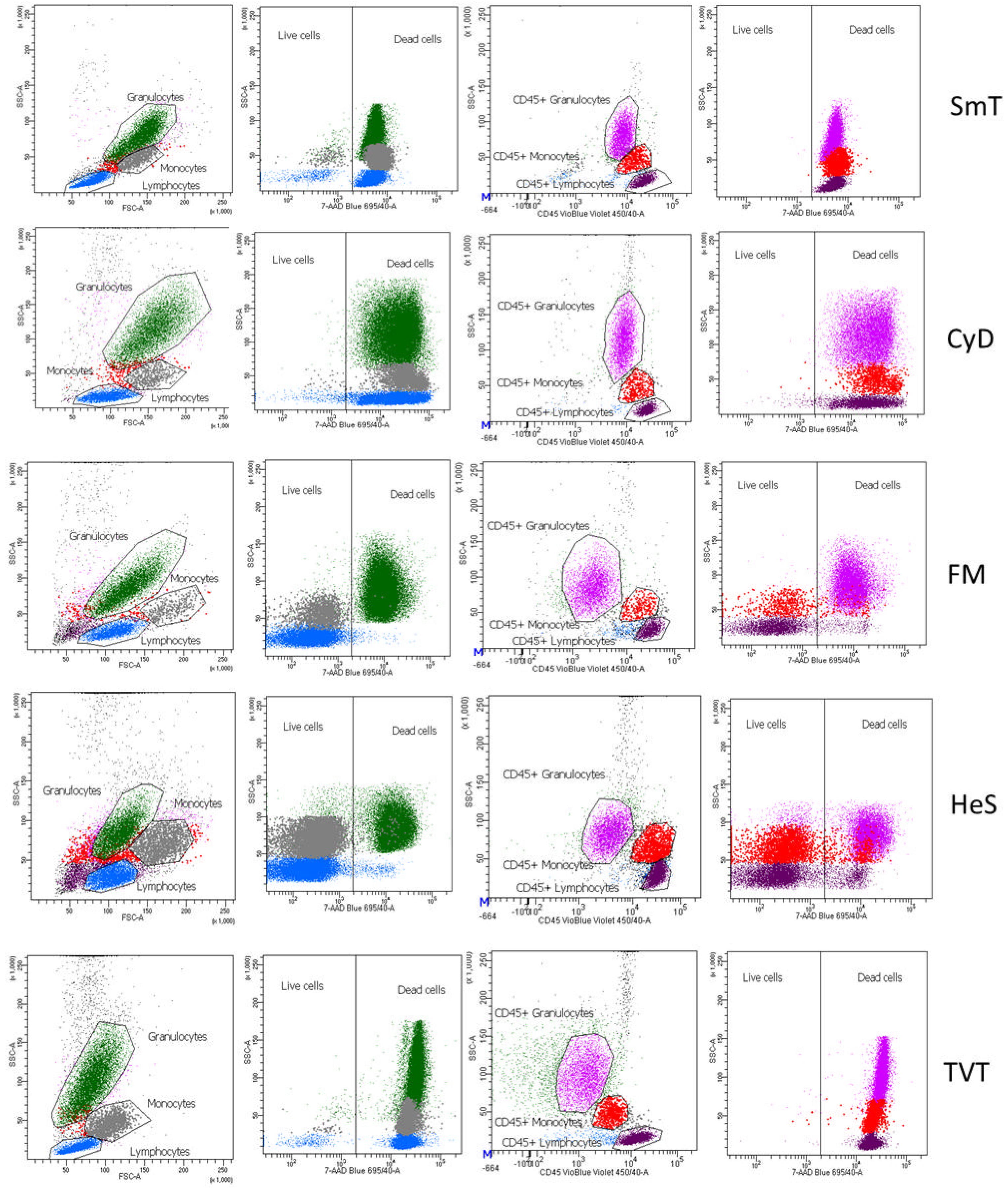
3.4. Leukocyte Viability
3.5. Further Immune Cell Characterization in FM Treated Bloods
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Orrù, V.; Steri, M.; Sole, G.; Sidore, C.; Virdis, F.; Dei, M.; Lai, S.; Zoledziewska, M.; Busonero, F.; Mulas, A.; et al. Genetic variants regulating immune cell levels in health and disease. Cell 2013, 155, 242–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patin, E.; Hasan, M.; Bergstedt, J.; Rouilly, V.; Libri, V.; Urrutia, A.; Alanio, C.; Scepanovic, P.; Hammer, C.; Jönsson, F.; et al. Natural variation in the parameters of innate immune cells is preferentially driven by genetic factors. Nat. Immunol. 2018, 19, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Orrù, V.; Steri, M.; Sidore, C.; Marongiu, M.; Serra, V.; Olla, S.; Sole, G.; Lai, S.; Dei, M.; Mulas, A.; et al. Complex genetic signatures in immune cells underlie autoimmunity and inform therapy. Nat. Genet. 2020, 52, 1266. [Google Scholar] [CrossRef] [PubMed]
- Roederer, M.; Quaye, L.; Mangino, M.; Beddall, M.H.; Mahnke, Y.; Chattopadhyay, P.; Tosi, I.; Napolitano, L.; Barberio, M.T.; Menni, C.; et al. The Genetic Architecture of the Human Immune System: A Bioresource for Autoimmunity and Disease Pathogenesis. Cell 2015, 161, 387–403. [Google Scholar] [CrossRef] [Green Version]
- Olin, A.; Henckel, E.; Chen, Y.; Lakshmikanth, T.; Pou, C.; Mikes, J.; Gustafsson, A.; Bernhardsson, A.K.; Zhang, C.; Bohlin, K.; et al. Stereotypic Immune System Development in Newborn Children. Cell 2018, 174, 1277–1292.e14. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.H.; Shannon, C.P.; Amenyogbe, N.; Bennike, T.B.; Diraj-Arce, J.; Idoko, O.T.; Gill, E.E.; Ben-Othman, R.; Pomat, W.S.; van Haren, S.D.; et al. Dynamic molecular changes during the first week of human life follow a robust developmental trajectory. Nat. Commun. 2019, 10, 1092. [Google Scholar] [CrossRef] [Green Version]
- Braudeau, C.; Guen, N.S.; Chevreuil, J.; Rimbert, M.; Martin, J.C.; Josien, R. An easy and reliable whole blood freezing method for flow cytometry immuno-phenotyping and functional analyses. Cytom. Part B Clin. Cytom. 2021, 100, 652–665. [Google Scholar] [CrossRef]
- Verschoor, C.P.; Kohli, V.; Balion, C. A comprehensive assessment of immunophenotyping performed in cryopreserved peripheral whole blood. Cytom. Part B Clin. Cytom. 2017, 94, 818–826. [Google Scholar] [CrossRef] [Green Version]
- Verschoor, C.P.; Kohli, V. Cryopreserved whole blood for the quantification of monocyte, T-cell and NK-cell subsets, and monocyte receptor expression by multi-color flow cytometry: A methodological study based on participants from the canadian longitudinal study on aging. Cytom. Part A 2018, 93, 548–555. [Google Scholar] [CrossRef] [Green Version]
- Peakman, T.C.; Elliott, P. The UK Biobank sample handling and storage validation studies. Int. J. Epidemiol. 2008, 37 (Suppl. 1), i2–i6. [Google Scholar] [CrossRef]
- Elliott, P.; Peakman, T.C.; Biobank, O.B.O.U. The UK Biobank sample handling and storage protocol for the collection, processing and archiving of human blood and urine. Int. J. Epidemiol. 2008, 37, 234–244. [Google Scholar] [CrossRef] [Green Version]
- Pilia, G.; Chen, W.-M.; Scuteri, A.; Orrù, M.; Albai, G.; Dei, M.; Lai, S.; Usala, G.; Lai, M.; Loi, P.; et al. Heritability of Cardiovascular and Personality Traits in 6,148 Sardinians. PLoS Genet. 2006, 2, e132. [Google Scholar] [CrossRef]
- Coupar, D.; Thompson, J.; Kaenzig, N. White Paper: Freezing Blood Samples for Deferred Immunophenotyping. Available online: https://www.cytomark.co.uk/downloads/WHITE_PAPER_Freezing_Blood_Samples_for_Deferred_Immunophenotyping_MARCH2019.pdf (accessed on 27 April 2022).
- Bohnhorst, J.; Bjørgan, M.B.; Thoen, J.E.; Natvig, J.B.; Thompson, K.M. Bm1–Bm5 Classification of Peripheral Blood B Cells Reveals Circulating Germinal Center Founder Cells in Healthy Individuals and Disturbance in the B Cell Subpopulations in Patients with Primary Sjögren’s Syndrome. J. Immunol. 2001, 167, 3610–3618. [Google Scholar] [CrossRef]
- Sallusto, F.; Lenig, D.; Forster, R.; Lipp, M.; Lanzavecchia, A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 1999, 401, 708–712. [Google Scholar] [CrossRef]
- Miyara, M.; Yoshioka, Y.; Kitoh, A.; Shima, T.; Wing, K.; Niwa, A.; Parizot, C.; Taflin, C.; Heike, T.; Valeyre, D.; et al. Functional Delineation and Differentiation Dynamics of Human CD4+ T Cells Expressing the FoxP3 Transcription Factor. Immunity 2009, 30, 899–911. [Google Scholar] [CrossRef] [Green Version]
- Perfetto, S.P.; Chattopadhyay, P.; Lamoreaux, L.; Nguyen, R.; Ambrozak, D.; Koup, R.A.; Roederer, M. Amine reactive dyes: An effective tool to discriminate live and dead cells in polychromatic flow cytometry. J. Immunol. Methods 2006, 313, 199–208. [Google Scholar] [CrossRef]
- Falzone, N.; Huyser, C.; Franken, D.R. Comparison between propidium iodide and 7-amino-actinomycin-D for viability assessment during flow cytometric analyses of the human sperm acrosome. Andrologia 2010, 42, 20–26. [Google Scholar] [CrossRef]
- Okada, R.; Kondo, T.; Matsuki, F.; Takata, H.; Takiguchi, M. Phenotypic classification of human CD4+ T cell subsets and their differentiation. Int. Immunol. 2008, 20, 1189–1199. [Google Scholar] [CrossRef]
- Cooper, M.; Fehniger, T.; Caligiuri, M.A. The biology of human natural killer-cell subsets. Trends Immunol. 2001, 22, 633–640. [Google Scholar] [CrossRef]
- Massey, F.J., Jr. The Kolmogorov–Smirnov Test for Goodness of Fit. J. Am. Stat. Assoc. 1951, 46, 68–78. [Google Scholar] [CrossRef]
- Tukey, J.W. Comparing individual means in the analysis of variance. Biometrics 1949, 5, 99–114. [Google Scholar] [CrossRef]
- Park, L.M.; Lannigan, J.; Jaimes, M.C. OMIP-069: Forty-Color Full Spectrum Flow Cytometry Panel for Deep Immunophenotyping of Major Cell Subsets in Human Peripheral Blood. Cytom. Part A 2020, 97, 1044–1051. [Google Scholar] [CrossRef]
- Diks, A.; Bonroy, C.; Teodosio, C.; Groenland, R.; de Mooij, B.; de Maertelaere, E.; Neirynck, J.; Philippé, J.; Orfao, A.; van Dongen, J.; et al. Impact of blood storage and sample handling on quality of high dimensional flow cytometric data in multicenter clinical research. J. Immunol. Methods 2019, 475, 112616. [Google Scholar] [CrossRef]
- Vera, E.J.; Chew, Y.V.; Nicholson, L.; Burns, H.; Anderson, P.; Chen, H.-T.; Williams, L.; Keung, K.; Zanjani, N.T.; Dervish, S.; et al. Standardisation of flow cytometry for whole blood immunophenotyping of islet transplant and transplant clinical trial recipients. PLoS ONE 2019, 14, e0217163. [Google Scholar] [CrossRef]
- Pinto, L.A.; Trivett, M.T.; Wallace, D.; Higgins, J.; Baseler, M.; Terabe, M.; Belyakov, I.M.; Berzofsky, J.A.; Hildesheim, A. Fixation and cryopreservation of whole blood and isolated mononuclear cells: Influence of different procedures on lymphocyte subset analysis by flow cytometry. Cytom. Part B Clin. Cytom. 2004, 63B, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Sakkestad, S.T.; Skavland, J.; Hanevik, K. Whole blood preservation methods alter chemokine receptor detection in mass cytometry experiments. J. Immunol. Methods 2019, 476, 112673. [Google Scholar] [CrossRef] [PubMed]
- Dzangué-Tchoupou, G.; Corneau, A.; Blanc, C.; Benveniste, O.; Allenbach, Y. Analysis of cell surface and intranuclear markers on non-stimulated human PBMC using mass cytometry. PLoS ONE 2018, 13, e0194593. [Google Scholar] [CrossRef]
- Amir, E.-A.D.; Lee, B.; Badoual, P.; Gordon, M.; Guo, X.V.; Merad, M.; Rahman, A.H. Development of a Comprehensive Antibody Staining Database Using a Standardized Analytics Pipeline. Front. Immunol. 2019, 10, 1315. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Methods | ||||||
|---|---|---|---|---|---|---|
| Individuals | Fresh | SmT | CyD | FM | HeS | TVT |
| 1 | B-CD4 | B-CD4 | B-CD4-Vi | B-CD4 | ||
| 2 | B-CD4 | B-CD4-Vi | B-CD4-Vi | B-CD4-Vi | ||
| 3 | B-CD4 | B-CD4 | B-CD4-Vi | B-CD4 | ||
| 4 | B-CD4 | B-CD4-Vi | B-CD4 | B-CD4-Vi | ||
| 5 | B-CD4 | B-CD4-Vi | B-CD4 | |||
| 6 | B-CD4 | B-CD4-Vi | ||||
| 7 | B-CD4 | B-CD4-Vi | ||||
| 8 | B-CD4 | B-CD4-Vi | ||||
| 9 | B-CD4 | B-CD4 | ||||
| 10 | B-CD4 | B-CD4 | ||||
| 11 | B-CD4 | B-CD4-Vi | ||||
| 12 | B-CD4 | B-CD4-Vi | ||||
| 13 | B-CD4 | B-CD4-Vi | ||||
| 14 | B-CD4 | |||||
| 15 | B-CD4 | |||||
| 16 | M8NK | B-M8NK | ||||
| 17 | M8NK | B-M8NK | ||||
| 18 | CD4 | CD4 | ||||
| 19 | CD4 | CD4 | ||||
| 20 | M8NK | M8NK | ||||
| 21 | M8NK | M8NK | ||||
| 22 | M8NK | M8NK | ||||
| Trait Name | Panel Name | rSmT | rCyD | rFM | rHeS | rTVTs | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mean | ±Std | Mean | ±Std | Mean | ±Std | Mean | ±Std | Mean | ±Std | ||
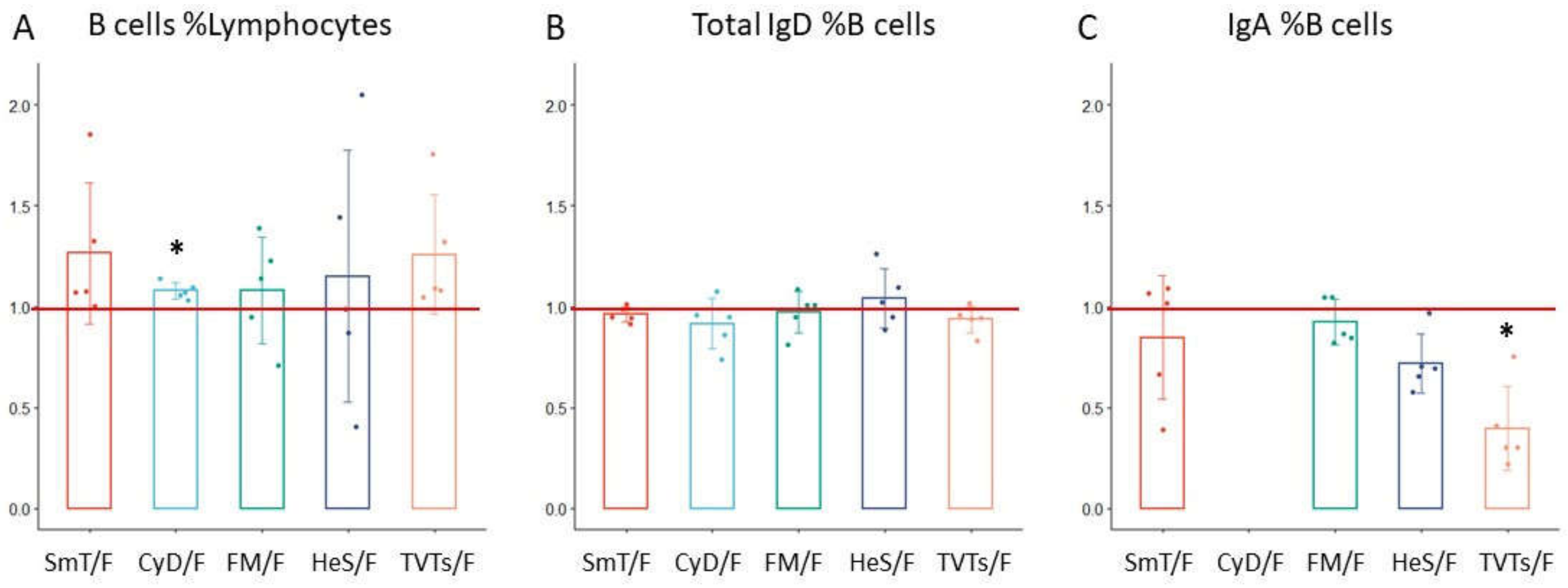
| B cells %Lymphocyte | B cell | 1.27 | 0.35 | 1.08 | 0.04 | 1.08 | 0.26 | 1.15 | 0.62 | 1.26 | 0.30 |
| Total IgD %B cell | B cell | 0.96 | 0.04 | 0.92 | 0.12 | 0.97 | 0.10 | 1.04 | 0.15 | 0.94 | 0.07 |
| IgD− CD38br %B cell | B cell | 0.56 | 0.15 | N/A | N/A | 1.24 | 0.78 | 2.25 | 1.06 | 0.54 | 0.33 |
| IgD− CD38dim %B cell | B cell | 1.57 | 0.33 | N/A | N/A | 0.97 | 0.49 | 1.12 | 0.45 | 2.65 | 0.34 |
| IgD− CD38− %B cell | B cell | 0.73 | 0.13 | N/A | N/A | 1.11 | 0.27 | 0.89 | 0.29 | 0.36 | 0.12 |
| IgD+ CD38br %B cell | B cell | 0.62 | 0.39 | N/A | N/A | 0.73 | 0.25 | 1.39 | 0.80 | 0.24 | 0.11 |
| IgD+ CD38dim %B cell | B cell | 1.31 | 0.07 | N/A | N/A | 0.90 | 0.30 | 1.16 | 0.38 | 1.47 | 0.11 |
| IgD+ CD38− %B cell | B cell | 0.75 | 0.20 | N/A | N/A | 1.30 | 0.42 | 0.82 | 0.35 | 0.27 | 0.12 |
| Naive %B cell | B cell | N/A | N/A | N/A | N/A | 0.92 | 0.15 | 1.03 | 0.19 | N/A | N/A |
| Not Sw Mem %B cell | B cell | N/A | N/A | N/A | N/A | 1.06 | 0.21 | 1.01 | 0.26 | N/A | N/A |
| IgD− CD27− %B cell | B cell | N/A | N/A | N/A | N/A | 1.11 | 0.29 | 0.81 | 0.25 | N/A | N/A |
| Sw Mem %B cells | B cell | N/A | N/A | N/A | N/A | 1.04 | 0.18 | 1.07 | 0.33 | N/A | N/A |
| Plasma cells %B cell | B cell | 0.90 | 0.96 | N/A | N/A | 2.52 | 1.00 | 2.26 | 1.22 | 1.58 | 1.04 |
| Memory %B cell | B cell | 0.67 | 0.19 | 0.82 | 0.10 | 0.79 | 0.14 | 0.89 | 0.20 | 0.49 | 0.07 |
| Naive-Mature %B cell | B cell | 1.22 | 0.06 | 1.37 | 0.53 | 1.13 | 0.09 | 0.99 | 0.12 | 1.37 | 0.15 |
| CD24+ CD27+ %B cell | B cell | N/A | N/A | N/A | N/A | 0.88 | 0.24 | 0.91 | 0.23 | N/A | N/A |
| Transitional %B cell | B cell | 0.85 | 0.26 | 0.78 | 0.03 | 0.95 | 0.18 | 1.66 | 0.11 | 0.90 | 0.24 |
| IgA %B cell | B cell | 0.85 | 0.31 | N/A | N/A | 0.93 | 0.11 | 0.72 | 0.15 | 0.40 | 0.21 |
| CD3+ %Lymphocyte | CD4 | 1.04 | 0.05 | 1.08 | 0.05 | 1.19 | 0.17 | 0.89 | 0.12 | 1.00 | 0.04 |
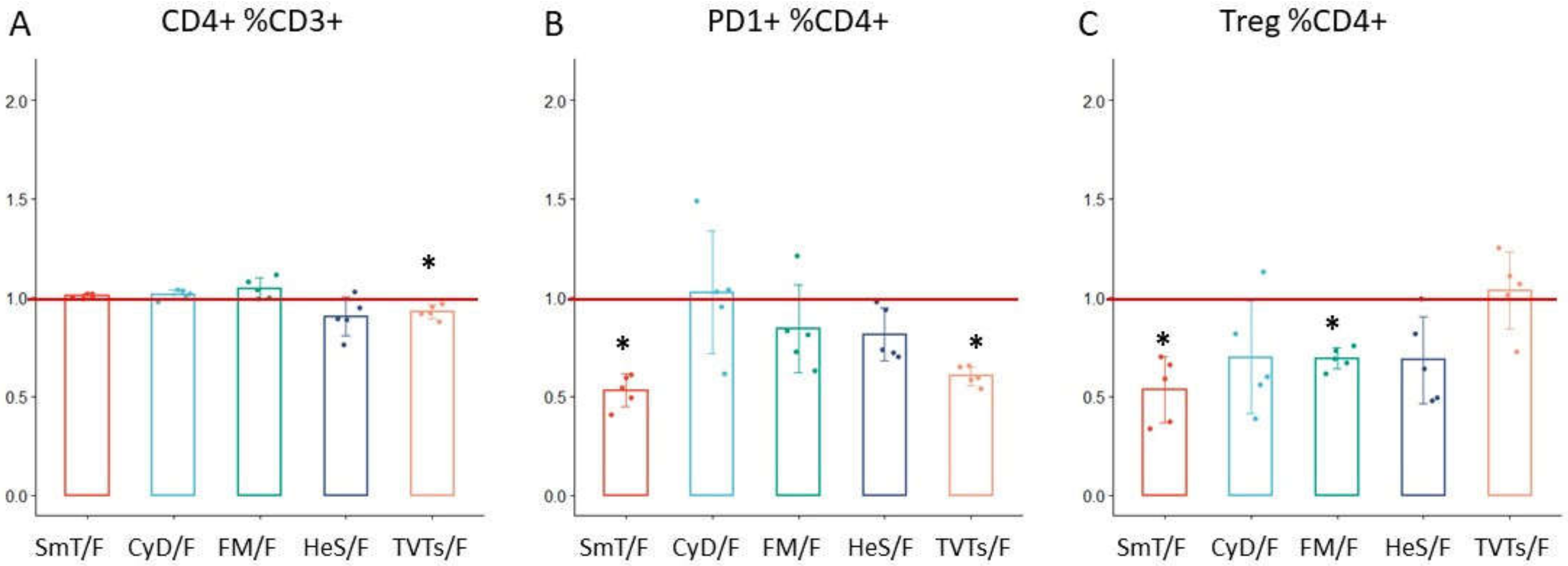
| CD4+ %CD3+ | CD4 | 1.01 | 0.01 | 1.02 | 0.02 | 1.05 | 0.05 | 0.90 | 0.10 | 0.93 | 0.04 |
| Treg %CD4+ | CD4 | 0.53 | 0.17 | 0.70 | 0.29 | 0.69 | 0.05 | 0.68 | 0.22 | 1.03 | 0.19 |
| resting %Treg | CD4 | 1.10 | 0.28 | 3.04 | 2.03 | 2.30 | 1.26 | 1.90 | 0.79 | N/A | N/A |
| activated %Treg | CD4 | 0.94 | 0.27 | 0.99 | 0.36 | 0.80 | 0.16 | 0.92 | 0.12 | N/A | N/A |
| secreting %Treg | CD4 | 1.08 | 0.25 | 0.86 | 0.13 | 1.11 | 0.10 | 1.00 | 0.11 | N/A | N/A |
| Th17 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 3.12 | 4.76 | N/A | N/A | N/A | N/A |
| Th1-Th17 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 0.93 | 1.13 | N/A | N/A | N/A | N/A |
| Th2 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 1.43 | 0.20 | N/A | N/A | N/A | N/A |
| Th1 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 0.23 | 0.28 | N/A | N/A | N/A | N/A |
| CXCR3 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 0.25 | 0.30 | N/A | N/A | N/A | N/A |
| CCR6 %CD4+ | CD4 | N/A | N/A | N/A | N/A | 1.92 | 3.41 | N/A | N/A | N/A | N/A |
| CCR4 %CD4+ | CD4 | 0.15 | 0.03 | 0.77 | 0.60 | 0.92 | 0.11 | 1.12 | 0.32 | 0.23 | 0.04 |
| CM %CD4+ | CD4 | 1.17 | 0.12 | 0.90 | 0.08 | 1.06 | 0.14 | 0.90 | 0.21 | N/A | N/A |
| Naive %CD4+ | CD4 | 1.06 | 0.03 | 1.35 | 0.12 | 1.29 | 0.22 | 1.39 | 0.27 | N/A | N/A |
| EM %CD4+ | CD4 | 0.76 | 0.07 | 0.80 | 0.15 | 0.69 | 0.14 | 0.84 | 0.32 | N/A | N/A |
| TD %CD4+ | CD4 | 1.12 | 0.26 | 1.09 | 0.33 | 1.01 | 0.55 | 1.00 | 0.28 | N/A | N/A |
| CXCR5 %CD4+ | CD4 | N/A | N/A | 0.36 | 0.28 | 0.81 | 0.57 | N/A | N/A | N/A | N/A |
| ICOS+ %CD4+ | CD4 | 0.56 | 0.21 | 0.47 | 0.15 | 0.79 | 0.36 | 0.97 | 0.39 | 0.68 | 0.18 |
| PD1+ %CD4+ | CD4 | 0.53 | 0.08 | 1.03 | 0.31 | 0.84 | 0.22 | 0.81 | 0.13 | 0.60 | 0.05 |
| CD161+ %CD4+ | CD4 | 0.44 | 0.12 | N/A | N/A | 0.74 | 0.23 | 0.68 | 0.08 | 0.95 | 0.07 |
| CD8+ %CD3+ | M8NK | - | - | - | - | 0.96 | 0.20 | - | - | - | - |
| Non classical %Monocytes | M8NK | - | - | - | - | 0.62 | 0.20 | - | - | - | - |
| Intermediate %Monocytes | M8NK | - | - | - | - | 0.52 | 0.43 | - | - | - | - |
| Classical %Monocytes | M8NK | - | - | - | - | 1.32 | 0.29 | - | - | - | - |
| EMRA CD8+ %CD8+ | M8NK | - | - | - | - | 1.18 | 0.45 | - | - | - | - |
| Naive CD8+ %CD8+ | M8NK | - | - | - | - | 1.06 | 0.25 | - | - | - | - |
| CM CD8+ %CD8+ | M8NK | - | - | - | - | 1.12 | 0.50 | - | - | - | - |
| EM CD8+ %CD8+ | M8NK | - | - | - | - | 1.07 | 0.51 | - | - | - | - |
| NK CD56br %NKs | M8NK | - | - | - | - | 1.05 | 0.90 | - | - | - | - |
| NK CD56dim %NKs | M8NK | - | - | - | - | 1.00 | 0.18 | - | - | - | - |
| NK CD56neg %NKs | M8NK | - | - | - | - | 1.26 | 0.65 | - | - | - | - |
| M/Ly | Vi | 1.19 | 0.05 | 1.20 | 0.19 | 0.96 | 0.35 | 1.06 | 0.13 | 0.95 | 0.04 |
| Gr/Ly | Vi | 1.23 | 0.18 | 1.17 | 0.05 | 1.06 | 0.22 | 0.35 | 0.12 | 0.78 | 0.33 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, V.; Orrù, V.; Lai, S.; Lobina, M.; Steri, M.; Cucca, F.; Fiorillo, E. Comparison of Whole Blood Cryopreservation Methods for Extensive Flow Cytometry Immunophenotyping. Cells 2022, 11, 1527. https://doi.org/10.3390/cells11091527
Serra V, Orrù V, Lai S, Lobina M, Steri M, Cucca F, Fiorillo E. Comparison of Whole Blood Cryopreservation Methods for Extensive Flow Cytometry Immunophenotyping. Cells. 2022; 11(9):1527. https://doi.org/10.3390/cells11091527
Chicago/Turabian StyleSerra, Valentina, Valeria Orrù, Sandra Lai, Monia Lobina, Maristella Steri, Francesco Cucca, and Edoardo Fiorillo. 2022. "Comparison of Whole Blood Cryopreservation Methods for Extensive Flow Cytometry Immunophenotyping" Cells 11, no. 9: 1527. https://doi.org/10.3390/cells11091527
APA StyleSerra, V., Orrù, V., Lai, S., Lobina, M., Steri, M., Cucca, F., & Fiorillo, E. (2022). Comparison of Whole Blood Cryopreservation Methods for Extensive Flow Cytometry Immunophenotyping. Cells, 11(9), 1527. https://doi.org/10.3390/cells11091527