
A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine–Chemokine Receptor Interactions


Abstract
:1. Introduction
2. Materials and Methods
2.1. Expression and Purification of ACKR3
2.2. Expression and Purification of CXCL12
2.3. Expression, Purification and Biotinylation of Membrane Scaffold Proteins
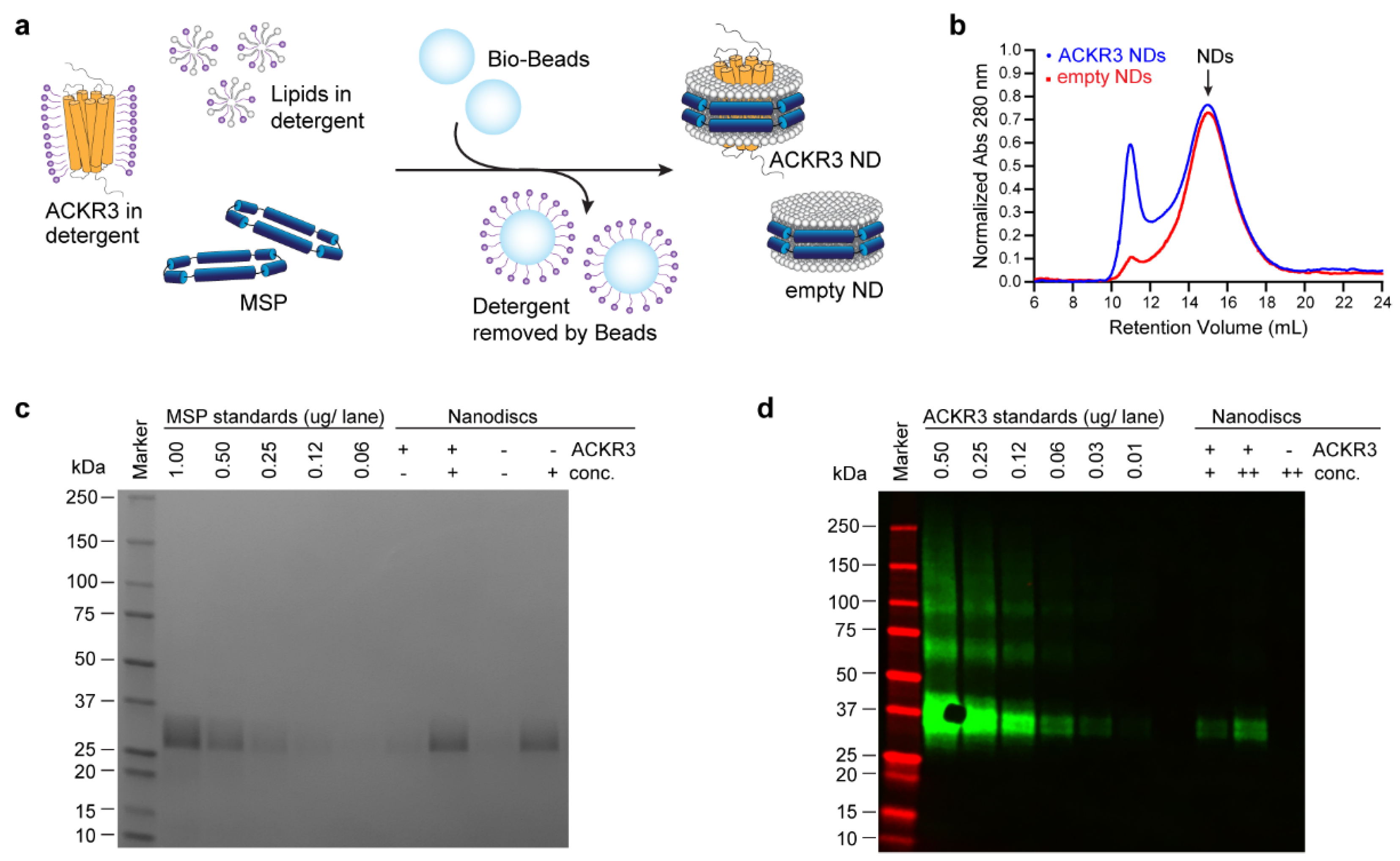
2.4. Reconstitution of ACKR3 into Nanodiscs
2.5. Nanodisc Analysis by Western Blot and SDS-PAGE
2.6. Scintillation Proximity Assay
2.7. Data Analysis
3. Results
3.1. Principle of the Assay
3.2. Sample Preparation
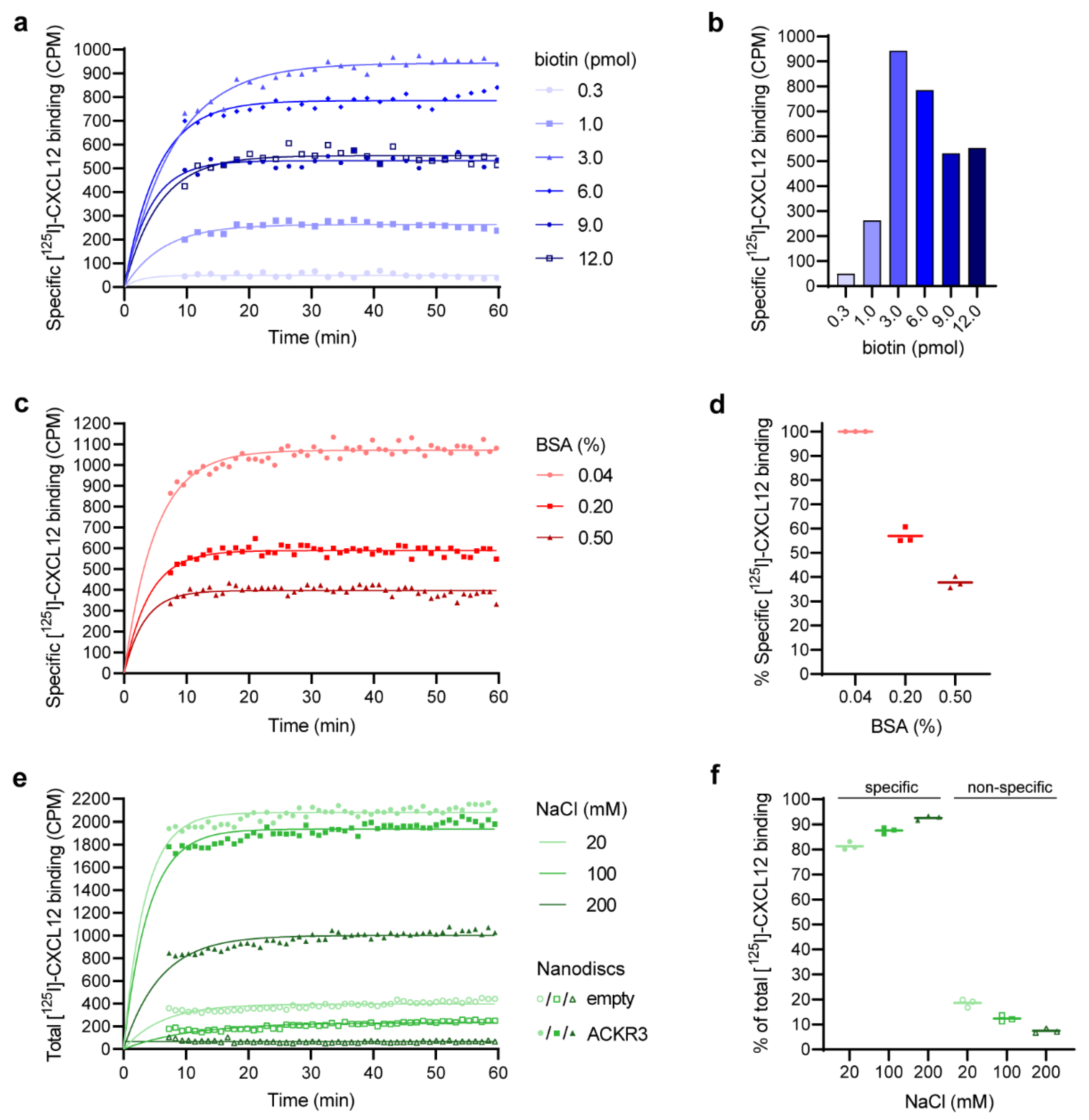
3.3. Determination of the Optimal Biotin–Beads Ratio
3.4. Reduction in Non-Specific Binding
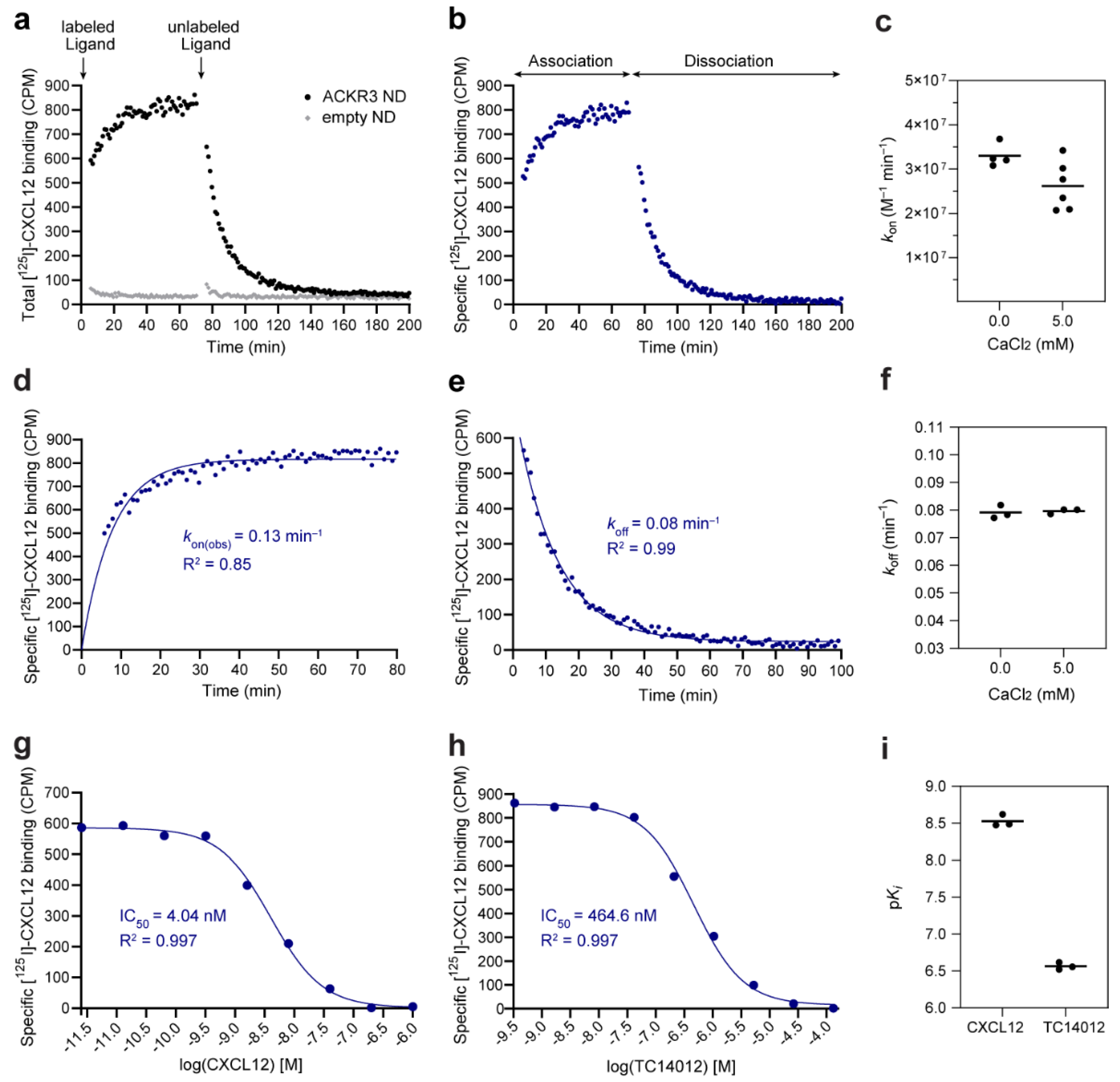
3.5. Assay Validation: Binding Kinetic Studies on the CXCL12–ACKR3 Interaction
3.6. Alternative Assay Application: Competition Binding Studies
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hauser, A.S.; Attwood, M.M.; Rask-Andersen, M.; Schiöth, H.B.; Gloriam, D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017, 16, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.G.; Schiöth, H.B. The G-Protein-Coupled Receptors in the Human Genome Form Five Main Families. Phylogenetic Analysis, Paralogon Groups, and Fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ollila, T.A.; Sahin, I.; Olszewski, A.J. Mogamulizumab: A New Tool for Management of Cutaneous T-Cell Lymphoma. OncoTargets Ther. 2019, 12, 1085–1094. [Google Scholar] [CrossRef] [Green Version]
- Qi, B.; Fang, Q.; Liu, S.; Hou, W.; Li, J.; Huang, Y.; Shi, J. Advances of CCR5 Antagonists: From Small Molecules to Macromolecules. Eur. J. Med. Chem. 2020, 208, 112819. [Google Scholar] [CrossRef]
- de Clercq, E. Mozobil® (Plerixafor, AMD3100), 10 Years after Its Approval by the US Food and Drug Administration. Antivir. Chem. Chemother. 2019, 27, 2040206619829382. [Google Scholar] [CrossRef] [Green Version]
- Hughes, C.E.; Nibbs, R.J.B. A Guide to Chemokines and Their Receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Stone, M.J.; Hayward, J.A.; Huang, C.; Huma, Z.E.; Sanchez, J. Mechanisms of Regulation of the Chemokine-Receptor Network. Int. J. Mol. Sci. 2017, 18, 342. [Google Scholar] [CrossRef] [Green Version]
- Chen, K.; Bao, Z.; Tang, P.; Gong, W.; Yoshimura, T.; Wang, J.M. Chemokines in Homeostasis and Diseases. Cell. Mol. Immunol. 2018, 15, 324–334. [Google Scholar] [CrossRef]
- Zhao, S.; Wu, B.; Stevens, R.C. Advancing Chemokine GPCR Structure Based Drug Discovery. Structure 2019, 27, 405–408. [Google Scholar] [CrossRef] [Green Version]
- Miao, M.; de Clercq, E.; Li, G. Clinical Significance of Chemokine Receptor Antagonists. Expert Opin. Drug Metab. Toxicol. 2020, 16, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Solari, R.; Pease, J.E.; Begg, M. Chemokine Receptors as Therapeutic Targets: Why Aren’t There More Drugs? Eur. J. Pharmacol. 2015, 746, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Nibbs, R.J.B.; Graham, G.J. Immune Regulation by Atypical Chemokine Receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Vacchini, A.; Locati, M.; Borroni, E.M. Overview and Potential Unifying Themes of the Atypical Chemokine Receptor Family. J. Leukoc. Biol. 2016, 99, 883–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyer, D.P.; Medina-Ruiz, L.; Bartolini, R.; Schuette, F.; Hughes, C.E.; Pallas, K.; Vidler, F.; Macleod, M.K.L.; Kelly, C.J.; Lee, K.M.; et al. Chemokine Receptor Redundancy and Specificity Are Context Dependent. Immunity 2019, 50, 378–389.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schall, T.J.; Proudfoot, A.E.I. Overcoming Hurdles in Developing Successful Drugs Targeting Chemokine Receptors. Nat. Rev. Immunol. 2011, 11, 355–363. [Google Scholar] [CrossRef]
- Horuk, R. Chemokine Receptor Antagonists: Overcoming Developmental Hurdles. Nat. Rev. Drug Discov. 2009, 8, 23–33. [Google Scholar] [CrossRef]
- Kufareva, I.; Gustavsson, M.; Zheng, Y.; Stephens, B.S.; Handel, T.M. What Do Structures Tell Us about Chemokine Receptor Function and Antagonism? Annu. Rev. Biophys. 2017, 46, 175–198. [Google Scholar] [CrossRef] [Green Version]
- Mabonga, L.; Kappo, A.P. Protein-Protein Interaction Modulators: Advances, Successes and Remaining Challenges. Biophys. Rev. 2019, 11, 559–581. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent Advances in the Development of Protein–Protein Interactions Modulators: Mechanisms and Clinical Trials. Signal Transduct. Target. Ther. 2020, 5, 213. [Google Scholar] [CrossRef]
- Gherbi, K.; Groenewoud, N.J.; Holliday, N.D.; Sengmany, K.; Charlton, S.J. Chapter 10-Kinetics of Ligand Binding and Signaling. GPCRs Struct. Funct. Drug Discov. 2020, 171–194. [Google Scholar] [CrossRef]
- IJzerman, A.P.; Guo, D. Drug-Target Association Kinetics in Drug Discovery. Trends Biochem. Sci. 2019, 44, 861–871. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.A. The Drug–Target Residence Time Model: A 10-Year Retrospective. Nat. Rev. Drug Discov. 2016, 15, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Neubig, R.R.; Spedding, M.; Kenakin, T.; Christopoulos, A. International Union of Pharmacology Committee on Receptor Nomenclature and Drug Classification. XXXVIII. Update on Terms and Symbols in Quantitative Pharmacology. Pharmacol. Rev. 2003, 55, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Copeland, R.A.; Pompliano, D.L.; Meek, T.D. Drug–Target Residence Time and Its Implications for Lead Optimization. Nat. Rev. Drug Discov. 2006, 5, 730–739. [Google Scholar] [CrossRef]
- Sykes, D.A.; Stoddart, L.A.; Kilpatrick, L.E.; Hill, S.J. Binding Kinetics of Ligands Acting at GPCRs. Mol. Cell. Endocrinol. 2019, 485, 9–19. [Google Scholar] [CrossRef]
- Hoare, S.R.J. Kinetics of Drug-Target Binding: A Guide for Drug Discovery. Ref. Modul. Biomed. Sci. 2021, 1–44. [Google Scholar] [CrossRef]
- Tonge, P.J. Drug-Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 2018, 9, 29–39. [Google Scholar] [CrossRef] [Green Version]
- Schuetz, D.A.; de Witte, W.E.A.; Wong, Y.C.; Knasmueller, B.; Richter, L.; Kokh, D.B.; Sadiq, S.K.; Bosma, R.; Nederpelt, I.; Heitman, L.H.; et al. Kinetics for Drug Discovery: An Industry-Driven Effort to Target Drug Residence Time. Drug Discov. Today 2017, 22, 896–911. [Google Scholar] [CrossRef] [Green Version]
- Hoare, S.R.J. The Problems of Applying Classical Pharmacology Analysis to Modern In Vitro Drug Discovery Assays: Slow Binding Kinetics and High Target Concentration. SLAS Discov. 2021, 26, 835–850. [Google Scholar] [CrossRef]
- Hoare, S.R.J.; Fleck, B.A.; Williams, J.P.; Grigoriadis, D.E. The Importance of Target Binding Kinetics for Measuring Target Binding Affinity in Drug Discovery: A Case Study from a CRF1 Receptor Antagonist Program. Drug Discov. Today 2020, 25, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Dowling, M.R.; Charlton, S.J. Quantifying the Association and Dissociation Rates of Unlabelled Antagonists at the Muscarinic M3 Receptor. Br. J. Pharmacol. 2006, 148, 927–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van der Velden, W.J.C.; Heitman, L.H.; Rosenkilde, M.M. Perspective: Implications of Ligand–Receptor Binding Kinetics for Therapeutic Targeting of G Protein-Coupled Receptors. ACS Pharmacol. Transl. Sci. 2020, 3, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Heitman, L.H.; Ijzerman, A.P. The Role of Target Binding Kinetics in Drug Discovery. ChemMedChem 2015, 10, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Nederpelt, I.; Bunnik, J.; IJzerman, A.P.; Heitman, L.H. Kinetic Profile of Neuropeptide-Receptor Interactions. Trends Neurosci. 2016, 39, 830–839. [Google Scholar] [CrossRef]
- Vauquelin, G. Effects of Target Binding Kinetics on in Vivo Drug Efficacy: Koff, Kon and Rebinding. Br. J. Pharmacol. 2016, 173, 2319–2334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vauquelin, G.; Charlton, S.J. Long-Lasting Target Binding and Rebinding as Mechanisms to Prolong in Vivo Drug Action. Br. J. Pharmacol. 2010, 161, 488–508. [Google Scholar] [CrossRef] [Green Version]
- Vauquelin, G. Rebinding: Or Why Drugs May Act Longer in Vivo than Expected from Their in Vitro Target Residence Time. Expert Opin. Drug Discov. 2010, 5, 927–941. [Google Scholar] [CrossRef]
- Gustavsson, M. New Insights into the Structure and Function of Chemokine Receptor:Chemokine Complexes from an Experimental Perspective. J. Leukoc. Biol. 2020, 107, 1115–1122. [Google Scholar] [CrossRef]
- Burns, J.M.; Summers, B.C.; Wang, Y.; Melikian, A.; Berahovich, R.; Miao, Z.; Penfold, M.E.T.; Sunshine, M.J.; Littman, D.R.; Kuo, C.J.; et al. A Novel Chemokine Receptor for SDF-1 and I-TAC Involved in Cell Survival, Cell Adhesion, and Tumor Development. J. Exp. Med. 2006, 203, 2201–2213. [Google Scholar] [CrossRef]
- Huynh, C.; Dingemanse, J.; Meyer zu Schwabedissen, H.E.; Sidharta, P.N. Relevance of the CXCR4/CXCR7-CXCL12 Axis and Its Effect in Pathophysiological Conditions. Pharmacol. Res. 2020, 161, 105092. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, M.; Wang, L.; van Gils, N.; Stephens, B.S.; Zhang, P.; Schall, T.J.; Yang, S.; Abagyan, R.; Chance, M.R.; Kufareva, I.; et al. Structural Basis of Ligand Interaction with Atypical Chemokine Receptor 3. Nat. Commun. 2017, 8, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wijtmans, M.; Maussang, D.; Sirci, F.; Scholten, D.J.; Canals, M.; Mujić-Delić, A.; Chong, M.; Chatalic, K.L.S.; Custers, H.; Janssen, E.; et al. Synthesis, Modeling and Functional Activity of Substituted Styrene-Amides as Small-Molecule CXCR7 Agonists. Eur. J. Med. Chem. 2012, 51, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, M.; Zheng, Y.; Handel, T.M. Production of Chemokine/Chemokine Receptor Complexes for Structural and Biophysical Studies. Methods Enzymol. 2016, 570, 233–260. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, T.K.; Grinkova, Y.v.; Bayburt, T.H.; Denisov, I.G.; Zolnerciks, J.K.; Atkins, W.M.; Sligar, S.G. Chapter 11-Reconstitution of Membrane Proteins in Phospholipid Bilayer Nanodiscs. Methods Enzymol. 2009, 464, 211–231. [Google Scholar] [CrossRef] [Green Version]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of Image Analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Gravel, S.; Malouf, C.; Boulais, P.E.; Berchiche, Y.A.; Oishi, S.; Fujii, N.; Leduc, R.; Sinnett, D.; Heveker, N. The Peptidomimetic CXCR4 Antagonist TC14012 Recruits Beta-Arrestin to CXCR7: Roles of Receptor Domains. J. Biol. Chem. 2010, 285, 37939–37943. [Google Scholar] [CrossRef] [Green Version]
- Gustavsson, M.; Dyer, D.P.; Zhao, C.; Handel, T.M. Kinetics of CXCL12 Binding to Atypical Chemokine Receptor 3 Reveal a Role for the Receptor N Terminus in Chemokine Binding. Sci. Signal. 2019, 12, 3657. [Google Scholar] [CrossRef]
- Cer, R.Z.; Mudunuri, U.; Stephens, R.; Lebeda, F.J. IC50-to-Ki: A Web-Based Tool for Converting IC50 to Ki Values for Inhibitors of Enzyme Activity and Ligand Binding. Nucleic Acids Res. 2009, 37, W441–W445. [Google Scholar] [CrossRef] [Green Version]
- Burlingham, B.T.; Widlanski, T.S. An Intuitive Look at the Relationship of Ki and IC50: A More General Use for the Dixon Plot. J. Chem. Educ. 2003, 80, 214–218. [Google Scholar] [CrossRef]
- Carter, C.M.S.; Leighton-Davies, J.R.; Charlton, S.J. Miniaturized Receptor Binding Assays: Complications Arising from Ligand Depletion. J. Biomol. Screen. 2007, 12, 255–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldstein, A.; Barrett, R.W. Ligand Dissociation Constants from Competition Binding Assays: Errors Associated with Ligand Depletion. Mol. Pharmacol. 1987, 31, 603–609. [Google Scholar] [PubMed]
- Cook, N.D. Scintillation Proximity Assay: A Versatile High-Throughput Screening Technology. Drug Discov. Today 1996, 1, 287–294. [Google Scholar] [CrossRef]
- Dealwis, C.; Fernandez, E.J.; Thompson, D.A.; Simon, R.J.; Siani, M.A.; Lolis, E. Crystal Structure of Chemically Synthesized [N33A] Stromal Cell-Derived Factor 1α, a Potent Ligand for the HIV-1 “Fusin” Coreceptor. Proc. Natl. Acad. Sci. USA 1998, 95, 6941–6946. [Google Scholar] [CrossRef] [Green Version]
- Denisov, I.G.; Sligar, S.G. Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev. 2017, 117, 4669–4713. [Google Scholar] [CrossRef]
- Liu, J.; Feldman, P.A.; Lippy, J.S.; Bobkova, E.; Kurilla, M.G.; Chung, T.D.Y. A Scintillation Proximity Assay for RNA Detection. Anal. Biochem. 2001, 289, 239–245. [Google Scholar] [CrossRef]
- Davenport, A.P. Receptor Binding Techniques. Methods Mol. Biol. 2012, 897, 93–94. [Google Scholar] [CrossRef] [Green Version]
- Nare, B.; Allocco, J.J.; Kuningas, R.; Galuska, S.; Myers, R.W.; Bednarek, M.A.; Schmatz, D.M. Development of a Scintillation Proximity Assay for Histone Deacetylase Using a Biotinylated Peptide Derived from Histone-H4. Anal. Biochem. 1999, 267, 390–396. [Google Scholar] [CrossRef]
- Schröder, J.-M. Purification of Chemokines from Natural Sources. In Chemokine Protocols; Proudfoot, A.E.I., Wells, T.N.C., Power, C.A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2000; pp. 1–10. [Google Scholar]
- Zarzycka, B.; Zaidi, S.A.; Roth, B.L.; Katritch, V. Harnessing Ion-Binding Sites for GPCR Pharmacology. Pharmacol. Rev. 2019, 71, 571–595. [Google Scholar] [CrossRef]
- Bisswanger, H. Enzyme Assays. Perspect. Sci. 2014, 1, 41–55. [Google Scholar] [CrossRef] [Green Version]
- Rathert, P.; Cheng, X.; Jeltsch, A. Continuous Enzymatic Assay for Histone Lysine Methyltransferases. Biotechniques 2007, 43, 602–608. [Google Scholar] [CrossRef] [PubMed]
- Balabanian, K.; Lagane, B.; Infantino, S.; Chow, K.Y.C.; Harriague, J.; Moepps, B.; Arenzana-Seisdedos, F.; Thelen, M.; Bachelerie, F. The Chemokine SDF-1/CXCL12 Binds to and Signals through the Orphan Receptor RDC1 in T Lymphocytes. J. Biol. Chem. 2005, 280, 35760–35766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drury, L.J.; Ziarek, J.J.; Gravel, S.; Veldkamp, C.T.; Takekoshi, T.; Hwang, S.T.; Heveker, N.; Volkman, B.F.; Dwinell, M.B. Monomeric and Dimeric CXCL12 Inhibit Metastasis through Distinct CXCR4 Interactions and Signaling Pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 17655–17660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canals, M.; Scholten, D.J.; de Munnik, S.; Han, M.K.L.; Smit, M.J.; Leurs, R. Ubiquitination of CXCR7 Controls Receptor Trafficking. PLoS ONE 2012, 7, e34192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanes, M.S.; Salanga, C.L.; Chowdry, A.B.; Comerford, I.; McColl, S.R.; Kufareva, I.; Handel, T.M. Dual Targeting of the Chemokine Receptors CXCR4 and ACKR3 with Novel Engineered Chemokines. J. Biol. Chem. 2015, 290, 22385–22397. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, F.; Müller, W.; Schütz, D.; Penfold, M.E.; Wong, Y.H.; Schulz, S.; Stumm, R. Rapid Uptake and Degradation of CXCL12 Depend on CXCR7 Carboxyl-Terminal Serine/Threonine Residues. J. Biol. Chem. 2012, 287, 28362–28377. [Google Scholar] [CrossRef] [Green Version]
- Szpakowska, M.; Nevins, A.M.; Meyrath, M.; Rhainds, D.; D’huys, T.; Guité-Vinet, F.; Dupuis, N.; Gauthier, P.A.; Counson, M.; Kleist, A.; et al. Different Contributions of Chemokine N-Terminal Features Attest to a Different Ligand Binding Mode and a Bias towards Activation of ACKR3/CXCR7 Compared with CXCR4 and CXCR3. Br. J. Pharmacol. 2018, 175, 1419–1438. [Google Scholar] [CrossRef]
- Montpas, N.; St-Onge, G.; Nama, N.; Rhainds, D.; Benredjem, B.; Girard, M.; Hickson, G.; Pons, V.; Heveker, N. Ligand-Specific Conformational Transitions and Intracellular Transport Are Required for Atypical Chemokine Receptor 3–Mediated Chemokine Scavenging. J. Biol. Chem. 2018, 293, 893–905. [Google Scholar] [CrossRef] [Green Version]
- Benredjem, B.; Girard, M.; Rhainds, D.; St-Onge, G.; Heveker, N. Mutational Analysis of Atypical Chemokine Receptor 3 (ACKR3/CXCR7) Interaction with Its Chemokine Ligands CXCL11 and CXCL12. J. Biol. Chem. 2017, 292, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Zabel, B.A.; Wang, Y.; Lewén, S.; Berahovich, R.D.; Penfold, M.E.T.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B.; et al. Elucidation of CXCR7-Mediated Signaling Events and Inhibition of CXCR4-Mediated Tumor Cell Transendothelial Migration by CXCR7 Ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Oishi, S.; Kuroyanagi, T.; Kubo, T.; Montpas, N.; Yoshikawa, Y.; Misu, R.; Kobayashi, Y.; Ohno, H.; Heveker, N.; Furuya, T.; et al. Development of Novel CXC Chemokine Receptor 7 (CXCR7) Ligands: Selectivity Switch from CXCR4 Antagonists with a Cyclic Pentapeptide Scaffold. J. Med. Chem. 2015, 58, 5218–5225. [Google Scholar] [CrossRef] [PubMed]
- Montpas, N.; Cabana, J.; St-Onge, G.; Gravel, S.; Morin, G.; Kuroyanagi, T.; Lavigne, P.; Fujii, N.; Oishi, S.; Heveker, N. Mode of Binding of the Cyclic Agonist Peptide TC14012 to CXCR7: Identification of Receptor and Compound Determinants. Biochemistry 2015, 54, 1505–1515. [Google Scholar] [CrossRef] [PubMed]
- Motulsky, H.J.; Mahan, L.C. The Kinetics of Competitive Radioligand Binding Predicted by the Law of Mass Action. Mol. Pharmacol. 1984, 25, 1–9. [Google Scholar] [PubMed]
- Bayburt, T.H.; Sligar, S.G. Membrane Protein Assembly into Nanodiscs. FEBS Lett. 2010, 584, 1721–1727. [Google Scholar] [CrossRef] [Green Version]
- Rouck, J.E.; Krapf, J.E.; Roy, J.; Huff, H.C.; Das, A. Recent Advances in Nanodisc Technology for Membrane Protein Studies (2012–2017). FEBS Lett. 2017, 591, 2057–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawaliby, R.; Trubbia, C.; Delporte, C.; Masureel, M.; van Antwerpen, P.; Kobilka, B.K.; Govaerts, C. Allosteric Regulation of G Protein-Coupled Receptor Activity by Phospholipids. Nat. Chem. Biol. 2016, 12, 35–39. [Google Scholar] [CrossRef]
- Oates, J.; Watts, A. Uncovering the Intimate Relationship between Lipids, Cholesterol and GPCR Activation. Curr. Opin. Struct. Biol. 2011, 21, 802–807. [Google Scholar] [CrossRef]
- Salanga, C.L.; Handel, T.M. Chemokine Oligomerization and Interactions with Receptors and Glycosaminoglycans: The Role of Structural Dynamics in Function. Exp. Cell Res. 2011, 317, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Monneau, Y.; Arenzana-Seisdedos, F.; Lortat-Jacob, H. The Sweet Spot: How GAGs Help Chemokines Guide Migrating Cells. J. Leukoc. Biol. 2016, 99, 935–953. [Google Scholar] [CrossRef] [Green Version]
- Proudfoot, A.E.I.; Handel, T.M.; Johnson, Z.; Lau, E.K.; LiWang, P.; Clark-Lewis, I.; Borlat, F.; Wells, T.N.C.; Kosco-Vilbois, M.H. Glycosaminoglycan Binding and Oligomerization Are Essential for the in Vivo Activity of Certain Chemokines. Proc. Natl. Acad. Sci. USA 2003, 100, 1885–1890. [Google Scholar] [CrossRef] [Green Version]
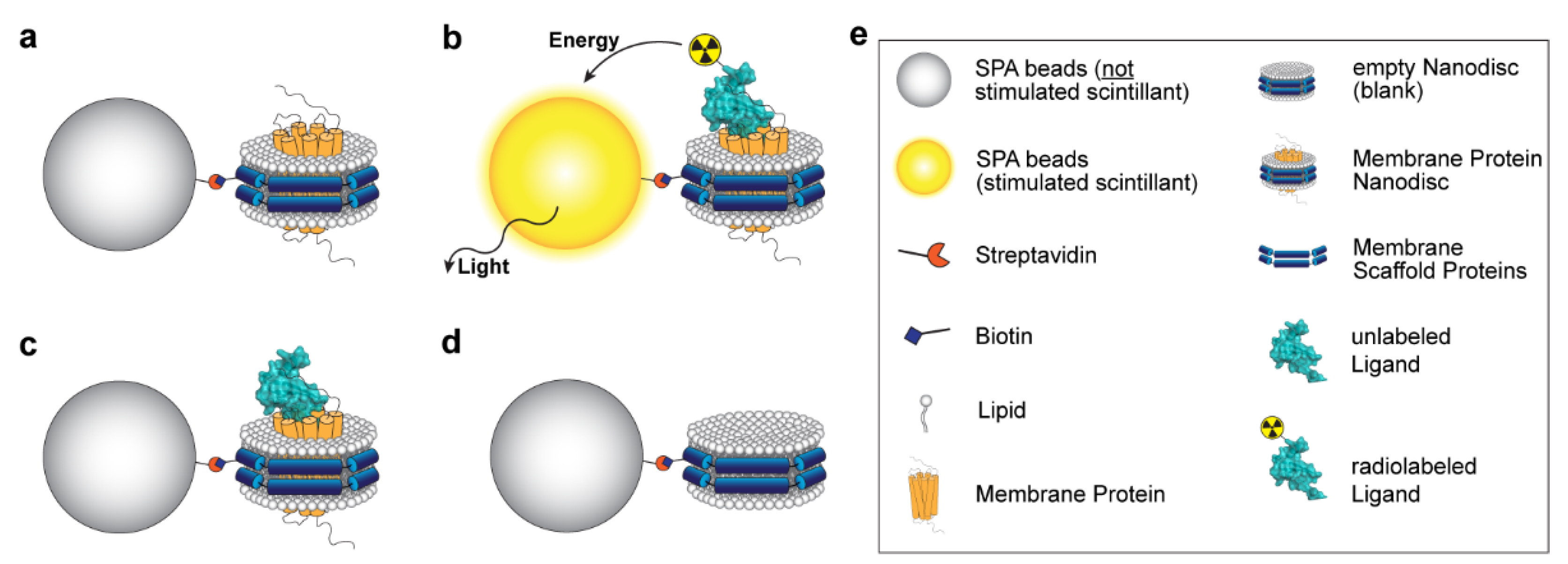



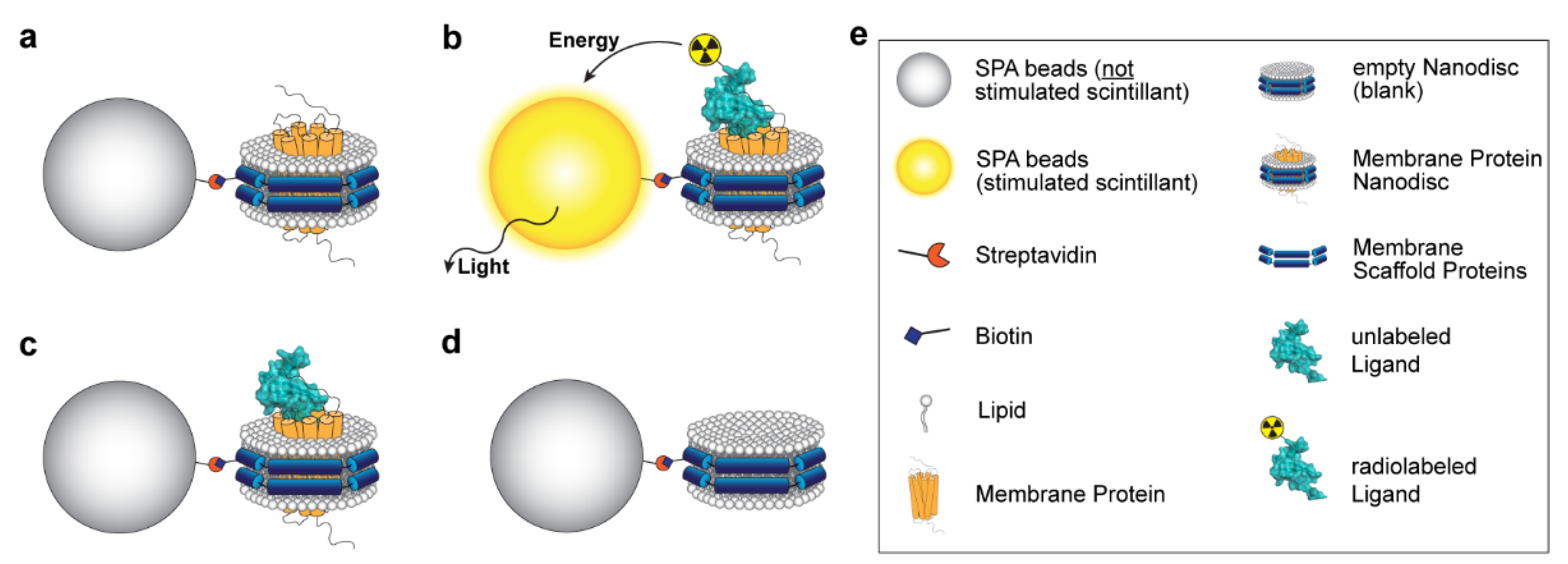{kind=link}
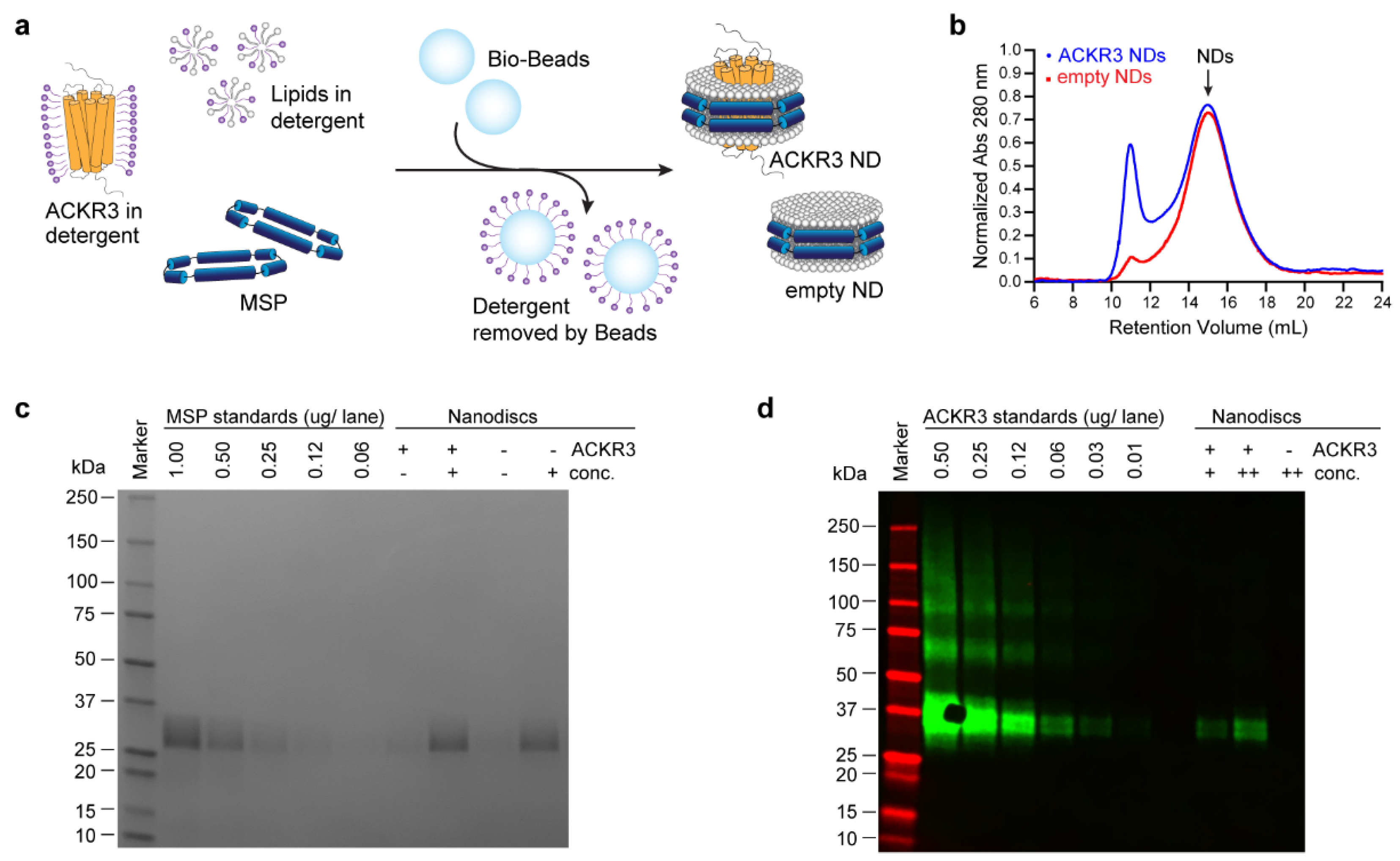{kind=link}
{kind=link}
{kind=link}
| SPA Experiment | Buffer Adjustment * |
|---|---|
| NSB: BSA Screen | 0.04–0.50% (w/v) BSA |
| NSB: NaCl Screen | 20–200 mM NaCl |
| Kinetic measurements | 0 or 5 mM CaCl2 |
| Competition binding | 5 mM CaCl2 |
| 0 mM CaCl2 | 5 mM CaCl2 | ||||
|---|---|---|---|---|---|
| Mean ± SEM | 95% CI | Mean ± SEM | 95% CI | ||
| kon (M−1 min−1) | (3.30 ± 0.13) × 107 | (3.04 to 3.55) × 107 | (2.62 ± 0.22) × 107 | (2.19 ± 3.06) × 107 | |
| koff (min−1) | 0.079 ± 0.001 | 0.076 to 0.082 | 0.080 ± 0.001 | 0.079 to 0.081 | |
| T1/2 (min) | 8.76 ± 0.15 | 8.47 to 9.06 | 8.70 ± 0.06 | 8.59 to 8.81 | |
| Kd (nM) * | 2.40 ± 0.10 | - | 3.04 ± 0.26 | - | |
| IC50 (nM) | |||||
| CXCL12 | - | - | 5.05 ± 0.51 | 4.06 to 6.04 | |
| TC14012 | - | - | 458.6 ± 29.0 | 401.8 to 515.4 | |
| Ki (nM) | |||||
| CXCL12 | - | - | 3.01 ± 0.30 | 2.42 to 3.60 | |
| TC14012 | - | - | 273.9 ± 17.3 | 240.0 to 307.9 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eberle, S.A.; Gustavsson, M. A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine–Chemokine Receptor Interactions. Cells 2022, 11, 1317. https://doi.org/10.3390/cells11081317
Eberle SA, Gustavsson M. A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine–Chemokine Receptor Interactions. Cells. 2022; 11(8):1317. https://doi.org/10.3390/cells11081317
Chicago/Turabian StyleEberle, Stefanie Alexandra, and Martin Gustavsson. 2022. "A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine–Chemokine Receptor Interactions" Cells 11, no. 8: 1317. https://doi.org/10.3390/cells11081317
APA StyleEberle, S. A., & Gustavsson, M. (2022). A Scintillation Proximity Assay for Real-Time Kinetic Analysis of Chemokine–Chemokine Receptor Interactions. Cells, 11(8), 1317. https://doi.org/10.3390/cells11081317