
Intestinal Epithelial AMPK Deficiency Causes Delayed Colonic Epithelial Repair in DSS-Induced Colitis

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents and Antibodies
2.2. Mice
2.3. DSS-Induced Acute Colitis
2.4. Histopathological Analysis
2.5. In Vivo Intestinal Permeability Assay
2.6. Quantification of Lipocalin-2
2.7. Cell Culture and Measure of TEER
2.8. Cell Adhesion Assay and xCELLigence Real-Time Cell Analysis
2.9. Scratch Wound Assay
2.10. Western Blotting
2.11. Quantitative Real-Time PCR Analysis
2.12. Statistical Analysis
3. Results
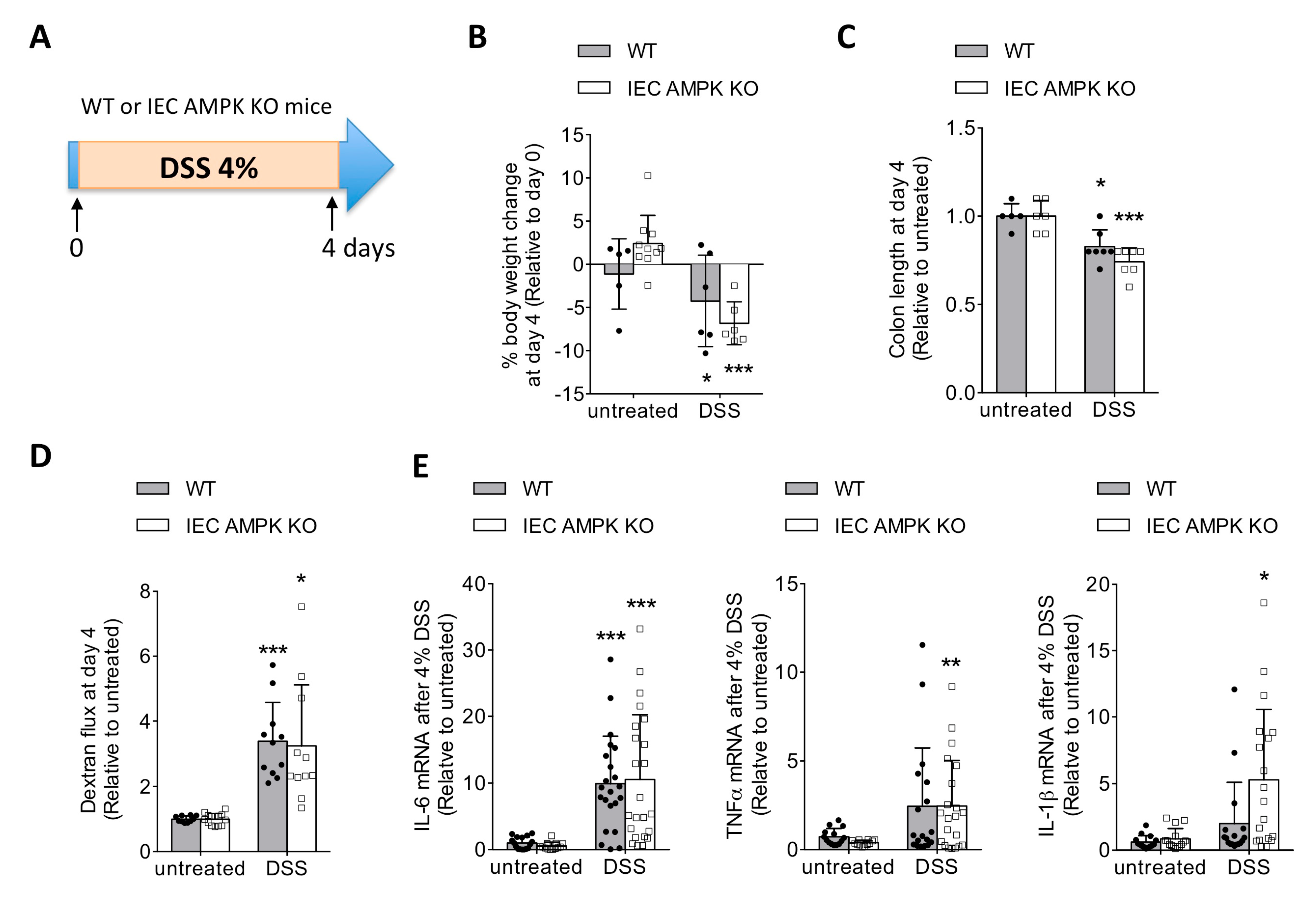
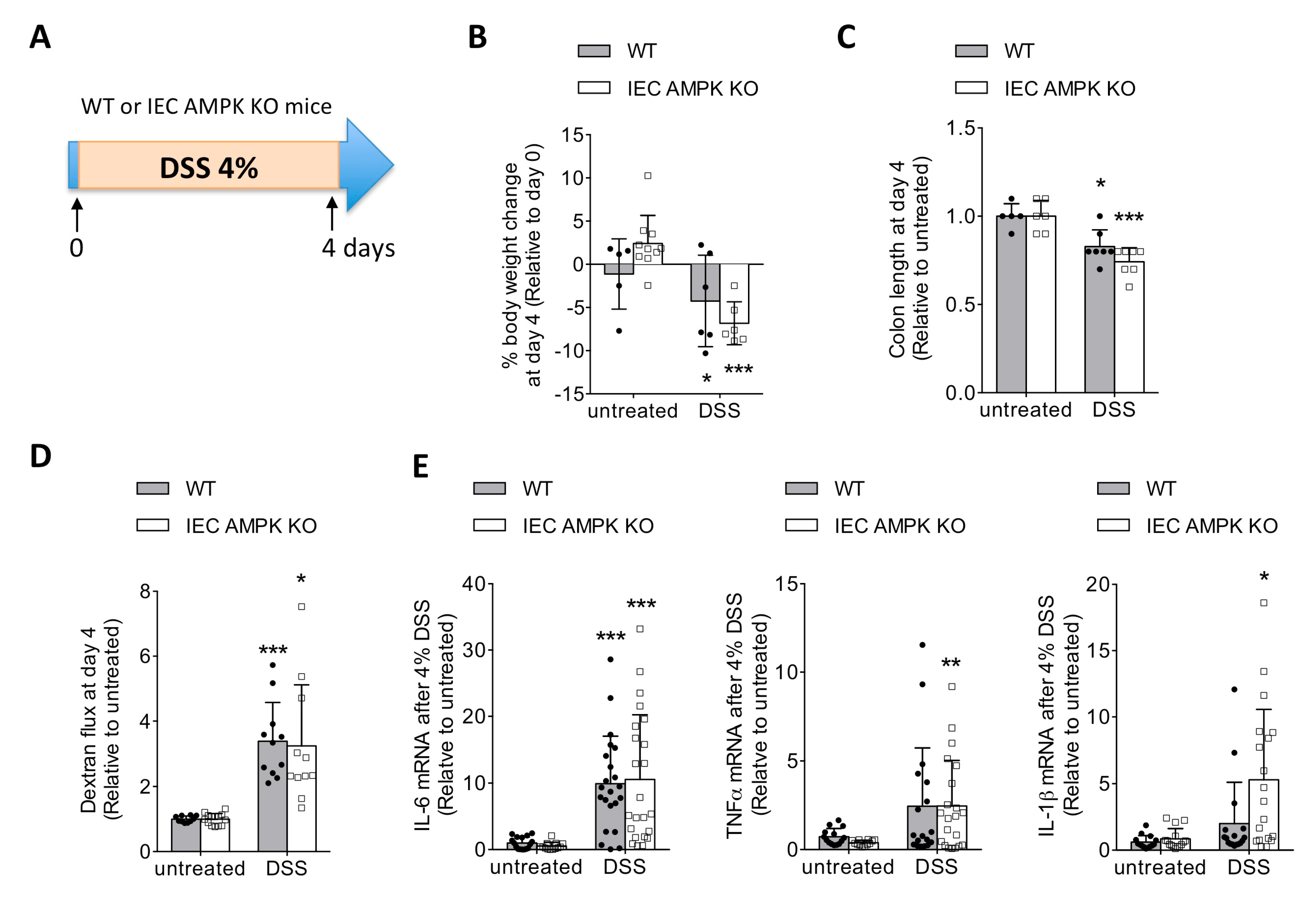
3.1. IEC-Specific AMPK α1/α2 Deficiency Influenced Intestinal Inflammation upon DSS-Induced Epithelial Injury
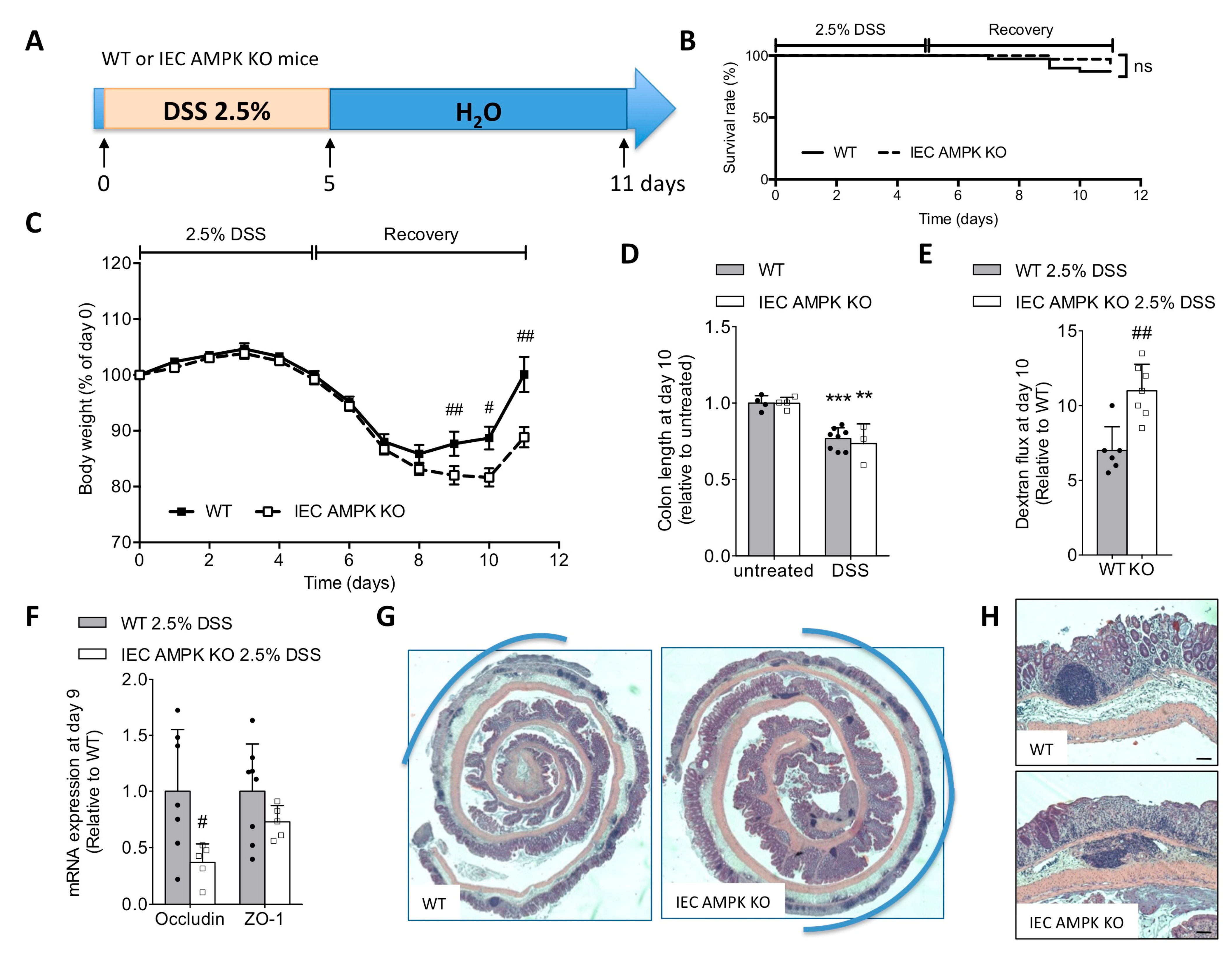
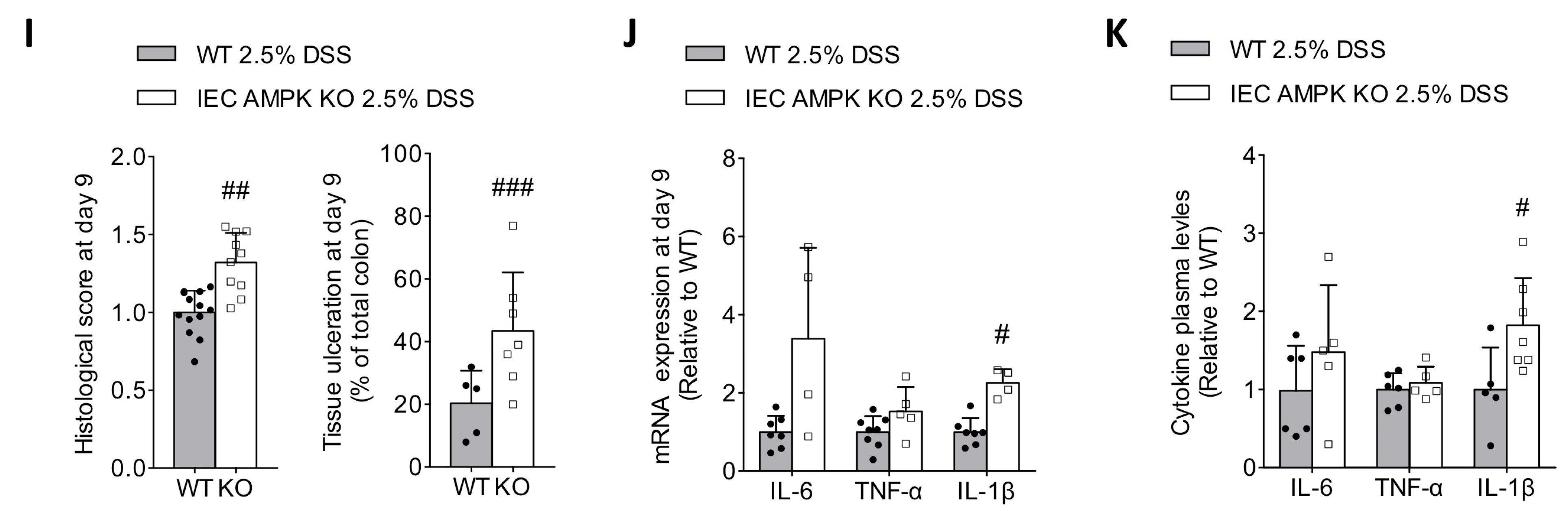
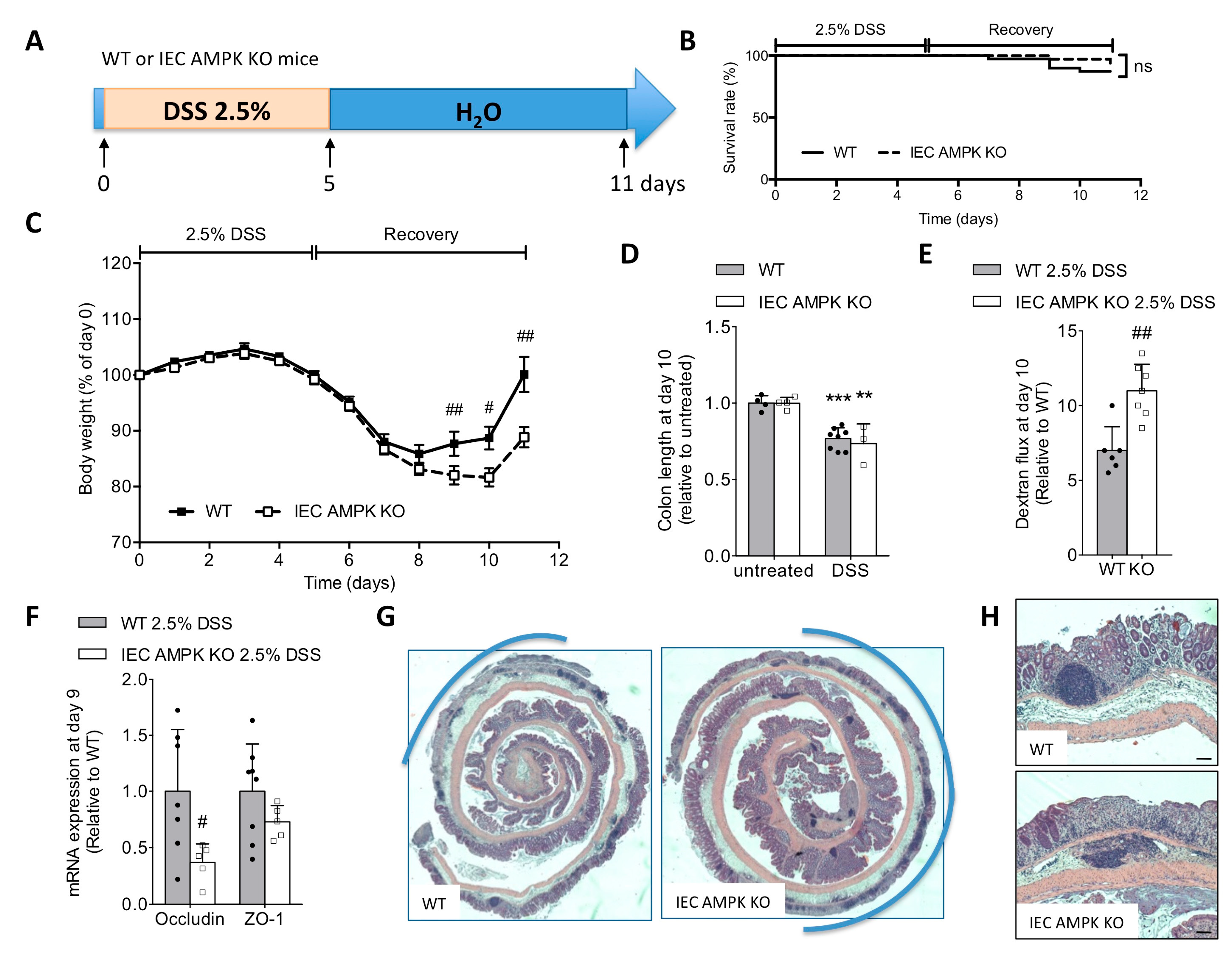
3.2. Loss of IEC AMPK α1/α2 Caused Impaired Recovery from DSS-Induced Epithelial Injury
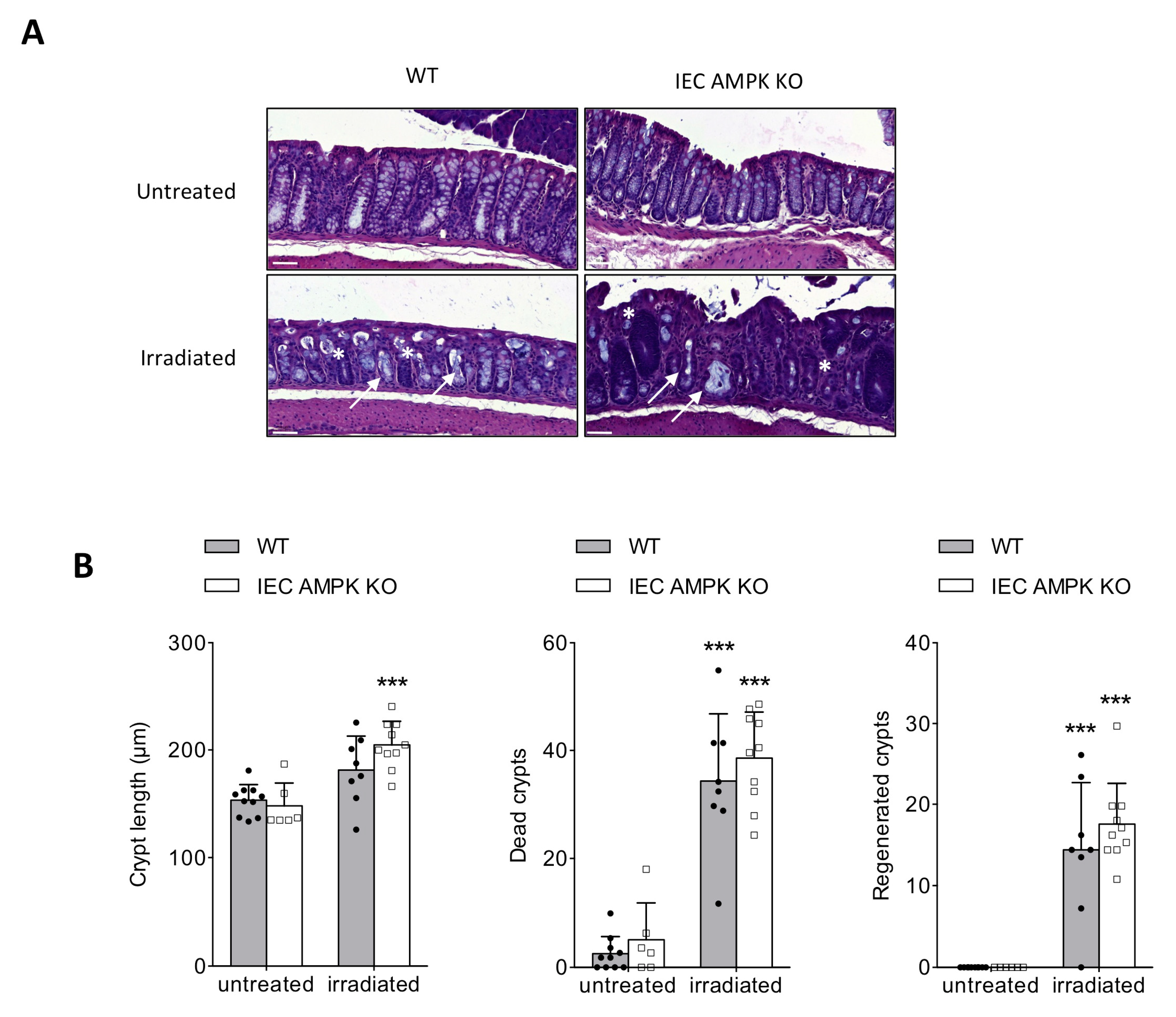
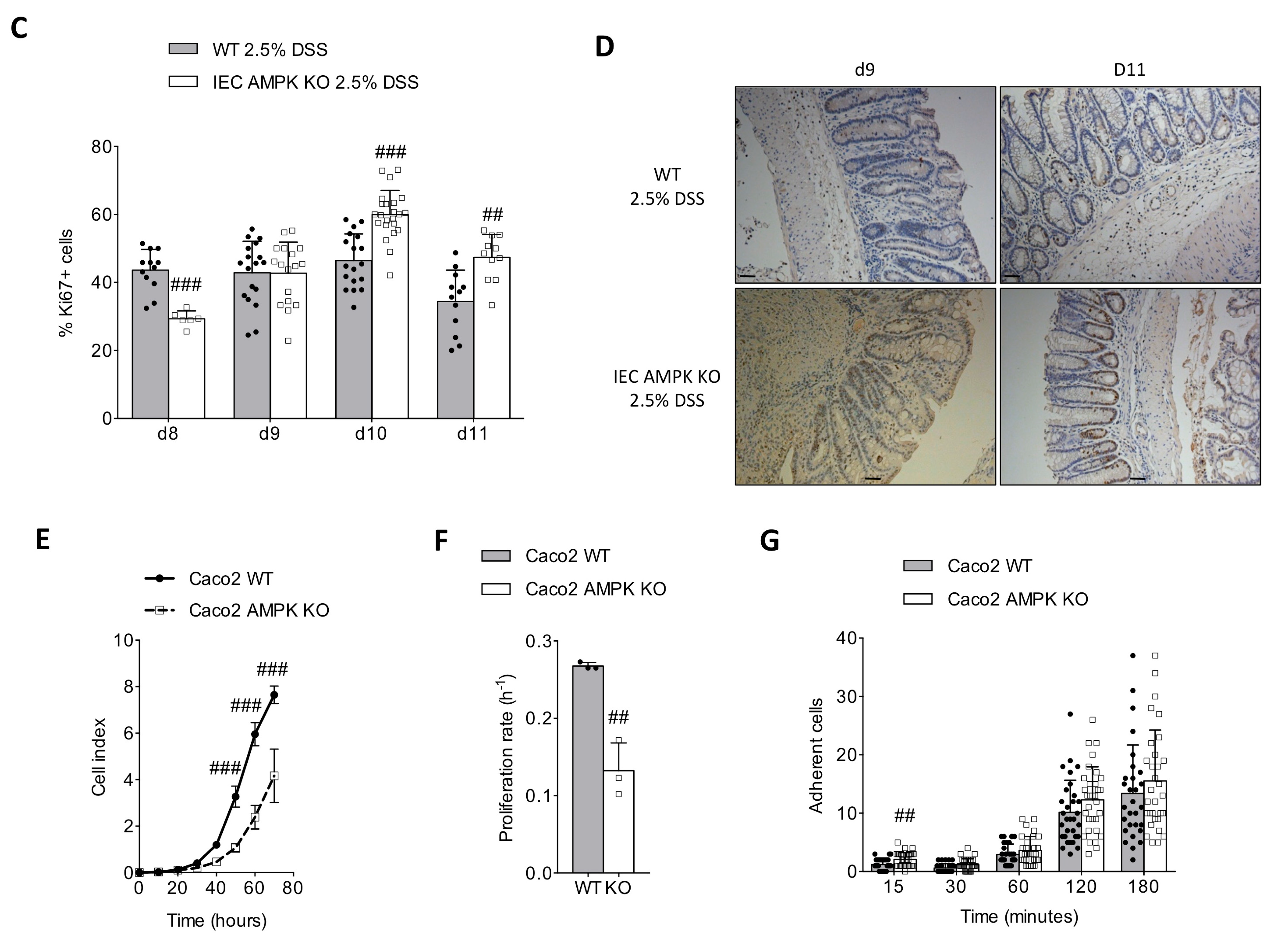
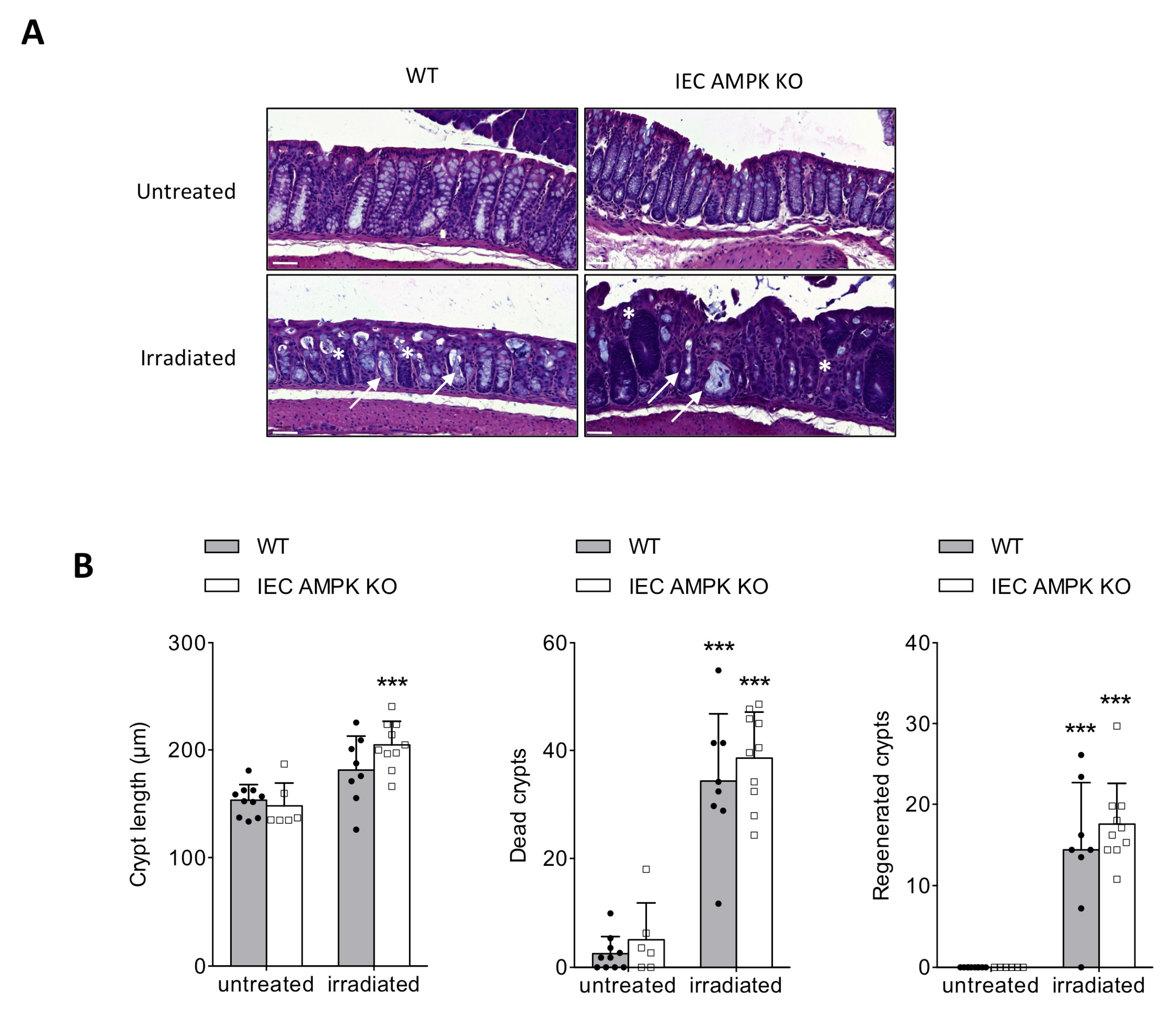
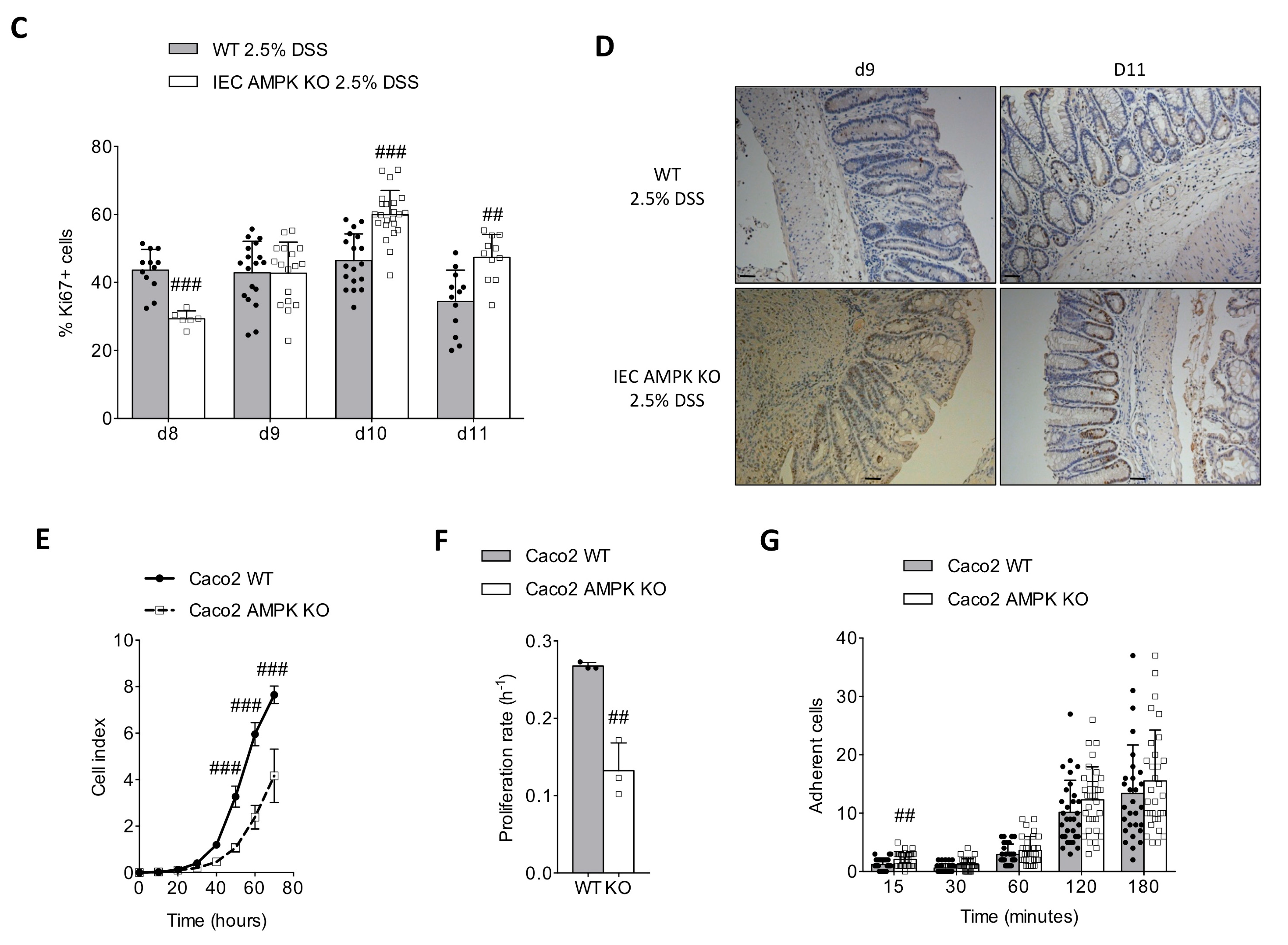
3.3. AMPK Signaling Supports Epithelial Proliferation Following DSS-Mediated Injury
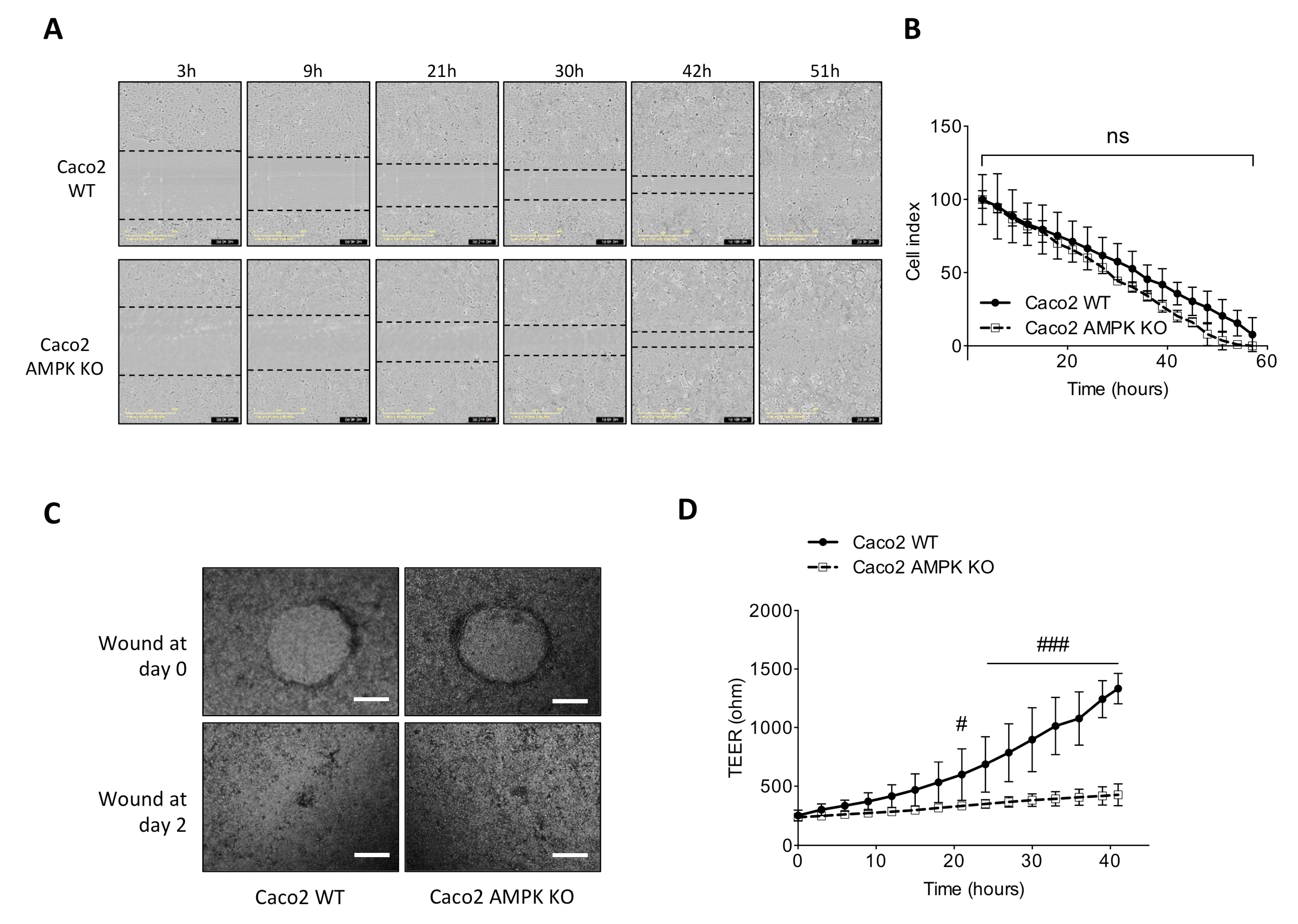
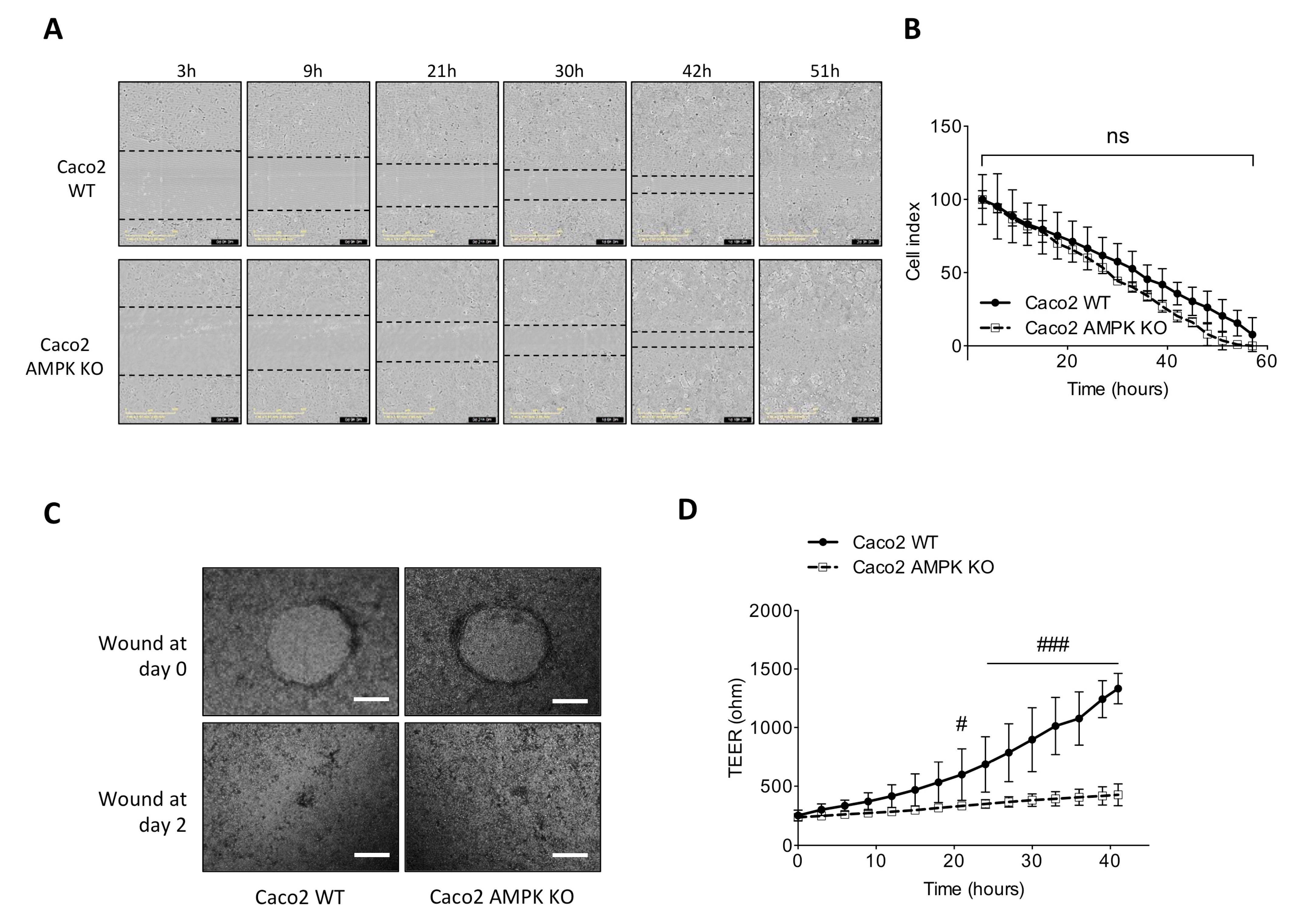
3.4. Absence of AMPK Is Not Detrimental for Cell Spreading Response in Caco2 Cells but Necessary for Mature Barrier Establishment
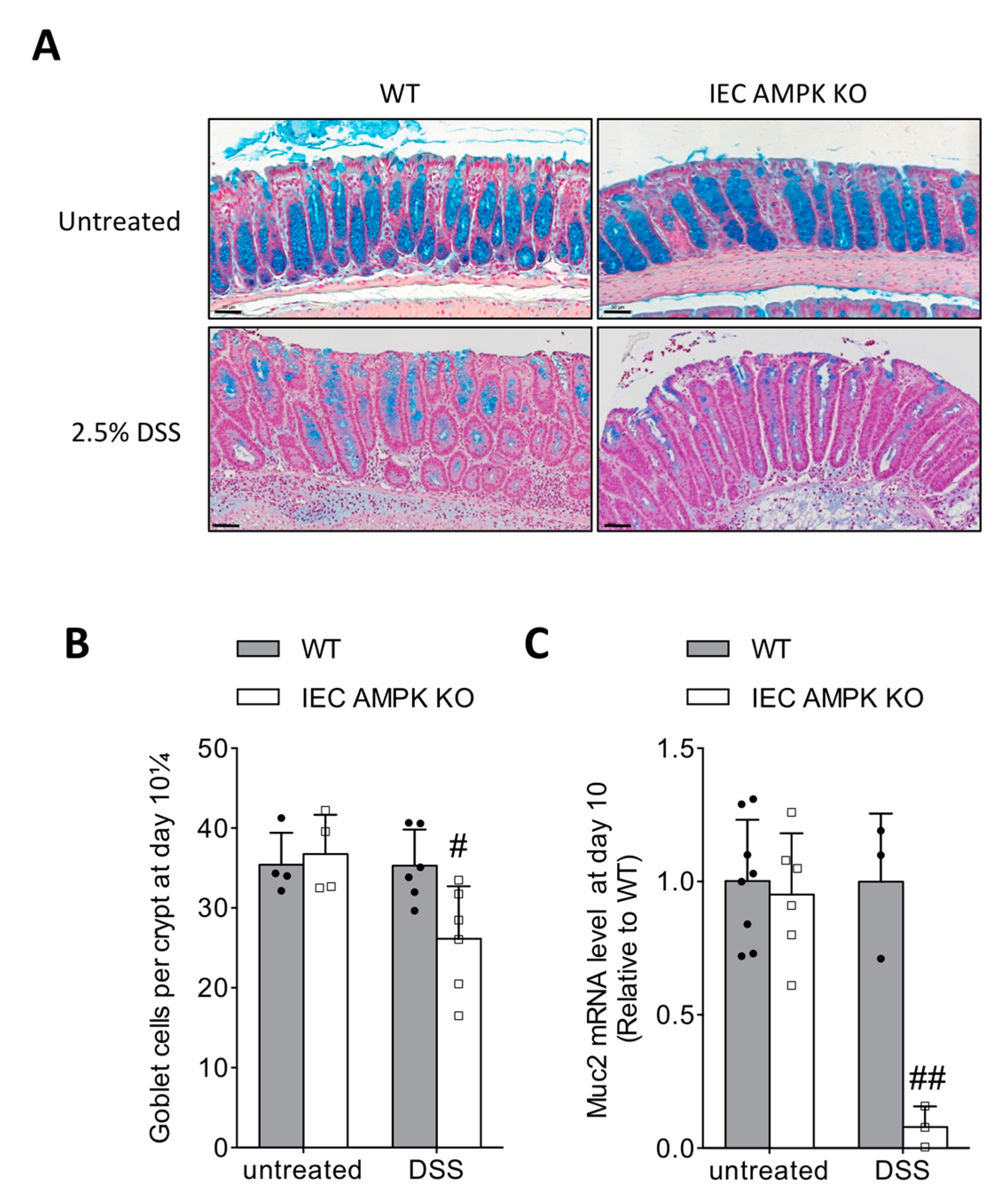
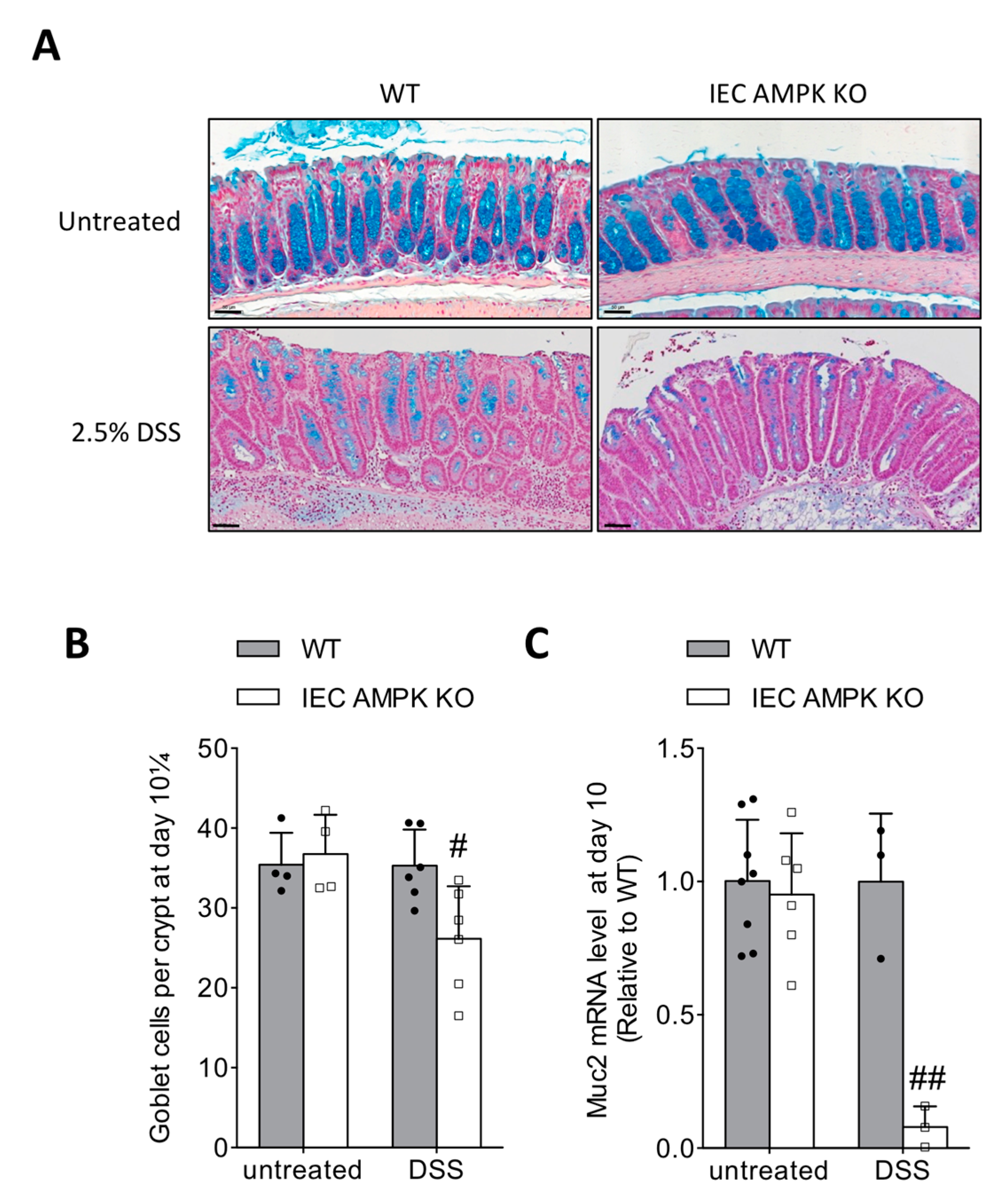
3.5. Impaired Restitution of Mucus-Producing Goblet Cells in the Colon of IEC AMPK KO Mice Following DSS-Induced Colitis
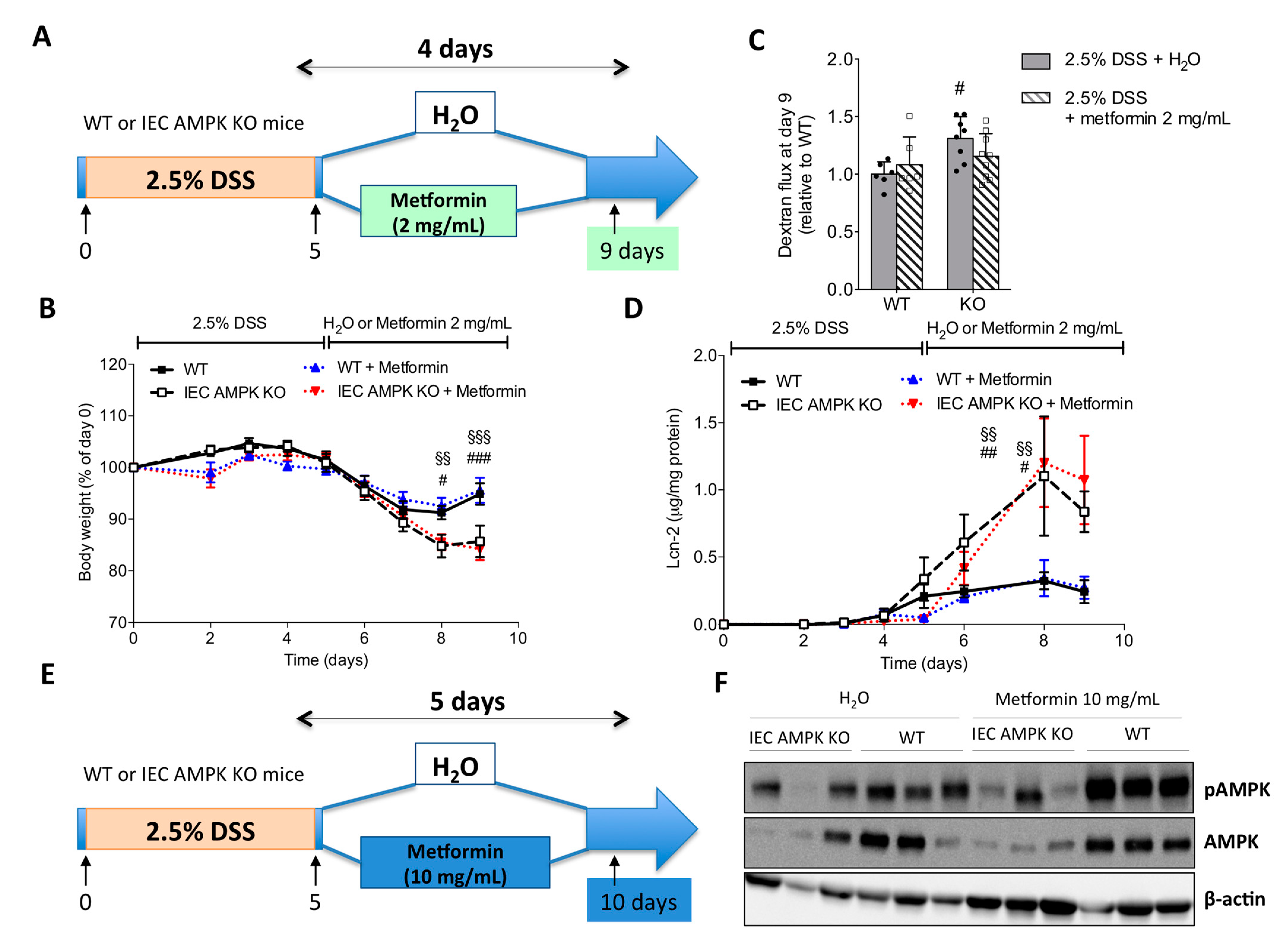
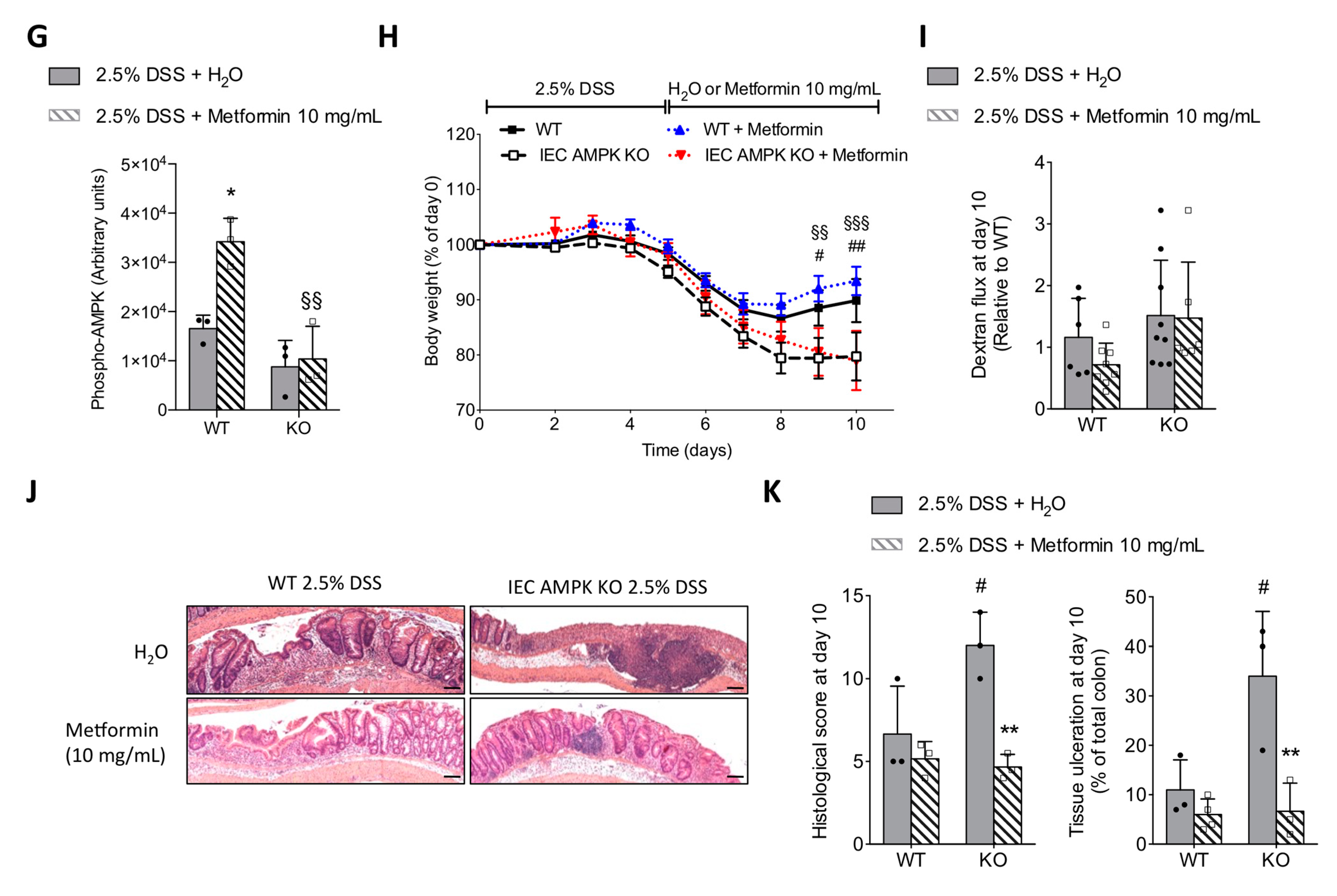
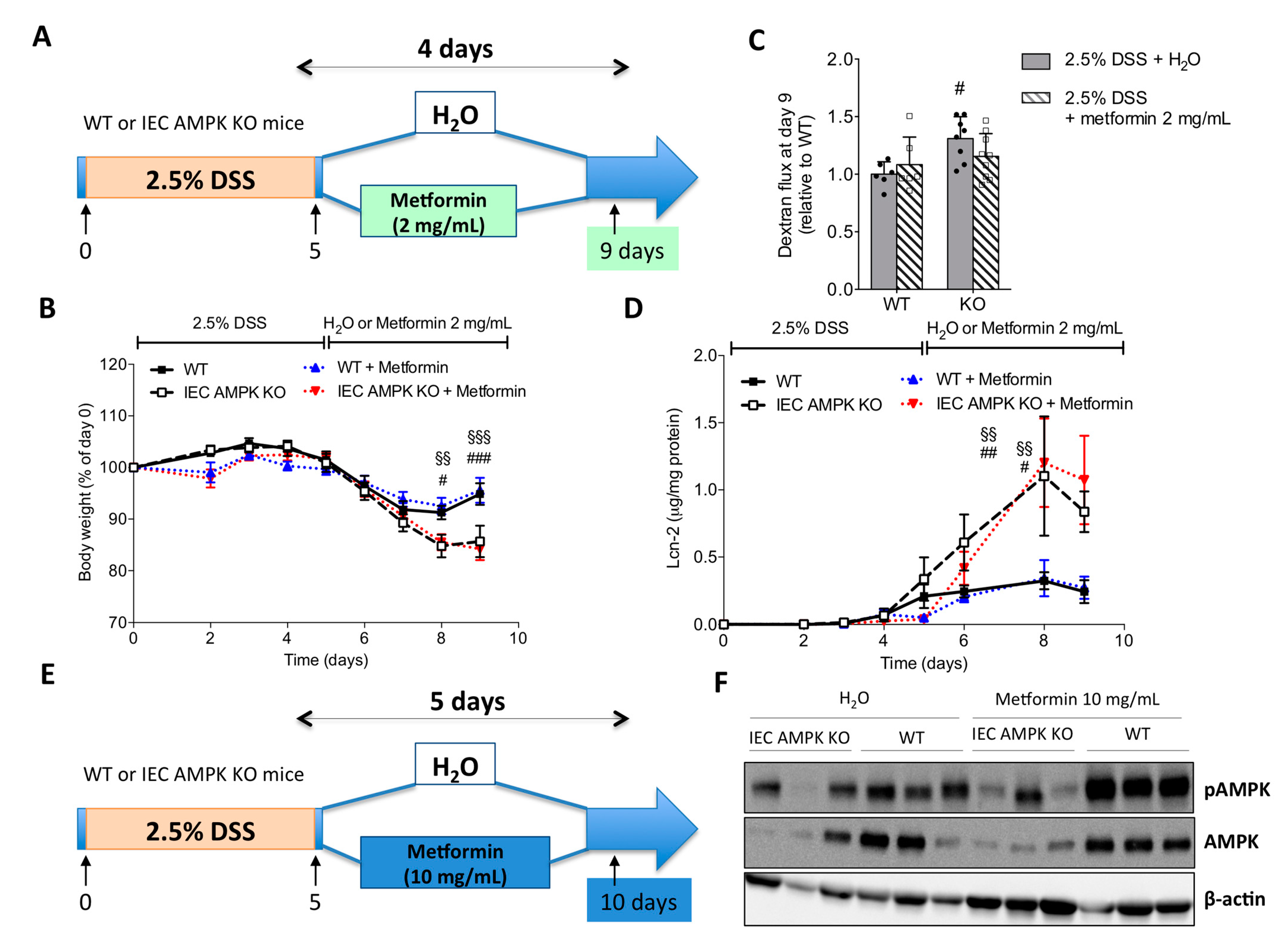
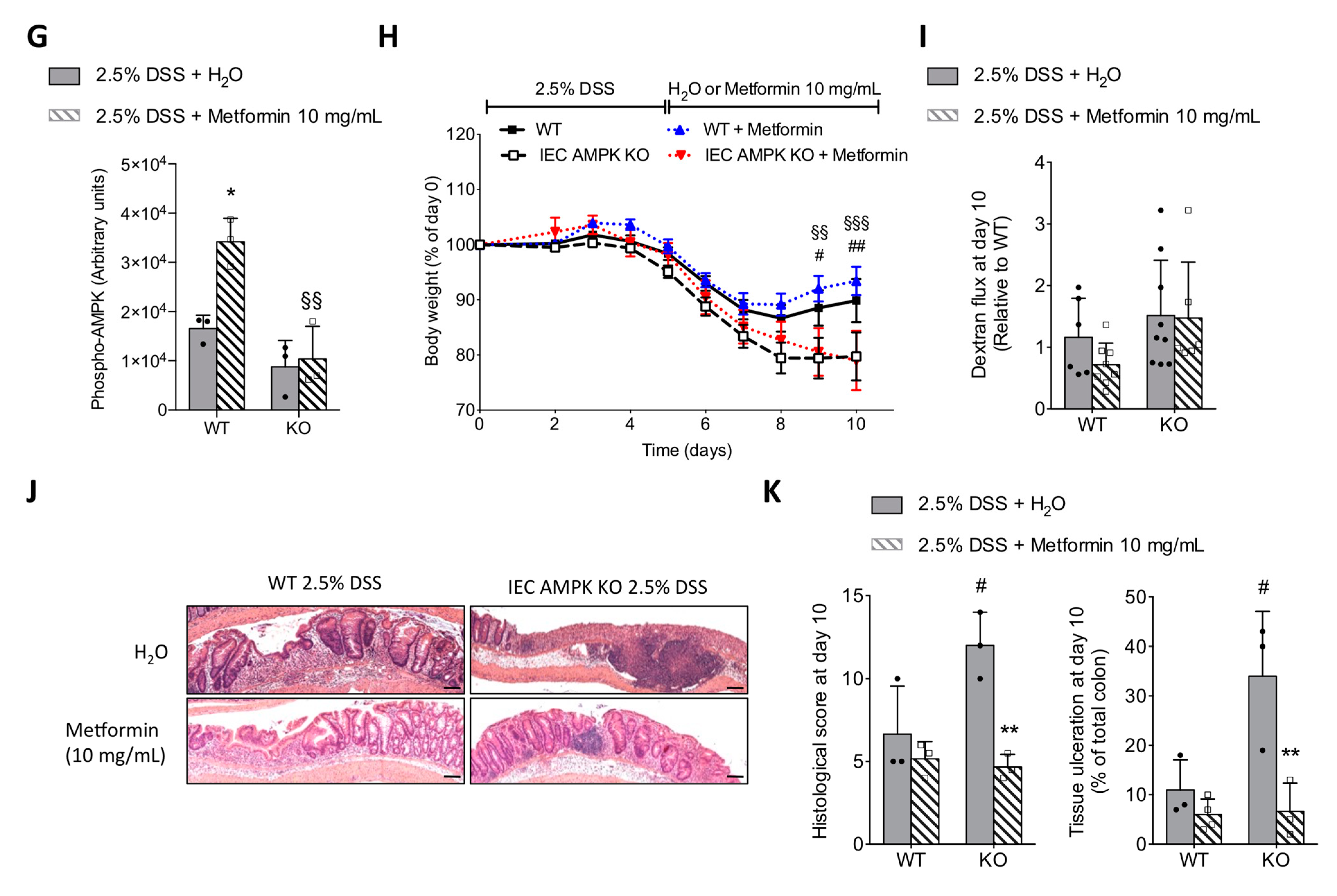
3.6. Metformin Ameliorated DSS-Induced Colitis in an IEC AMPK-Independent Manner
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; Angelis, M.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.; Di Ciaula, A. Intestinal Barrier and Permeability in Health, Obesity and NAFLD. Biomedicines 2021, 10, 83. [Google Scholar] [CrossRef]
- Hardie, D.G.; Lin, S.C. AMP-activated protein kinase—Not just an energy sensor. F1000Res 2017, 6, 1724. [Google Scholar] [CrossRef]
- Zhang, L.; Li, J.; Young, L.H.; Caplan, M.J. AMP-activated protein kinase regulates the assembly of epithelial tight junctions. Proc. Natl. Acad. Sci. USA 2006, 103, 17272–17277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Cantley, L.C. Regulation of epithelial tight junction assembly and disassembly by AMP-activated protein kinase. Proc. Natl. Acad. Sci. USA 2007, 104, 819–822. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, P. The stress polarity pathway: AMPK ‘GIV’-es protection against metabolic insults. Aging 2017, 9, 303–314. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Wang, C.; Yan, J.; Li, X.; Wen, J.; Hu, C. Curcumin ameliorates oxidative stress-induced intestinal barrier injury and mitochondrial damage by promoting Parkin dependent mitophagy through AMPK-TFEB signal pathway. Free Radic. Biol. Med. 2020, 147, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wu, Z.; Ji, Y.; Sun, K.; Dai, Z.; Wu, G. L-Glutamine Enhances Tight Junction Integrity by Activating CaMK Kinase 2-AMP-Activated Protein Kinase Signaling in Intestinal Porcine Epithelial Cells. J. Nutr. 2016, 146, 501–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, L.; Li, Z.R.; Green, R.S.; Holzman, I.R.; Lin, J. Butyrate enhances the intestinal barrier by facilitating tight junction assembly via activation of AMP-activated protein kinase in Caco-2 cell monolayers. J. Nutr. 2009, 139, 1619–1625. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.J.; Sun, X.; Du, M. AMPK in regulation of apical junctions and barrier function of intestinal epithelium. Tissue Barriers 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Sun, X.; Yang, Q.; Rogers, C.J.; Du, M.; Zhu, M.J. AMPK improves gut epithelial differentiation and barrier function via regulating Cdx2 expression. Cell Death Differ. 2017, 24, 819–831. [Google Scholar] [CrossRef]
- Olivier, S.; Leclerc, J.; Grenier, A.; Foretz, M.; Tamburini, J.; Viollet, B. AMPK Activation Promotes Tight Junction Assembly in Intestinal Epithelial Caco-2 Cells. Int. J. Mol. Sci. 2019, 20, 5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the glucoregulatory mechanisms of metformin in type 2 diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Wang, J.; You, Q.; He, S.; Meng, Q.; Gao, J.; Wu, X.; Shen, Y.; Sun, Y.; Wu, X.; et al. Activating AMPK to Restore Tight Junction Assembly in Intestinal Epithelium and to Attenuate Experimental Colitis by Metformin. Front. Pharm. 2018, 9, 761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, J.; Zeng, L.; Lai, X.; Li, J.; Liu, L.; Lin, Q.; Chen, Y. Metformin protects against intestinal barrier dysfunction via AMPKα1-dependent inhibition of JNK signalling activation. J. Cell Mol. Med. 2018, 22, 546–557. [Google Scholar] [CrossRef] [Green Version]
- Di Fusco, D.; Dinallo, V.; Monteleone, I.; Laudisi, F.; Marafini, I.; Franzè, E.; Di Grazia, A.; Dwairi, R.; Colantoni, A.; Ortenzi, A.; et al. Metformin inhibits inflammatory signals in the gut by controlling AMPK and p38 MAP kinase activation. Clin. Sci. 2018, 132, 1155–1168. [Google Scholar] [CrossRef] [PubMed]
- Koh, S.J.; Kim, J.M.; Kim, I.K.; Ko, S.H.; Kim, J.S. Anti-inflammatory mechanism of metformin and its effects in intestinal inflammation and colitis-associated colon cancer. J. Gastroenterol. Hepatol. 2014, 29, 502–510. [Google Scholar] [CrossRef]
- Olivier, S.; Pochard, C.; Diounou, H.; Castillo, V.; Divoux, J.; Alcantara, J.; Leclerc, J.; Guilmeau, S.; Huet, C.; Charifi, W.; et al. Deletion of intestinal epithelial AMP-activated protein kinase alters distal colon permeability but not glucose homeostasis. Mol. Metab. 2021, 47, 101183. [Google Scholar] [CrossRef]
- Chassaing, B.; Aitken, J.D.; Malleshappa, M.; Vijay-Kumar, M. Dextran sulfate sodium (DSS)-induced colitis in mice. Curr. Protoc. Immunol. 2014, 104, 15–25. [Google Scholar] [CrossRef]
- Grenier, A.; Sujobert, P.; Olivier, S.; Guermouche, H.; Mondésir, J.; Kosmider, O.; Viollet, B.; Tamburini, J. Knockdown of Human AMPK Using the CRISPR/Cas9 Genome-Editing System. Methods Mol. Biol. 2018, 1732, 171–194. [Google Scholar] [CrossRef] [Green Version]
- Basak, O.; van de Born, M.; Korving, J.; Beumer, J.; van der Elst, S.; van Es, J.H.; Clevers, H. Mapping early fate determination in Lgr5+ crypt stem cells using a novel Ki67-RFP allele. Embo J. 2014, 33, 2057–2068. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Shi, H.Y.; Hamidi, N.; Underwood, F.E.; Tang, W.; Benchimol, E.I.; Panaccione, R.; Ghosh, S.; Wu, J.C.Y.; Chan, F.K.L.; et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: A systematic review of population-based studies. Lancet 2017, 390, 2769–2778. [Google Scholar] [CrossRef]
- Lee, J.Y.; Wasinger, V.C.; Yau, Y.Y.; Chuang, E.; Yajnik, V.; Leong, R.W. Molecular Pathophysiology of Epithelial Barrier Dysfunction in Inflammatory Bowel Diseases. Proteomes 2018, 6, 17. [Google Scholar] [CrossRef] [Green Version]
- Mankertz, J.; Schulzke, J.D. Altered permeability in inflammatory bowel disease: Pathophysiology and clinical implications. Curr. Opin. Gastroenterol. 2007, 23, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Pochard, C.; Gonzales, J.; Bessard, A.; Mahe, M.M.; Bourreille, A.; Cenac, N.; Jarry, A.; Coron, E.; Podevin, J.; Meurette, G.; et al. PGI(2) Inhibits Intestinal Epithelial Permeability and Apoptosis to Alleviate Colitis. Cell Mol. Gastroenterol. Hepatol. 2021, 12, 1037–1060. [Google Scholar] [CrossRef]
- Sun, X.; Zhu, M.J. AMP-activated protein kinase: A therapeutic target in intestinal diseases. Open Biol. 2017, 7, 170104. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Pellegrini, C.; Fornai, M.; Benvenuti, L.; D'Antongiovanni, V.; Colucci, R.; Bertani, L.; Di Salvo, C.; Semeghini, G.; La Motta, C.; et al. Preclinical Development of FA5, a Novel AMP-Activated Protein Kinase (AMPK) Activator as an Innovative Drug for the Management of Bowel Inflammation. Int. J. Mol. Sci. 2021, 22, 6325. [Google Scholar] [CrossRef] [PubMed]
- Aznar, N.; Patel, A.; Rohena, C.C.; Dunkel, Y.; Joosen, L.P.; Taupin, V.; Kufareva, I.; Farquhar, M.G.; Ghosh, P. AMP-activated protein kinase fortifies epithelial tight junctions during energetic stress via its effector GIV/Girdin. Elife 2016, 5, e20795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, T.; Matsui, T.; Tamura, A.; Uji, M.; Tsukita, S. The association of microtubules with tight junctions is promoted by cingulin phosphorylation by AMPK. J. Cell Biol. 2013, 203, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Fu, X.; Du, M.; Zhu, M.J. Ex vivo gut culture for studying differentiation and migration of small intestinal epithelial cells. Open Biol. 2018, 8, 170256. [Google Scholar] [CrossRef] [Green Version]
- Sturm, A.; Dignass, A.U. Epithelial restitution and wound healing in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Leoni, G.; Neumann, P.A.; Sumagin, R.; Denning, T.L.; Nusrat, A. Wound repair: Role of immune-epithelial interactions. Mucosal. Immunol. 2015, 8, 959–968. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Tsukamoto, O.; Nakano, A.; Kato, H.; Kioka, H.; Ito, N.; Higo, S.; Yamazaki, S.; Shintani, Y.; Matsuoka, K.; et al. Augmented AMPK activity inhibits cell migration by phosphorylating the novel substrate Pdlim5. Nat. Commun. 2015, 6, 6137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakano, A.; Kato, H.; Watanabe, T.; Min, K.D.; Yamazaki, S.; Asano, Y.; Seguchi, O.; Higo, S.; Shintani, Y.; Asanuma, H.; et al. AMPK controls the speed of microtubule polymerization and directional cell migration through CLIP-170 phosphorylation. Nat. Cell Biol. 2010, 12, 583–590. [Google Scholar] [CrossRef]
- Mizoguchi, E.; Xavier, R.J.; Reinecker, H.C.; Uchino, H.; Bhan, A.K.; Podolsky, D.K.; Mizoguchi, A. Colonic epithelial functional phenotype varies with type and phase of experimental colitis. Gastroenterology 2003, 125, 148–161. [Google Scholar] [CrossRef]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 2008, 118, 534–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zindl, C.L.; Lai, J.F.; Lee, Y.K.; Maynard, C.L.; Harbour, S.N.; Ouyang, W.; Chaplin, D.D.; Weaver, C.T. IL-22-producing neutrophils contribute to antimicrobial defense and restitution of colonic epithelial integrity during colitis. Proc. Natl. Acad. Sci. USA 2013, 110, 12768–12773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McElrath, C.; Espinosa, V.; Lin, J.D.; Peng, J.; Sridhar, R.; Dutta, O.; Tseng, H.C.; Smirnov, S.V.; Risman, H.; Sandoval, M.J.; et al. Critical role of interferons in gastrointestinal injury repair. Nat. Commun. 2021, 12, 2624. [Google Scholar] [CrossRef]
- Pu, Z.; Che, Y.; Zhang, W.; Sun, H.; Meng, T.; Xie, H.; Cao, L.; Hao, H. Dual roles of IL-18 in colitis through regulation of the function and quantity of goblet cells. Int. J. Mol. Med. 2019, 43, 2291–2302. [Google Scholar] [CrossRef]
- Grondin, J.A.; Kwon, Y.H.; Far, P.M.; Haq, S.; Khan, W.I. Mucins in Intestinal Mucosal Defense and Inflammation: Learning From Clinical and Experimental Studies. Front. Immunol. 2020, 11, 2054. [Google Scholar] [CrossRef] [PubMed]
- Taupin, D.; Podolsky, D.K. Trefoil factors: Initiators of mucosal healing. Nat. Rev. Mol. Cell Biol. 2003, 4, 721–732. [Google Scholar] [CrossRef]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Büller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- McCormick, D.A.; Horton, L.W.; Mee, A.S. Mucin depletion in inflammatory bowel disease. J. Clin. Pathol. 1990, 43, 143–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johansson, M.E.; Gustafsson, J.K.; Holmén-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjövall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Håkansson, Å.; Tormo-Badia, N.; Baridi, A.; Xu, J.; Molin, G.; Hagslätt, M.L.; Karlsson, C.; Jeppsson, B.; Cilio, C.M.; Ahrné, S. Immunological alteration and changes of gut microbiota after dextran sulfate sodium (DSS) administration in mice. Clin. Exp. Med. 2015, 15, 107–120. [Google Scholar] [CrossRef] [Green Version]
- Nagalingam, N.A.; Kao, J.Y.; Young, V.B. Microbial ecology of the murine gut associated with the development of dextran sodium sulfate-induced colitis. Inflamm. Bowel. Dis. 2011, 17, 917–926. [Google Scholar] [CrossRef]
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar] [CrossRef] [Green Version]
- Packey, C.D.; Sartor, R.B. Interplay of commensal and pathogenic bacteria, genetic mutations, and immunoregulatory defects in the pathogenesis of inflammatory bowel diseases. J. Intern. Med. 2008, 263, 597–606. [Google Scholar] [CrossRef]
- Hernández-Chirlaque, C.; Aranda, C.J.; Ocón, B.; Capitán-Cañadas, F.; Ortega-González, M.; Carrero, J.J.; Suárez, M.D.; Zarzuelo, A.; Sánchez de Medina, F.; Martínez-Augustin, O. Germ-free and Antibiotic-treated Mice are Highly Susceptible to Epithelial Injury in DSS Colitis. J. Crohns Colitis 2016, 10, 1324–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heimesaat, M.M.; Fischer, A.; Siegmund, B.; Kupz, A.; Niebergall, J.; Fuchs, D.; Jahn, H.K.; Freudenberg, M.; Loddenkemper, C.; Batra, A.; et al. Shift towards pro-inflammatory intestinal bacteria aggravates acute murine colitis via Toll-like receptors 2 and 4. PLoS ONE 2007, 2, e662. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Wu, Y.; Hu, Y.; Zhao, L.; Zhang, C. Initial gut microbiota structure affects sensitivity to DSS-induced colitis in a mouse model. Sci. China Life Sci. 2018, 61, 762–769. [Google Scholar] [CrossRef]
- Spalinger, M.R.; Schmidt, T.S.; Schwarzfischer, M.; Hering, L.; Atrott, K.; Lang, S.; Gottier, C.; Geirnaert, A.; Lacroix, C.; Dai, X.; et al. Protein tyrosine phosphatase non-receptor type 22 modulates colitis in a microbiota-dependent manner. J. Clin. Invest. 2019, 129, 2527–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, A.; Ma, A.G.; Yong, M.; Weiss, C.R.; Ma, Y.; Guan, Q.; Bernstein, C.N.; Peng, Z. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem. Pharm. 2010, 80, 1708–1717. [Google Scholar] [CrossRef]
- Bai, A.; Yong, M.; Ma, A.G.; Ma, Y.; Weiss, C.R.; Guan, Q.; Bernstein, C.N.; Peng, Z. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J. Pharm. Exp. Ther. 2010, 333, 717–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.Y.; Lee, S.H.; Yang, E.J.; Kim, E.K.; Kim, J.K.; Shin, D.Y.; Cho, M.L. Metformin Ameliorates Inflammatory Bowel Disease by Suppression of the STAT3 Signaling Pathway and Regulation of the between Th17/Treg Balance. PLoS ONE 2015, 10, e0135858. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.H. Metformin Use Is Associated with a Lower Risk of Inflammatory Bowel Disease in Patients with Type 2 Diabetes Mellitus. J. Crohns Colitis 2021, 15, 64–73. [Google Scholar] [CrossRef]
- Banskota, S.; Wang, H.; Kwon, Y.H.; Gautam, J.; Gurung, P.; Haq, S.; Hassan, F.M.N.; Bowdish, D.M.; Kim, J.A.; Carling, D.; et al. Salicylates Ameliorate Intestinal Inflammation by Activating Macrophage AMPK. Inflamm. Bowel. Dis. 2021, 27, 914–926. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Alkhouri, N.; Harrison, S.A.; Fouqueray, P.; Moller, D.E.; Hallakou-Bozec, S.; Bolze, S.; Grouin, J.M.; Jeannin Megnien, S.; Dubourg, J.; et al. Efficacy and safety of PXL770, a direct AMP kinase activator, for the treatment of non-alcoholic fatty liver disease (STAMP-NAFLD): A randomised, double-blind, placebo-controlled, phase 2a study. Lancet Gastroenterol. Hepatol. 2021, 6, 889–902. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Olivier, S.; Diounou, H.; Pochard, C.; Frechin, L.; Durieu, E.; Foretz, M.; Neunlist, M.; Rolli-Derkinderen, M.; Viollet, B. Intestinal Epithelial AMPK Deficiency Causes Delayed Colonic Epithelial Repair in DSS-Induced Colitis. Cells 2022, 11, 590. https://doi.org/10.3390/cells11040590
Olivier S, Diounou H, Pochard C, Frechin L, Durieu E, Foretz M, Neunlist M, Rolli-Derkinderen M, Viollet B. Intestinal Epithelial AMPK Deficiency Causes Delayed Colonic Epithelial Repair in DSS-Induced Colitis. Cells. 2022; 11(4):590. https://doi.org/10.3390/cells11040590
Chicago/Turabian StyleOlivier, Séverine, Hanna Diounou, Camille Pochard, Lisa Frechin, Emilie Durieu, Marc Foretz, Michel Neunlist, Malvyne Rolli-Derkinderen, and Benoit Viollet. 2022. "Intestinal Epithelial AMPK Deficiency Causes Delayed Colonic Epithelial Repair in DSS-Induced Colitis" Cells 11, no. 4: 590. https://doi.org/10.3390/cells11040590
APA StyleOlivier, S., Diounou, H., Pochard, C., Frechin, L., Durieu, E., Foretz, M., Neunlist, M., Rolli-Derkinderen, M., & Viollet, B. (2022). Intestinal Epithelial AMPK Deficiency Causes Delayed Colonic Epithelial Repair in DSS-Induced Colitis. Cells, 11(4), 590. https://doi.org/10.3390/cells11040590