
Modern Metaproteomics: A Unique Tool to Characterize the Active Microbiome in Health and Diseases, and Pave the Road towards New Biomarkers—Example of Crohn’s Disease and Ulcerative Colitis Flare-Ups

 , , ,
, , ,
Abstract
: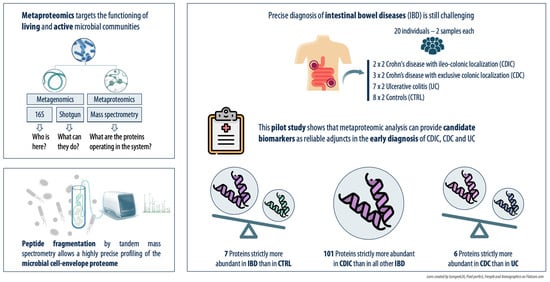
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Volunteers and Sample Collection
2.2. Sample Preparation
2.3. LC-MS/MS Analyses
2.4. Statistics
2.5. Search for Contrasts
2.6. Taxonomic and Functional Annotation
3. Results and Discussion
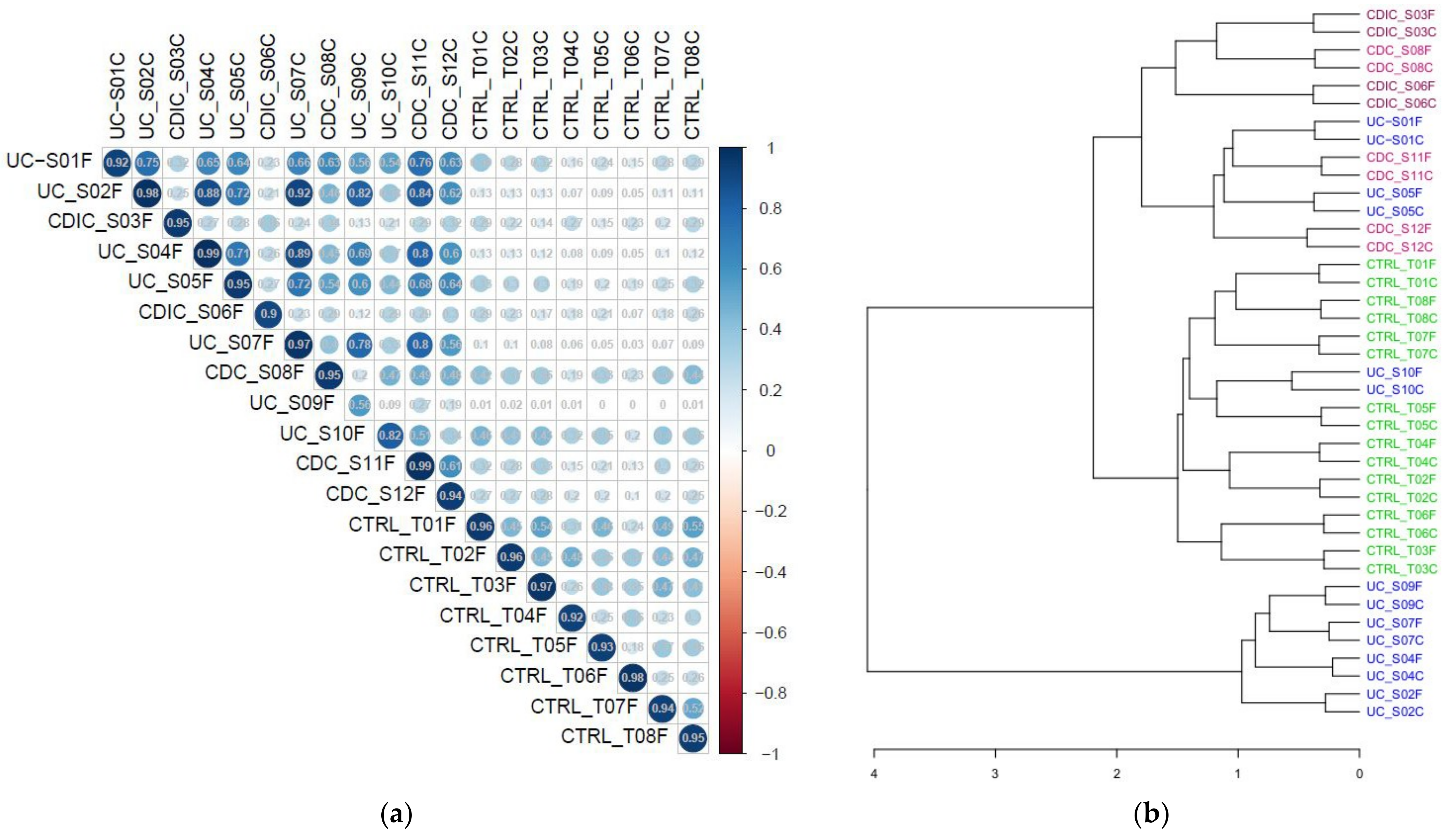
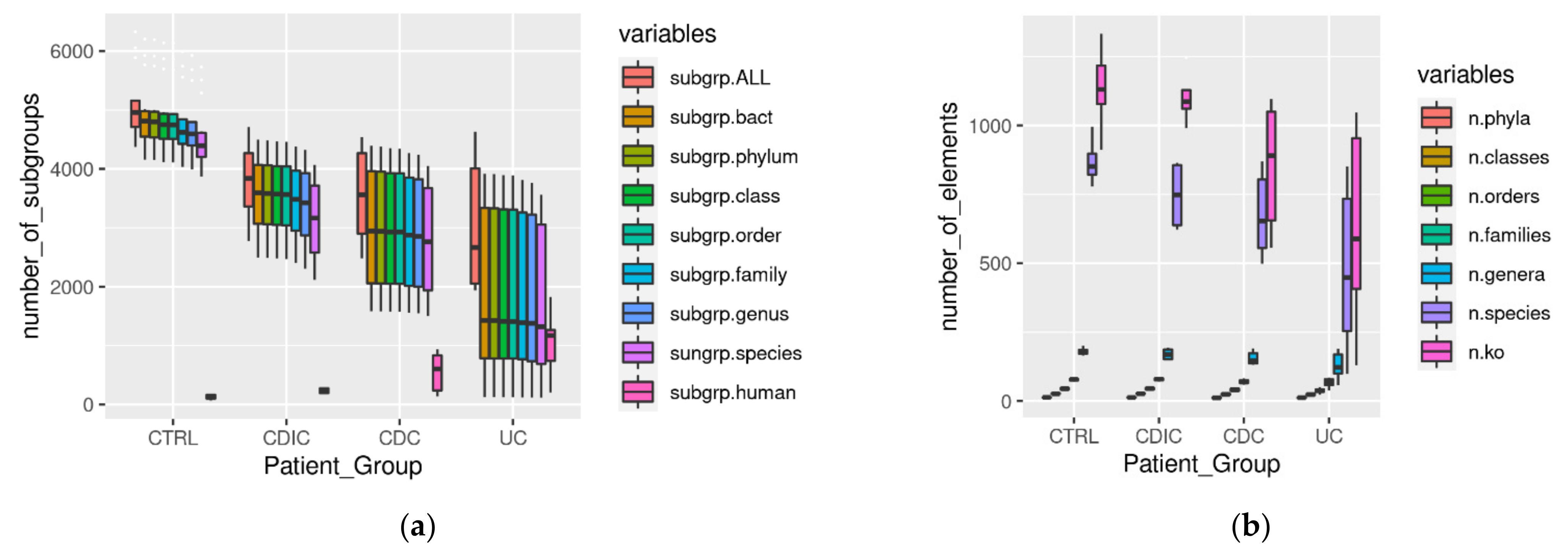
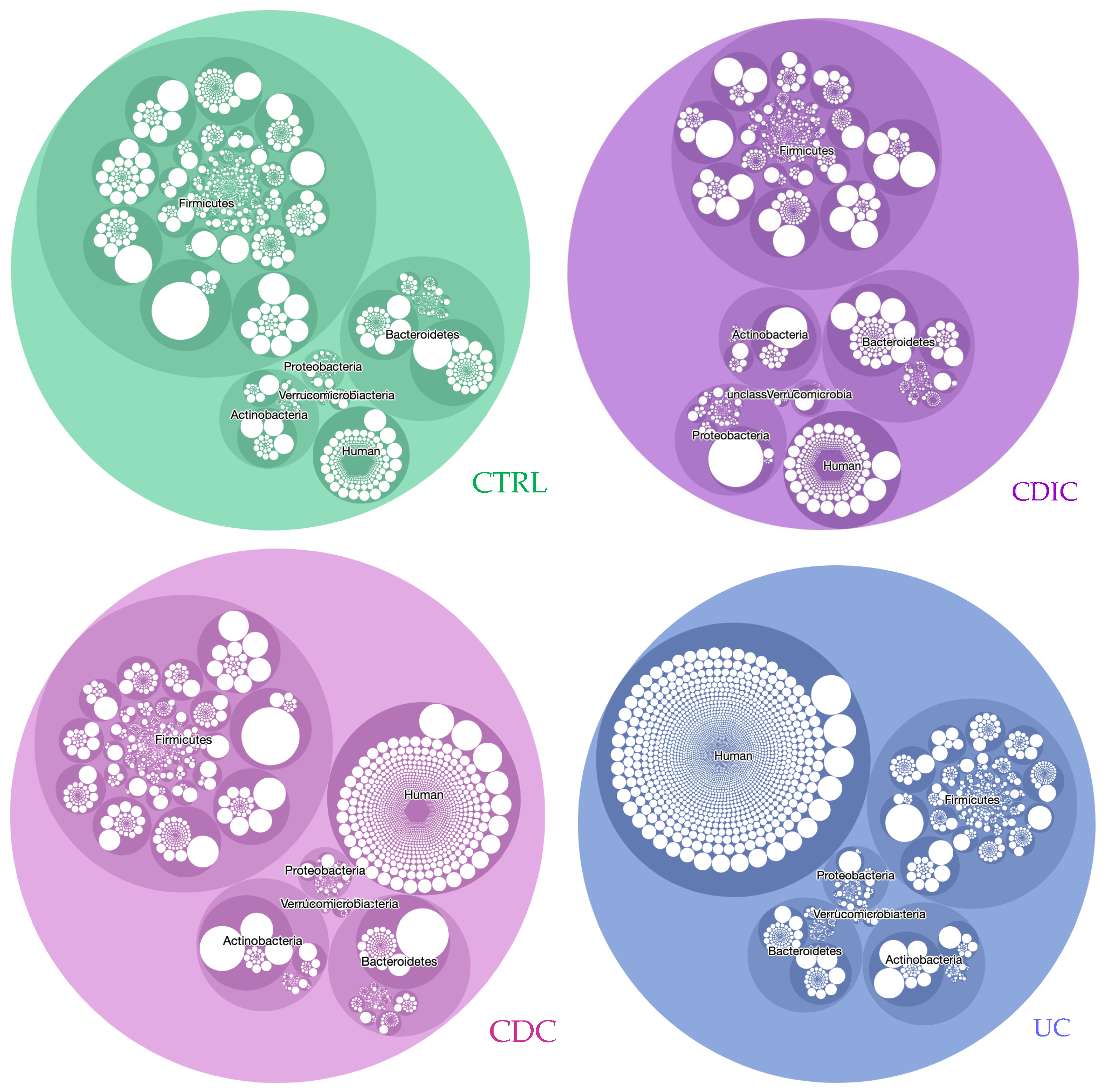
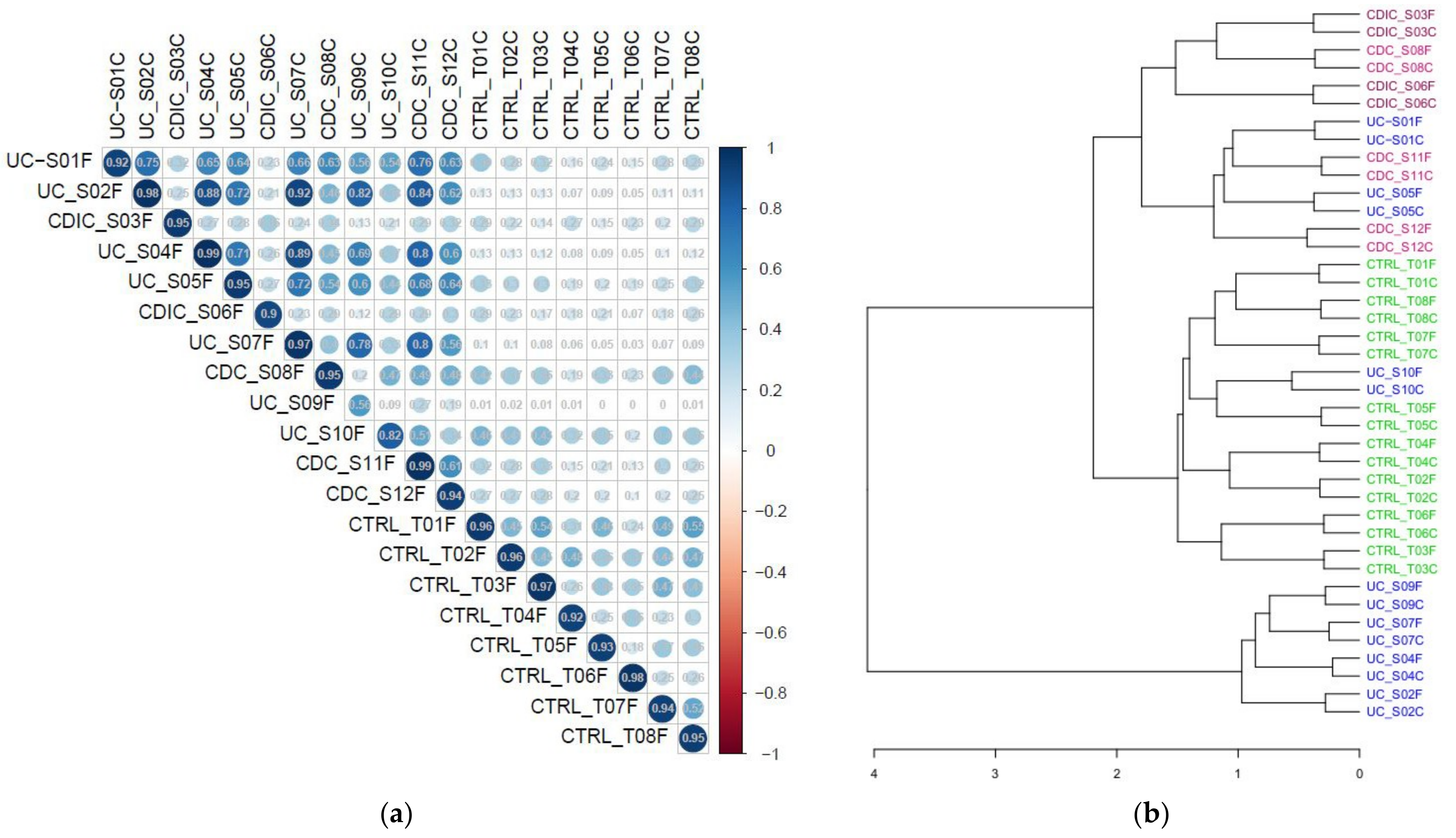
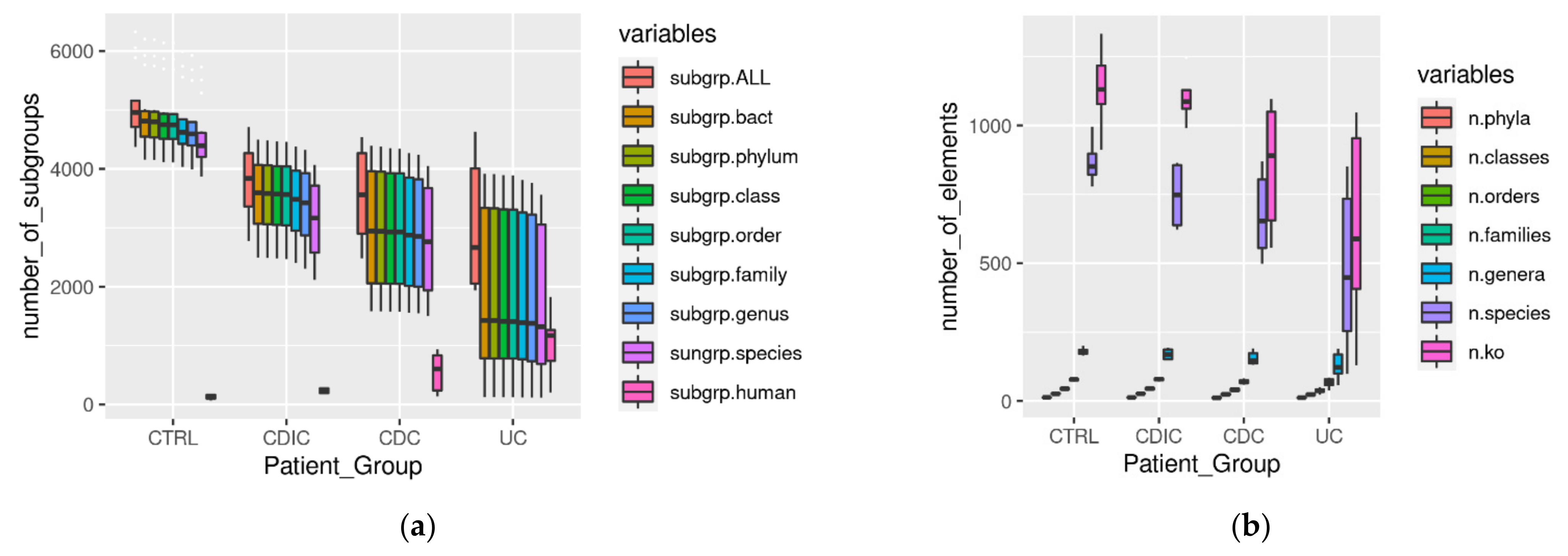
3.1. Metaproteomic Profiling of Stool Samples
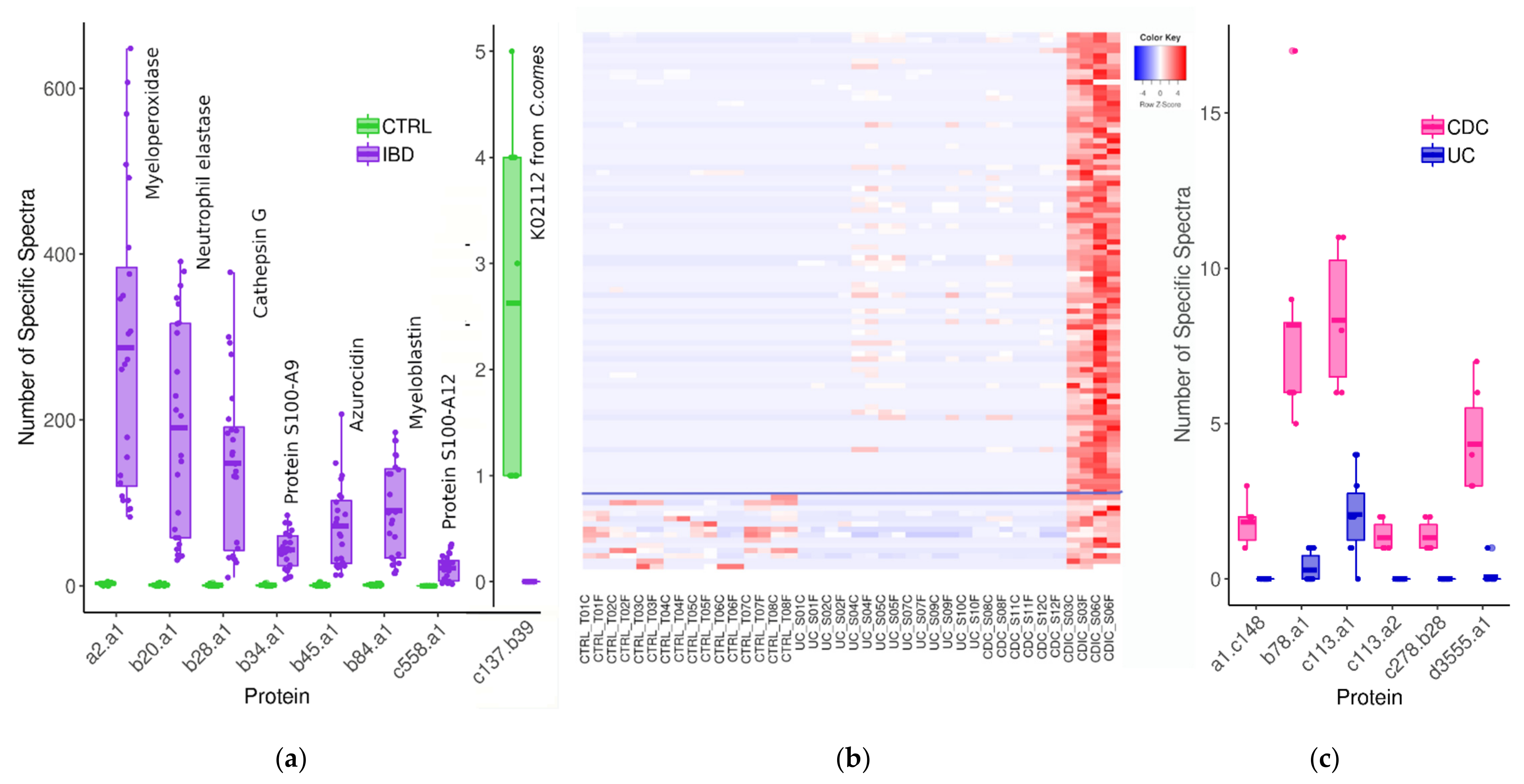
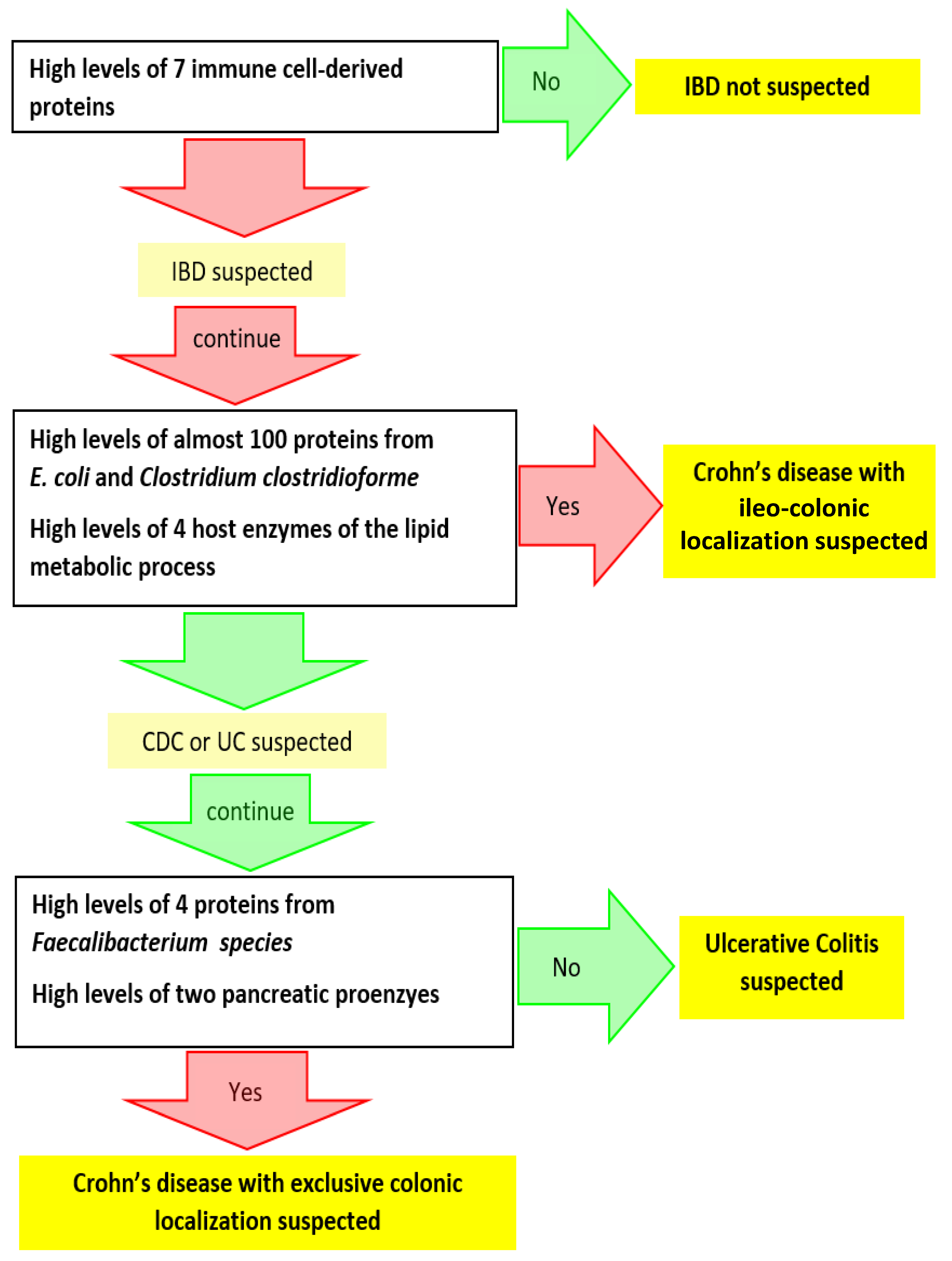
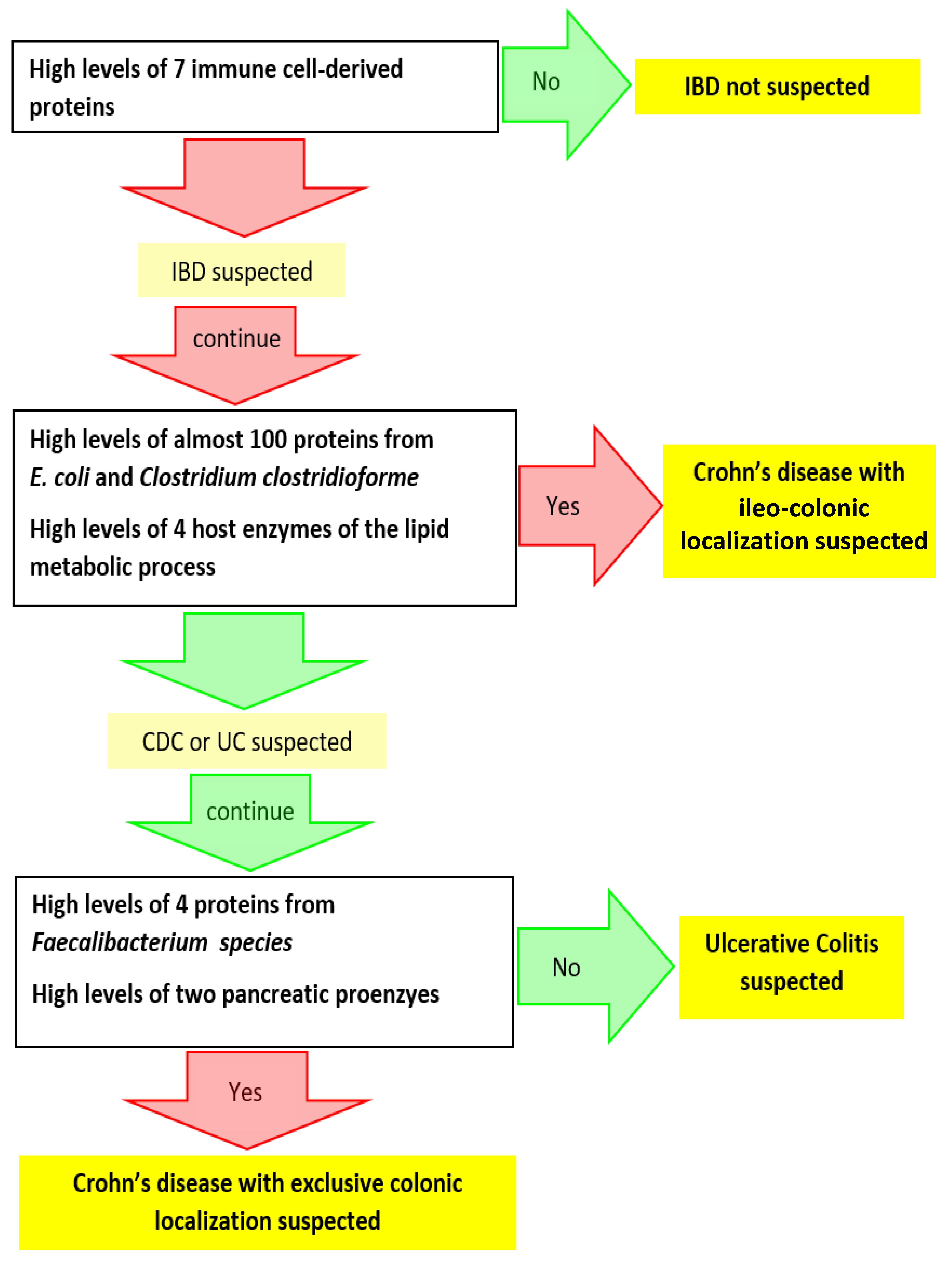
3.2. Search for Metaproteomic Signatures Specific of IBD Phenotypes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- GBD 2017 Inflammatory Bowel Disease Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the global burden of diseases Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Jairath, V.; Feagan, B.G. Global burden of inflammatory bowel disease. Lancet Gastroenterol. Hepatol. 2020, 5, 2–3. [Google Scholar] [CrossRef] [Green Version]
- Baumgart, D.C. The diagnosis and treatment of Crohn’s disease and ulcerative colitis. Dtsch. Arztebl. Int. 2009, 106, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Spekhorst, L.M.; Visschedijk, M.C.; Alberts, R.; Festen, E.A.; van der Wouden, E.J.; Dijkstra, G.; Weersma, R.K.; Dutch Initiative on Crohn and Colitis. Performance of the Montreal classification for inflammatory bowel diseases. World J. Gastroenterol. 2014, 20, 15374–15381. [Google Scholar] [CrossRef]
- De Mattos, B.R.; Garcia, M.P.; Nogueira, J.B.; Paiatto, L.N.; Albuquerque, C.G.; Souza, C.L.; Fernandes, L.G.; Tamashiro, W.M.; Simioni, P.U. Inflammatory Bowel Disease: An overview of immune mechanisms and biological treatments. Mediators Inflamm. 2015, 2015, 493012. [Google Scholar] [CrossRef]
- Rath, H.C.; Herfarth, H.H.; Ikeda, J.S.; Grenther, W.B.; Hamm, T.E., Jr.; Balish, E.; Taurog, J.D.; Hammer, R.E.; Wilson, K.H.; Sartor, R.B. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Investig. 1996, 98, 945–953. [Google Scholar] [CrossRef] [Green Version]
- D’Haens, G.R.; Geboes, K.; Peeters, M.; Baert, F.; Penninckx, F.; Rutgeerts, P. Early lesions of recurrent Crohn’s disease caused by infusion of intestinal contents in excluded ileum. Gastroenterology 1998, 114, 262–267. [Google Scholar] [CrossRef]
- Rutgeerts, P.; Hiele, M.; Geboes, K.; Peeters, M.; Penninckx, F.; Aerts, R.; Kerremans, R. Controlled trial of metronidazole treatment for prevention of Crohn’s recurrence after ileal resection. Gastroenterology 1995, 108, 1617–1621. [Google Scholar] [CrossRef]
- Mathew, C.G. New links to the pathogenesis of Crohn disease provided by genome-wide association scans. Nat. Rev. Genet. 2008, 9, 9–14. [Google Scholar] [CrossRef]
- Cho, J.H. The genetics and immunopathogenesis of inflammatory bowel disease. Nat. Rev. Immunol. 2008, 8, 458–466. [Google Scholar] [CrossRef]
- Cleynen, I.; González, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlík, M.; Crusius, B.J.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory Bowel Disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef] [PubMed]
- Lichtenstein, G.R.; Loftus, E.V.; Isaacs, K.L.; Regueiro, M.D.; Gerson, L.B.; Sands, B.E. ACG Clinical Guideline: Management of Crohn’s Disease in adults. Am. J. Gastroenterol. 2018, 113, 481–517. [Google Scholar] [CrossRef] [PubMed]
- Rubin, D.T.; Ananthakrishnan, A.N.; Siegel, C.A.; Sauer, B.G.; Long, M.D. ACG Clinical Guideline: Ulcerative Colitis in adults. Am. J. Gastroenterol. 2019, 114, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Oh, G.M.; Moon, W.; Seo, K.I.; Jung, K.; Kim, J.H.; Kim, S.E.; Park, M.I.; Park, S.J. Therapeutic potential of Escherichia coli Nissle 1917 in clinically remission-attained Ulcerative Colitis patients: A hospital-based cohort study. Korean J. Gastroenterol. 2021, 77, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Nishida, A.; Nishino, K.; Sakai, K.; Owaki, Y.; Noda, Y.; Imaeda, H. Can control of gut microbiota be a future therapeutic option for inflammatory bowel disease? World J. Gastroenterol. 2021, 27, 3317–3326. [Google Scholar] [CrossRef]
- Almeida, D.; Machado, D.; Andrade, J.C.; Mendo, S.; Gomes, A.M.; Freitas, A.C. Evolving trends in next-generation probiotics: A 5W1H perspective. Crit. Rev. Food Sci. Nutr. 2020, 60, 1783–1796. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Escher, J.; Hébuterne, X.; Kłęk, S.; Krznaric, Z.; Schneider, S.; Shamir, R.; Stardelova, K.; Wierdsma, N.; Wiskin, A.E.; et al. ESPEN practical guideline: Clinical Nutrition in inflammatory bowel disease. Clin. Nutr. 2020, 39, 632–653. [Google Scholar] [CrossRef] [Green Version]
- Żółkiewicz, J.; Marzec, A.; Ruszczyński, M.; Feleszko, W. Postbiotics-A step beyond pre- and probiotics. Nutrients 2020, 12, 2189. [Google Scholar] [CrossRef]
- Sokol, H.; Landman, C.; Seksik, P.; Berard, L.; Montil, M.; Nion-Larmurier, I.; Bourrier, A.; Le Gall, G.; Lalande, V.; De Rougemont, A.; et al. Fecal microbiota transplantation to maintain remission in Crohn’s disease: A pilot randomized controlled study. Microbiome 2020, 8, 12. [Google Scholar] [CrossRef]
- Mills, R.H.; Vázquez-Baeza, Y.; Zhu, Q.; Jiang, L.; Gaffney, J.; Humphrey, G.; Smarr, L.; Knight, R.; Gonzalez, D.J. Evaluating metagenomic prediction of the metaproteome in a 4.5-year study of a patient with Crohn’s disease. mSystems 2019, 4, e00337-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sato, K.; Kumita, W.; Ode, T.; Ichinose, S.; Ando, A.; Fujiyama, Y.; Chida, T.; Okamura, N. OmpA variants affecting the adherence of ulcerative colitis-derived Bacteroides Vulgatus. J. Med. Dent. Sci. 2010, 57, 55–64. [Google Scholar] [PubMed]
- Erickson, A.R.; Cantarel, B.L.; Lamendella, R.; Darzi, Y.; Mongodin, E.F.; Pan, C.; Shah, M.; Halfvarson, J.; Tysk, C.; Henrissat, B.; et al. Integrated metagenomics/metaproteomics reveals human host-microbiota signatures of Crohn’s disease. PLoS ONE 2012, 7, e49138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juste, C.; Kreil, D.; Beauvallet, C.; Guillot, A.; Vaca, S.; Carapito, C.; Mondot, S.; Sykacek, P.; Sokol, H.; Blon, F.; et al. Bacterial protein signals are associated with Crohn’s disease. Gut 2014, 63, 1566–1577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Deeke, S.A.; Ning, Z.; Starr, A.E.; Butcher, J.; Li, J.; Mayne, J.; Cheng, K.; Liao, B.; Li, L.; et al. Metaproteomics reveals associations between microbiome and intestinal extracellular vesicle proteins in pediatric inflammatory bowel disease. Nat. Commun. 2018, 9, 2873. [Google Scholar] [CrossRef]
- Lehmann, T.; Schallert, K.; Vilchez-Vargas, R.; Benndorf, D.; Püttker, S.; Sydor, S.; Schulz, C.; Bechmann, L.; Canbay, A.; Heidrich, B.; et al. Metaproteomics of fecal samples of Crohn’s disease and ulcerative colitis. J. Proteomics. 2019, 201, 93–103. [Google Scholar] [CrossRef]
- Thuy-Boun, P.S.; Wang, A.Y.; Crissien-Martinez, A.; Xu, J.H.; Chatterjee, S.; Stupp, G.S.; Su, A.I.; Coyle, W.J.; Wolan, D.W. Quantitative metaproteomics and activity-based protein profiling of patient fecal microbiome identifies host and microbial serine-type endopeptidase activity associated with ulcerative colitis. Mol. Cell. Proteomics 2022, 21, 1000197. [Google Scholar] [CrossRef]
- Lai, L.A.; Tong, Z.; Chen, R.; Pan, S. Metaproteomics study of the gut microbiome. Methods Mol. Biol. 2019, 1871, 123–132. [Google Scholar]
- Issa Isaac, N.; Philippe, D.; Nicholas, A.; Raoult, D.; Eric, C. Metaproteomics of the human gut microbiota: Challenges and contributions to other OMICS. Clin. Mass Spectrom. 2019, 14 Pt A, 18–30. [Google Scholar] [CrossRef]
- Lacroix, V.; Cassard, A.; Mas, E.; Barreau, F. Multi-Omics analysis of gut microbiota in inflammatory bowel diseases: What benefits for diagnostic, prognostic and therapeutic tools? Int. J. Mol. Sci. 2021, 22, 11255. [Google Scholar] [CrossRef]
- Metaproteomics Initiative. Available online: http://metaproteomics.org (accessed on 10 April 2022).
- Van Den Bossche, T.; Arntzen, M.Ø.; Becher, D.; Benndorf, D.; Eijsink, V.G.H.; Henry, C.; Jagtap, P.D.; Jehmlich, N.; Juste, C.; Kunath, B.J.; et al. The Metaproteomics Initiative: A coordinated approach for propelling the functional characterization of microbiomes. Microbiome 2021, 9, 243. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bossche, T.; Kunath, B.J.; Schallert, K.; Schäpe, S.S.; Abraham, P.E.; Armengaud, J.; Arntzen, M.Ø.; Bassignani, A.; Benndorf, D.; Fuchs, S.; et al. Critical Assessment of MetaProteome Investigation (CAMPI): A multi-laboratory comparison of established workflows. Nat. Commun. 2021, 12, 7305. [Google Scholar] [CrossRef] [PubMed]
- Bassignani, A.; Plancade, S.; Berland, M.; Blein-Nicolas, M.; Guillot, A.; Chevret, D.; Moritz, C.; Huet, S.; Rizkalla, S.; Clément, K.; et al. Benefits of iterative searches of large databases to interpret large human gut metaproteomic data sets. J. Proteome Res. 2021, 20, 1522–1534. [Google Scholar] [CrossRef]
- Langella, O.; Valot, B.; Balliau, T.; Blein-Nicolas, M.; Bonhomme, L.; Zivy, M. X!TandemPipeline: A tool to manage sequence redundancy for protein inference and phosphosite identification. J. Proteome Res. 2017, 2, 494–503. [Google Scholar] [CrossRef] [PubMed]
- PAPPSO, Plateforme d’Analyse Protéomique de Paris Sud-Ouest. Available online: http://pappso.inrae.fr/ (accessed on 10 April 2022).
- Taxonomic and Functional Annotations of the Integrated Non-Redundant Gene Catalog 9.9. Available online: https://zenodo.org/record/3997093#.YlQepNPP1EY (accessed on 11 April 2022).
- Bassignani, A. Metaproteomics analysis to study functionalities of the gut microbiota in large cohorts. Ph.D. Thesis, Sorbonne University, Paris, France, Thesis Defended. 30 September 2019. [Google Scholar]
- Imhann, F.; Vich Vila, A.; Bonder, M.J.; Fu, J.; Gevers, D.; Visschedijk, M.C.; Spekhorst, L.M.; Alberts, R.; Franke, L.; van Dullemen, H.M.; et al. Interplay of host genetics and gut microbiota underlying the onset and clinical presentation of inflammatory bowel disease. Gut 2018, 67, 108–119. [Google Scholar] [CrossRef]
- Halfvarson, J.; Brislawn, C.J.; Lamendella, R.; Vázquez-Baeza, Y.; Walters, W.A.; Bramer, L.M.; D’Amato, M.; Bonfiglio, F.; McDonald, D.; Gonzalez, A.; et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017, 2, 17004. [Google Scholar] [CrossRef] [Green Version]
- Franzosa, E.A.; Sirota-Madi, A.; Avila-Pacheco, J.; Fornelos, N.; Haiser, H.J.; Reinker, S.; Vatanen, T.; Hall, A.B.; Mallick, H.; McIver, L.J.; et al. Gut microbiome structure and metabolic activity in inflammatory bowel disease. Nat. Microbiol. 2019, 4, 293–305. [Google Scholar] [CrossRef]
- Becker, K.; Niederau, C.; Frieling, T. Fecal excretion of alpha 2-macroglobulin: A novel marker for disease activity in patients with inflammatory bowel disease. Z. Gastroenterol. 1999, 37, 597–605. [Google Scholar]
- Fournier, B.M.; Parkos, C.A. The role of neutrophils during intestinal inflammation. Mucosal Immunol. 2012, 5, 354–366. [Google Scholar] [CrossRef]
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [Green Version]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 proteins as an important regulator of macrophage inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foell, D.; Kucharzik, T.; Kraft, M.; Vogl, T.; Sorg, C.; Domschke, W.; Roth, J. Neutrophil derived human S100A12 (EN-RAGE) is strongly expressed during chronic active inflammatory bowel disease. Gut 2003, 52, 847–853. [Google Scholar] [CrossRef] [Green Version]
- Rawat, K.; Syeda, S.; Shrivastava, A. Neutrophil-derived granule cargoes: Paving the way for tumor growth and progression. Cancer Metastasis Rev. 2021, 40, 221–244. [Google Scholar] [CrossRef] [PubMed]
- Hansberry, D.R.; Shah, K.; Agarwal, P.; Agarwal, N. Fecal myeloperoxidase as a biomarker for inflammatory bowel disease. Cureus 2017, 9, e1004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seishima, J.; Iida, N.; Kitamura, K.; Yutani, M.; Wang, Z.; Seki, A.; Yamashita, T.; Sakai, Y.; Honda, M.; Yamshita, T.; et al. Gut-derived Enterococcus faecium from ulcerative colitis patients promotes colitis in a genetically susceptible mouse host. Genome Biol. 2019, 20, 252. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazenberg, M.P.; van de Merwe, J.P.; Peña, A.S.; Pennock-Schröder, A.M.; van Lieshout, L.M. Antibodies to Coprococcus comes in sera of patients with Crohn’s disease. Isolation and purification of the agglutinating antigen tested with an ELISA technique. J. Clin. Lab. Immunol. 1987, 23, 143–148. [Google Scholar] [PubMed]
- Edelblum, K.L.; Washington, M.K.; Koyama, T.; Robine, S.; Baccarini, M.; Polk, D.B. Raf protects against colitis by promoting mouse colon epithelial cell survival through NF-kappaB. Gastroenterology 2008, 135, 539–551. [Google Scholar] [CrossRef] [Green Version]
- He, Q.; Gao, Y.; Jie, Z.; Yu, X.; Laursen, J.M.; Xiao, L.; Li, Y.; Li, L.; Zhang, F.; Feng, Q.; et al. Two distinct metacommunities characterize the gut microbiota in Crohn’s disease patients. GigaScience 2017, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- MassIVE, mass Spectrometry Interactive Virtual Environment. Available online: https://massive.ucsd.edu/ProteoSAFe/static/massive.jsp (accessed on 11 April 2022).
- INRAE, Institut National de Recherche pour L’agriculture, L’alimentation et L’environnement. Available online: https://www.inrae.fr/ (accessed on 10 April 2022).
- Région îledefrance, Éducation-Recherche. Available online: https://www.iledefrance.fr/education-recherche (accessed on 10 April 2022).
- IBiSA, Infrastructures en Biologie Santé et Agronomie. Available online: https://www.ibisa.net/ (accessed on 10 April 2022).
- CNRS, Centre National de la Recherche Scientifique. Available online: http://www.cnrs.fr (accessed on 10 April 2022).
- ProteomeXchange Consortium. Available online: http://proteomecentral.proteomexchange.org (accessed on 10 April 2022).
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Henry, C.; Bassignani, A.; Berland, M.; Langella, O.; Sokol, H.; Juste, C. Modern Metaproteomics: A Unique Tool to Characterize the Active Microbiome in Health and Diseases, and Pave the Road towards New Biomarkers—Example of Crohn’s Disease and Ulcerative Colitis Flare-Ups. Cells 2022, 11, 1340. https://doi.org/10.3390/cells11081340
Henry C, Bassignani A, Berland M, Langella O, Sokol H, Juste C. Modern Metaproteomics: A Unique Tool to Characterize the Active Microbiome in Health and Diseases, and Pave the Road towards New Biomarkers—Example of Crohn’s Disease and Ulcerative Colitis Flare-Ups. Cells. 2022; 11(8):1340. https://doi.org/10.3390/cells11081340
Chicago/Turabian StyleHenry, Céline, Ariane Bassignani, Magali Berland, Olivier Langella, Harry Sokol, and Catherine Juste. 2022. "Modern Metaproteomics: A Unique Tool to Characterize the Active Microbiome in Health and Diseases, and Pave the Road towards New Biomarkers—Example of Crohn’s Disease and Ulcerative Colitis Flare-Ups" Cells 11, no. 8: 1340. https://doi.org/10.3390/cells11081340
APA StyleHenry, C., Bassignani, A., Berland, M., Langella, O., Sokol, H., & Juste, C. (2022). Modern Metaproteomics: A Unique Tool to Characterize the Active Microbiome in Health and Diseases, and Pave the Road towards New Biomarkers—Example of Crohn’s Disease and Ulcerative Colitis Flare-Ups. Cells, 11(8), 1340. https://doi.org/10.3390/cells11081340