
Multi-Omics Approach Reveals Dysregulation of Protein Phosphorylation Correlated with Lipid Metabolism in Mouse Non-Alcoholic Fatty Liver

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
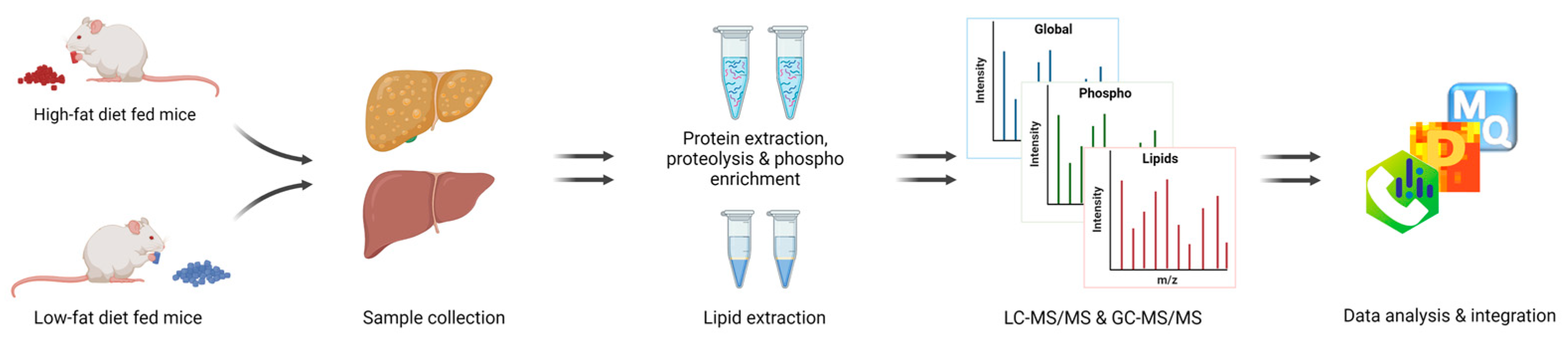
2. Materials and Methods
2.1. Mouse Husbandry and Diets
2.2. Liver Tissue Preparation for Proteomics
2.3. Mass Spectrometry Analysis of Liver Proteome
2.4. Data Analysis
2.5. Lipid Extraction and Measurement
3. Results
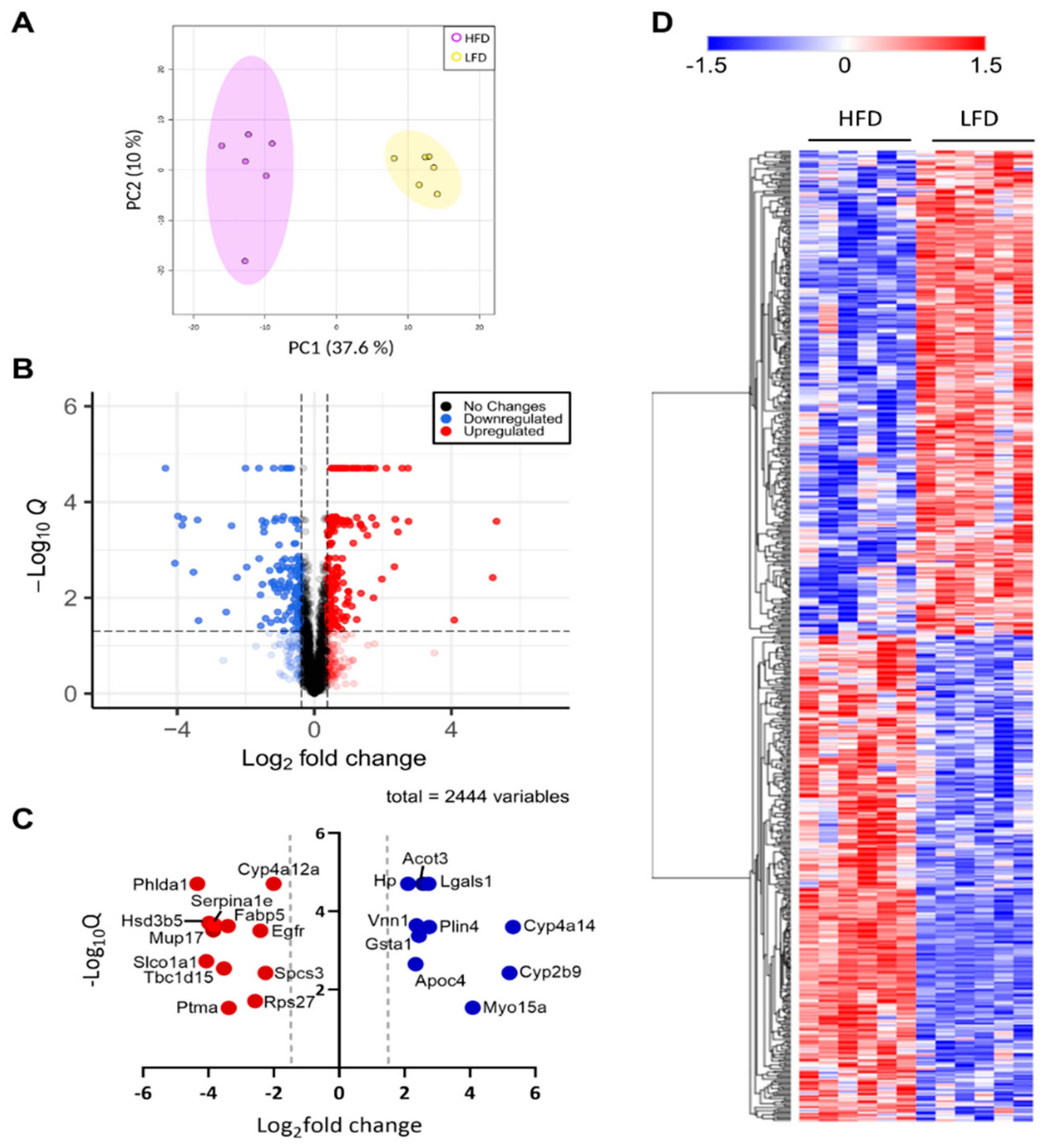
3.1. Mice Fed a Chronic High-Fat Diet Develop Obesity
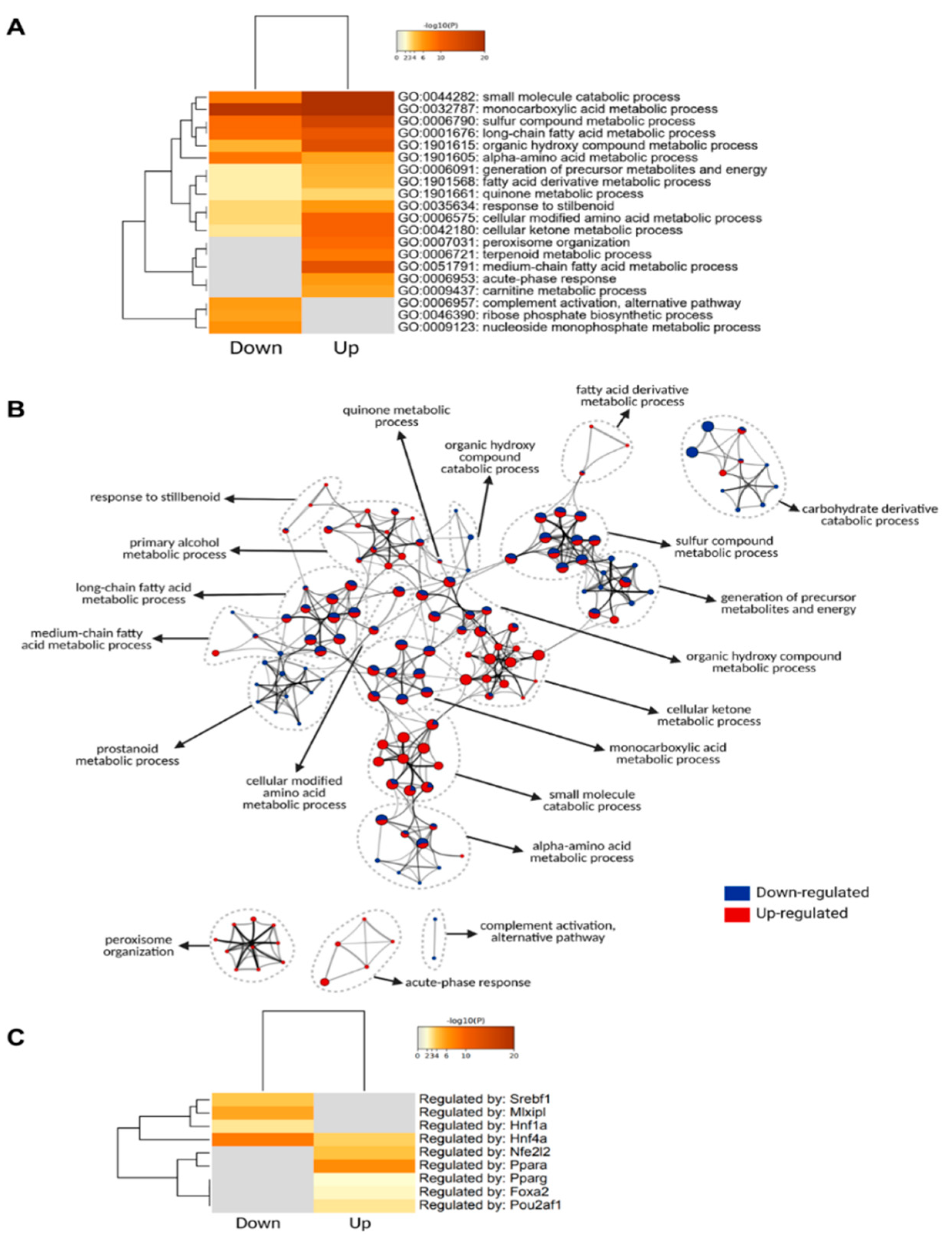
3.2. Liver Proteome Profile Change upon Diet-Induced Obesity
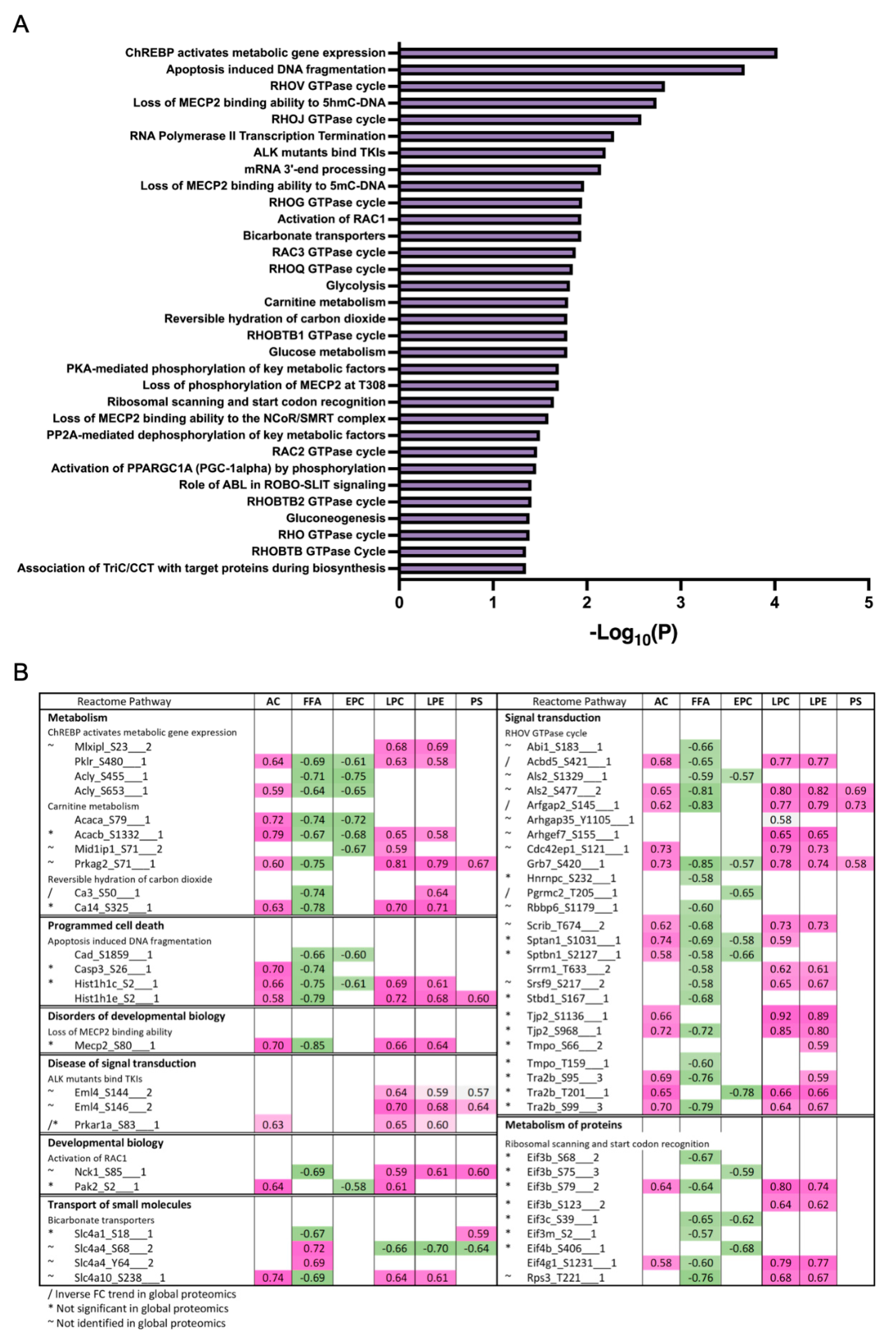
3.3. Differentially Expressed Phosphoproteins in the Liver from HFD and LFD Fed Mice
3.4. Differentially Expressed Lipids in the Liver from HFD and LFD Fed Mice
4. Discussion
4.1. Altered Proteomes Related to Lipoprotein Assembly, Lipoprotein Uptake, De Novo Lipogenesis (DNL) and Fatty Acid Uptake
4.2. Increased Mitochondrial Fatty Acid β-Oxidation, Ketone Body Formation
4.3. Increased Abundance of Proteins Involved in Peroxisomal β-Oxidation and Microsomal ω-Oxidation
4.4. Decreased Glycolysis and Increased Gluconeogenesis
4.5. Altered Abundance of Proteins Involved in ROS Scavenging
4.6. Regulation of PML-NB Proteins under HFD
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations for Lipid Classes
References
- Younossi, Z.M. Non-alcoholic fatty liver disease—A global public health perspective. J. Hepatol. 2019, 70, 531–544. [Google Scholar] [CrossRef] [PubMed]
- Utzschneider, K.M.; Kahn, S.E. Review: The Role of Insulin Resistance in Nonalcoholic Fatty Liver Disease. J. Clin. Endocrinol. Metab. 2006, 91, 4753–4761. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.G.; Roelstraete, B.; Khalili, H.; Hagström, H.; Ludvigsson, J.F. Mortality in biopsy-confirmed nonalcoholic fatty liver disease: Results from a nationwide cohort. Gut 2021, 70, 1375–1382. [Google Scholar] [CrossRef]
- Lionetti, L.; Mollica, M.P.; Lombardi, A.; Cavaliere, G.; Gifuni, G.; Barletta, A. From chronic overnutrition to insulin resistance: The role of fat-storing capacity and inflammation. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 146–152. [Google Scholar] [CrossRef]
- Jahn, D.; Kircher, S.; Hermanns, H.M.; Geier, A. Animal models of NAFLD from a hepatologist’s point of view. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2019, 1865, 943–953. [Google Scholar] [CrossRef]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high-fat diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Zembroski, A.S.; Buhman, K.K.; Aryal, U.K. Proteome and phosphoproteome characterization of liver in the postprandial state from diet-induced obese and lean mice. J. Proteom. 2021, 232, 104072. [Google Scholar] [CrossRef]
- Krahmer, N.; Najafi, B.; Schueder, F.; Quagliarini, F.; Steger, M.; Seitz, S.; Kasper, R.; Salinas, F.; Cox, J.; Uhlenhaut, H.; et al. Organellar Proteomics and Phospho-Proteomics Reveal Subcellular Reorganization in Diet-Induced Hepatic Steatosis. Dev. Cell 2018, 47, 205–221.e7. [Google Scholar] [CrossRef]
- Sabidó, E.; Wu, Y.; Bautista, L.; Porstmann, T.; Chang, C.; Vitek, O.; Stoffel, M.; Aebersold, R. Targeted proteomics reveals strain-specific changes in the mouse insulin and central metabolic pathways after a sustained high-fat diet. Mol. Syst. Biol. 2013, 9, 681. [Google Scholar] [CrossRef]
- Li, Z.; Lai, Z.W.; Christiano, R.; Gazos-Lopes, F.; Walther, T.C.; Farese, R.V., Jr. Global Analyses of Selective Insulin Resistance in Hepatocytes Caused by Palmitate Lipotoxicity. Mol. Cell. Proteom. 2018, 17, 836–849. [Google Scholar] [CrossRef] [PubMed]
- Nam, M.; Choi, M.-S.; Jung, S.; Jung, Y.; Choi, J.-Y.; Ryu, D.H.; Hwang, G.-S. Lipidomic Profiling of Liver Tissue from Obesity-Prone and Obesity-Resistant Mice Fed a High Fat Diet. Sci. Rep. 2015, 5, 16984. [Google Scholar] [CrossRef] [PubMed]
- Mohallem, R.; Aryal, U.K. Regulators of TNFα mediated insulin resistance elucidated by quantitative proteomics. Sci. Rep. 2020, 10, 20878. [Google Scholar] [CrossRef] [PubMed]
- Hedrick, V.E.; LaLand, M.N.; Nakayasu, E.S.; Paul, L.N. Digestion, Purification, and Enrichment of Protein Samples for Mass Spectrometry. Curr. Protoc. Chem. Biol. 2015, 7, 201–222. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Cox, J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016, 11, 2301–2319. [Google Scholar] [CrossRef]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. 2020. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 15 February 2022).
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef]
- Tsugawa, H.; Ikeda, K.; Takahashi, M.; Satoh, A.; Mori, Y.; Uchino, H.; Okahashi, N.; Yamada, Y.; Tada, I.; Bonini, P.; et al. A lipidome atlas in MS-DIAL 4. Nat. Biotechnol. 2020, 38, 1159–1163. [Google Scholar] [CrossRef]
- Klähn, S.; Mikkat, S.; Riediger, M.; Georg, J.; Hess, W.R.; Hagemann, M. Integrative analysis of the salt stress response in cyanobacteria. Biol. Direct 2021, 16, 26. [Google Scholar] [CrossRef]
- Li, J.; Paulo, J.A.; Nusinow, D.P.; Huttlin, E.L.; Gygi, S.P. Investigation of Proteomic and Phosphoproteomic Responses to Signaling Network Perturbations Reveals Functional Pathway Organizations in Yeast. Cell Rep. 2019, 29, 2092–2104.e4. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Sun, F.; Forghani, P.; Armand, L.C.; Rampoldi, A.; Li, D.; Wu, R.; Xu, C. Proteomic Profiling Reveals Roles of Stress Response, Ca2+ Transient Dysregulation, and Novel Signaling Pathways in Alcohol-Induced Cardiotoxicity. Alcohol. Clin. Exp. Res. 2020, 44, 2187–2199. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.D.; Roschitzki, B.; Snoek, L.B.; Grossmann, J.; Zheng, X.; Elvin, M.; Kamkina, P.; Schrimpf, S.P.; Poulin, G.B.; Kammenga, J.E.; et al. Natural Genetic Variation Influences Protein Abundances in C. elegans Developmental Signalling Pathways. PLoS ONE 2016, 11, e0149418. [Google Scholar] [CrossRef] [PubMed]
- Sheng, H.; Guo, Y.; Zhang, L.; Zhang, J.; Miao, M.; Tan, H.; Hu, D.; Li, X.; Ding, X.; Li, G.; et al. Proteomic Studies on the Mechanism of Myostatin Regulating Cattle Skeletal Muscle Development. Front. Genet. 2021, 12, 752129. [Google Scholar] [CrossRef] [PubMed]
- Kahle, M.; Horsch, M.; Fridrich, B.; Seelig, A.; Schultheiß, J.; Leonhardt, J.; Irmler, M.; Beckers, J.; Rathkolb, B.; Wolf, E.; et al. Phenotypic comparison of common mouse strains developing high-fat diet-induced hepatosteatosis. Mol. Metab. 2013, 2, 435–446. [Google Scholar] [CrossRef]
- Rommelaere, S.; Millet, V.; Gensollen, T.; Bourges, C.; Eeckhoute, J.; Hennuyer, N.; Baugé, E.; Chasson, L.; Cacciatore, I.; Staels, B.; et al. PPARalpha regulates the production of serum Vanin-1 by liver. FEBS Lett. 2013, 587, 3742–3748. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, W.; Tang, C.; Tang, X.; Liu, L.; Liu, C. Vanin-1 Is a Key Activator for Hepatic Gluconeogenesis. Diabetes 2014, 63, 2073–2085. [Google Scholar] [CrossRef]
- Baek, J.-H.; Kim, D.-H.; Lee, J.; Kim, S.-J.; Chun, K.-H. Galectin-1 accelerates high-fat diet-induced obesity by activation of peroxisome proliferator-activated receptor gamma (PPARγ) in mice. Cell Death Dis. 2021, 12, 66. [Google Scholar] [CrossRef]
- Mukherjee, R.; Yun, J.W. Pharmacological inhibition of galectin-1 by lactulose alleviates weight gain in diet-induced obese rats. Life Sci. 2016, 148, 112–117. [Google Scholar] [CrossRef]
- Mukherjee, R.; Kim, S.W.; Park, T.; Choi, M.S.; Yun, J.W. Targeted inhibition of galectin 1 by thiodigalactoside dramatically reduces body weight gain in diet-induced obese rats. Int. J. Obes. 2015, 39, 1349–1358. [Google Scholar] [CrossRef]
- Yu, H.; Yang, F.; Zhong, W.; Jiang, X.; Zhang, F.; Ji, X.; Xue, M.; Qiu, Y.; Yu, J.; Hu, X.; et al. Secretory Galectin-3 promotes hepatic steatosis via regulation of the PPARγ/CD36 signaling pathway. Cell. Signal. 2021, 84, 110043. [Google Scholar] [CrossRef] [PubMed]
- Nomoto, K.; Tsuneyama, K.; Aziz, H.A.; Takahashi, H.; Murai, Y.; Cui, Z.-G.; Fujimoto, M.; Kato, I.; Hiraga, K.; Hsu, D.; et al. Disrupted galectin-3 causes non-alcoholic fatty liver disease in male mice. J. Pathol. 2006, 210, 469–477. [Google Scholar] [CrossRef]
- Suppli, M.P.; Rigbolt, K.T.G.; Veidal, S.S.; Heebøll, S.; Eriksen, P.L.; Demant, M.; Bagger, J.I.; Nielsen, J.C.; Oró, D.; Thrane, S.W.; et al. Hepatic transcriptome signatures in patients with varying degrees of nonalcoholic fatty liver disease compared with healthy normal-weight individuals. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G462–G472. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Cho, J.-W.; Lee, S.-Y.; Yun, A.; Kim, H.; Bae, D.; Yang, S.; Kim, C.Y.; Lee, M.; Kim, E.; et al. TRRUST v2: An expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2018, 46, D380–D386. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Jensen, O.N. Proteomic analysis of post-translational modifications. Nat. Biotechnol. 2003, 21, 255–261. [Google Scholar] [CrossRef]
- Xu, Y.; Chou, K.-C. Recent Progress in Predicting Posttranslational Modification Sites in Proteins. Curr. Top. Med. Chem. 2016, 16, 591–603. [Google Scholar] [CrossRef]
- Shaik, A.A.; Qiu, B.; Wee, S.; Choi, H.; Gunaratne, J.; Tergaonkar, V. Phosphoprotein network analysis of white adipose tissues unveils deregulated pathways in response to high-fat diet. Sci. Rep. 2016, 6, 25844. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Sun, K.; Meng, Z.; Chen, L. The SLC transporter in nutrient and metabolic sensing, regulation, and drug development. J. Mol. Cell Biol. 2019, 11, 1–13. [Google Scholar] [CrossRef]
- Kim, M.K.; Yang, S.; Lee, K.-H.; Um, J.-H.; Liu, M.; Kang, H.; Park, S.J.; Chung, J.H. Promyelocytic leukemia inhibits adipogenesis, and loss of promyelocytic leukemia results in fat accumulation in mice. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1130–E1142. [Google Scholar] [CrossRef][Green Version]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef]
- Ivanschitz, L.; De Thé, H.; Le Bras, M. PML, SUMOylation, and Senescence. Front. Oncol. 2013, 3, 171. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.-H.; Bernardi, R.; Pandolfi, P.P.; Benveniste, E.N. The promyelocytic leukemia protein functions as a negative regulator of IFN-γ signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 18715–18720. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Guo, S.; Liu, Y.; Chu, H.; Hakimi, P.; Berger, N.A.; Hanson, R.W.; Kao, H.-Y. Ablation of Promyelocytic Leukemia Protein (PML) Re-patterns Energy Balance and Protects Mice from Obesity Induced by a Western Diet. J. Biol. Chem. 2013, 288, 29746–29759. [Google Scholar] [CrossRef] [PubMed]
- Molenaar, M.R.; Jeucken, A.; Wassenaar, T.A.; Van De Lest, C.H.A.; Brouwers, J.F.; Helms, J.B. LION/web: A web-based ontology enrichment tool for lipidomic data analysis. GigaScience 2019, 8, giz061. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, M.Z.; Arcuri, J.; Park, K.K.; Zafar, M.K.; Fatmi, R.; Hackam, A.S.; Yin, Y.; Benowitz, L.; Goldberg, J.L.; Samarah, M.; et al. Multi-Omic Analyses of Growth Cones at Different Developmental Stages Provides Insight into Pathways in Adult Neuroregeneration. iScience 2020, 23, 100836. [Google Scholar] [CrossRef]
- Zhou, J.; Febbraio, M.; Wada, T.; Zhai, Y.; Kuruba, R.; He, J.; Lee, J.H.; Khadem, S.; Ren, S.; Li, S.; et al. Hepatic Fatty Acid Transporter Cd36 Is a Common Target of LXR, PXR, and PPARγ in Promoting Steatosis. Gastroenterology 2008, 134, 556–567.e1. [Google Scholar] [CrossRef]
- Potapova, I.A.; El-Maghrabi, M.R.; Doronin, S.V.; Benjamin, W.B. Phosphorylation of Recombinant Human ATP:Citrate Lyase by cAMP-Dependent Protein Kinase Abolishes Homotropic Allosteric Regulation of the Enzyme by Citrate and Increases the Enzyme Activity. Allosteric Activation of ATP:Citrate Lyase by Phosphorylated Sugars. Biochemistry 2000, 39, 1169–1179. [Google Scholar] [CrossRef]
- Lally, J.S.; Ghoshal, S.; DePeralta, D.K.; Moaven, O.; Wei, L.; Masia, R.; Erstad, D.J.; Fujiwara, N.; Leong, V.; Houde, V.P.; et al. Inhibition of Acetyl-CoA Carboxylase by Phosphorylation or the Inhibitor ND-654 Suppresses Lipogenesis and Hepatocellular Carcinoma. Cell Metab. 2019, 29, 174–182.e5. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Ishii, S.; Iizuka, K.; Miller, B.C.; Uyeda, K. Carbohydrate response element binding protein directly promotes lipogenic enzyme gene transcription. Proc. Natl. Acad. Sci. USA 2004, 101, 15597–15602. [Google Scholar] [CrossRef]
- Griffett, K.; Solt, L.A.; Elgendy, B.l.-D.; Kamenecka, T.M.; Burris, T.P. A Liver-Selective LXR Inverse Agonist That Suppresses Hepatic Steatosis. ACS Chem. Biol. 2012, 8, 559–567. [Google Scholar] [CrossRef]
- Gavrilova, O.; Haluzik, M.; Matsusue, K.; Cutson, J.J.; Johnson, L.; Dietz, K.R.; Nicol, C.J.; Vinson, C.; Gonzalez, F.J.; Reitman, M.L. Liver Peroxisome Proliferator-activated Receptor γ Contributes to Hepatic Steatosis, Triglyceride Clearance, and Regulation of Body Fat Mass. J. Biol. Chem. 2003, 278, 34268–34276. [Google Scholar] [CrossRef]
- Rusu, V.; Hoch, E.; Mercader, J.M.; Tenen, D.E.; Gymrek, M.; Hartigan, C.R.; DeRan, M.; Von Grotthuss, M.; Fontanillas, P.; Spooner, A.; et al. Type 2 Diabetes Variants Disrupt Function of SLC16A11 through Two Distinct Mechanisms. Cell 2017, 170, 199–212.e20. [Google Scholar] [CrossRef] [PubMed]
- Guillam, M.T.; Hummler, E.; Schaerer, E.; Yeh, J.I.; Birnbaum, M.J.; Beermann, F.; Schmidt, A.; Deriaz, N.; Thorens, B. Early diabetes and abnormal postnatal pancreatic islet development in mice lacking Glut-2. Nat. Genet. 1997, 17, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Kuwata, H.; Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat. 2019, 144, 106363. [Google Scholar] [CrossRef]
- Kan, C.F.K.; Singh, A.B.; Stafforini, D.M.; Azhar, S.; Liu, J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J. Lipid Res. 2014, 55, 1657–1667. [Google Scholar] [CrossRef] [PubMed]
- Yoshimura, Y.; Araki, A.; Maruta, H.; Takahashi, Y.; Yamashita, H. Molecular cloning of ratacss3and characterization of mammalian propionyl-CoA synthetase in the liver mitochondrial matrix. J. Biochem. 2017, 161, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Carrer, A.; Parris, J.L.D.; Trefely, S.; Henry, R.A.; Montgomery, D.C.; Torres, A.; Viola, J.M.; Kuo, Y.-M.; Blair, I.A.; Meier, J.L. Impact of a high-fat diet on tissue Acyl-CoA and histone acetylation levels. J. Biol. Chem. 2017, 292, 3312–3322. [Google Scholar] [CrossRef]
- Huang, Z.; Zhang, M.; Plec, A.A.; Estill, S.J.; Cai, L.; Repa, J.J.; McKnight, S.L.; Tu, B.P. ACSS2 promotes systemic fat storage and utilization through selective regulation of genes involved in lipid metabolism. Proc. Natl. Acad. Sci. USA 2018, 115, E9499–E9506. [Google Scholar] [CrossRef]
- Wanders, R.J.A.; Waterham, H.R.; Ferdinandusse, S. Metabolic Interplay between Peroxisomes and Other Subcellular Organelles Including Mitochondria and the Endoplasmic Reticulum. Front. Cell Dev. Biol. 2015, 3, 83. [Google Scholar] [CrossRef]
- Kurland, I.J.; El-Maghrabi, M.R.; Correia, J.J.; Pilkis, S.J. Rat liver 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase. Properties of phospho- and dephospho- forms and of two mutants in which Ser32 has been changed by site-directed mu-tagenesis. J. Biol. Chem. 1992, 267, 4416–4423. [Google Scholar] [CrossRef]
- Yugi, K.; Kubota, H.; Toyoshima, Y.; Noguchi, R.; Kawata, K.; Komori, Y.; Uda, S.; Kunida, K.; Tomizawa, Y.; Funato, Y.; et al. Reconstruction of Insulin Signal Flow from Phosphoproteome and Metabolome Data. Cell Rep. 2014, 8, 1171–1183. [Google Scholar] [CrossRef] [PubMed]
- Yang, A.-Q.; Li, D.; Chi, L.; Ye, X.-S. Validation, Identification, and Biological Consequences of the Site-specific. Mol. Cell. Proteom. 2017, 16, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef]
- Chowdhry, S.; Nazmy, M.H.; Meakin, P.J.; Dinkova-Kostova, A.T.; Walsh, S.V.; Tsujita, T.; Dillon, J.F.; Ashford, M.L. Loss of Nrf2 markedly exacerbates nonalcoholic steatohepatitis. Free Radic. Biol. Med. 2010, 48, 357–371. [Google Scholar] [CrossRef]
- Carracedo, A.; Rousseau, D.; Douris, N.; Fernández-Ruiz, S.; Martin-Martin, N.; Weiss, O.; Webster, K.; Adams, A.C.; Vázquez-Chantada, M.; Martínez-Chantar, M.L.; et al. The Promyelocytic Leukemia Protein Is Upregulated in Conditions of Obesity and Liver Steatosis. Int. J. Biol. Sci. 2015, 11, 629–632. [Google Scholar] [CrossRef]
- Ito, K.; Bernardi, R.; Morotti, A.; Matsuoka, S.; Saglio, G.; Ikeda, Y.; Rosenblatt, J.; Avigan, D.E.; Teruya-Feldstein, J.; Pandolfi, P.P. PML targeting eradicates quiescent leukaemia-initiating cells. Nature 2008, 453, 1072–1078. [Google Scholar] [CrossRef]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef]
- Cheng, X.; Kao, H.-Y. Post-translational modifications of PML: Consequences and implications. Front. Oncol. 2012, 2, 210. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Grishina, I. Regulation of the tumor suppressor PML by sequential post-translational modifications. Front. Oncol. 2012, 2, 204. [Google Scholar] [CrossRef][Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.Q.; Mohallem, R.; Franco, J.; Buhman, K.K.; Kim, K.-H.; Aryal, U.K. Multi-Omics Approach Reveals Dysregulation of Protein Phosphorylation Correlated with Lipid Metabolism in Mouse Non-Alcoholic Fatty Liver. Cells 2022, 11, 1172. https://doi.org/10.3390/cells11071172
Kim SQ, Mohallem R, Franco J, Buhman KK, Kim K-H, Aryal UK. Multi-Omics Approach Reveals Dysregulation of Protein Phosphorylation Correlated with Lipid Metabolism in Mouse Non-Alcoholic Fatty Liver. Cells. 2022; 11(7):1172. https://doi.org/10.3390/cells11071172
Chicago/Turabian StyleKim, Sora Q., Rodrigo Mohallem, Jackeline Franco, Kimberly K. Buhman, Kee-Hong Kim, and Uma K. Aryal. 2022. "Multi-Omics Approach Reveals Dysregulation of Protein Phosphorylation Correlated with Lipid Metabolism in Mouse Non-Alcoholic Fatty Liver" Cells 11, no. 7: 1172. https://doi.org/10.3390/cells11071172
APA StyleKim, S. Q., Mohallem, R., Franco, J., Buhman, K. K., Kim, K.-H., & Aryal, U. K. (2022). Multi-Omics Approach Reveals Dysregulation of Protein Phosphorylation Correlated with Lipid Metabolism in Mouse Non-Alcoholic Fatty Liver. Cells, 11(7), 1172. https://doi.org/10.3390/cells11071172