
The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Reagents, Plasmids, and Cell Lines
2.2. β-arrestins BRET Assay
2.3. Subcellular Localization and Trafficking
2.4. BRET Proximity Assay
2.5. Chemerin Scavenging
2.6. MAP Kinase Assay
2.7. Statistical Analysis
3. Results
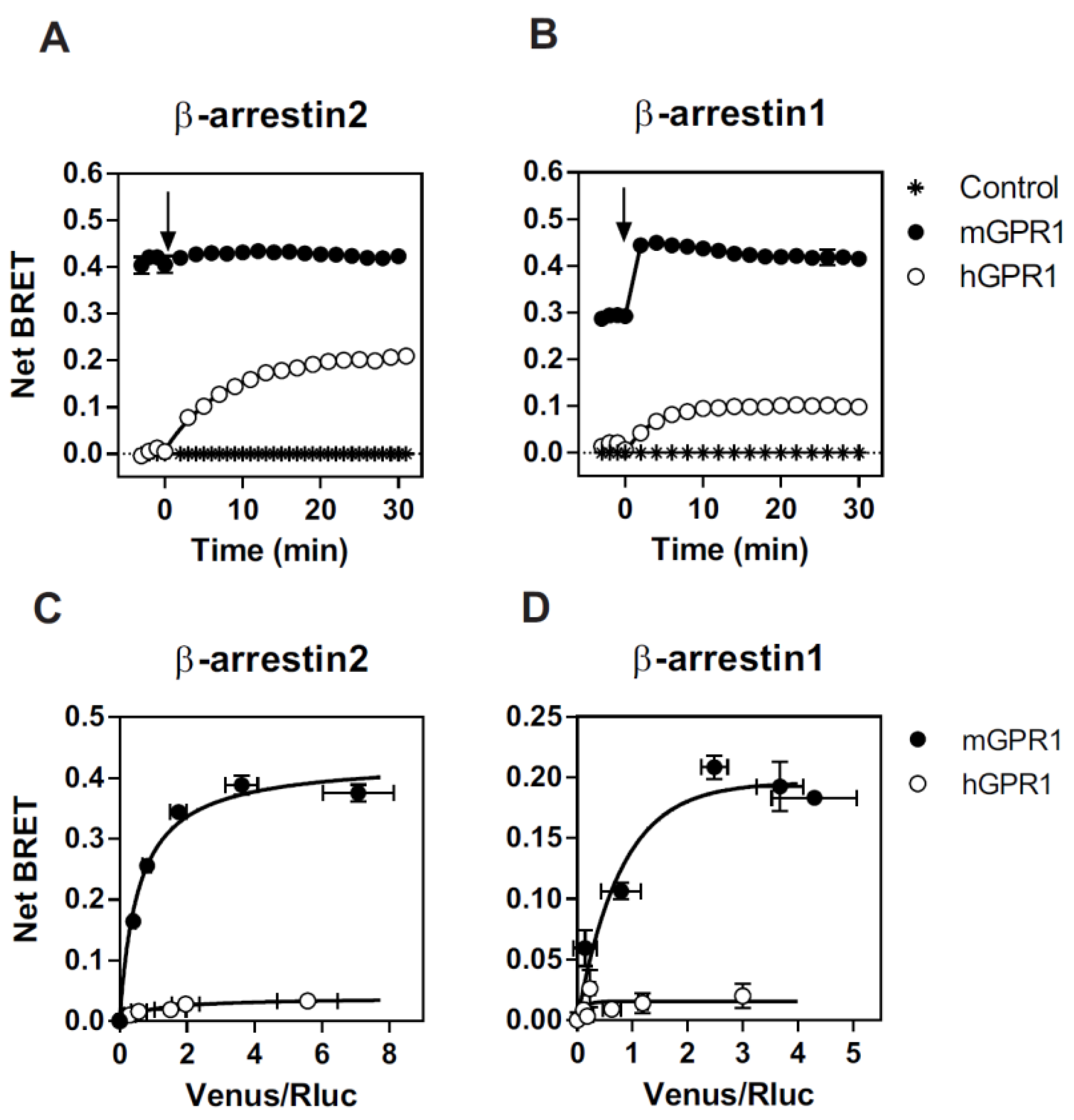
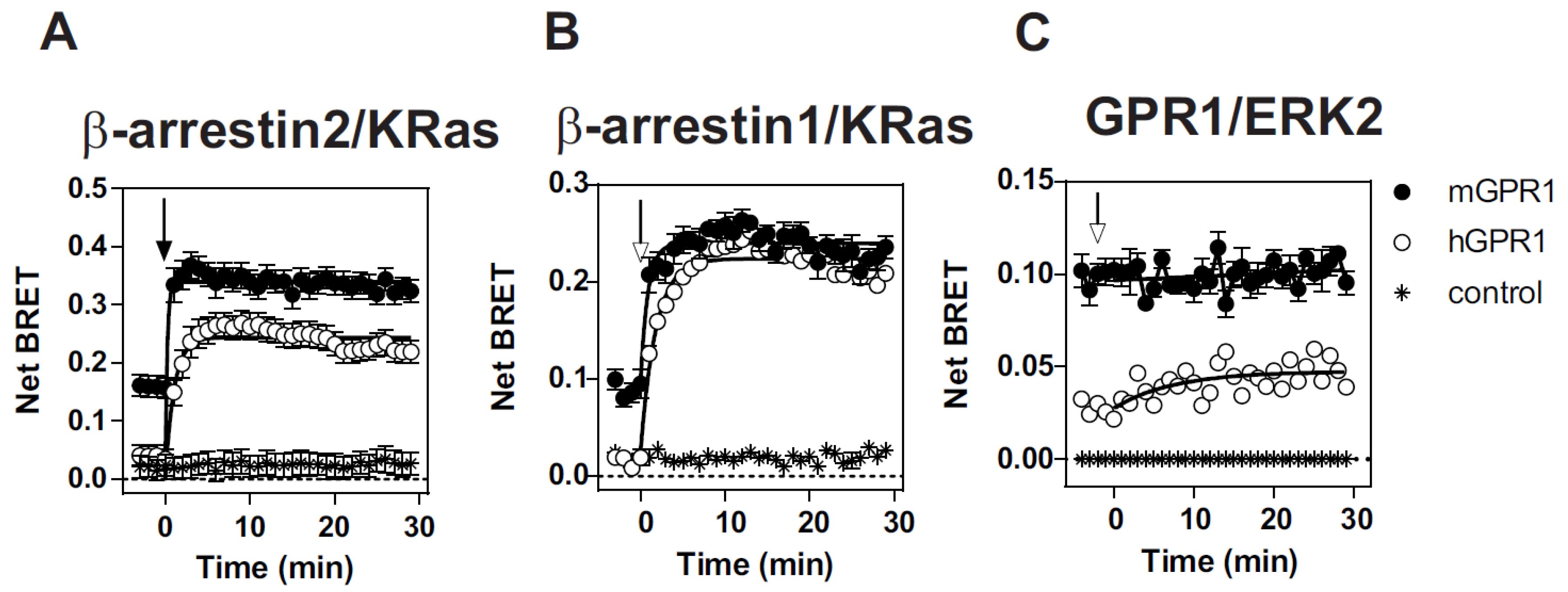
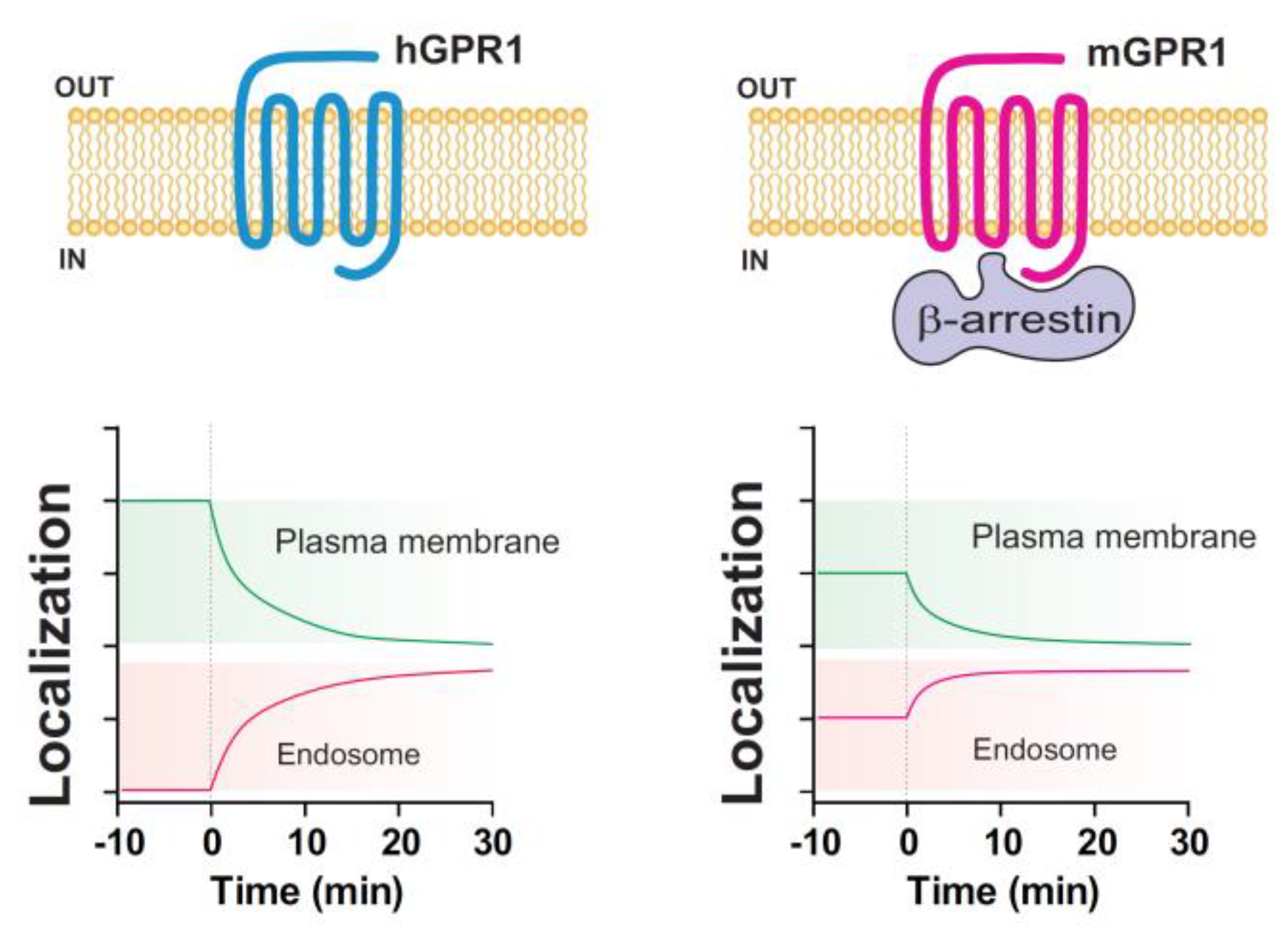
3.1. mGPR1 Constitutively Interacts with β-Arrestins 1 and 2
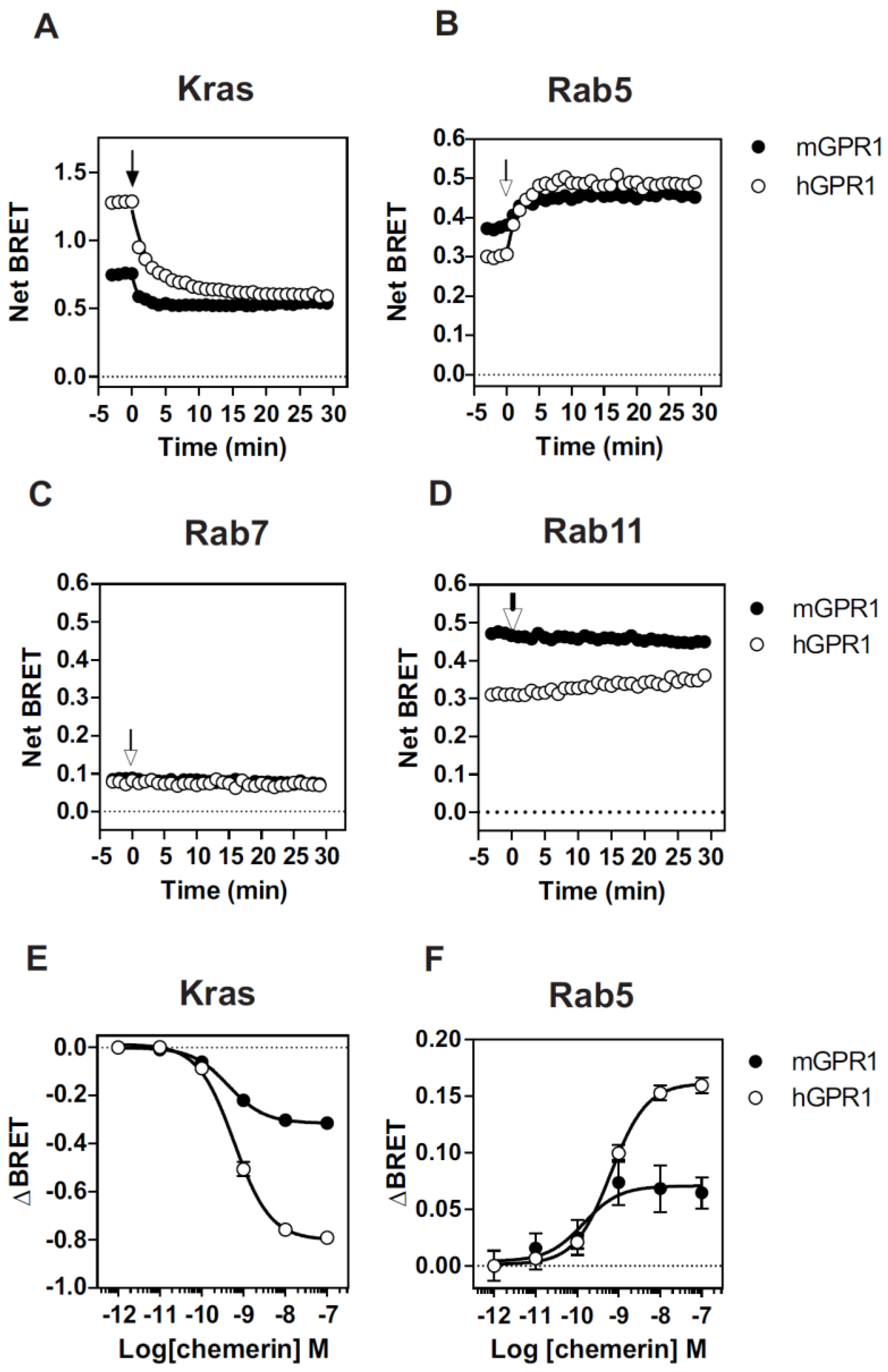
3.2. mGPR1 Partially Localizes in Early and Recycling Endosomes
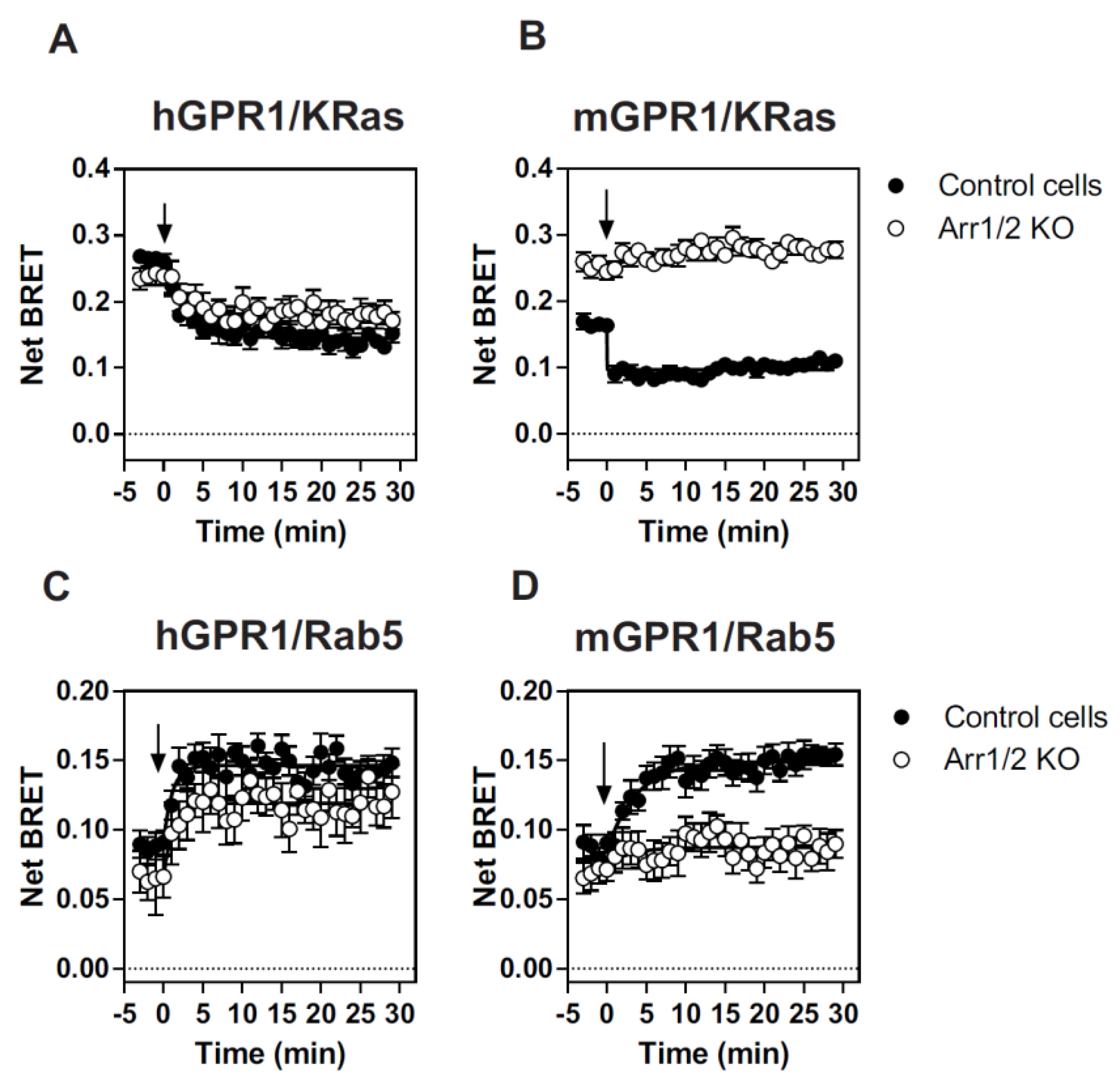
3.3. β-Arrestins Differentially Contribute to the Cellular Distribution and Trafficking of hGPR1 and mGPR1
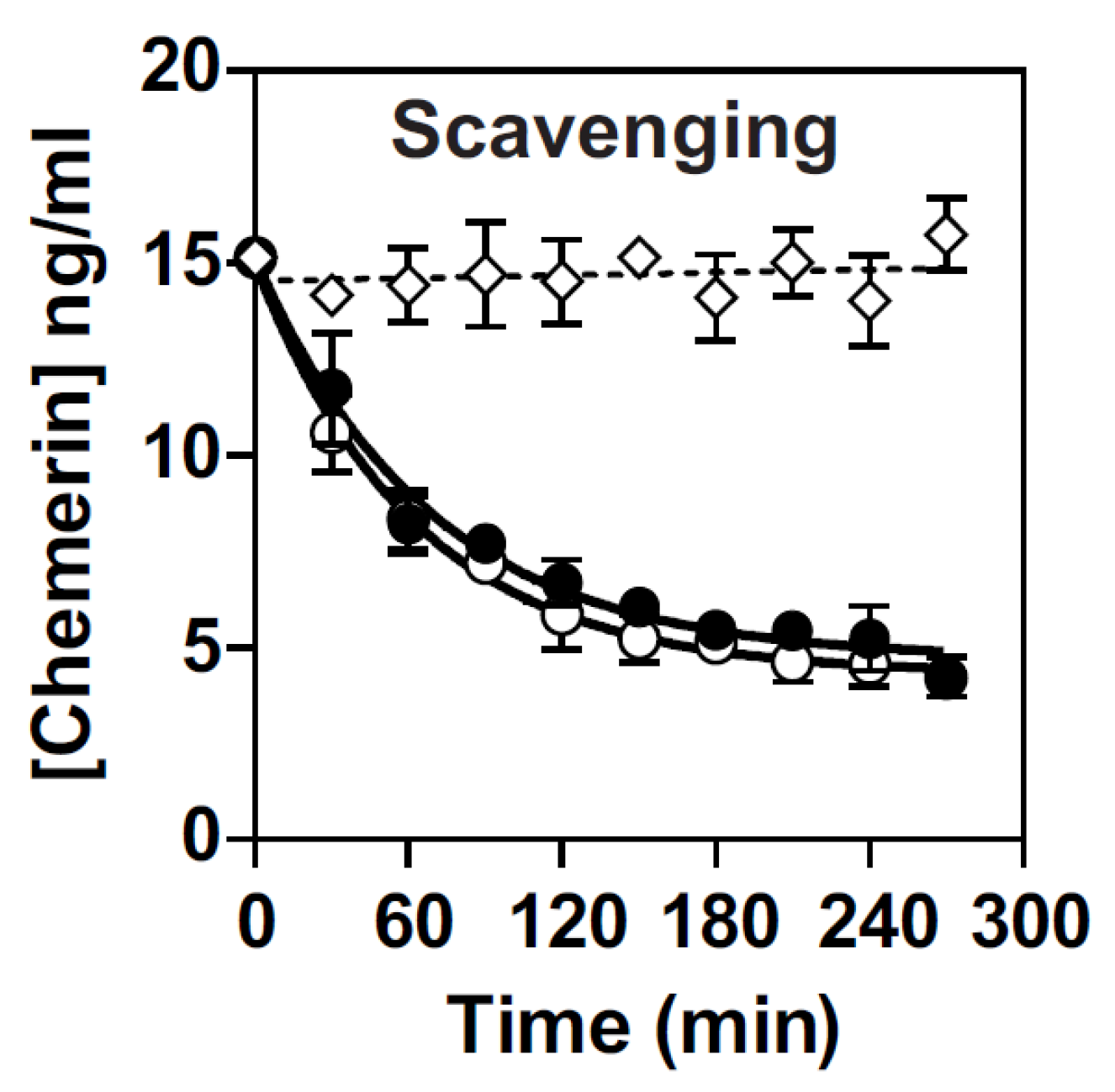
3.4. Both hGPR1 and mGPR1 Scavenge Chemerin
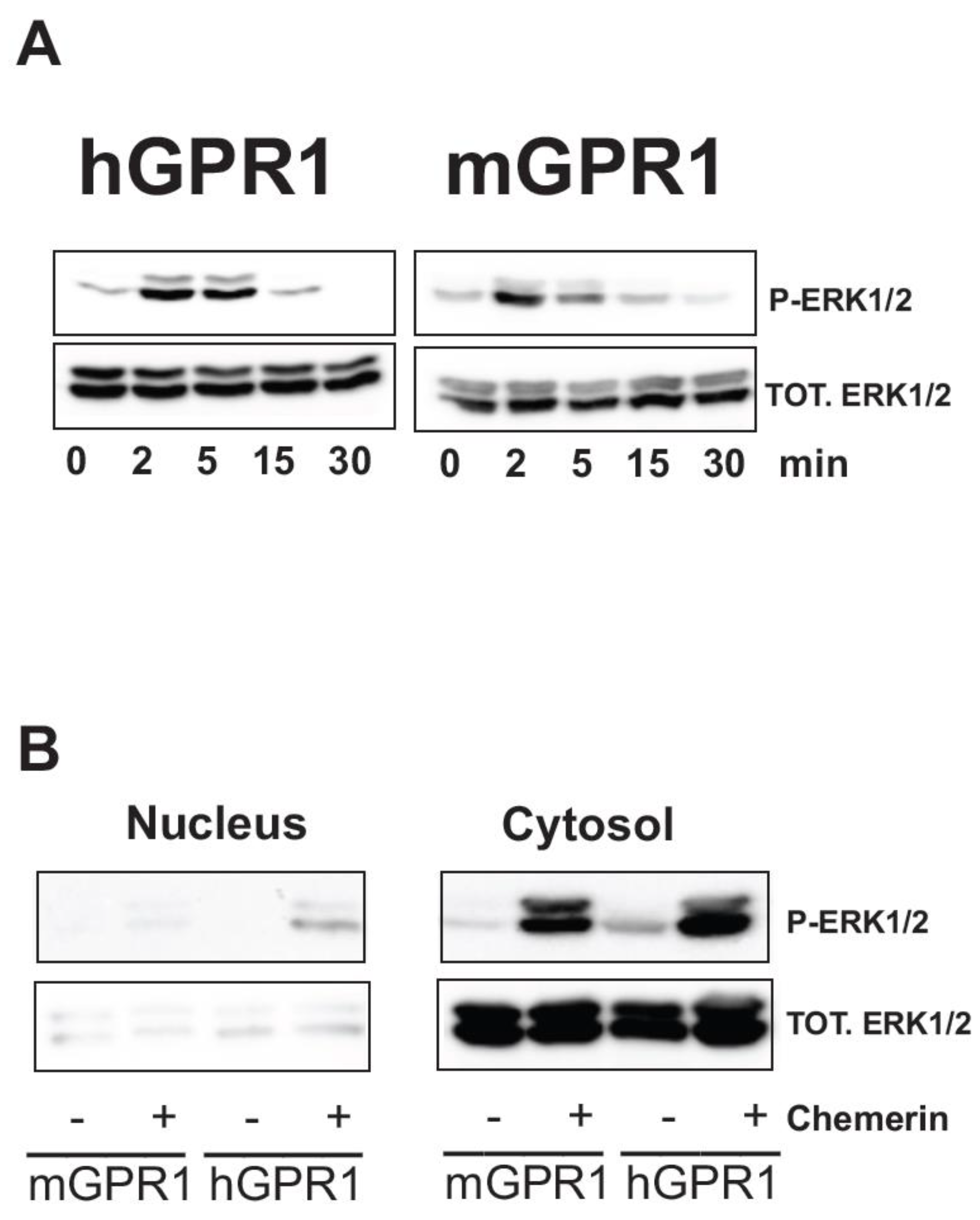
3.5. Both hGPR1 and mGPR1 Activate MAP Kinases ERK1/2
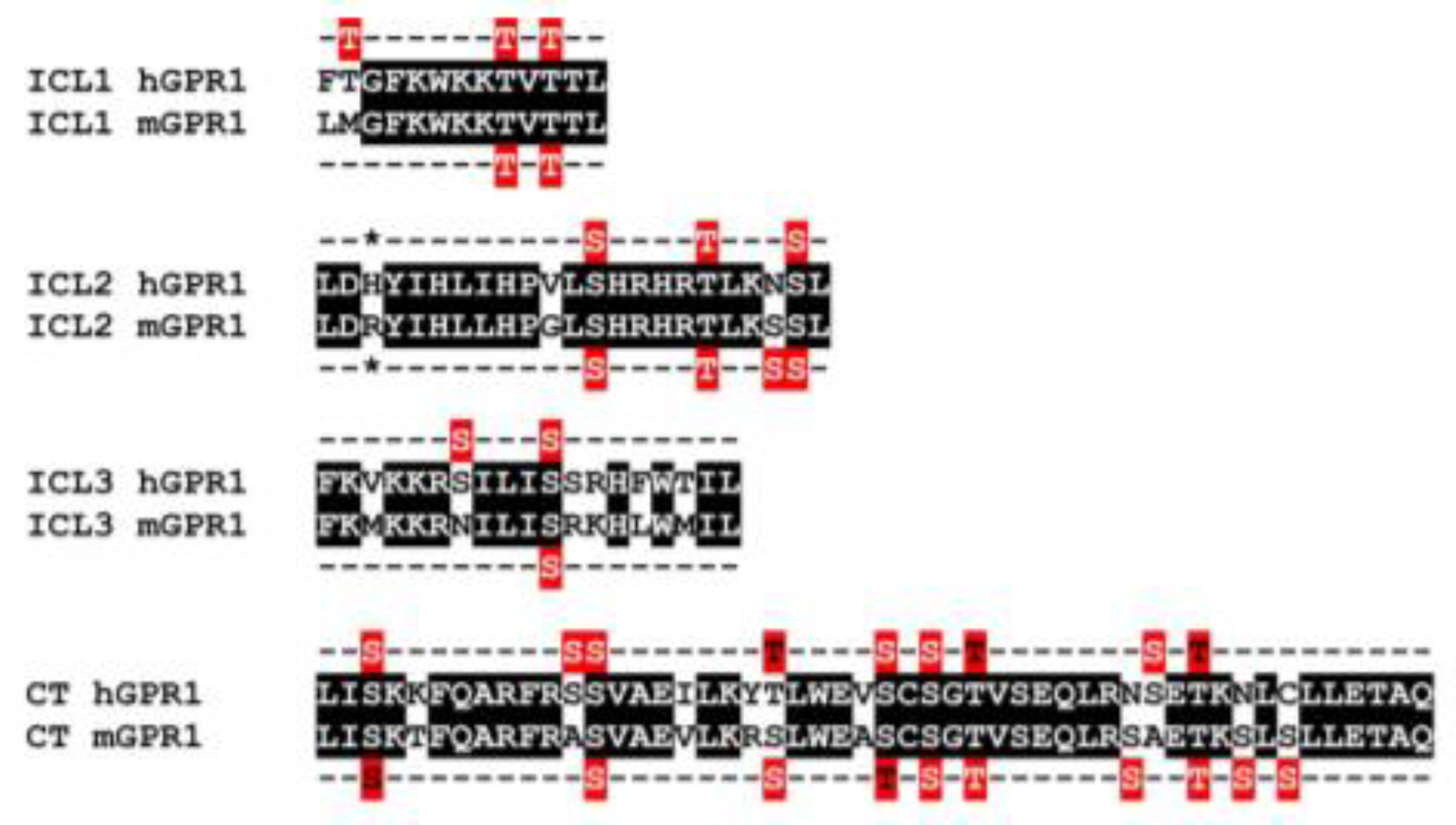
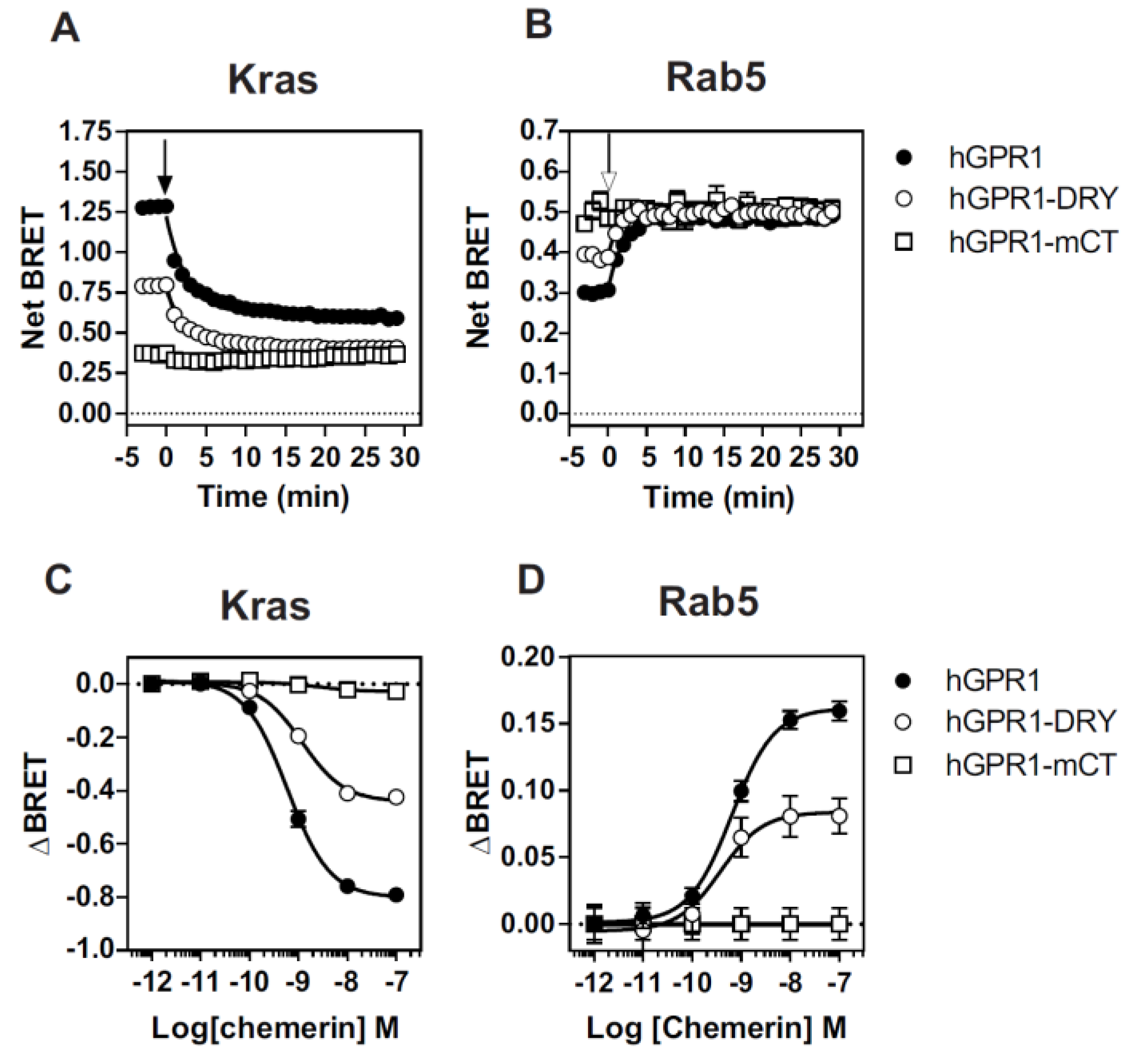
3.6. The Constitutive Interaction of mGPR1 with β-Arrestins Involves the Receptor C-Terminus and R3.50
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bachelerie, F.; Ben-Baruch, A.; Burkhardt, A.M.; Combadiere, C.; Farber, J.M.; Graham, G.J.; Horuk, R.; Sparre-Ulrich, A.H.; Locati, M.; Luster, A.D.; et al. International Union of Pharmacology. LXXXIX. Update on the Extended Family of Chemokine Receptors and Introducing a New Nomenclature for Atypical Chemokine Receptors. Pharmacol. Rev. 2014, 66, 1–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nibbs, R.J.B.; Graham, G.J. Immune Regulation by Atypical Chemokine Receptors. Nat. Rev. Immunol. 2013, 13, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Bonecchi, R.; Graham, G.J. Atypical Chemokine Receptors and Their Roles in the Resolution of the Inflammatory Response. Front. Immunol. 2016, 7, 224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliera, E.; Jala, V.R.; Trent, J.O.; Bonecchi, R.; Signorelli, P.; Lefkowitz, R.J.; Mantovani, A.; Locati, M.; Haribabu, B. Beta-Arrestin-Dependent Constitutive Internalization of the Human Chemokine Decoy Receptor D6. J. Biol. Chem. 2004, 279, 25590–25597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luker, K.E.; Steele, J.M.; Mihalko, L.A.; Ray, P.; Luker, G.D. Constitutive and Chemokine-Dependent Internalization and Recycling of CXCR7 in Breast Cancer Cells to Degrade Chemokine Ligands. Oncogene 2010, 29, 4599–4610. [Google Scholar] [CrossRef] [Green Version]
- Décaillot, F.M.; Kazmi, M.A.; Lin, Y.; Ray-Saha, S.; Sakmar, T.P.; Sachdev, P. CXCR7/CXCR4 Heterodimer Constitutively Recruits Beta-Arrestin to Enhance Cell Migration. J. Biol. Chem. 2011, 286, 32188–32197. [Google Scholar] [CrossRef] [Green Version]
- Zabel, B.A.; Wang, Y.; Lewén, S.; Berahovich, R.D.; Penfold, M.E.T.; Zhang, P.; Powers, J.; Summers, B.C.; Miao, Z.; Zhao, B.; et al. Elucidation of CXCR7-Mediated Signaling Events and Inhibition of CXCR4-Mediated Tumor Cell Transendothelial Migration by CXCR7 Ligands. J. Immunol. 2009, 183, 3204–3211. [Google Scholar] [CrossRef] [Green Version]
- Rajagopal, S.; Kim, J.; Ahn, S.; Craig, S.; Lam, C.M.; Gerard, N.P.; Gerard, C.; Lefkowitz, R.J. Beta-Arrestin- but Not G Protein-Mediated Signaling by the “Decoy” Receptor CXCR7. Proc. Natl. Acad. Sci. USA 2010, 107, 628–632. [Google Scholar] [CrossRef] [Green Version]
- Vacchini, A.; Busnelli, M.; Chini, B.; Locati, M.; Borroni, E.M. Analysis of G Protein and β-Arrestin Activation in Chemokine Receptors Signaling. Methods Enzymol. 2016, 570, 421–440. [Google Scholar] [CrossRef]
- Borroni, E.M.; Cancellieri, C.; Vacchini, A.; Benureau, Y.; Lagane, B.; Bachelerie, F.; Arenzana-Seisdedos, F.; Mizuno, K.; Mantovani, A.; Bonecchi, R.; et al. β-Arrestin-Dependent Activation of the Cofilin Pathway Is Required for the Scavenging Activity of the Atypical Chemokine Receptor D6. Sci. Signal. 2013, 6, ra30. [Google Scholar] [CrossRef]
- Ahn, S.; Shenoy, S.K.; Luttrell, L.M.; Lefkowitz, R.J. SnapShot: β-Arrestin Functions. Cell 2020, 182, 1362. [Google Scholar] [CrossRef] [PubMed]
- Luttrell, L.M.; Roudabush, F.L.; Choy, E.W.; Miller, W.E.; Field, M.E.; Pierce, K.L.; Lefkowitz, R.J. Activation and Targeting of Extracellular Signal-Regulated Kinases by Beta-Arrestin Scaffolds. Proc. Natl. Acad. Sci. USA 2001, 98, 2449–2454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonald, P.H.; Chow, C.W.; Miller, W.E.; Laporte, S.A.; Field, M.E.; Lin, F.T.; Davis, R.J.; Lefkowitz, R.J. Beta-Arrestin 2: A Receptor-Regulated MAPK Scaffold for the Activation of JNK3. Science 2000, 290, 1574–1577. [Google Scholar] [CrossRef] [PubMed]
- De Henau, O.; Degroot, G.-N.; Imbault, V.; Robert, V.; De Poorter, C.; Mcheik, S.; Galés, C.; Parmentier, M.; Springael, J.-Y. Signaling Properties of Chemerin Receptors CMKLR1, GPR1 and CCRL2. PLoS ONE 2016, 11, e0164179. [Google Scholar] [CrossRef]
- Wittamer, V.; Bondue, B.; Guillabert, A.; Vassart, G.; Parmentier, M.; Communi, D. Neutrophil-Mediated Maturation of Chemerin: A Link between Innate and Adaptive Immunity. J. Immunol. 2005, 175, 487–493. [Google Scholar] [CrossRef]
- Bondue, B.; Wittamer, V.; Parmentier, M. Chemerin and Its Receptors in Leukocyte Trafficking, Inflammation and Metabolism. Cytokine Growth Factor Rev. 2011, 22, 331–338. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.J.; Davenport, A.P. International Union of Basic and Clinical Pharmacology CIII: Chemerin Receptors CMKLR1 (Chemerin1) and GPR1 (Chemerin2) Nomenclature, Pharmacology, and Function. Pharmacol. Rev. 2018, 70, 174–196. [Google Scholar] [CrossRef] [Green Version]
- Helfer, G.; Wu, Q.-F. Chemerin: A Multifaceted Adipokine Involved in Metabolic Disorders. J. Endocrinol. 2018, 238, R79–R94. [Google Scholar] [CrossRef]
- Wittamer, V.; Franssen, J.-D.; Vulcano, M.; Mirjolet, J.-F.; Le Poul, E.; Migeotte, I.; Brézillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific Recruitment of Antigen-Presenting Cells by Chemerin, a Novel Processed Ligand from Human Inflammatory Fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef]
- Zabel, B.A.; Nakae, S.; Zúñiga, L.; Kim, J.-Y.; Ohyama, T.; Alt, C.; Pan, J.; Suto, H.; Soler, D.; Allen, S.J.; et al. Mast Cell–Expressed Orphan Receptor CCRL2 Binds Chemerin and Is Required for Optimal Induction of IgE-Mediated Passive Cutaneous Anaphylaxis. J. Exp. Med. 2008, 205, 2207–2220. [Google Scholar] [CrossRef]
- Barnea, G.; Strapps, W.; Herrada, G.; Berman, Y.; Ong, J.; Kloss, B.; Axel, R.; Lee, K.J. The Genetic Design of Signaling Cascades to Record Receptor Activation. Proc. Natl. Acad. Sci. USA 2008, 105, 64–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, T.F.; Czerniak, A.S.; Weiß, T.; Schoeder, C.T.; Wolf, P.; Seitz, O.; Meiler, J.; Beck-Sickinger, A.G. Ligand-Binding and -Scavenging of the Chemerin Receptor GPR1. Cell. Mol. Life Sci. 2021, 78, 6265–6281. [Google Scholar] [CrossRef] [PubMed]
- Grundmann, M.; Merten, N.; Malfacini, D.; Inoue, A.; Preis, P.; Simon, K.; Rüttiger, N.; Ziegler, N.; Benkel, T.; Schmitt, N.K.; et al. Lack of Beta-Arrestin Signaling in the Absence of Active G Proteins. Nat. Commun. 2018, 9, 341. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Pack, T.F.; Inoue, A.; Lee, C.; Zheng, K.; Choi, I.; Eiger, D.S.; Warman, A.; Xiong, X.; Ma, Z.; et al. Noncanonical Scaffolding of Gαi and β-Arrestin by G Protein-Coupled Receptors. Science 2021, 371, eaay1833. [Google Scholar] [CrossRef]
- Li, J.; Xiang, L.; Jiang, X.; Teng, B.; Sun, Y.; Chen, G.; Chen, J.; Zhang, J.V.; Ren, P.-G. Investigation of Bioeffects of G Protein-Coupled Receptor 1 on Bone Turnover in Male Mice. J. Orthop. Transl. 2017, 10, 42–51. [Google Scholar] [CrossRef]
- Rourke, J.L.; Muruganandan, S.; Dranse, H.J.; McMullen, N.M.; Sinal, C.J. Gpr1 Is an Active Chemerin Receptor Influencing Glucose Homeostasis in Obese Mice. J. Endocrinol. 2014, 222, 201–215. [Google Scholar] [CrossRef]
- Storez, H.; Scott, M.G.H.; Issafras, H.; Burtey, A.; Benmerah, A.; Muntaner, O.; Piolot, T.; Tramier, M.; Coppey-Moisan, M.; Bouvier, M.; et al. Homo- and Hetero-Oligomerization of Beta-Arrestins in Living Cells. J. Biol. Chem. 2005, 280, 40210–40215. [Google Scholar] [CrossRef] [Green Version]
- Rubinfeld, H.; Hanoch, T.; Seger, R. Identification of a cytoplasmic-retention sequence in ERK2. J. Biol. Chem. 1999, 274, 30349–30352. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.R.; Lambert, N.A. Activated G Protein Gαs Samples Multiple Endomembrane Compartments. J. Biol. Chem. 2016, 291, 20295–20302. [Google Scholar] [CrossRef] [Green Version]
- O’Hayre, M.; Eichel, K.; Avino, S.; Zhao, X.; Steffen, D.J.; Feng, X.; Kawakami, K.; Aoki, J.; Messer, K.; Sunahara, R.; et al. Genetic Evidence That β-Arrestins Are Dispensable for the Initiation of Β2-Adrenergic Receptor Signaling to ERK. Sci. Signal. 2017, 10, eaal3395. [Google Scholar] [CrossRef] [Green Version]
- Corbisier, J.; Galès, C.; Huszagh, A.; Parmentier, M.; Springael, J.-Y. Biased Signaling at Chemokine Receptors. J. Biol. Chem. 2015, 290, 9542–9554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohgo, A.; Choy, E.W.; Gesty-Palmer, D.; Pierce, K.L.; Laporte, S.; Oakley, R.H.; Caron, M.G.; Lefkowitz, R.J.; Luttrell, L.M. The Stability of the G Protein-Coupled Receptor-β-Arrestin Interaction Determines the Mechanism and Functional Consequence of ERK Activation. J. Biol. Chem. 2003, 278, 6258–6267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tohgo, A.; Pierce, K.L.; Choy, E.W.; Lefkowitz, R.J.; Luttrell, L.M. β-Arrestin Scaffolding of the ERK Cascade Enhances Cytosolic ERK Activity but Inhibits ERK-Mediated Transcription Following Angiotensin AT1a Receptor Stimulation. J. Biol. Chem. 2002, 277, 9429–9436. [Google Scholar] [CrossRef] [Green Version]
- Marchese, A.; Docherty, J.M.; Nguyen, T.; Heiber, M.; Cheng, R.; Heng, H.H.Q.; Tsui, L.-C.; Shi, X.; George, S.R.; O’Dowd, B.F. Cloning of Human Genes Encoding Novel G Protein-Coupled Receptors. Genomics 1994, 23, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Wilbanks, A.M.; Laporte, S.A.; Bohn, L.M.; Barak, L.S.; Caron, M.G. Apparent Loss-of-Function Mutant GPCRs Revealed as Constitutively Desensitized Receptors. Biochemistry 2002, 41, 11981–11989. [Google Scholar] [CrossRef] [PubMed]
- Barak, L.S.; Oakley, R.H.; Laporte, S.A.; Caron, M.G. Constitutive Arrestin-Mediated Desensitization of a Human Vasopressin Receptor Mutant Associated with Nephrogenic Diabetes Insipidus. Proc. Natl. Acad. Sci. USA 2001, 98, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Matti, C.; Salnikov, A.; Artinger, M.; D’Agostino, G.; Kindinger, I.; Uguccioni, M.; Thelen, M.; Legler, D.F. ACKR4 Recruits GRK3 Prior to β-Arrestins but Can Scavenge Chemokines in the Absence of β-Arrestins. Front. Immunol. 2020, 11, 720. [Google Scholar] [CrossRef] [Green Version]
- Liggett, S.B. Phosphorylation Barcoding as a Mechanism of Directing GPCR Signaling. Sci. Signal. 2011, 4, pe36. [Google Scholar] [CrossRef]
- Nobles, K.N.; Xiao, K.; Ahn, S.; Shukla, A.K.; Lam, C.M.; Rajagopal, S.; Strachan, R.T.; Huang, T.-Y.; Bressler, E.A.; Hara, M.R.; et al. Distinct Phosphorylation Sites on the β(2)-Adrenergic Receptor Establish a Barcode that Encodes Differential Functions of β-Arrestin. Sci. Signal. 2011, 4, ra51. [Google Scholar] [CrossRef] [Green Version]
- Ostermaier, M.K.; Schertler, G.F.X.; Standfuss, J. Molecular Mechanism of Phosphorylation-Dependent Arrestin Activation. Curr. Opin. Struct. Biol. 2014, 29, 143–151. [Google Scholar] [CrossRef]
- Reiter, E.; Ahn, S.; Shukla, A.K.; Lefkowitz, R.J. Molecular Mechanism of β-Arrestin-Biased Agonism at Seven-Transmembrane Receptors. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 179–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seyedabadi, M.; Gharghabi, M.; Gurevich, E.V.; Gurevich, V.V. Receptor-Arrestin Interactions: The GPCR Perspective. Biomolecules 2021, 11, 218. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degroot, G.-N.; Lepage, V.; Parmentier, M.; Springael, J.-Y. The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking. Cells 2022, 11, 1037. https://doi.org/10.3390/cells11061037
Degroot G-N, Lepage V, Parmentier M, Springael J-Y. The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking. Cells. 2022; 11(6):1037. https://doi.org/10.3390/cells11061037
Chicago/Turabian StyleDegroot, Gaetan-Nagim, Valentin Lepage, Marc Parmentier, and Jean-Yves Springael. 2022. "The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking" Cells 11, no. 6: 1037. https://doi.org/10.3390/cells11061037
APA StyleDegroot, G.-N., Lepage, V., Parmentier, M., & Springael, J.-Y. (2022). The Atypical Chemerin Receptor GPR1 Displays Different Modes of Interaction with β-Arrestins in Humans and Mice with Important Consequences on Subcellular Localization and Trafficking. Cells, 11(6), 1037. https://doi.org/10.3390/cells11061037