
The Ash2l SDI Domain Is Required to Maintain the Stability and Binding of DPY30

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cell Culture
2.3. Tissue Section and Immunofluorescence
2.4. Plasmid Construction
2.5. RT-qPCR
2.6. Western Blot and Coimmunoprecipitation
2.7. RNA-Seq and ChIP-Seq Analysis
3. Results
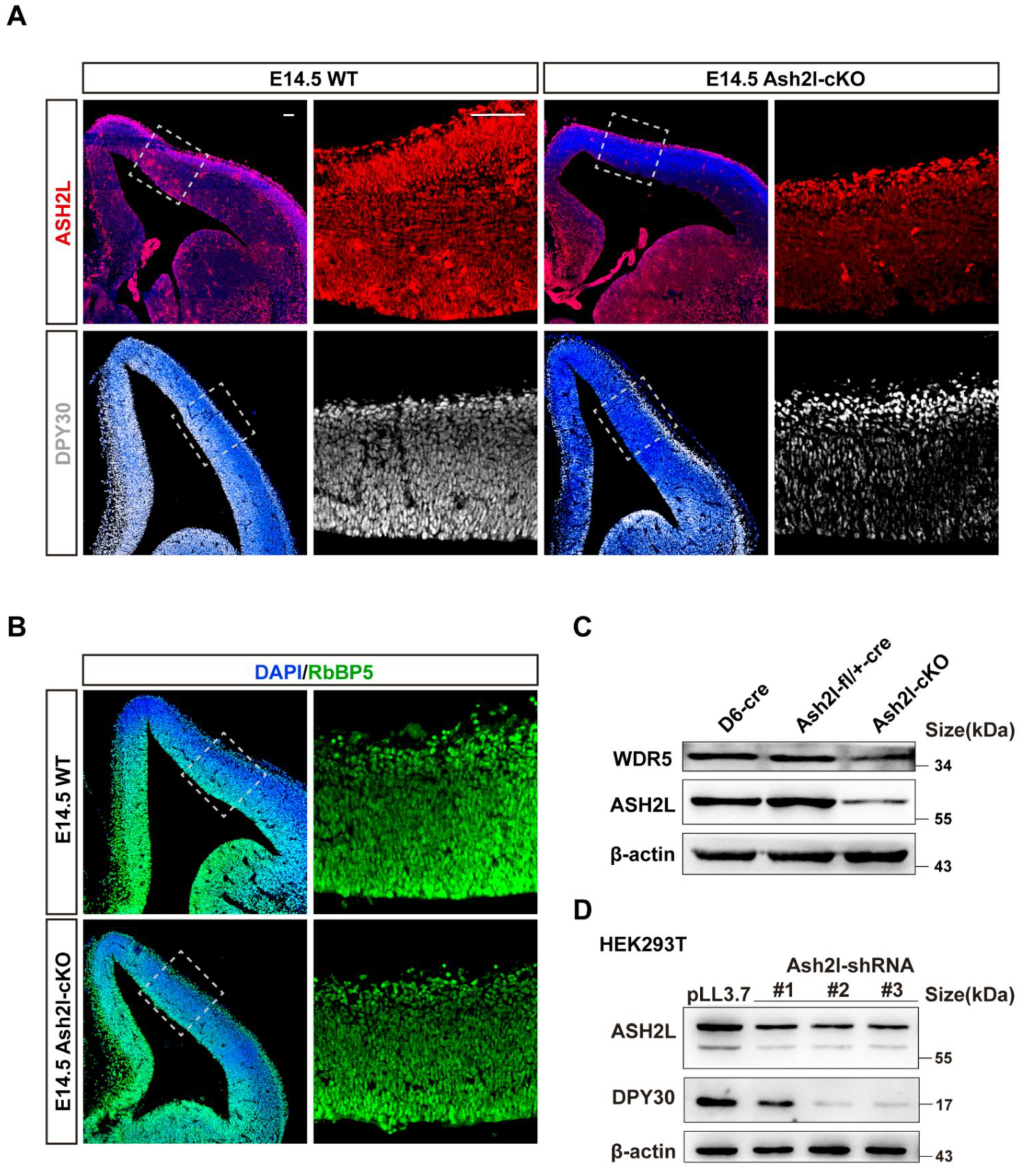
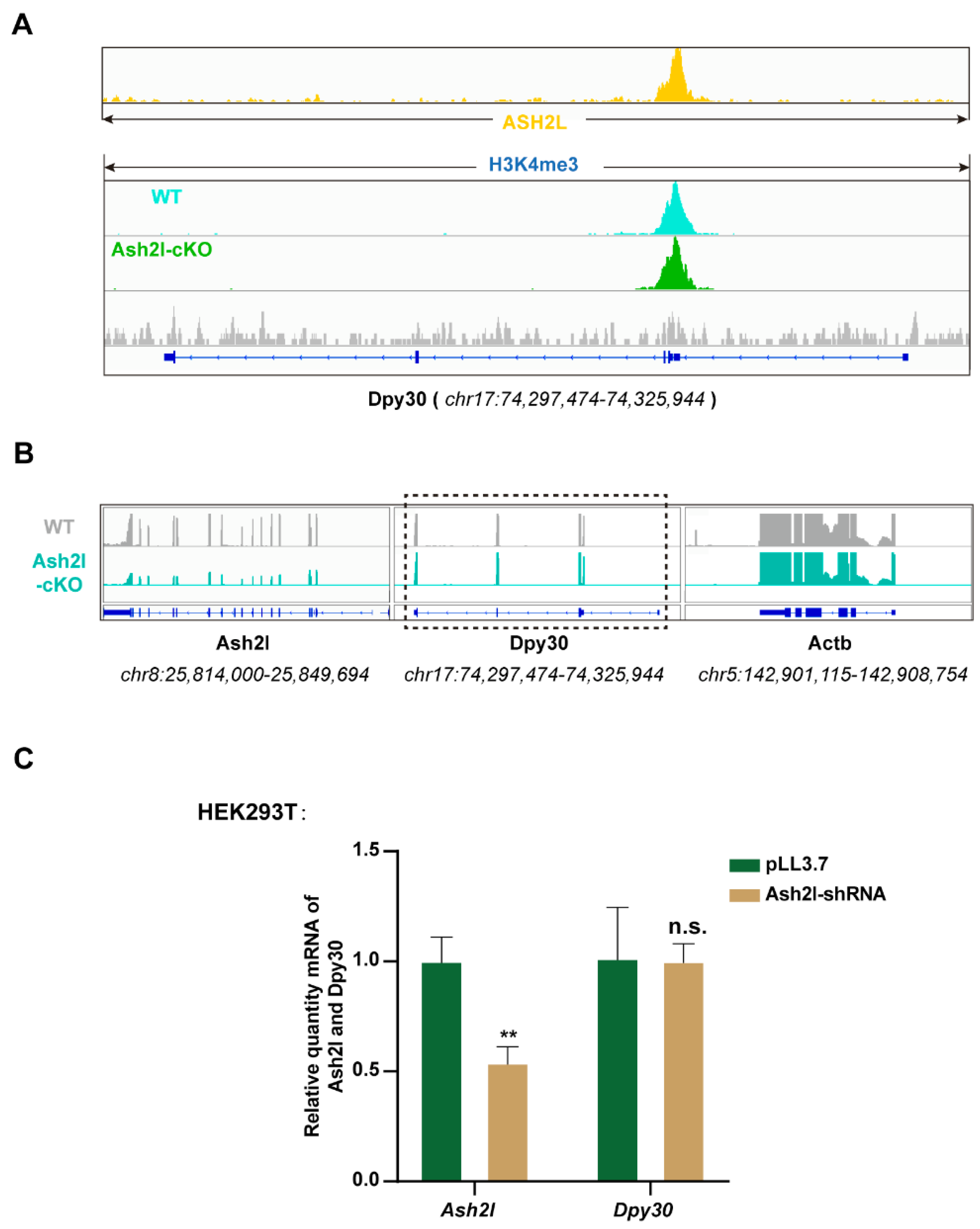
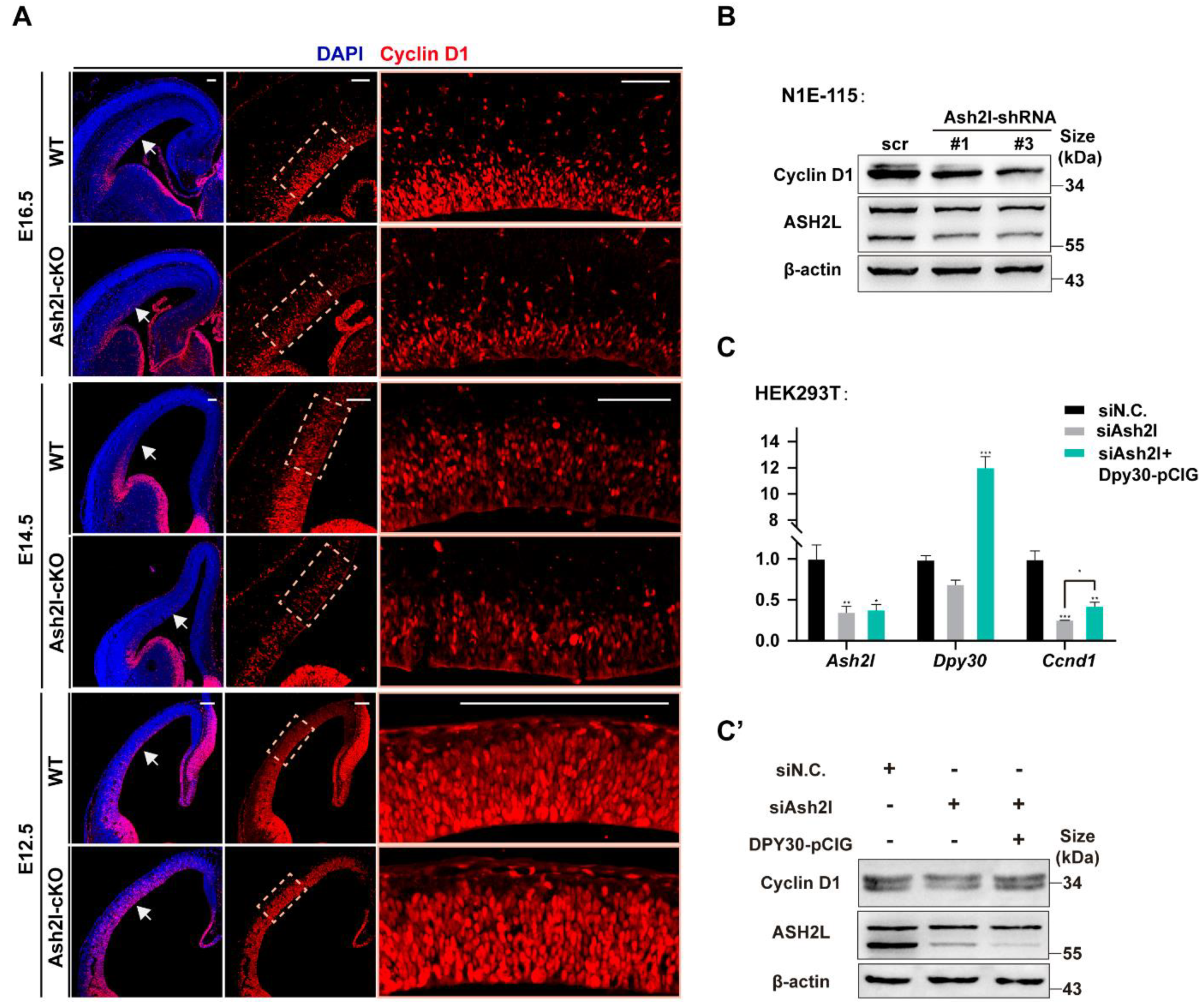
3.1. ASH2L Is Necessary for the Expression of DPY30
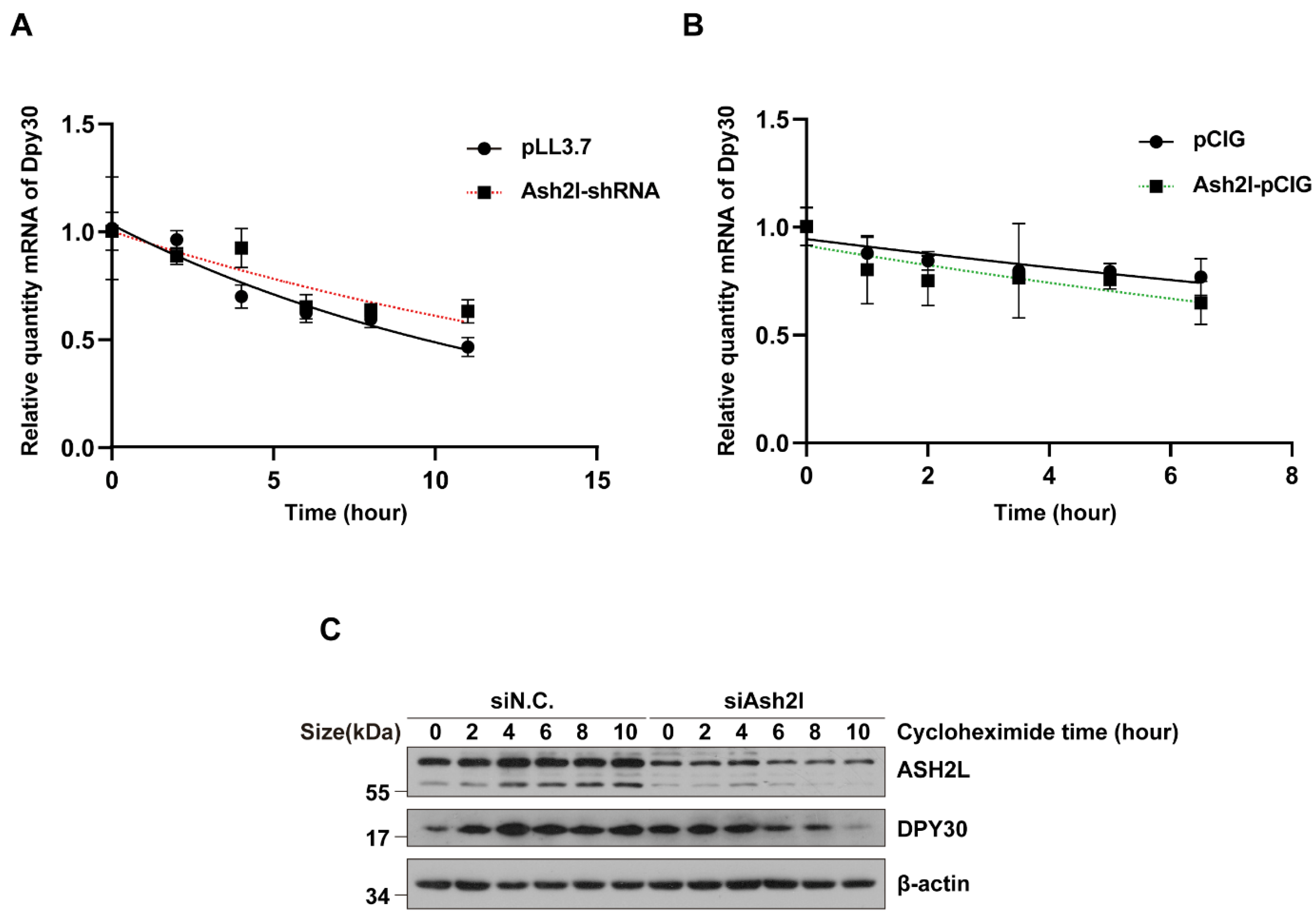
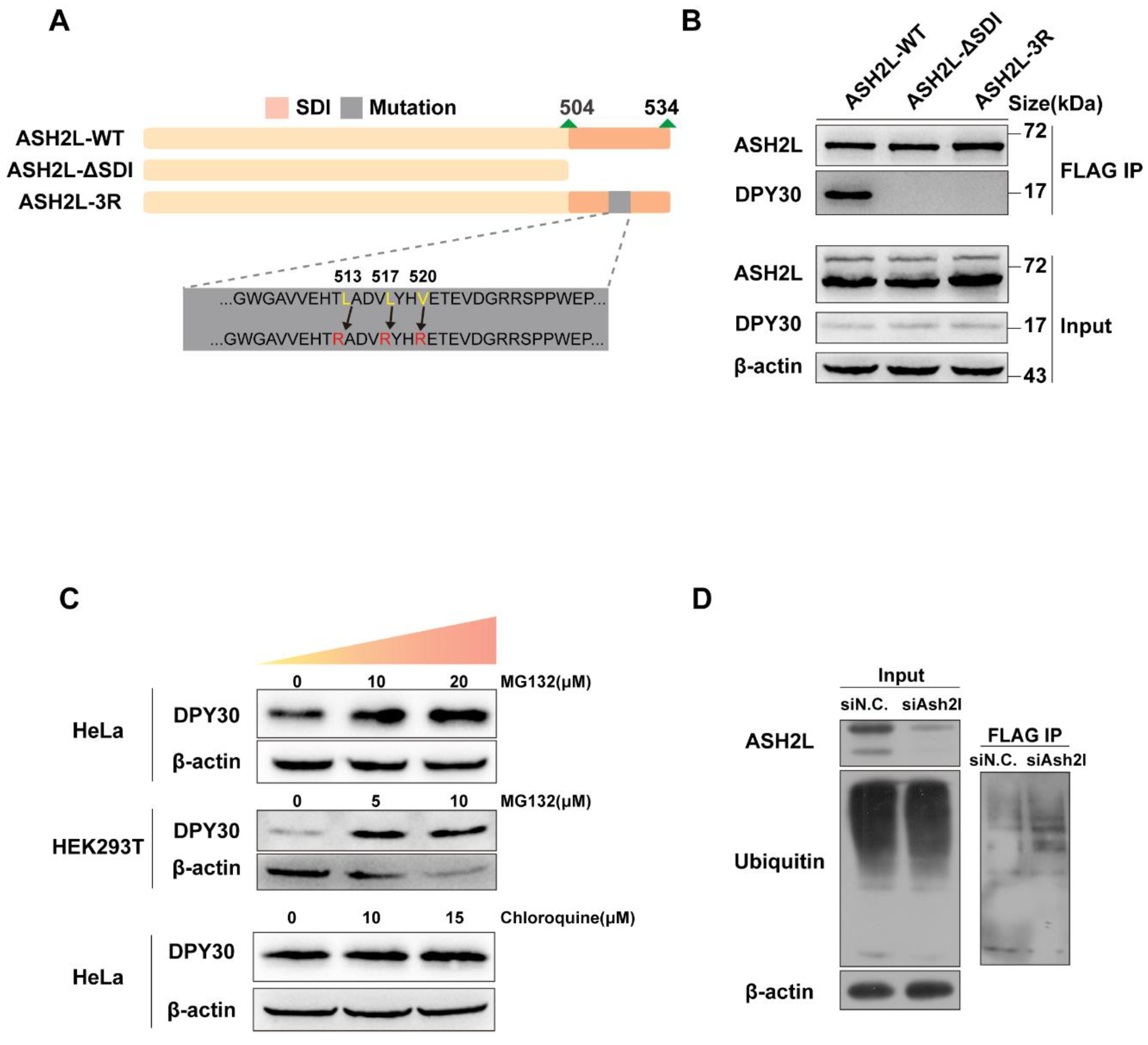
3.2. The Stability of DPY30 Relies on Direct Binding with ASH2L
3.3. The Degradation of DPY30 Relies on the Ubiquitin-Proteasome System
3.4. DPY30 Is Capable of Ameliorating the Outcome of ASH2L Depletion
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ringrose, L.; Paro, R. Epigenetic regulation of cellular memory by the Polycomb and Trithorax group proteins. Annu. Rev. Genet. 2004, 38, 413–443. [Google Scholar] [CrossRef] [PubMed]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; et al. New Nomenclature for Chromatin-Modifying Enzymes. Cell 2007, 131, 633–636. [Google Scholar] [CrossRef] [PubMed]
- Dou, Y.; Milne, T.A.; Ruthenburg, A.J.; Lee, S.; Lee, J.W.; Verdine, G.L.; Allis, C.D.; Roeder, R.G. Regulation of MLL1 H3K4 methyltransferase activity by its core components. Nat. Struct. Mol. Biol. 2006, 13, 713–719. [Google Scholar] [CrossRef]
- Schuetz, A.; Allali-Hassani, A.; Martín, F.; Loppnau, P.; Vedadi, M.; Bochkarev, A.; Plotnikov, A.N.; Arrowsmith, C.; Min, J. Structural basis for molecular recognition and presentation of histone H3 by WDR5. EMBO J. 2006, 25, 4245–4252. [Google Scholar] [CrossRef]
- Steward, M.M.; Lee, J.-S.; O’Donovan, A.; Wyatt, M.; Bernstein, B.E.; Shilatifard, A. Molecular regulation of H3K4 trimethylation by ASH2L, a shared subunit of MLL complexes. Nat. Struct. Mol. Biol. 2006, 13, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Chen, Y.; Cierpicki, T.; Liu, Y.; Basrur, V.; Lei, M.; Dou, Y. An Ash2L/RbBP5 heterodimer stimulates the MLL1 methyltransferase activity through coordinated substrate interactions with the MLL1 SET domain. PLoS ONE 2010, 5, e14102. [Google Scholar] [CrossRef]
- Nagy, P.L.; Griesenbeck, J.; Kornberg, R.D.; Cleary, M.L. A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc. Natl. Acad. Sci. USA 2002, 99, 90–94. [Google Scholar] [CrossRef]
- Shilatifard, A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu. Rev. Biochem. 2012, 81, 65–95. [Google Scholar] [CrossRef]
- Patel, A.; Dharmarajan, V.; Vought, V.E.; Cosgrove, M.S. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J. Biol. Chem. 2009, 284, 24242–24256. [Google Scholar] [CrossRef]
- Chen, Y.; Cao, F.; Wan, B.; Dou, Y.; Lei, M. Structure of the SPRY domain of human Ash2L and its interactions with RbBP5 and DPY30. Cell Res. 2012, 22, 598–602. [Google Scholar] [CrossRef]
- Lee, Y.T.; Ayoub, A.; Park, S.-H.; Sha, L.; Xu, J.; Mao, F.; Zheng, W.; Zhang, Y.; Cho, U.-S.; Dou, Y. Mechanism for DPY30 and ASH2L intrinsically disordered regions to modulate the MLL/SET1 activity on chromatin. Nat. Commun. 2021, 12, 2953. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, V.; Zhang, P.; Chaturvedi, C.-P.; Thornton, J.; Brunzelle, J.S.; Skiniotis, G.; Shilatifard, A.; Brand, M.; Couture, J.-F. Molecular basis for DPY-30 association to COMPASS-like and NURF complexes. Structure 2014, 22, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H. The complex activities of the SET1/MLL complex core subunits in development and disease. Biochim. Biophys. Acta Gene Regul. Mech. 2020, 1863, 194560. [Google Scholar] [CrossRef] [PubMed]
- Schneider, R.; Bannister, A.J.; Myers, F.A.; Thorne, A.W.; Crane-Robinson, C.; Kouzarides, T. Histone H3 lysine 4 methylation patterns in higher eukaryotic genes. Nat. Cell Biol. 2004, 6, 73–77. [Google Scholar] [CrossRef]
- Ruthenburg, A.J.; Allis, C.D.; Wysocka, J. Methylation of lysine 4 on histone H3: Intricacy of writing and reading a single epigenetic mark. Mol. Cell 2007, 25, 15–30. [Google Scholar] [CrossRef]
- Blobel, G.A.; Kadauke, S.; Wang, E.; Lau, A.W.; Zuber, J.; Chou, M.M.; Vakoc, C.R. A reconfigured pattern of MLL occupancy within mitotic chromatin promotes rapid transcriptional reactivation following mitotic exit. Mol. Cell 2009, 36, 970–983. [Google Scholar] [CrossRef]
- Soares, L.M.; He, P.C.; Chun, Y.; Suh, H.; Kim, T.; Buratowski, S. Determinants of Histone H3K4 Methylation Patterns. Mol. Cell 2017, 68, 773–785.e6. [Google Scholar] [CrossRef]
- Ning, B.; Li, W.; Zhao, W.; Wang, R. Targeting Epigenetic Regulations in Cancer. Acta Biochim. Biophys. Sin. (Shanghai) 2016, 48, 97–109. [Google Scholar] [CrossRef]
- Landgrave-Gómez, J.; Mercado-Gómez, O.; Guevara-Guzmán, R. Epigenetic mechanisms in neurological and neurodegenerative diseases. Front. Cell. Neurosci. 2015, 9, 58. [Google Scholar]
- Lim, D.A.; Huang, Y.-C.; Swigut, T.; Mirick, A.L.; García-Verdugo, J.M.; Wysocka, J.; Ernst, P.; Alvarez-Buylla, A. Chromatin remodelling factor Mll1 is essential for neurogenesis from postnatal neural stem cells. Nature 2009, 458, 529–533. [Google Scholar] [CrossRef]
- Lavery, W.J.; Barski, A.; Wiley, S.; Schorry, E.K.; Lindsley, A.W. KMT2C/D COMPASS complex-associated diseases [KCDCOM-ADs]: An emerging class of congenital regulopathies. Clin. Epigenet. 2020, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Koemans, T.S.; Kleefstra, T.; Chubak, M.C.; Stone, M.H.; Reijnders, M.R.F.; De Munnik, S.; Willemsen, M.H.; Fenckova, M.; Stumpel, C.T.R.M.; Bok, L.A.; et al. Functional convergence of histone methyltransferases EHMT1 and KMT2C involved in intellectual disability and autism spectrum disorder. PLoS Genet. 2017, 13, e1006864. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Ruan, X.; Wen, C.; Chen, P.; Liu, W.; Zhu, L.; Xiang, P.; Zhang, X.; Wei, Q.; Hou, L.; et al. The COMPASS Family Protein ASH2L Mediates Corticogenesis via Transcriptional Regulation of Wnt Signaling. Cell Rep. 2019, 28, 698–711.e5. [Google Scholar] [CrossRef] [PubMed]
- Beilharz, T.H.; Harrison, P.F.; Miles, D.M.; See, M.M.; Le, U.M.; Kalanon, M.; Curtis, M.J.; Hasan, Q.; Saksouk, J.; Margaritis, T.; et al. Coordination of Cell Cycle Progression and Mitotic Spindle Assembly Involves Histone H3 Lysine 4 Methylation by Set1/COMPASS. Genetics 2017, 205, 185–199. [Google Scholar] [CrossRef]
- Yang, Z.; Augustin, J.; Chang, C.; Hu, J.; Shah, K.; Chang, C.W.; Townes, T.; Jiang, H. The DPY30 subunit in SET1/MLL complexes regulates the proliferation and differentiation of hematopoietic progenitor cells. Blood 2014, 124, 2025–2033. [Google Scholar] [CrossRef]
- Ali, A.; Veeranki, S.N.; Tyagi, S. A SET-domain-independent role of WRAD complex in cell-cycle regulatory function of mixed lineage leukemia. Nucleic Acids Res. 2014, 42, 7611–7624. [Google Scholar] [CrossRef]
- Luscher-Firzlaff, J.; Chatain, N.; Kuo, C.-C.; Braunschweig, T.; Bochyńska, A.; Ullius, A.; Denecke, B.; Costa, I.G.; Koschmieder, S.; Lüscher, B. Hematopoietic stem and progenitor cell proliferation and differentiation requires the trithorax protein Ash2l. Sci. Rep. 2019, 9, 8262. [Google Scholar] [CrossRef]
- Campbell, S.A.; McDonald, C.L.; Krentz, N.; Lynn, F.; Hoffman, B.G. TrxG Complex Catalytic and Non-catalytic Activity Play Distinct Roles in Pancreas Progenitor Specification and Differentiation. Cell Rep. 2019, 28, 1830–1844.e6. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, S.; Li, A.; Zhang, A.; Zhang, S.; Chen, L. DPY30 is required for the enhanced proliferation, motility and epithelial-mesenchymal transition of epithelial ovarian cancer cells. Int. J. Mol. Med. 2018, 42, 3065–3072. [Google Scholar] [CrossRef]
- Machon, O.; Bout, C.V.D.; Backman, M.; Røsok, Ø.; Caubit, X.; Fromm, S.; Geronimo, B.; Krauss, S. Forebrain-specific promoter/enhancer D6 derived from the mouse Dach1 gene controls expression in neural stem cells. Neuroscience 2002, 112, 951–966. [Google Scholar] [CrossRef]
- Jiang, H.; Shukla, A.; Wang, X.; Chen, W.Y.; Bernstein, B.E.; Roeder, R.G. Role for Dpy-30 in ES cell-fate specification by regulation of H3K4 methylation within bivalent domains. Cell 2011, 144, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; Ayoub, A.; Lee, Y.-T.; Xu, J.; Kim, H.; Zheng, W.; Zhang, B.; Sha, L.; An, S.; Zhang, Y.; et al. Cryo-EM structure of the human MLL1 core complex bound to the nucleosome. Nat. Commun. 2019, 10, 5540. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Peng, Y.; Peng, Y.; Xu, F.; He, X.; Wang, F.; Peng, X.; Qiang, B.; Yuan, J.; Rao, Z.; et al. Characterization and crystallization of human DPY-30-like protein, an essential component of dosage compensation complex. Biochim. Biophys. Acta 2005, 1753, 257–262. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Goldberg, A.L. Proteasome inhibitors: Valuable new tools for cell biologists. Trends Cell Biol. 1998, 8, 397–403. [Google Scholar] [CrossRef]
- Nguyen, V.N.; Huang, K.-Y.; Weng, J.T.-Y.; Lai, K.R.; Lee, T.-Y. UbiNet: An online resource for exploring the functional associations and regulatory networks of protein ubiquitylation. Database (Oxf.) 2016, 2016, baw054. [Google Scholar] [CrossRef]
- Wang, X.; Wang, C.; Yan, G.; Kang, Y.; Sun, G.; Wang, S.; Zou, R.; Sun, H.; Zeng, K.; Song, H.; et al. BAP18 is involved in upregulation of CCND1/2 transcription to promote cell growth in oral squamous cell carcinoma. EBioMedicine 2020, 53, 102685. [Google Scholar] [CrossRef]
- Mattiroli, F.; Sixma, T.K. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat. Struct. Mol. Biol. 2014, 21, 308–316. [Google Scholar] [CrossRef]
- Cowan, A.D.; Ciulli, A. Driving E3 Ligase Substrate Specificity for Targeted Protein Degradation: Lessons from Nature and the Laboratory. Annu. Rev. Biochem. 2022, 91, 8–27. [Google Scholar] [CrossRef]
- Shah, K.K.; Whitaker, R.H.; Busby, T.; Hu, J.; Shi, B.; Wang, Z.; Zang, C.; Placzek, W.J.; Jiang, H. Specific inhibition of DPY30 activity by ASH2L-derived peptides suppresses blood cancer cell growth. Exp. Cell Res. 2019, 382, 111485. [Google Scholar] [CrossRef]
- Vermeulen, M.; Whitaker, R.H.; Busby, T.; Hu, J.; Shi, B.; Wang, Z.; Zang, C.; Placzek, W.J.; Jiang, H. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell 2010, 142, 967–980. [Google Scholar] [CrossRef]
- Sun, G.; Wang, C.; Wang, S.; Sun, H.; Zeng, K.; Zou, R.; Lin, L.; Liu, W.; Zhou, T.; Jin, F.; et al. An H3K4me3 reader, BAP18 as an adaptor of COMPASS-like core subunits co-activates ERalpha action and associates with the sensitivity of antiestrogen in breast cancer. Nucleic Acids Res. 2020, 48, 10768–10784. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Zhong, X.; Wang, C.; Sun, H.; Wang, S.; Zhou, T.; Zou, R.; Lin, L.; Sun, N.; Sun, G.; et al. BAP18 coactivates androgen receptor action and promotes prostate cancer progression. Nucleic Acids Res. 2016, 44, 8112–8128. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence |
|---|---|
| hAsh2l-F | ATGGATACTCAGGCGGGCTC |
| hAsh2l-R | TCAGGGTTCCCATGGGGG |
| hAsh2l-ΔSDI-F | ATGGATACTCAGGCGGGCTC |
| hAsh2l-ΔSDI-R | CATGTCACTCATAGGGCGGTAA |
| hAsh2l-3E-F | ACCCTTGCTGACGTCCTCTATCACCTCGAGACAGAAGTGGATGGGAGGCG |
| hAsh2l-3E-R | CTCGAGGTGATAGAGGACGTCAGCAAGGGTGTGCTCTACCACGGCGCC |
| Ash2l-qPCR-F | CCTACTTTCTCCGGAAGCAAG |
| Ash2l-qPCR-R | GACAATGTTATTGGGCCAAGTC |
| Dpy30-qPCR-F | AACGCAGGTTGCAGA AAATCCT |
| Dpy30-qPCR-R | TCTGATCCAGGTAGGCACGAG |
| Ccnd1-qPCR-F | CTGATTGGACAGGCATGGGT |
| Ccnd1-qPCR-R | GTGCCTGGAAGTCAACGGTA |
| Gapdh-qPCR-F | GGTCATCCATGACAACTTTGG |
| Gapdh-qPCR-R | GGCCATCACGCCACAG |
| siAsh2l-1# | CAGTAAAGATCCAGAAGAA |
| siAsh2l-2# | GGTGCCTGGTATTTTGAAA |
| siAsh2l-3# | CGAAGACAATGTTCTCCAA |
| mAsh2l-shRNA-1 | CAAGAAGGCCAGAAGTGATCCTTTATTCAAGAGATAAAGGATCACTTCTGGCCTTCTTGTTTTTT |
| mAsh2l-shRNA-3 | CCTGTGTCTGTGTGTTCCAAATTCAAGAGATTTGGAACACACAGACACAGGTTTTTT |
| hAsh2l-shRNA-1 | CCGGGTGACTTGTTATCCTACTATACTCGAGTATAGTAGGATAACAAGTCACTTTTT |
| hAsh2l-shRNA-2 | CCGGCCGAAGACAATGTTCTCCAAACTCGAGTTTGGAGAACATTGTCTTCGGTTTTT |
| hAsh2l-shRNA-3 | CCGGCCTGCTTGTATGAACGGGTTTCTCGAGAAACCCGTTCATACAAGCAGGTTTTT |
| Name | Resource |
|---|---|
| Anti-Ash2 | BETHYL (Waltham, MA, USA, #A300–112A) |
| Anti-DPY30 | Sigma (St. Louis, MO, USA, #HPA043761) |
| Anti-WDR5 | R&D system (Minneapolis, MN, USA, #AF5810) |
| Anti-RbBP5 | Abcam (Cambridge, UK, #ab52084) |
| Anti-Cyclin D1 | Abcam (Cambridge, UK, #ab16663) |
| Anti-FLAG | ABclonal (Cambridge, UK, #WH166500) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, M.; Zhou, J.; Ma, Z.; Chen, H.; Li, L.; Hou, L.; Yin, B.; Qiang, B.; Shu, P.; Peng, X. The Ash2l SDI Domain Is Required to Maintain the Stability and Binding of DPY30. Cells 2022, 11, 1450. https://doi.org/10.3390/cells11091450
Ma M, Zhou J, Ma Z, Chen H, Li L, Hou L, Yin B, Qiang B, Shu P, Peng X. The Ash2l SDI Domain Is Required to Maintain the Stability and Binding of DPY30. Cells. 2022; 11(9):1450. https://doi.org/10.3390/cells11091450
Chicago/Turabian StyleMa, Mengjie, Jiafeng Zhou, Zhihua Ma, Hanxue Chen, Liang Li, Lin Hou, Bin Yin, Boqin Qiang, Pengcheng Shu, and Xiaozhong Peng. 2022. "The Ash2l SDI Domain Is Required to Maintain the Stability and Binding of DPY30" Cells 11, no. 9: 1450. https://doi.org/10.3390/cells11091450
APA StyleMa, M., Zhou, J., Ma, Z., Chen, H., Li, L., Hou, L., Yin, B., Qiang, B., Shu, P., & Peng, X. (2022). The Ash2l SDI Domain Is Required to Maintain the Stability and Binding of DPY30. Cells, 11(9), 1450. https://doi.org/10.3390/cells11091450