
Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition

 , , ,
, , ,  ,
,  ,
,  and
and 
Abstract
1. Introduction
1.1. Polypharmacology
1.2. Endocannabinoid System, Endocannabinoids, and Exogenous Cannabinoids
1.2.1. Cannabinoid Receptors
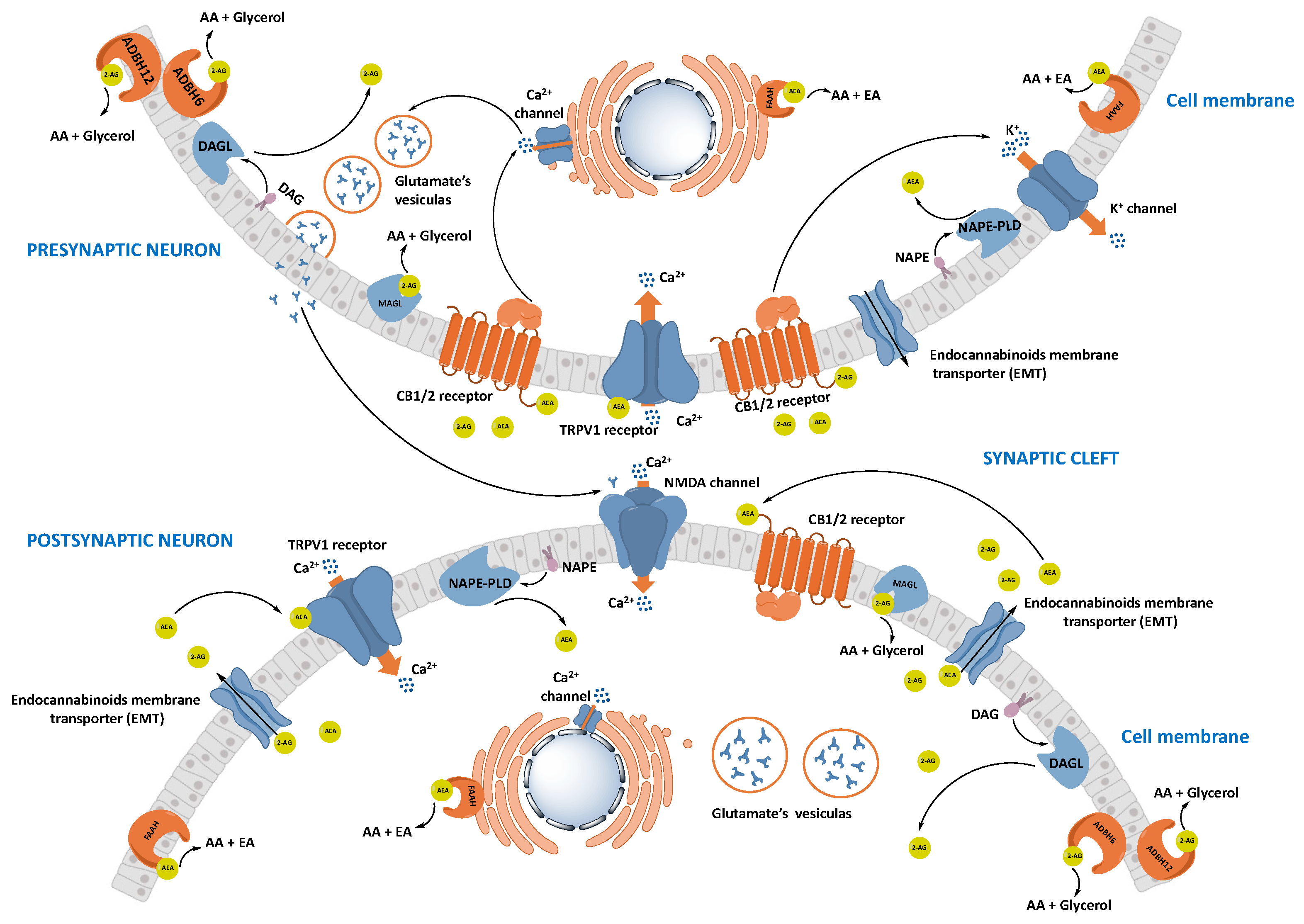
1.2.2. Synthesis and Transport of Endocannabinoids
1.2.3. Degradation of Endocannabinoids
Fatty Acid Amide Hydrolase (FAAH)
Monoacylglycerol Lipase (MAGL)
1.3. Interconnections between ECS and Neurotransmitter Systems
ECS Relevance to CNS Diseases
2. Multitarget Approaches Involving FAAH and MAGL Inhibitors
2.1. Dual FAAH-MAGL Inhibitors
2.2. FAAH Polypharmacology
2.2.1. FAAH/COX Dualism
2.2.2. Hybrid FAAH/Soluble Epoxide Hydrolase (sEH) Inhibitors
2.2.3. Hybrid FAAH/MAGL and Cholinesterase Inhibitors
2.2.4. Hybrid FAAH/Dopaminergic System Modulators
2.2.5. Hybrid FAAH/Melatonin Receptors Ligands
2.2.6. Dual FAAH/TRPV1 Inhibitors
3. Conclusions and Future Perspectives
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Di Marzo, V.; Stella, N.; Zimmer, A. Endocannabinoid Signalling and the Deteriorating Brain. Nat. Rev. Neurosci. 2015, 16, 30–42. [Google Scholar] [CrossRef] [PubMed]
- Stella, N. Cannabinoid and Cannabinoid-like Receptors in Microglia, Astrocytes, and Astrocytomas. Glia 2010, 58, 1017–1030. [Google Scholar] [CrossRef]
- Ren, S.; Wang, Z.; Zhang, Y.; Chen, N. Potential Application of Endocannabinoid System Agents in Neuropsychiatric and Neurodegenerative Diseases—Focusing on FAAH/MAGL Inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef] [PubMed]
- van Egmond, N.; Straub, V.M.; van der Stelt, M. Targeting Endocannabinoid Signaling: FAAH and MAG Lipase Inhibitors. Annu. Rev. Pharmacol. Toxicol. 2021, 61, 441–463. [Google Scholar] [CrossRef]
- Albertini, C.; Salerno, A.; de Sena Murteira Pinheiro, P.; Bolognesi, M.L. From Combinations to Multitarget-Directed Ligands: A Continuum in Alzheimer’s Disease Polypharmacology. Med. Res. Rev. 2021, 41, 2606–2633. [Google Scholar] [CrossRef] [PubMed]
- Boran, A.D.; Iyengar, R. Systems Approaches to Polypharmacology and Drug Discovery. Curr. Opin. Drug Discov. Devel. 2010, 13, 297–309. [Google Scholar]
- Mestres, J.; Gregori-Puigjané, E. Conciliating Binding Efficiency and Polypharmacology. Trends Pharmacol. Sci. 2009, 30, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.-U. Polypharmacology—Foe or Friend? J. Med. Chem. 2013, 56, 8955–8971. [Google Scholar] [CrossRef]
- Anighoro, A.; Bajorath, J.; Rastelli, G. Polypharmacology: Challenges and Opportunities in Drug Discovery. J. Med. Chem. 2014, 57, 7874–7887. [Google Scholar] [CrossRef]
- Merk, L.G. and D. Therapeutic Potential of Peroxisome Proliferator-Activated Receptor Modulation in Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis. Nucl. Recept. Res. 2017, 4, 101310. [Google Scholar] [CrossRef]
- Cavalli, A.; Bolognesi, M.L.; Minarini, A.; Rosini, M.; Tumiatti, V.; Recanatini, M.; Melchiorre, C. Multi-Target-Directed Ligands To Combat Neurodegenerative Diseases. J. Med. Chem. 2008, 51, 347–372. [Google Scholar] [CrossRef] [PubMed]
- Antolin, A.; Workman, P.; Mestres, J.; Al-Lazikani, B. Polypharmacology in Precision Oncology: Current Applications and Future Prospects. Curr. Pharm. Des. 2016, 22, 6935–6945. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-J.; Pan, W.; Hu, Y.-J.; Wang, Y.-T. Multi-Target Drugs: The Trend of Drug Research and Development. PLoS ONE 2012, 7, e40262. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Giulianotti, M.A.; Welmaker, G.S.; Houghten, R.A. Shifting from the Single- to the Multitarget Paradigm in Drug Discovery. Drug Discov. Today 2013, 18, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Butini, S.; Nikolic, K.; Kassel, S.; Brückmann, H.; Filipic, S.; Agbaba, D.; Gemma, S.; Brogi, S.; Brindisi, M.; Campiani, G.; et al. Polypharmacology of Dopamine Receptor Ligands. Prog. Neurobiol. 2016, 142, 68–103. [Google Scholar] [CrossRef]
- Lötsch, J.; Geisslinger, G. Low-Dose Drug Combinations along Molecular Pathways Could Maximize Therapeutic Effectiveness While Minimizing Collateral Adverse Effects. Drug Discov. Today 2011, 16, 1001–1006. [Google Scholar] [CrossRef]
- Valant, C.; Sexton, P.M.; Christopoulos, A. Orthosteric/Allosteric Bitopic Ligands. Mol. Interv. 2009, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Fronik, P.; Gaiser, B.I.; Sejer Pedersen, D. Bitopic Ligands and Metastable Binding Sites: Opportunities for G Protein-Coupled Receptor (GPCR) Medicinal Chemistry. J. Med. Chem. 2017, 60, 4126–4134. [Google Scholar] [CrossRef]
- Schmidt, A.P.; Böhmer, A.E.; Antunes, C.; Schallenberger, C.; Porciúncula, L.O.; Elisabetsky, E.; Lara, D.R.; Souza, D.O. Anti-Nociceptive Properties of the Xanthine Oxidase Inhibitor Allopurinol in Mice: Role of A1 Adenosine Receptors. Br. J. Pharmacol. 2009, 156, 163–172. [Google Scholar] [CrossRef]
- Dolles, D.; Hoffmann, M.; Gunesch, S.; Marinelli, O.; Möller, J.; Santoni, G.; Chatonnet, A.; Lohse, M.J.; Wittmann, H.-J.; Strasser, A.; et al. Structure–Activity Relationships and Computational Investigations into the Development of Potent and Balanced Dual-Acting Butyrylcholinesterase Inhibitors and Human Cannabinoid Receptor 2 Ligands with Pro-Cognitive In Vivo Profiles. J. Med. Chem. 2018, 61, 1646–1663. [Google Scholar] [CrossRef]
- Hu, Y.; Bajorath, J. High-Resolution View of Compound Promiscuity. F1000Research 2013, 2, 144. [Google Scholar] [CrossRef] [PubMed]
- Talevi, A. Multi-Target Pharmacology: Possibilities and Limitations of the “Skeleton Key Approach” from a Medicinal Chemist Perspective. Front. Pharmacol. 2015, 6, 205. [Google Scholar] [CrossRef] [PubMed]
- Morphy, R.; Kay, C.; Rankovic, Z. From Magic Bullets to Designed Multiple Ligands. Drug Discov. Today 2004, 9, 641–651. [Google Scholar] [CrossRef]
- Espinoza-Fonseca, L.M. The Benefits of the Multi-Target Approach in Drug Design and Discovery. Bioorg. Med. Chem. 2006, 14, 896–897. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Pei, J.; Lai, L. Computational Multitarget Drug Design. J. Chem. Inf. Model. 2017, 57, 403–412. [Google Scholar] [CrossRef]
- Millan, M.J. On ‘Polypharmacy’ and Multi-Target Agents, Complementary Strategies for Improving the Treatment of Depression: A Comparative Appraisal. Int. J. Neuropsychopharmacol. 2014, 17, 1009–1037. [Google Scholar] [CrossRef]
- Proschak, E.; Stark, H.; Merk, D. Polypharmacology by Design: A Medicinal Chemist’s Perspective on Multitargeting Compounds. J. Med. Chem. 2019, 62, 420–444. [Google Scholar] [CrossRef]
- Benek, O.; Korabecny, J.; Soukup, O. A Perspective on Multi-Target Drugs for Alzheimer’s Disease. Trends Pharmacol. Sci. 2020, 41, 434–445. [Google Scholar] [CrossRef]
- de Lera, A.R.; Ganesan, A. Epigenetic Polypharmacology: From Combination Therapy to Multitargeted Drugs. Clin. Epigenetics 2016, 8, 105. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. An Introduction to the Endogenous Cannabinoid System. Biol. Psychiatry 2016, 79, 516–525. [Google Scholar] [CrossRef]
- Lu, H.-C.; Mackie, K. Review of the Endocannabinoid System. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2021, 6, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Hillard, C.J. Circulating Endocannabinoids: From Whence Do They Come and Where Are They Going? Neuropsychopharmacology 2018, 43, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Grabiec, U.; Dehghani, F. N-Arachidonoyl Dopamine: A Novel Endocannabinoid and Endovanilloid with Widespread Physiological and Pharmacological Activities. Cannabis Cannabinoid Res. 2017, 2, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and Endocannabinoid-Related Mediators: Targets, Metabolism and Role in Neurological Disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. New Approaches and Challenges to Targeting the Endocannabinoid System. Nat. Rev. Drug Discov. 2018, 17, 623–639. [Google Scholar] [CrossRef] [PubMed]
- Morales, P.; Reggio, P.H. An Update on Non-CB 1, Non-CB 2 Cannabinoid Related G-Protein-Coupled Receptors. Cannabis Cannabinoid Res. 2017, 2, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Morrison, B.E.; Blankman, J.L.; Long, J.Z.; Kinsey, S.G.; Marcondes, M.C.G.; Ward, A.M.; Hahn, Y.K.; Lichtman, A.H.; Conti, B.; et al. Endocannabinoid Hydrolysis Generates Brain Prostaglandins That Promote Neuroinflammation. Science 2011, 334, 809–813. [Google Scholar] [CrossRef] [PubMed]
- Crocq, M.-A. History of Cannabis and the Endocannabinoid System. Dialogues Clin. Neurosci. 2020, 22, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Parker, L.A. The Endocannabinoid System and the Brain. Annu. Rev. Psychol. 2013, 64, 21–47. [Google Scholar] [CrossRef]
- Pertwee, R. Receptors and Channels Targeted by Synthetic Cannabinoid Receptor Agonists and Antagonists. Curr. Med. Chem. 2010, 17, 1360–1381. [Google Scholar] [CrossRef]
- Mechoulam, R.; Ben-Shabat, S.; Hanus, L.; Ligumsky, M.; Kaminski, N.E.; Schatz, A.R.; Gopher, A.; Almog, S.; Martin, B.R.; Compton, D.R.; et al. Identification of an Endogenous 2-Monoglyceride, Present in Canine Gut, That Binds to Cannabinoid Receptors. Biochem. Pharmacol. 1995, 50, 83–90. [Google Scholar] [CrossRef]
- Baggelaar, M.P.; Maccarrone, M.; van der Stelt, M. 2-Arachidonoylglycerol: A Signaling Lipid with Manifold Actions in the Brain. Prog. Lipid Res. 2018, 71, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Elphick, M.R. The Evolution and Comparative Neurobiology of Endocannabinoid Signalling. Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3201–3215. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; De Petrocellis, L. Why Do Cannabinoid Receptors Have More than One Endogenous Ligand? Philos. Trans. R. Soc. B Biol. Sci. 2012, 367, 3216–3228. [Google Scholar] [CrossRef] [PubMed]
- Castillo, P.E.; Younts, T.J.; Chávez, A.E.; Hashimotodani, Y. Endocannabinoid Signaling and Synaptic Function. Neuron 2012, 76, 70–81. [Google Scholar] [CrossRef]
- Alger, B.E. Endocannabinoids at the Synapse a Decade after the Dies Mirabilis (29 March 2001): What We Still Do Not Know: Endocannabinoids at the Synapse. J. Physiol. 2012, 590, 2203–2212. [Google Scholar] [CrossRef]
- Talwar, R.; Potluri, V.K. Cannabinoid 1 (CB1) Receptor--Pharmacology, Role in Pain and Recent Developments in Emerging CB1 Agonists. CNS Neurol. Disord. Drug Targets 2011, 10, 536–544. [Google Scholar] [CrossRef]
- Matias, I.; Di Marzo, V. Endocannabinoids and the Control of Energy Balance. Trends Endocrinol. Metab. 2007, 18, 27–37. [Google Scholar] [CrossRef]
- Staiano, R.I.; Loffredo, S.; Borriello, F.; Iannotti, F.A.; Piscitelli, F.; Orlando, P.; Secondo, A.; Granata, F.; Lepore, M.T.; Fiorelli, A.; et al. Human Lung-Resident Macrophages Express CB1 and CB2 Receptors Whose Activation Inhibits the Release of Angiogenic and Lymphangiogenic Factors. J. Leukoc. Biol. 2016, 99, 531–540. [Google Scholar] [CrossRef]
- Nagy, I.; Friston, D.; Valente, J.S.; Perez, J.V.T.; Andreou, A.P. Pharmacology of the Capsaicin Receptor, Transient Receptor Potential Vanilloid Type-1 Ion Channel. In Capsaicin as a Therapeutic Molecule; Abdel-Salam, O.M.E., Ed.; Springer: Basel, Switzerland, 2014; pp. 39–76. ISBN 978-3-0348-0827-9. [Google Scholar]
- Julius, D. TRP Channels and Pain. Annu. Rev. Cell Dev. Biol. 2013, 29, 355–384. [Google Scholar] [CrossRef]
- Oka, S.; Nakajima, K.; Yamashita, A.; Kishimoto, S.; Sugiura, T. Identification of GPR55 as a Lysophosphatidylinositol Receptor. Biochem. Biophys. Res. Commun. 2007, 362, 928–934. [Google Scholar] [CrossRef] [PubMed]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The Endocannabinoids Anandamide and Virodhamine Modulate the Activity of the Candidate Cannabinoid Receptor GPR55. J. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [PubMed]
- Scotter, E.L.; Abood, M.E.; Glass, M. The Endocannabinoid System as a Target for the Treatment of Neurodegenerative Disease: Endocannabinoids in Neurodegenerative Disease. Br. J. Pharmacol. 2010, 160, 480–498. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Ovejero, D.; Arevalo-Martin, A.; Petrosino, S.; Docagne, F.; Hagen, C.; Bisogno, T.; Watanabe, M.; Guaza, C.; Di Marzo, V.; Molina-Holgado, E. The Endocannabinoid System Is Modulated in Response to Spinal Cord Injury in Rats. Neurobiol. Dis. 2009, 33, 57–71. [Google Scholar] [CrossRef]
- Chicca, A.; Marazzi, J.; Nicolussi, S.; Gertsch, J. Evidence for Bidirectional Endocannabinoid Transport across Cell Membranes*. J. Biol. Chem. 2012, 287, 34660–34682. [Google Scholar] [CrossRef]
- Tripathi, R.K.P. A Perspective Review on Fatty Acid Amide Hydrolase (Faah) Inhibitors as Potential Therapeutic Agents. Eur. J. Med. Chem. 2020, 188, 111953. [Google Scholar] [CrossRef]
- Deng, H.; Li, W. Monoacylglycerol Lipase Inhibitors: Modulators for Lipid Metabolism in Cancer Malignancy, Neurological and Metabolic Disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef]
- Bedse, G.; Romano, A.; Lavecchia, A.M.; Cassano, T.; Gaetani, S. The Role of Endocannabinoid Signaling in the Molecular Mechanisms of Neurodegeneration in Alzheimer’s Disease. J. Alzheimers Dis. 2015, 43, 1115–1136. [Google Scholar] [CrossRef]
- Khanna, I.; Alexander, C. Fatty Acid Amide Hydrolase Inhibitors-Progress and Potential. CNS Neurol. Disord.-Drug Targets-CNS Neurol. Disord. 2011, 10, 545–558. [Google Scholar] [CrossRef]
- Maccarrone, M.; Bari, M.; Lorenzon, T.; Bisogno, T.; Di Marzo, V.; Finazzi-Agrò, A. Anandamide Uptake by Human Endothelial Cells and Its Regulation by Nitric Oxide. J. Biol. Chem. 2000, 275, 13484–13492. [Google Scholar] [CrossRef]
- Gulyas, A.I.; Cravatt, B.F.; Bracey, M.H.; Dinh, T.P.; Piomelli, D.; Boscia, F.; Freund, T.F. Segregation of Two Endocannabinoid-Hydrolyzing Enzymes into Pre- and Postsynaptic Compartments in the Rat Hippocampus, Cerebellum and Amygdala. Eur. J. Neurosci. 2004, 20, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Sałaga, M.; Sobczak, M.; Fichna, J. Inhibition of Fatty Acid Amide Hydrolase (FAAH) as a Novel Therapeutic Strategy in the Treatment of Pain and Inflammatory Diseases in the Gastrointestinal Tract. Eur. J. Pharm. Sci. 2014, 52, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Kinsey, S.G.; Liu, Q.; Hruba, L.; McMahon, L.R.; Grim, T.W.; Merritt, C.R.; Wise, L.E.; Abdullah, R.A.; Selley, D.E.; et al. Full Fatty Acid Amide Hydrolase Inhibition Combined with Partial Monoacylglycerol Lipase Inhibition: Augmented and Sustained Antinociceptive Effects with Reduced Cannabimimetic Side Effects in Mice. J. Pharmacol. Exp. Ther. 2015, 354, 111–120. [Google Scholar] [CrossRef]
- Huang, W.-J.; Chen, W.-W.; Zhang, X. Endocannabinoid System: Role in Depression, Reward and Pain Control (Review). Mol. Med. Rep. 2016, 14, 2899–2903. [Google Scholar] [CrossRef]
- Panlilio, L.V.; Justinova, Z.; Goldberg, S.R. Inhibition of FAAH and Activation of PPAR: New Approaches to the Treatment of Cognitive Dysfunction and Drug Addiction. Pharmacol. Ther. 2013, 138, 84–102. [Google Scholar] [CrossRef]
- Berardi, A.; Schelling, G.; Campolongo, P. The Endocannabinoid System and Post Traumatic Stress Disorder (PTSD): From Preclinical Findings to Innovative Therapeutic Approaches in Clinical Settings. Pharmacol. Res. 2016, 111, 668–678. [Google Scholar] [CrossRef]
- Justinova, Z.; Panlilio, L.V.; Moreno-Sanz, G.; Redhi, G.H.; Auber, A.; Secci, M.E.; Mascia, P.; Bandiera, T.; Armirotti, A.; Bertorelli, R.; et al. Effects of Fatty Acid Amide Hydrolase (FAAH) Inhibitors in Non-Human Primate Models of Nicotine Reward and Relapse. Neuropsychopharmacology 2015, 40, 2185–2197. [Google Scholar] [CrossRef]
- Sloan, M.E.; Gowin, J.L.; Yan, J.; Schwandt, M.L.; Spagnolo, P.A.; Sun, H.; Hodgkinson, C.A.; Goldman, D.; Ramchandani, V.A. Severity of Alcohol Dependence Is Associated with the Fatty Acid Amide Hydrolase Pro129Thr Missense Variant: FAAH and Alcohol Dependence. Addict. Biol. 2018, 23, 474–484. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Zanaboni, A.M.; Piomelli, D.; Tassorelli, C. Endocannabinoid System and Migraine Pain: An Update. Front. Neurosci. 2018, 12, 172. [Google Scholar] [CrossRef]
- Blankman, J.L.; Simon, G.M.; Cravatt, B.F. A Comprehensive Profile of Brain Enzymes That Hydrolyze the Endocannabinoid 2-Arachidonoylglycerol. Chem. Biol. 2007, 14, 1347–1356. [Google Scholar] [CrossRef]
- Dinh, T.P.; Kathuria, S.; Piomelli, D. RNA Interference Suggests a Primary Role for Monoacylglycerol Lipase in the Degradation of the Endocannabinoid 2-Arachidonoylglycerol. Mol. Pharmacol. 2004, 66, 1260–1264. [Google Scholar] [CrossRef] [PubMed]
- Savinainen, J.R.; Kansanen, E.; Pantsar, T.; Navia-Paldanius, D.; Parkkari, T.; Lehtonen, M.; Laitinen, T.; Nevalainen, T.; Poso, A.; Levonen, A.-L.; et al. Robust Hydrolysis of Prostaglandin Glycerol Esters by Human Monoacylglycerol Lipase (MAGL). Mol. Pharmacol. 2014, 86, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.; Taschler, U.; Radulovic, M.; Aschauer, P.; Eichmann, T.O.; Grond, S.; Wolinski, H.; Oberer, M.; Zechner, R.; Kohlwein, S.D.; et al. Monoacylglycerol Lipases Act as Evolutionarily Conserved Regulators of Non-Oxidative Ethanol Metabolism. J. Biol. Chem. 2016, 291, 11865–11875. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Lombardi, D.P.; Chang, J.W.; Niessen, S.; Ward, A.M.; Long, J.Z.; Hoover, H.H.; Cravatt, B.F. Monoacylglycerol Lipase Exerts Dual Control over Endocannabinoid and Fatty Acid Pathways to Support Prostate Cancer. Chem. Biol. 2011, 18, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Long, J.Z.; Li, W.; Booker, L.; Burston, J.J.; Kinsey, S.G.; Schlosburg, J.E.; Pavón, F.J.; Serrano, A.M.; Selley, D.E.; Parsons, L.H.; et al. Selective Blockade of 2-Arachidonoylglycerol Hydrolysis Produces Cannabinoid Behavioral Effects. Nat. Chem. Biol. 2009, 5, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Chanda, P.K.; Gao, Y.; Mark, L.; Btesh, J.; Strassle, B.W.; Lu, P.; Piesla, M.J.; Zhang, M.-Y.; Bingham, B.; Uveges, A.; et al. Monoacylglycerol Lipase Activity Is a Critical Modulator of the Tone and Integrity of the Endocannabinoid System. Mol. Pharmacol. 2010, 78, 996–1003. [Google Scholar] [CrossRef]
- Schlosburg, J.E.; Blankman, J.L.; Long, J.Z.; Nomura, D.K.; Pan, B.; Kinsey, S.G.; Nguyen, P.T.; Ramesh, D.; Booker, L.; Burston, J.J.; et al. Chronic Monoacylglycerol Lipase Blockade Causes Functional Antagonism of the Endocannabinoid System. Nat. Neurosci. 2010, 13, 1113–1119. [Google Scholar] [CrossRef]
- Turcotte, C.; Chouinard, F.; Lefebvre, J.S.; Flamand, N. Regulation of Inflammation by Cannabinoids, the Endocannabinoids 2-Arachidonoyl-Glycerol and Arachidonoyl-Ethanolamide, and Their Metabolites. J. Leukoc. Biol. 2015, 97, 1049–1070. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Khan, M.; Fraser, A. Cox-2 Inhibitors and the Risk of Cardiovascular Thrombotic Events. Ir. Med. J. 2012, 105, 119–121. [Google Scholar]
- Klein, T.W. Cannabinoid-Based Drugs as Anti-Inflammatory Therapeutics. Nat. Rev. Immunol. 2005, 5, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Benjamin, D.I.; Duerr, J.M.; Pi, Y.; Gonzales, C.; Wood, K.M.; Schwartz, J.W.; Nomura, D.K.; Samad, T.A. A Dysregulated Endocannabinoid-Eicosanoid Network Supports Pathogenesis in a Mouse Model of Alzheimer’s Disease. Cell Rep. 2012, 1, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting Effects of Selective MAGL and FAAH Inhibition on Dopamine Depletion and GDNF Expression in a Chronic MPTP Mouse Model of Parkinson’s Disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Suárez, D.; Celorrio, M.; Riezu-Boj, J.I.; Ugarte, A.; Pacheco, R.; González, H.; Oyarzabal, J.; Hillard, C.J.; Franco, R.; Aymerich, M.S. The Monoacylglycerol Lipase Inhibitor JZL184 Is Neuroprotective and Alters Glial Cell Phenotype in the Chronic MPTP Mouse Model. Neurobiol. Aging 2014, 35, 2603–2616. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Maramai, S.; Gemma, S.; Brogi, S.; Grillo, A.; Di Cesare Mannelli, L.; Gabellieri, E.; Lamponi, S.; Saponara, S.; Gorelli, B.; et al. Development and Pharmacological Characterization of Selective Blockers of 2-Arachidonoyl Glycerol Degradation with Efficacy in Rodent Models of Multiple Sclerosis and Pain. J. Med. Chem. 2016, 59, 2612–2632. [Google Scholar] [CrossRef]
- Llorente-Berzal, A.; Terzian, A.L.B.; di Marzo, V.; Micale, V.; Viveros, M.P.; Wotjak, C.T. 2-AG Promotes the Expression of Conditioned Fear via Cannabinoid Receptor Type 1 on GABAergic Neurons. Psychopharmacology 2015, 232, 2811–2825. [Google Scholar] [CrossRef]
- Vitale, R.M.; Iannotti, F.A.; Amodeo, P. The (Poly)Pharmacology of Cannabidiol in Neurological and Neuropsychiatric Disorders: Molecular Mechanisms and Targets. Int. J. Mol. Sci. 2021, 22, 4876. [Google Scholar] [CrossRef]
- Klein, M.O.; Battagello, D.S.; Cardoso, A.R.; Hauser, D.N.; Bittencourt, J.C.; Correa, R.G. Dopamine: Functions, Signaling, and Association with Neurological Diseases. Cell. Mol. Neurobiol. 2019, 39, 31–59. [Google Scholar] [CrossRef]
- Terzian, A.L.; Drago, F.; Wotjak, C.; Micale, V. The Dopamine and Cannabinoid Interaction in the Modulation of Emotions and Cognition: Assessing the Role of Cannabinoid CB1 Receptor in Neurons Expressing Dopamine D1 Receptors. Front. Behav. Neurosci. 2011, 5, 49. [Google Scholar] [CrossRef]
- Micale, V.; Stepan, J.; Jurik, A.; Pamplona, F.A.; Marsch, R.; Drago, F.; Eder, M.; Wotjak, C.T. Extinction of Avoidance Behavior by Safety Learning Depends on Endocannabinoid Signaling in the Hippocampus. J. Psychiatr. Res. 2017, 90, 46–59. [Google Scholar] [CrossRef]
- Covey, D.P.; Mateo, Y.; Sulzer, D.; Cheer, J.F.; Lovinger, D.M. Endocannabinoid Modulation of Dopamine Neurotransmission. Neuropharmacology 2017, 124, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Lovinger, D.M. Retrograde Endocannabinoid Signaling at Striatal Synapses Requires a Regulated Postsynaptic Release Step. Proc. Natl. Acad. Sci. USA 2007, 104, 20564–20569. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Talani, G.; Lovinger, D.M. Endocannabinoid-Dependent Plasticity at GABAergic and Glutamatergic Synapses in the Striatum Is Regulated by Synaptic Activity. Eur. J. Neurosci. 2009, 29, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Riegel, A.C.; Lupica, C.R. Independent Presynaptic and Postsynaptic Mechanisms Regulate Endocannabinoid Signaling at Multiple Synapses in the Ventral Tegmental Area. J. Neurosci. 2004, 24, 11070–11078. [Google Scholar] [CrossRef] [PubMed]
- Sagheddu, C.; Muntoni, A.L.; Pistis, M.; Melis, M. Endocannabinoid Signaling in Motivation, Reward, and Addiction: Influences on Mesocorticolimbic Dopamine Function. Int. Rev. Neurobiol. 2015, 125, 257–302. [Google Scholar] [CrossRef]
- Laksmidewi, A.A.A.P.; Soejitno, A. Endocannabinoid and Dopaminergic System: The Pas de Deux Underlying Human Motivation and Behaviors. J. Neural Transm. 2021, 128, 615–630. [Google Scholar] [CrossRef]
- Colangeli, R.; Teskey, G.C.; Di Giovanni, G. Endocannabinoid-Serotonin Systems Interaction in Health and Disease. Prog. Brain Res. 2021, 259, 83–134. [Google Scholar] [CrossRef]
- Wang, Z.; Kai, L.; Day, M.; Ronesi, J.; Yin, H.H.; Ding, J.; Tkatch, T.; Lovinger, D.M.; Surmeier, D.J. Dopaminergic Control of Corticostriatal Long-Term Synaptic Depression in Medium Spiny Neurons Is Mediated by Cholinergic Interneurons. Neuron 2006, 50, 443–452. [Google Scholar] [CrossRef]
- Narushima, M.; Uchigashima, M.; Fukaya, M.; Matsui, M.; Manabe, T.; Hashimoto, K.; Watanabe, M.; Kano, M. Tonic Enhancement of Endocannabinoid-Mediated Retrograde Suppression of Inhibition by Cholinergic Interneuron Activity in the Striatum. J. Neurosci. 2007, 27, 496–506. [Google Scholar] [CrossRef]
- Farran, B. An Update on the Physiological and Therapeutic Relevance of GPCR Oligomers. Pharmacol. Res. 2017, 117, 303–327. [Google Scholar] [CrossRef]
- Franco, R.; Martínez-Pinilla, E.; Lanciego, J.L.; Navarro, G. Basic Pharmacological and Structural Evidence for Class A G-Protein-Coupled Receptor Heteromerization. Front. Pharmacol. 2016, 7, 76. [Google Scholar] [CrossRef]
- Viñals, X.; Moreno, E.; Lanfumey, L.; Cordomí, A.; Pastor, A.; Torre, R.D.L.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9- Tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194. [Google Scholar] [CrossRef]
- Rozenfeld, R.; Gupta, A.; Gagnidze, K.; Lim, M.P.; Gomes, I.; Lee-Ramos, D.; Nieto, N.; Devi, L.A. AT1R-CB1 R Heteromerization Reveals a New Mechanism for the Pathogenic Properties of Angiotensin II. EMBO J. 2011, 30, 2350–2363. [Google Scholar] [CrossRef]
- Bushlin, I.; Gupta, A.; Stockton, S.D.; Miller, L.K.; Devi, L.A. Dimerization with Cannabinoid Receptors Allosterically Modulates Delta Opioid Receptor Activity during Neuropathic Pain. PLoS ONE 2012, 7, e49789. [Google Scholar] [CrossRef]
- Fujita, W.; Gomes, I.; Devi, L.A. International Union Of Basic And Clinical Pharmacology Review Revolution in GPCR Signalling: Opioid Receptor Heteromers as Novel Therapeutic Targets: IUPHAR Review 10. Wwwbrjpharmacolorg Br. J. Pharmacol. 2014, 171, 4155. [Google Scholar] [CrossRef]
- Hojo, M.; Sudo, Y.; Ando, Y.; Minami, K.; Takada, M.; Matsubara, T.; Kanaide, M.; Taniyama, K.; Sumikawa, K.; Uezono, Y. μ-Opioid Receptor Forms a Functional Heterodimer with Cannabinoid CB1 Receptor: Electrophysiological and Fret Assay Analysis. J. Pharmacol. Sci. 2008, 108, 308–319. [Google Scholar] [CrossRef]
- Martínez-Pinilla, E.; Reyes-Resina, I.; Oñatibia-Astibia, A.; Zamarbide, M.; Ricobaraza, A.; Navarro, G.; Moreno, E.; Dopeso-Reyes, I.G.; Sierra, S.; Rico, A.J.; et al. CB1 and GPR55 Receptors Are Co-Expressed and Form Heteromers in Rat and Monkey Striatum. Exp. Neurol. 2014, 261, 44–52. [Google Scholar] [CrossRef]
- Zou, S.; Somvanshi, R.K.; Kumar, U. Somatostatin Receptor 5 Is a Prominent Regulator of Signaling Pathways in Cells with Coexpression of Cannabinoid Receptors 1. Neuroscience 2017, 340, 218–231. [Google Scholar] [CrossRef]
- Jäntti, M.H.; Mandrika, I.; Kukkonen, J.P. Human Orexin/Hypocretin Receptors Form Constitutive Homo- and Heteromeric Complexes with Each Other and with Human CB1 Cannabinoid Receptors. Biochem. Biophys. Res. Commun. 2014, 445, 486–490. [Google Scholar] [CrossRef]
- Ward, R.J.; Pediani, J.D.; Milligan, G. Heteromultimerization of Cannabinoid CB 1 Receptor and Orexin OX 1 Receptor Generates a Unique Complex in Which Both Protomers Are Regulated by Orexin A. J. Biol. Chem. 2011, 286, 37414–37428. [Google Scholar] [CrossRef]
- Glass, M.; Felder, C.C. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Augments CAMP Accumulation in Striatal Neurons: Evidence for a Gs Linkage to the CB1 Receptor. J Neurosci 1997, 17, 5327–5333. [Google Scholar] [CrossRef]
- Kearn, C.S.; Blake-Palmer, K.; Daniel, E.; Mackie, K.; Glass, M. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Enhances Heterodimer Formation: A Mechanism for Receptor Cross-Talk? Mol. Pharmacol. 2005, 67, 1697–1704. [Google Scholar] [CrossRef]
- Przybyla, J.A.; Watts, V.J. Ligand-Induced Regulation and Localization of Cannabinoid CB1 and Dopamine D2L Receptor Heterodimers. J. Pharmacol. Exp. Ther. 2010, 332, 710–719. [Google Scholar] [CrossRef]
- Moreno, E.; Chiarlone, A.; Medrano, M.; Puigdellívol, M.; Bibic, L.; Howell, L.A.; Resel, E.; Puente, N.; Casarejos, M.J.; Perucho, J.; et al. Singular Location and Signaling Profile of Adenosine A2A-Cannabinoid CB1 Receptor Heteromers in the Dorsal Striatum. Neuropsychopharmacology 2018, 43, 964–977. [Google Scholar] [CrossRef]
- Callén, L.; Moreno, E.; Barroso-Chinea, P.; Moreno-Delgado, D.; Cortés, A.; Mallol, J.; Casadó, V.; Lanciego, J.L.; Franco, R.; Lluis, C.; et al. Cannabinoid Receptors CB1 and CB2 Form Functional Heteromers in Brain. J. Biol. Chem. 2012, 287, 20851–20865. [Google Scholar] [CrossRef]
- Balenga, N.A.; Martínez-Pinilla, E.; Kargl, J.; Schröder, R.; Peinhaupt, M.; Platzer, W.; Bálint, Z.; Zamarbide, M.; Dopeso-Reyes, I.G.; Ricobaraza, A.; et al. Heteromerization of GPR55 and Cannabinoid CB2 Receptors Modulates Signalling. Br. J. Pharmacol. 2014, 171, 5387–5406. [Google Scholar] [CrossRef]
- Moreno, E.; Andradas, C.; Medrano, M.; Caffarel, M.M.; Pérez-Gómez, E.; Blasco-Benito, S.; Gómez-Cañas, M.; Pazos, M.R.; Irving, A.J.; Lluís, C.; et al. Targeting CB2-GPR55 Receptor Heteromers Modulates Cancer Cell Signaling. J. Biol. Chem. 2014, 289, 21960–21972. [Google Scholar] [CrossRef]
- Pazos, M.R.; Mohammed, N.; Lafuente, H.; Santos, M.; Martínez-Pinilla, E.; Moreno, E.; Valdizan, E.; Romero, J.; Pazos, A.; Franco, R.; et al. Mechanisms of Cannabidiol Neuroprotection in Hypoxic-Ischemic Newborn Pigs: Role of 5HT1A and CB2 Receptors. Neuropharmacology 2013, 71, 282–291. [Google Scholar] [CrossRef]
- Coke, C.J.; Scarlett, K.A.; Chetram, M.A.; Jones, K.J.; Sandifer, B.J.; Davis, A.S.; Marcus, A.I.; Hinton, C.V. Simultaneous Activation of Induced Heterodimerization between CXCR4 Chemokine Receptor and Cannabinoid Receptor 2 (CB2) Reveals a Mechanism for Regulation of Tumor Progression. J. Biol. Chem. 2016, 291, 9991–10005. [Google Scholar] [CrossRef]
- Galve-Roperh, I.; Palazuelos, J.; Aguado, T.; Guzmán, M. The Endocannabinoid System and the Regulation of Neural Development: Potential Implications in Psychiatric Disorders. Eur. Arch. Psychiatry Clin. Neurosci. 2009, 259, 371–382. [Google Scholar] [CrossRef]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The Endocannabinoid System Promotes Astroglial Differentiation by Acting on Neural Progenitor Cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef]
- Berghuis, P.; Dobszay, M.B.; Wang, X.; Spano, S.; Ledda, F.; Sousa, K.M.; Schulte, G.; Ernfors, P.; Mackie, K.; Paratcha, G.; et al. Endocannabinoids Regulate Interneuron Migration and Morphogenesis by Transactivating the TrkB Receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 19115–19120. [Google Scholar] [CrossRef]
- Mulder, J.; Aguado, T.; Keimpema, E.; Barabás, K.; Ballester Rosado, C.J.; Nguyen, L.; Monory, K.; Marsicano, G.; Di Marzo, V.; Hurd, Y.L.; et al. Endocannabinoid Signaling Controls Pyramidal Cell Specification and Long-Range Axon Patterning. Proc. Natl. Acad. Sci. USA 2008, 105, 8760–8765. [Google Scholar] [CrossRef]
- Berghuis, P.; Rajnicek, A.M.; Morozov, Y.M.; Ross, R.A.; Mulder, J.; Urbán, G.M.; Monory, K.; Marsicano, G.; Matteoli, M.; Canty, A.; et al. Hardwiring the Brain: Endocannabinoids Shape Neuronal Connectivity. Science 2007, 316, 1212–1216. [Google Scholar] [CrossRef]
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid System in Neurodegenerative Disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef]
- Micale, V.; Di Marzo, V.; Sulcova, A.; Wotjak, C.T.; Drago, F. Endocannabinoid System and Mood Disorders: Priming a Target for New Therapies. Pharmacol. Ther. 2013, 138, 18–37. [Google Scholar] [CrossRef]
- Hill, M.N.; Patel, S.; Carrier, E.J.; Rademacher, D.J.; Ormerod, B.K.; Hillard, C.J.; Gorzalka, B.B. Downregulation of Endocannabinoid Signaling in the Hippocampus Following Chronic Unpredictable Stress. Neuropsychopharmacology 2005, 30, 508–515. [Google Scholar] [CrossRef]
- Micale, V.; Tabiova, K.; Kucerova, J.; Drago, F. Role of the Endocannabinoid System in Depression: From Preclinical to Clinical Evidence. In Cannabinoid Modulation of Emotion, Memory, and Motivation; Campolongo, P., Fattore, L., Eds.; Springer: New York, NY, USA, 2015; pp. 97–129. ISBN 978-1-4939-2294-9. [Google Scholar]
- Micale, V.; Drago, F. Endocannabinoid System, Stress and HPA Axis. Eur. J. Pharmacol. 2018, 834, 230–239. [Google Scholar] [CrossRef]
- Hill, M.N.; Miller, G.E.; Ho, W.S.V.; Gorzalka, B.B.; Hillard, C.J. Serum Endocannabinoid Content Is Altered in Females with Depressive Disorders: A Preliminary Report. Pharmacopsychiatry 2008, 41, 48–53. [Google Scholar] [CrossRef]
- Mayo, L.M.; Asratian, A.; Lindé, J.; Morena, M.; Haataja, R.; Hammar, V.; Augier, G.; Hill, M.N.; Heilig, M. Elevated Anandamide, Enhanced Recall of Fear Extinction, and Attenuated Stress Responses Following Inhibition of Fatty Acid Amide Hydrolase: A Randomized, Controlled Experimental Medicine Trial. Biol. Psychiatry 2020, 87, 538–547. [Google Scholar] [CrossRef]
- Mayo, L.M.; Asratian, A.; Lindé, J.; Holm, L.; Nätt, D.; Augier, G.; Stensson, N.; Vecchiarelli, H.A.; Balsevich, G.; Aukema, R.J.; et al. Protective Effects of Elevated Anandamide on Stress and Fear-Related Behaviors: Translational Evidence from Humans and Mice. Mol. Psychiatry 2020, 25, 993–1005. [Google Scholar] [CrossRef]
- van der Stelt, M.; Mazzola, C.; Esposito, G.; Matias, I.; Petrosino, S.; Filippis, D.D.; Micale, V.; Steardo, L.; Drago, F.; Iuvone, T.; et al. Endocannabinoids and β-Amyloid-Induced Neurotoxicity in Vivo: Effect of Pharmacological Elevation of Endocannabinoid Levels. Cell. Mol. Life Sci. CMLS 2006, 63, 1410–1424. [Google Scholar] [CrossRef]
- Ramírez, B.G.; Blázquez, C.; del Pulgar, T.G.; Guzmán, M.; de Ceballos, M.L. Prevention of Alzheimer’s Disease Pathology by Cannabinoids: Neuroprotection Mediated by Blockade of Microglial Activation. J. Neurosci. 2005, 25, 1904–1913. [Google Scholar] [CrossRef]
- Tolón, R.M.; Núñez, E.; Pazos, M.R.; Benito, C.; Castillo, A.I.; Martínez-Orgado, J.A.; Romero, J. The Activation of Cannabinoid CB2 Receptors Stimulates in Situ and in Vitro Beta-Amyloid Removal by Human Macrophages. Brain Res. 2009, 1283, 148–154. [Google Scholar] [CrossRef]
- Ehrhart, J.; Obregon, D.; Mori, T.; Hou, H.; Sun, N.; Bai, Y.; Klein, T.; Fernandez, F.; Tan, J.; Shytle, R.D. Stimulation of Cannabinoid Receptor 2 (CB2) Suppresses Microglial Activation. J. Neuroinflam. 2005, 2, 29. [Google Scholar] [CrossRef]
- Benito, C.; Núñez, E.; Tolón, R.M.; Carrier, E.J.; Rábano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 Receptors and Fatty Acid Amide Hydrolase Are Selectively Overexpressed in Neuritic Plaque-Associated Glia in Alzheimer’s Disease Brains. J Neurosci 2003, 23, 11136–11141. [Google Scholar] [CrossRef]
- Manuel, I.; Román, E.G.D.S.; Giralt, M.T.; Ferrer, I.; Rodríguez-Puertas, R. Type-1 Cannabinoid Receptor Activity during Alzheimer’s Disease Progression. J. Alzheimers Dis. 2014, 42, 761–766. [Google Scholar] [CrossRef]
- Micale, V.; Mazzola, C.; Drago, F. Endocannabinoids and Neurodegenerative Diseases. Pharmacol. Res. 2007, 56, 382–392. [Google Scholar] [CrossRef]
- Marsicano, G.; Goodenough, S.; Monory, K.; Hermann, H.; Eder, M.; Cannich, A.; Azad, S.C.; Cascio, M.G.; Gutiérrez, S.O.; van der Stelt, M.; et al. CB1 Cannabinoid Receptors and On-Demand Defense against Excitotoxicity. Science 2003, 302, 84–88. [Google Scholar] [CrossRef]
- Colangeli, R.; Morena, M.; Pittman, Q.J.; Hill, M.N.; Teskey, G.C. Anandamide Signaling Augmentation Rescues Amygdala Synaptic Function and Comorbid Emotional Alterations in a Model of Epilepsy. J. Neurosci. 2020, 40, 6068–6081. [Google Scholar] [CrossRef]
- Grillo, A.; Chemi, G.; Brogi, S.; Brindisi, M.; Relitti, N.; Fezza, F.; Fazio, D.; Castelletti, L.; Perdona, E.; Wong, A.; et al. Development of Novel Multipotent Compounds Modulating Endocannabinoid and Dopaminergic Systems. Eur. J. Med. Chem. 2019, 183, 111674. [Google Scholar] [CrossRef]
- Butini, S.; Brindisi, M.; Gemma, S.; Minetti, P.; Cabri, W.; Gallo, G.; Vincenti, S.; Talamonti, E.; Borsini, F.; Caprioli, A.; et al. Discovery of Potent Inhibitors of Human and Mouse Fatty Acid Amide Hydrolases. J. Med. Chem. 2012, 55, 6898–6915. [Google Scholar] [CrossRef]
- Colangeli, R.; Pierucci, M.; Benigno, A.; Campiani, G.; Butini, S.; Di Giovanni, G. The FAAH Inhibitor URB597 Suppresses Hippocampal Maximal Dentate Afterdischarges and Restores Seizure-Induced Impairment of Short and Long-Term Synaptic Plasticity. Sci. Rep. 2017, 7, 11152. [Google Scholar] [CrossRef]
- von Rüden, E.L.; Bogdanovic, R.M.; Wotjak, C.T.; Potschka, H. Inhibition of Monoacylglycerol Lipase Mediates a Cannabinoid 1-Receptor Dependent Delay of Kindling Progression in Mice. Neurobiol. Dis. 2015, 77, 238–245. [Google Scholar] [CrossRef]
- Wei, D.; Dinh, D.; Lee, D.; Li, D.; Anguren, A.; Moreno-Sanz, G.; Gall, C.M.; Piomelli, D. Enhancement of Anandamide-Mediated Endocannabinoid Signaling Corrects Autism-Related Social Impairment. Cannabis Cannabinoid Res. 2016, 1, 81–89. [Google Scholar] [CrossRef]
- Litvin, Y.; Phan, A.; Hill, M.N.; Pfaff, D.W.; Mcewen, B.S. CB1 Receptor Signaling Regulates Social Anxiety and Memory. Genes Brain Behav. 2013, 12, 479–489. [Google Scholar] [CrossRef]
- Terzian, A.L.B.; Micale, V.; Wotjak, C.T. Cannabinoid Receptor Type 1 Receptors on GABAergic vs. Glutamatergic Neurons Differentially Gate Sex-Dependent Social Interest in Mice. Eur. J. Neurosci. 2014, 40, 2293–2298. [Google Scholar] [CrossRef]
- Cassano, T.; Gaetani, S.; MacHeda, T.; Laconca, L.; Romano, A.; Morgese, M.G.; Cimmino, C.S.; Chiarotti, F.; Bambico, F.R.; Gobbi, G.; et al. Evaluation of the Emotional Phenotype and Serotonergic Neurotransmission of Fatty Acid Amide Hydrolase-Deficient Mice. Psychopharmacology 2011, 214, 465–476. [Google Scholar] [CrossRef]
- Trezza, V.; Damsteegt, R.; Manduca, A.; Petrosino, S.; van Kerkhof, L.W.M.; Pasterkamp, R.J.; Zhou, Y.; Campolongo, P.; Cuomo, V.; Marzo, V.D.; et al. Endocannabinoids in Amygdala and Nucleus Accumbens Mediate Social Play Reward in Adolescent Rats. J. Neurosci. 2012, 32, 14899–14908. [Google Scholar] [CrossRef]
- Wei, D.; Lee, D.Y.; Cox, C.D.; Karsten, C.A.; Penagarikano, O.; Geschwind, D.H.; Gall, C.M.; Piomelli, D. Endocannabinoid Signaling Mediates Oxytocin-Driven Social Reward. Proc. Natl. Acad. Sci. USA 2015, 112, 14084–14089. [Google Scholar] [CrossRef]
- Lim, J.; Igarashi, M.; Jung, K.M.; Butini, S.; Campiani, G.; Piomelli, D. Endocannabinoid Modulation of Predator Stress-Induced Long-Term Anxiety in Rats. Neuropsychopharmacology 2016, 41, 1329–1339. [Google Scholar] [CrossRef]
- Grillo, A.; Fezza, F.; Chemi, G.; Colangeli, R.; Brogi, S.; Fazio, D.; Federico, S.; Papa, A.; Relitti, N.; Di Maio, R.; et al. Selective Fatty Acid Amide Hydrolase Inhibitors as Potential Novel Antiepileptic Agents. ACS Chem. Neurosci. 2021, 12, 1716–1736. [Google Scholar] [CrossRef]
- Brindisi, M.; Borrelli, G.; Brogi, S.; Grillo, A.; Maramai, S.; Paolino, M.; Benedusi, M.; Pecorelli, A.; Valacchi, G.; Di Cesare Mannelli, L.; et al. Development of Potent Inhibitors of Fatty Acid Amide Hydrolase Useful for the Treatment of Neuropathic Pain. ChemMedChem 2018, 13, 2090–2103. [Google Scholar] [CrossRef]
- Butini, S.; Gemma, S.; Campiani, G. Chapter 8: Natural Compounds and Synthetic Drugs to Target FAAH Enzyme. In New Tools to Interrogate Endocannabinoid Signalling: From Natural Compounds to Synthetic Drugs; Royal Society of Chemistry: Cambridge, UK, 2020; pp. 337–384. [Google Scholar] [CrossRef]
- Anderson, W.B.; Gould, M.J.; Torres, R.D.; Mitchell, V.A.; Vaughan, C.W. Actions of the Dual FAAH/MAGL Inhibitor JZL195 in a Murine Inflammatory Pain Model. Neuropharmacology 2014, 81, 224–230. [Google Scholar] [CrossRef]
- Sakin, Y.S.; Tanoğlu, A.; Gülşen, M. Dual FAAH and MAGL Inhibition Might Play a Key Role in Visceral Pain. Turk. J. Gastroenterol. 2018, 29, 625–626. [Google Scholar] [CrossRef]
- Ramesh, D.; Gamage, T.F.; Vanuytsel, T.; Owens, R.A.; Abdullah, R.A.; Niphakis, M.J.; Shea-Donohue, T.; Cravatt, B.F.; Lichtman, A.H. Dual Inhibition of Endocannabinoid Catabolic Enzymes Produces Enhanced Antiwithdrawal Effects in Morphine-Dependent Mice. Neuropsychopharmacology 2013, 38, 1039–1049. [Google Scholar] [CrossRef]
- Long, J.Z.; Nomura, D.K.; Vann, R.E.; Walentiny, D.M.; Booker, L.; Jin, X.; Burston, J.J.; Sim-Selley, L.J.; Lichtman, A.H.; Wiley, J.L.; et al. Dual Blockade of FAAH and MAGL Identifies Behavioral Processes Regulated by Endocannabinoid Crosstalk In Vivo. Proc. Natl. Acad. Sci. USA 2009, 106, 20270–20275. [Google Scholar] [CrossRef]
- Seillier, A.; Dominguez Aguilar, D.; Giuffrida, A. The Dual FAAH/MAGL Inhibitor JZL195 Has Enhanced Effects on Endocannabinoid Transmission and Motor Behavior in Rats as Compared to Those of the MAGL Inhibitor JZL184. Pharmacol. Biochem. Behav. 2014, 124, 153–159. [Google Scholar] [CrossRef]
- Greco, R.; Demartini, C.; Francavilla, M.; Zanaboni, A.M.; Tassorelli, C. Dual Inhibition of Faah and Magl Counteracts Migraine-like Pain and Behavior in an Animal Model of Migraine. Cells 2021, 10, 2543. [Google Scholar] [CrossRef]
- Adamson Barnes, N.S.; Mitchell, V.A.; Kazantzis, N.P.; Vaughan, C.W. Actions of the Dual FAAH/MAGL Inhibitor JZL195 in a Murine Neuropathic Pain Model. Br. J. Pharmacol. 2016, 173, 77–87. [Google Scholar] [CrossRef]
- Niphakis, M.J.; Johnson, D.S.; Ballard, T.E.; Stiff, C.; Cravatt, B.F. O-Hydroxyacetamide Carbamates as a Highly Potent and Selective Class of Endocannabinoid Hydrolase Inhibitors. ACS Chem Neurosci 2012, 3, 418–426. [Google Scholar] [CrossRef]
- Wilkerson, J.L.; Ghosh, S.; Mustafa, M.; Abdullah, R.A.; Niphakis, M.J.; Cabrera, R.; Maldonado, R.; Cravatt, B.F.; Lichtman, A.H. The Endocannabinoid Hydrolysis Inhibitor SA-57: Intrinsic Antinociceptive Effects, Augmented Morphine-Induced Antinociception, and Attenuated Heroin Seeking Behavior in Mice. Neuropharmacology 2017, 114, 156–167. [Google Scholar] [CrossRef]
- Bedse, G.; Bluett, R.J.; Patrick, T.A.; Romness, N.K.; Gaulden, A.D.; Kingsley, P.J.; Plath, N.; Marnett, L.J.; Patel, S. Therapeutic Endocannabinoid Augmentation for Mood and Anxiety Disorders: Comparative Profiling of FAAH, MAGL and Dual Inhibitors. Transl. Psychiatry 2018, 8, 92. [Google Scholar] [CrossRef]
- Gonul, A.S.; Akdeniz, F.; Taneli, F.; Donat, O.; Eker, Ç.; Vahip, S. Effect of Treatment on Serum Brain-Derived Neurotrophic Factor Levels in Depressed Patients. Eur. Arch. Psychiatry Clin. Neurosci. 2005, 255, 381–386. [Google Scholar] [CrossRef]
- Dong, B.; Shilpa, B.M.; Shah, R.; Goyal, A.; Xie, S.; Bakalian, M.J.; Suckow, R.F.; Cooper, T.B.; Mann, J.J.; Arango, V.; et al. Dual Pharmacological Inhibitor of Endocannabinoid Degrading Enzymes Reduces Depressive-like Behavior in Female Rats. J. Psychiatr. Res. 2020, 120, 103–112. [Google Scholar] [CrossRef]
- Wise, L.E.; Long, K.A.; Abdullah, R.A.; Long, J.Z.; Cravatt, B.F.; Lichtman, A.H. Dual Fatty Acid Amide Hydrolase and Monoacylglycerol Lipase Blockade Produces THC-like Morris Water Maze Deficits in Mice. ACS Chem. Neurosci. 2012, 3, 369–378. [Google Scholar] [CrossRef]
- Yesilyurt, O.; Cayirli, M.; Sakin, Y.S.; Seyrek, M.; Akar, A.; Dogrul, A. Systemic and Spinal Administration of FAAH, MAGL Inhibitors and Dual FAAH/MAGL Inhibitors Produce Antipruritic Effect in Mice. Arch. Dermatol. Res. 2016, 308, 335–345. [Google Scholar] [CrossRef]
- Butini, S.; Gemma, S.; Brindisi, M.; Maramai, S.; Minetti, P.; Celona, D.; Napolitano, R.; Borsini, F.; Cabri, W.; Fezza, F.; et al. Identification of a Novel Arylpiperazine Scaffold for Fatty Acid Amide Hydrolase Inhibition with Improved Drug Disposition Properties. Bioorg. Med. Chem. Lett. 2013, 23, 492–495. [Google Scholar] [CrossRef]
- Cisneros, J.A.; Björklund, E.; González-Gil, I.; Hu, Y.; Canales, Á.; Medrano, F.J.; Romero, A.; Ortega-Gutiérrez, S.; Fowler, C.J.; López-Rodríguez, M.L. Structure-Activity Relationship of a New Series of Reversible Dual Monoacylglycerol Lipase/Fatty Acid Amide Hydrolase Inhibitors. J. Med. Chem. 2012, 55, 824–836. [Google Scholar] [CrossRef]
- Korhonen, J.; Kuusisto, A.; Van Bruchem, J.; Patel, J.Z.; Laitinen, T.; Navia-Paldanius, D.; Laitinen, J.T.; Savinainen, J.R.; Parkkari, T.; Nevalainen, T.J. Piperazine and Piperidine Carboxamides and Carbamates as Inhibitors of Fatty Acid Amide Hydrolase (FAAH) and Monoacylglycerol Lipase (MAGL). Bioorg. Med. Chem. 2014, 22, 6694–6705. [Google Scholar] [CrossRef]
- Chang, J.W.; Niphakis, M.J.; Lum, K.M.; Cognetta, A.B.; Wang, C.; Matthews, M.L.; Niessen, S.; Buczynski, M.W.; Parsons, L.H.; Cravatt, B.F. Highly Selective Inhibitors of Monoacylglycerol Lipase Bearing a Reactive Group That Is Bioisosteric with Endocannabinoid Substrates. Chem. Biol. 2012, 19, 579–588. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Brogi, S.; Maramai, S.; Grillo, A.; Borrelli, G.; Butini, S.; Novellino, E.; Allarà, M.; Ligresti, A.; Campiani, G.; et al. Harnessing the Pyrroloquinoxaline Scaffold for FAAH and MAGL Interaction: Definition of the Structural Determinants for Enzyme Inhibition. RSC Adv. 2016, 6, 64651–64664. [Google Scholar] [CrossRef]
- Gühring, H.; Hamza, M.; Sergejeva, M.; Ates, M.; Kotalla, C.E.; Ledent, C.; Brune, K. A Role for Endocannabinoids in Indomethacin-Induced Spinal Antinociception. Eur. J. Pharmacol. 2002, 454, 153–163. [Google Scholar] [CrossRef]
- Cipriano, M.; Björklund, E.; Wilson, A.A.; Congiu, C.; Onnis, V.; Fowler, C.J. Inhibition of Fatty Acid Amide Hydrolase and Cyclooxygenase by the N-(3-Methylpyridin-2-Yl)Amide Derivatives of Flurbiprofen and Naproxen. Eur. J. Pharmacol. 2013, 720, 383–390. [Google Scholar] [CrossRef]
- Sasso, O.; Wagner, K.; Morisseau, C.; Inceoglu, B.; Hammock, B.D.; Piomelli, D. Peripheral FAAH and Soluble Epoxide Hydrolase Inhibitors Are Synergistically Antinociceptive. Pharmacol. Res. 2015, 97, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Rampa, A.; Bartolini, M.; Bisi, A.; Belluti, F.; Gobbi, S.; Andrisano, V.; Ligresti, A.; Di Marzo, V. The First Dual ChE/FAAH Inhibitors: New Perspectives for Alzheimer’s Disease? ACS Med. Chem. Lett. 2012, 3, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Parsons, L.H.; Hurd, Y.L. Endocannabinoid Signalling in Reward and Addiction. Nat. Rev. Neurosci. 2015, 16, 579–594. [Google Scholar] [CrossRef]
- Spadoni, G.; Bedini, A.; Furiassi, L.; Mari, M.; Mor, M.; Scalvini, L.; Lodola, A.; Ghidini, A.; Lucini, V.; Dugnani, S.; et al. Identification of Bivalent Ligands with Melatonin Receptor Agonist and Fatty Acid Amide Hydrolase (FAAH) Inhibitory Activity That Exhibit Ocular Hypotensive Effect in the Rabbit. J. Med. Chem. 2018, 61, 7902–7916. [Google Scholar] [CrossRef] [PubMed]
- Blobaum, A.L.; Marnett, L.J. Structural and Functional Basis of Cyclooxygenase Inhibition. J. Med. Chem. 2007, 50, 1425–1441. [Google Scholar] [CrossRef]
- Aiello, F.; Carullo, G.; Badolato, M.; Brizzi, A. TRPV1–FAAH–COX: The Couples Game in Pain Treatment. ChemMedChem 2016, 1, 1686–1694. [Google Scholar] [CrossRef]
- Morphy, R.; Rankovic, Z. Designed Multiple Ligands. An Emerging Drug Discovery Paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Lehár, J.; Keith, C.T. Multi-Target Therapeutics: When the Whole Is Greater than the Sum of the Parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
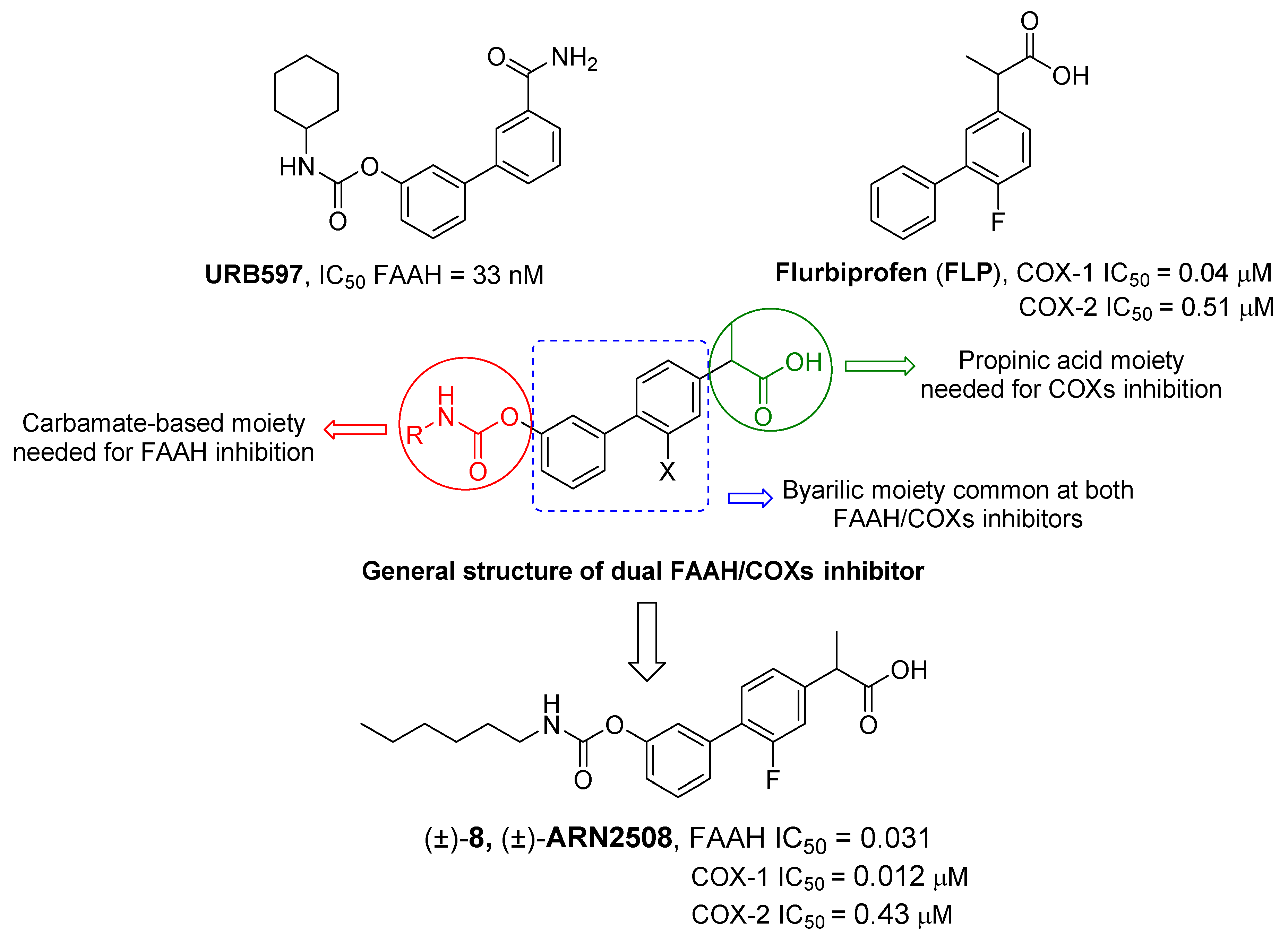
- Bertolacci, L.; Romeo, E.; Veronesi, M.; Magotti, P.; Albani, C.; Dionisi, M.; Lambruschini, C.; Scarpelli, R.; Cavalli, A.; De Vivo, M.; et al. A Binding Site for Nonsteroidal Anti-Inflammatory Drugs in Fatty Acid Amide Hydrolase. J. Am. Chem. Soc. 2013, 135, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.D.; Habrant, D.; Scarpelli, R.; Migliore, M.; Albani, C.; Bertozzi, S.M.; Dionisi, M.; Tarozzo, G.; Piomelli, D.; Cavalli, A.; et al. Identification and Characterization of Carprofen as a Multitarget Fatty Acid Amide Hydrolase/Cyclooxygenase Inhibitor. J. Med. Chem. 2012, 55, 8809–8826. [Google Scholar] [CrossRef]
- Sasso, O.; Migliore, M.; Habrant, D.; Armirotti, A.; Albani, C.; Summa, M.; Moreno-Sanz, G.; Scarpelli, R.; Piomelli, D. Multitarget Fatty Acid Amide Hydrolase/Cyclooxygenase Blockade Suppresses Intestinal Inflammation and Protects against Nonsteroidal Anti-Inflammatory Drug-Dependent Gastrointestinal Damage. FASEB J. 2015, 29, 2616–2627. [Google Scholar] [CrossRef]
- Seierstad, M.; Breitenbucher, J.G. Discovery and Development of Fatty Acid Amide Hydrolase (FAAH) Inhibitors. J. Med. Chem. 2008, 51, 7327–7343. [Google Scholar] [CrossRef]
- Bhattacharyya, D.K.; Lecomte, M.; Rieke, C.J.; Garavito, R.M.; Smith, W.L. Involvement of Arginine 120, Glutamate 524, and Tyrosine 355 in the Binding of Arachidonate and 2-Phenylpropionic Acid Inhibitors to the Cyclooxygenase Active Site of Ovine Prostaglandin Endoperoxide H Synthase-1. J. Biol. Chem. 1996, 271, 2179–2184. [Google Scholar] [CrossRef]
- Palermo, G.; Favia, A.D.; Convertino, M.; De Vivo, M. The Molecular Basis for Dual Fatty Acid Amide Hydrolase (FAAH)/Cyclooxygenase (COX) Inhibition. ChemMedChem 2016, 11, 1252–1258. [Google Scholar] [CrossRef]
- Migliore, M.; Habrant, D.; Sasso, O.; Albani, C.; Bertozzi, S.M.; Armirotti, A.; Piomelli, D.; Scarpelli, R. Potent Multitarget FAAH-COX Inhibitors: Design and Structure-Activity Relationship Studies. Eur. J. Med. Chem. 2016, 109, 216–237. [Google Scholar] [CrossRef]
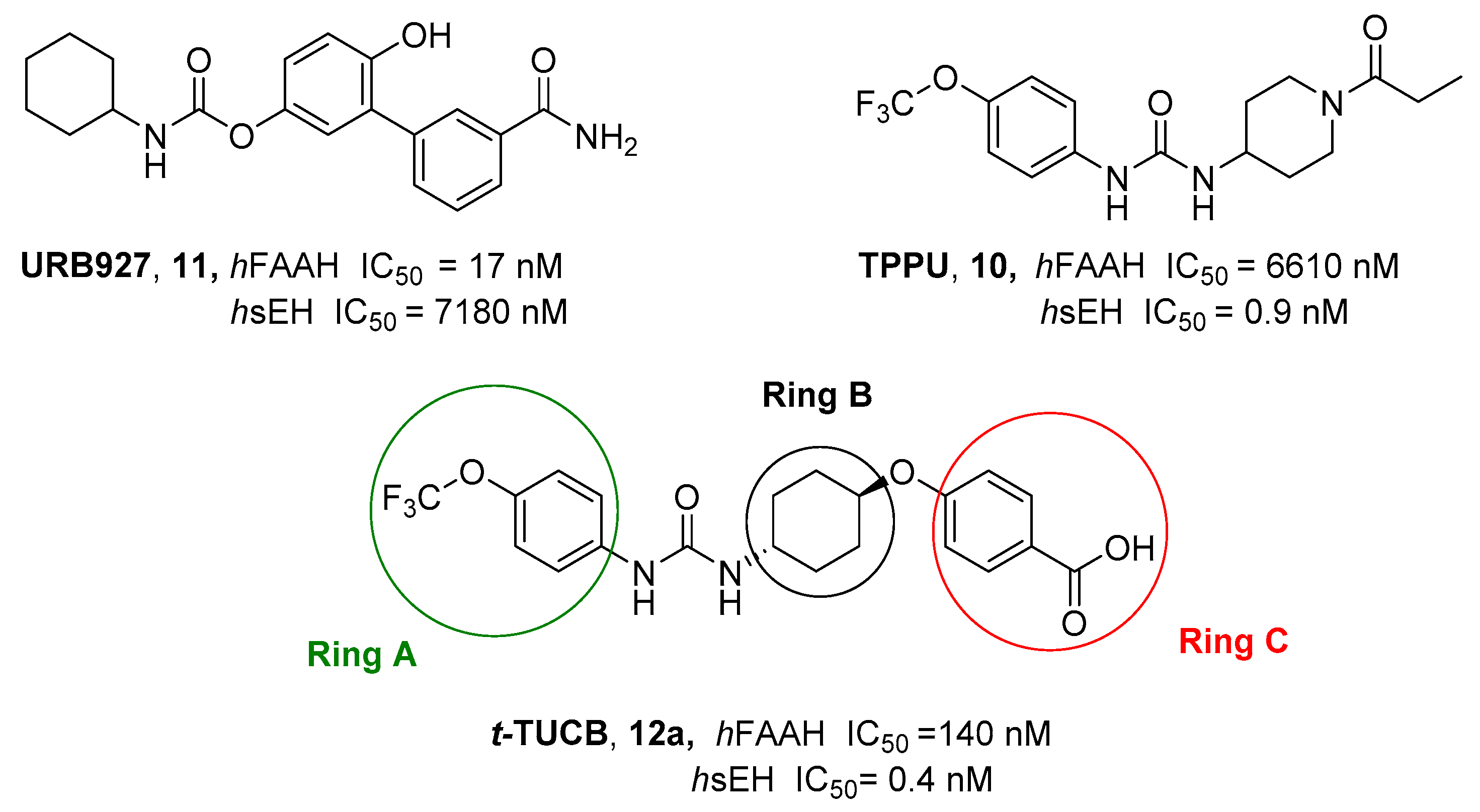
- Morisseau, C.; Hammock, B.D. Impact of Soluble Epoxide Hydrolase and Epoxyeicosanoids on Human Health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef]
- Zhang, G.; Kodani, S.; Hammock, B.D. Stabilized Epoxygenated Fatty Acids Regulate Inflammation, Pain, Angiogenesis and Cancer. Prog. Lipid Res. 2014, 53, 108–123. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Hohmann, A.G. Cannabinoid CB2 Receptors: A Therapeutic Target for the Treatment of Inflammatory and Neuropathic Pain. Br. J. Pharmacol. 2008, 153, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Kodani, S.D.; Bhakta, S.; Hwang, S.H.; Pakhomova, S.; Newcomer, M.E.; Morisseau, C.; Hammock, B.D. Identification and Optimization of Soluble Epoxide Hydrolase Inhibitors with Dual Potency towards Fatty Acid Amide Hydrolase. Bioorg. Med. Chem. Lett. 2018, 28, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Kodani, S.D.; Wan, D.; Wagner, K.M.; Hwang, S.H.; Morisseau, C.; Hammock, B.D. Design and Potency of Dual Soluble Epoxide Hydrolase/Fatty Acid Amide Hydrolase Inhibitors. ACS Omega 2018, 3, 14076–14086. [Google Scholar] [CrossRef] [PubMed]
- Weinstock, M. Selectivity of Cholinesterase Inhibition. CNS Drugs 1999, 12, 307–323. [Google Scholar] [CrossRef]
- Greig, N.H.; Utsuki, T.; Ingram, D.K.; Wang, Y.; Pepeu, G.; Scali, C.; Yu, Q.-S.; Mamczarz, J.; Holloway, H.W.; Giordano, T.; et al. Selective Butyrylcholinesterase Inhibition Elevates Brain Acetylcholine, Augments Learning and Lowers Alzheimer β-Amyloid Peptide in Rodent. Proc. Natl. Acad. Sci. USA 2005, 102, 17213–17218. [Google Scholar] [CrossRef]
- Fernández, J.A.; Rojo, L.E.; Kulji\vs, R.O.; Maccioni, R.B. The Damage Signals Hypothesis of Alzheimer’s Disease Pathogenesis. J. Alzheimers Dis. JAD 2008, 14, 329–333. [Google Scholar] [CrossRef]
- Jung, K.-M.; Astarita, G.; Yasar, S.; Vasilevko, V.; Cribbs, D.H.; Head, E.; Cotman, C.W.; Piomelli, D. An Amyloid Β42-Dependent Deficit in Anandamide Mobilization Is Associated with Cognitive Dysfunction in Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 1522–1532. [Google Scholar] [CrossRef]
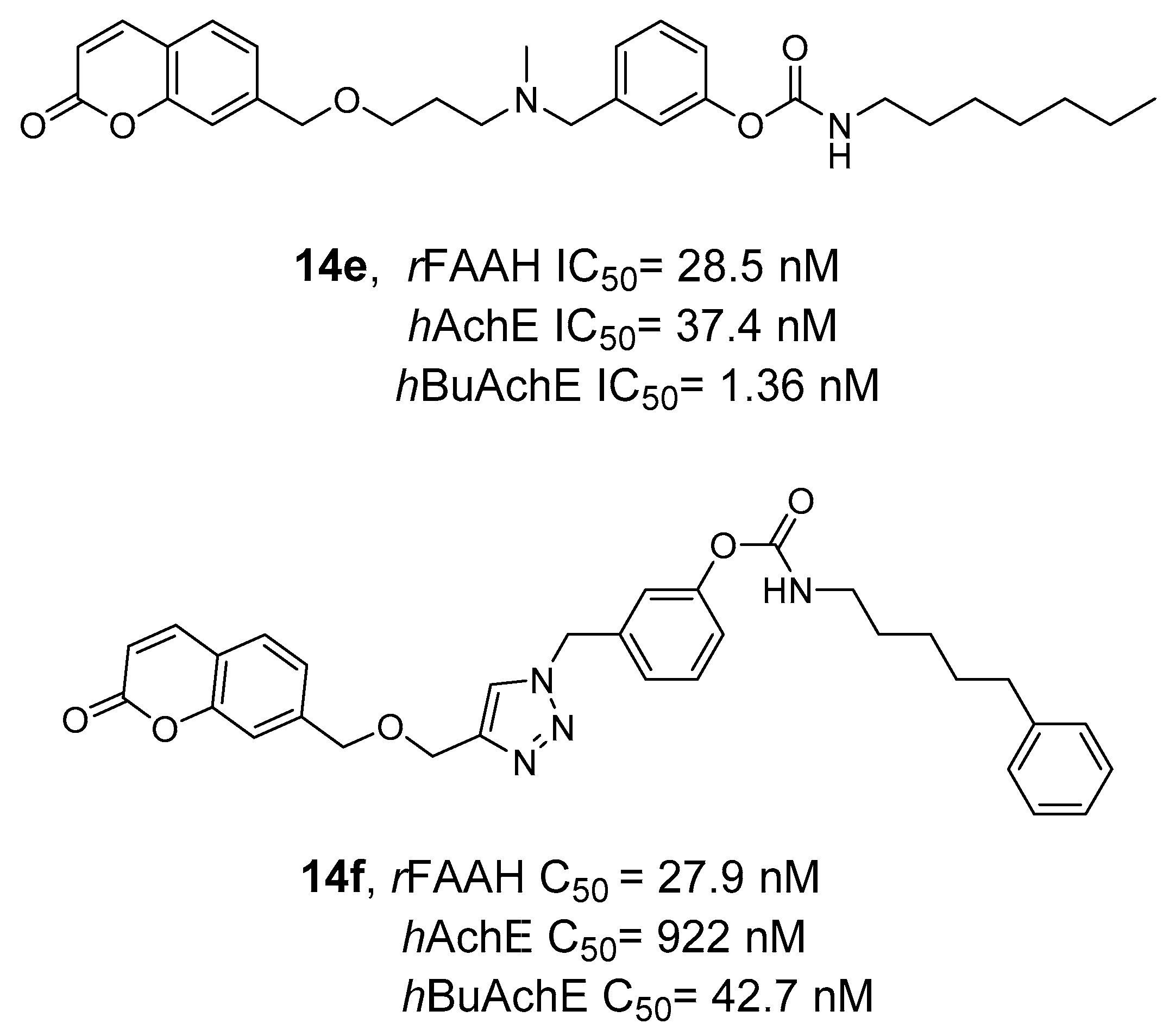
- Montanari, S.; Scalvini, L.; Bartolini, M.; Belluti, F.; Gobbi, S.; Andrisano, V.; Ligresti, A.; Di Marzo, V.; Rivara, S.; Mor, M.; et al. Fatty Acid Amide Hydrolase (FAAH), Acetylcholinesterase (AChE), and Butyrylcholinesterase (BuChE): Networked Targets for the Development of Carbamates as Potential Anti-Alzheimer’s Disease Agents. J. Med. Chem. 2016, 59, 6387–6406. [Google Scholar] [CrossRef]
- Rudolph, S.; Dahlhaus, H.; Hanekamp, W.; Albers, C.; Barth, M.; Michels, G.; Friedrich, D.; Lehr, M. Aryl N-[ω-(6-Fluoroindol-1-Yl)Alkyl]Carbamates as Inhibitors of Fatty Acid Amide Hydrolase, Monoacylglycerol Lipase, and Butyrylcholinesterase: Structure-Activity Relationships and Hydrolytic Stability. ACS Omega 2021, 6, 13466–13483. [Google Scholar] [CrossRef]
- Solinas, M.; Justinova, Z.; Goldberg, S.R.; Tanda, G. Anandamide Administration Alone and after Inhibition of Fatty Acid Amide Hydrolase (FAAH) Increases Dopamine Levels in the Nucleus Accumbens Shell in Rats. J. Neurochem. 2006, 98, 408–419. [Google Scholar] [CrossRef] [PubMed]
- Okita, K.; Mandelkern, M.A.; London, E.D. Cigarette Use and Striatal Dopamine D2/3 Receptors: Possible Role in the Link between Smoking and Nicotine Dependence. Int. J. Neuropsychopharmacol. 2016, 19, pyw074. [Google Scholar] [CrossRef] [PubMed]
- De Simone, A.; Ruda, G.F.; Albani, C.; Tarozzo, G.; Piomelli, D.; Cavalli, A.; Bottegoni, G. Applying a Multitarget Rational Drug Design Strategy: The First Set of Modulators with Potent and Balanced Activity toward Dopamine D3 Receptor and Fatty Acid Amide Hydrolase. Chem. Commun. 2014, 50, 4904–4907. [Google Scholar] [CrossRef] [PubMed]
- Brindisi, M.; Butini, S.; Franceschini, S.; Brogi, S.; Trotta, F.; Ros, S.; Cagnotto, A.; Salmona, M.; Casagni, A.; Andreassi, M.; et al. Targeting Dopamine D3 and Serotonin 5-HT1A and 5-HT2A Receptors for Developing Effective Antipsychotics: Synthesis, Biological Characterization, and Behavioral Studies. J. Med. Chem. 2014, 57, 9578–9597. [Google Scholar] [CrossRef] [PubMed]
- Vargas, H.O.; Nunes, S.O.V.; de Castro, M.R.P.; Vargas, M.M.; Barbosa, D.S.; Bortolasci, C.C.; Venugopal, K.; Dodd, S.; Berk, M. Oxidative Stress and Inflammatory Markers Are Associated with Depression and Nicotine Dependence. Neurosci. Lett. 2013, 544, 136–140. [Google Scholar] [CrossRef]
- Porcella, A.; Maxia, C.; Gessa, G.L.; Pani, L. The Synthetic Cannabinoid WIN55212-2 Decreases the Intraocular Pressure in Human Glaucoma Resistant to Conventional Therapies. Eur. J. Neurosci. 2001, 13, 409–412. [Google Scholar] [CrossRef]
- Jockers, R.; Delagrange, P.; Dubocovich, M.L.; Markus, R.P.; Renault, N.; Tosini, G.; Cecon, E.; Zlotos, D.P. Update on Melatonin Receptors: IUPHAR Review 20. Br. J. Pharmacol. 2016, 173, 2702–2725. [Google Scholar] [CrossRef]
- Rubino, T.; Realini, N.; Castiglioni, C.; Guidali, C.; Viganó, D.; Marras, E.; Petrosino, S.; Perletti, G.; Maccarrone, M.; Di Marzo, V.; et al. Role in Anxiety Behavior of the Endocannabinoid System in the Prefrontal Cortex. Cereb. Cortex 2008, 18, 1292–1301. [Google Scholar] [CrossRef]
- Ho, K.W.; Ward, N.J.; Calkins, D.J. TRPV1: A Stress Response Protein in the Central Nervous System. Am. J. Neurodegener. Dis. 2012, 1, 1–14. [Google Scholar]
- Maione, S.; De Petrocellis, L.; de Novellis, V.; Moriello, A.S.; Petrosino, S.; Palazzo, E.; Rossi, F.S.; Woodward, D.F.; Di Marzo, V. Analgesic Actions of N-Arachidonoyl-Serotonin, a Fatty Acid Amide Hydrolase Inhibitor with Antagonistic Activity at Vanilloid TRPV1 Receptors. Br. J. Pharmacol. 2007, 150, 766–781. [Google Scholar] [CrossRef]
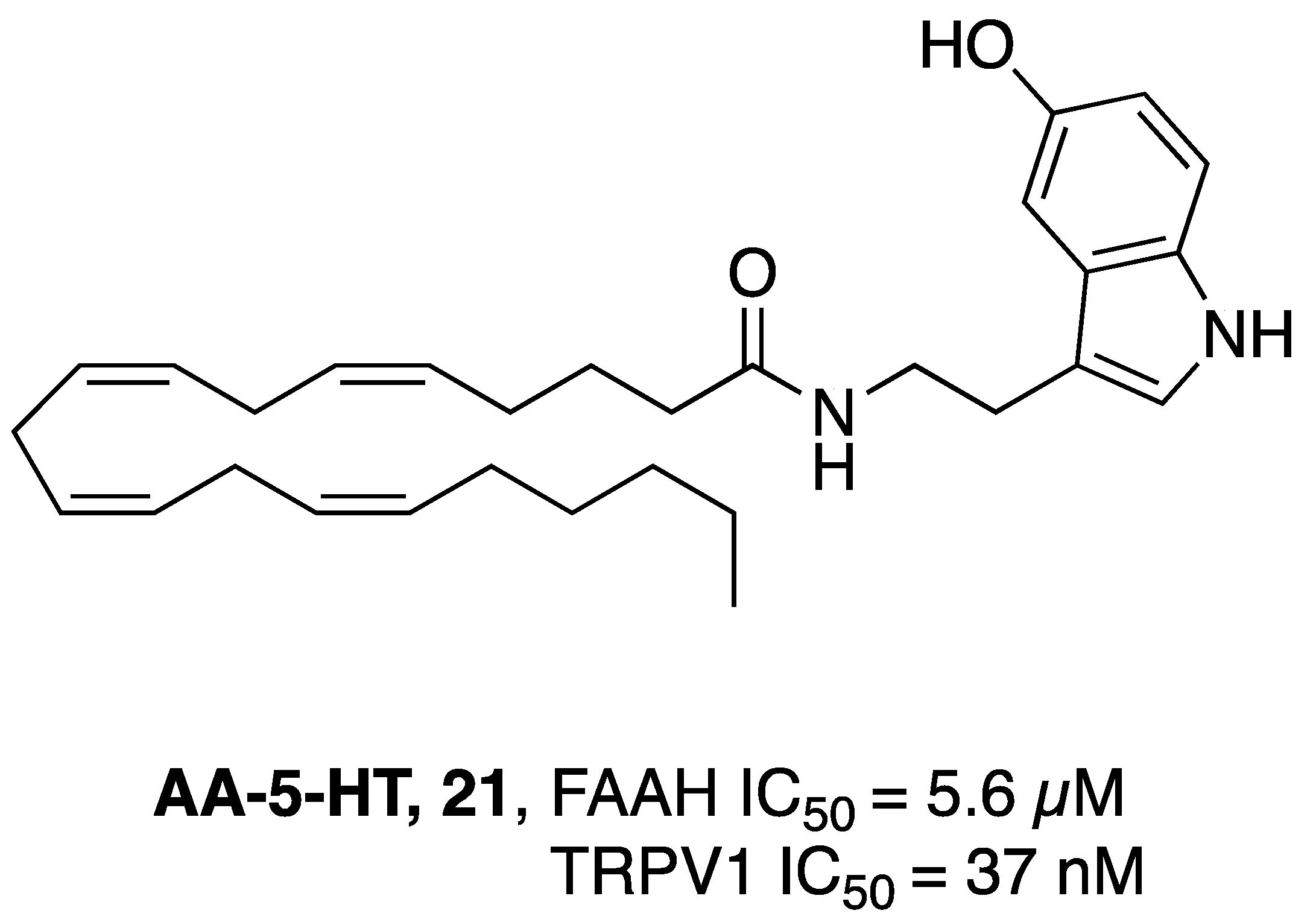
- Micale, V.; Cristino, L.; Tamburella, A.; Petrosino, S.; Leggio, G.M.; Drago, F.; Di Marzo, V. Anxiolytic Effects in Mice of a Dual Blocker of Fatty Acid Amide Hydrolase and Transient Receptor Potential Vanilloid Type-1 Channels. Neuropsychopharmacology 2009, 34, 593–606. [Google Scholar] [CrossRef] [PubMed]
- Navarria, A.; Tamburella, A.; Iannotti, F.A.; Micale, V.; Camillieri, G.; Gozzo, L.; Verde, R.; Imperatore, R.; Leggio, G.M.; Drago, F.; et al. The Dual Blocker of FAAH/TRPV1 N-Arachidonoylserotonin Reverses the Behavioral Despair Induced by Stress in Rats and Modulates the HPA-Axis. Pharmacol. Res. 2014, 87, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Micale, V.; Cristino, L.; Tamburella, A.; Petrosino, S.; Leggio, G.M.; Drago, F.; Di Marzo, V. Altered Responses of Dopamine D3 Receptor Null Mice to Excitotoxic or Anxiogenic Stimuli: Possible Involvement of the Endocannabinoid and Endovanilloid Systems. Neurobiol. Dis. 2009, 36, 70–80. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| General Structure A | General Structure B | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
 |  | ||||||||||
| Cpds | Spacer | R1 | Hydrolysis Inhibition (µM) | Cpds | Spacer | R1 | Hydrolysis Inhibition (µM) | ||||
| hrMAGL | 2-OG | AEA | hrMAGL | 2-OG | AEA | ||||||
| (±)-1 | (CH2)5 |  | 4.31 | 1.8 | 5.1 | (±)-2 | (CH2)2 |  | 16 | 10 | 0.28 |
| (R)-1a | (CH2)5 |  | 33 ± 5% | 4.9 | 3.9 | (R)-2a | (CH2)2 |  | 2.4 | 0.68 | 0.29 |
| (S)-1b | (CH2)5 |  | (45 ± 5%) a | 5.1 | 4.5 | (S)-2b | (CH2)2 |  | n.i b | 70 | 0.34 |
 | |||||
|---|---|---|---|---|---|
| Cpds | R1 | R2 | X | MAGL IC50 (nM) a | hrFAAH IC50 (nM) b |
| 3a |  |  | N | 7.8 | 89 |
| 3b |  |  | N | 5.5 | 23 |
| 3c |  |  | N | 74 | 76 c |
| 3d |  |  | N | 660 | 3.4 |
| 3e |  |  | CH | 0.7 | 622 |
 | |||
|---|---|---|---|
| Cpds | R1 | MAGL a IC50 (nM) | FAAH b IC50 (nM) |
| 4a | H | 37.0 | 44.7 |
| 4b | 7-F | 10.7 | 49.9 |
| 4c | 7-Cl | 32.4 | 95.5 |
| 4d | 7,8-diMe | 32.4 | 80.1 |
 | ||||
|---|---|---|---|---|
| Compounds | R1 | IC50 (µM) ± SD | ||
| FAAH | COX-1 | COX-2 | ||
| Carprofen | H | 76.6 ± 19.7 | 22.3 ± 6.6 | 3.9 ± 1.0 |
| (S)-(+)–Carprofen | H | 64.2 ± 3.6 | 5.6 ± 0.1 | 5.3 ± 3.0 |
| (R)-(−)–Carprofen | H | >100 | >100 | >100 |
| (±)-6 |  | 22.0 ± 4.2 | 74.3 ± 28.0 | 72.3 ± 28.0 |
| (R)-(−)–6a |  | 14.9 ± 1.6 | >100 | >100 |
| (S)-(+)–6b |  | >100 | 45.0 ± 0.3 | 46.5 ± 4.3 |
| (±)-7 |  | 84.8 ± 10.6 | 30.0 ± 12.1 | 27.8 ± 9.7 |
| (R)-(−)–7a |  | 53.2 ± 22.6 | >100 | >100 |
| (S)-(+)–7b |  | >100 | 4.1 ± 2.8 | 2.5 ± 1.4 |
 | |||||
|---|---|---|---|---|---|
| Cpds | R1 | R2 | IC50 (µM) ± SD | ||
| FAAH | COX-1 | COX-2 | |||
| (±)-8 |  | -F | 0.031 ± 0.002 | 0.012 ± 0.002 | 0.43 ± 0.025 |
| 9a |  | -F | 0.063 ± 0.010 | 2.1 ± 0.1 | 0.24 ± 0.04 |
| 9b |  | -Cl | 0.023 ± 0.008 | 0.009 ± 0.001 | 0.73 ± 0.21 |
| 9c |  | -CH3 | 0.010 ± 0.001 | 0.011 ± 0.001 | 0.40 ± 0.31 |
| 9d |  | -CF3 | 0.005 ± 0.001 | 0.01 ± 0.003 | 0.2 ± 0.08 |
| (−)-8 |  | -F | 0.0099 ± 0.002 | 4.0 ± 1.3 | 22.8 ± 8.7 |
| (+)-8 |  | -F | 0.0094 ± 0.003 | 0.00029 ± 0.0004 | 0.050 ± 0.012 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpds | Ring B | R1 | IC50 (nM) | |||
| hFAAH | hsEP | mFAAH | rFAAH | |||
| t-TUCB (12a) |  | -OH | 0.8 | 140 | - | - |
| 12b |  | -OH | 7 | 170 | n.t | n.t |
| 12c |  | -OMe | 7 | 35 | n.t | n.t |
| 12d |  | -OBn | 3 | 24 | 510 | >10,000 |
| 12e |  | -NHCH2CO2Me | 3 | 30 | n.t | n.t |
 | |||||||
|---|---|---|---|---|---|---|---|
| Cpds | R | IC50 (nM) | |||||
| hFAAH a | hsEP a | mFAAH a | mFAAH b | mFAAH a | rFAAH b | ||
| 13a |  | 8 | 5 | 1400 | 66 | >10,000 | 330 |
| 13b |  | 8 | 7 | >10,000 | 290 | >10,000 | 710 |
| 13c |  | 3 | 60 | 560 | 28 | >10,000 | 110 |
| 13d |  | 3 | 9 | >10,000 | 340 | >10,000 | 1100 |
 | |||||||
|---|---|---|---|---|---|---|---|
| Cpds | Ar | R1 | IC50 (nM) | ||||
| rFAAH a | rFAAH b | hrFAAH b | hrAChE | hrBuChE | |||
| 14a | A |  | 280 | 50 | 2260 | 6647.9 | 1.57 |
| 14b | A |  | 5590 | 1820 | 14,840 | 119 | 11.2 |
| 14c | B |  | 370 | 40 | 520 | 89.5 | 1.71 |
| 14d | B |  | 4310 | 2710 | 39,040 | 139 | 27.6 |
 | ||||||
|---|---|---|---|---|---|---|
| Cpds | R | IC50 (µM) | Stability (%) a | |||
| hrFAAH | hrMAGL | hrBuChE | PBS Buffer (pH 7.4) | Porcine Blood Plasma | ||
| 15a |  | 0.029 | n.a. | 4.3 | >95 | 83 ± 9 |
| 15b |  | 0.038 | 0.038 | 2.3 | 73 ± 6 | 28 ± 4 |
| 15c |  | 0.18 | n.a. | 0.55 | >95 | 80 ± 3 |
 | |||||||
|---|---|---|---|---|---|---|---|
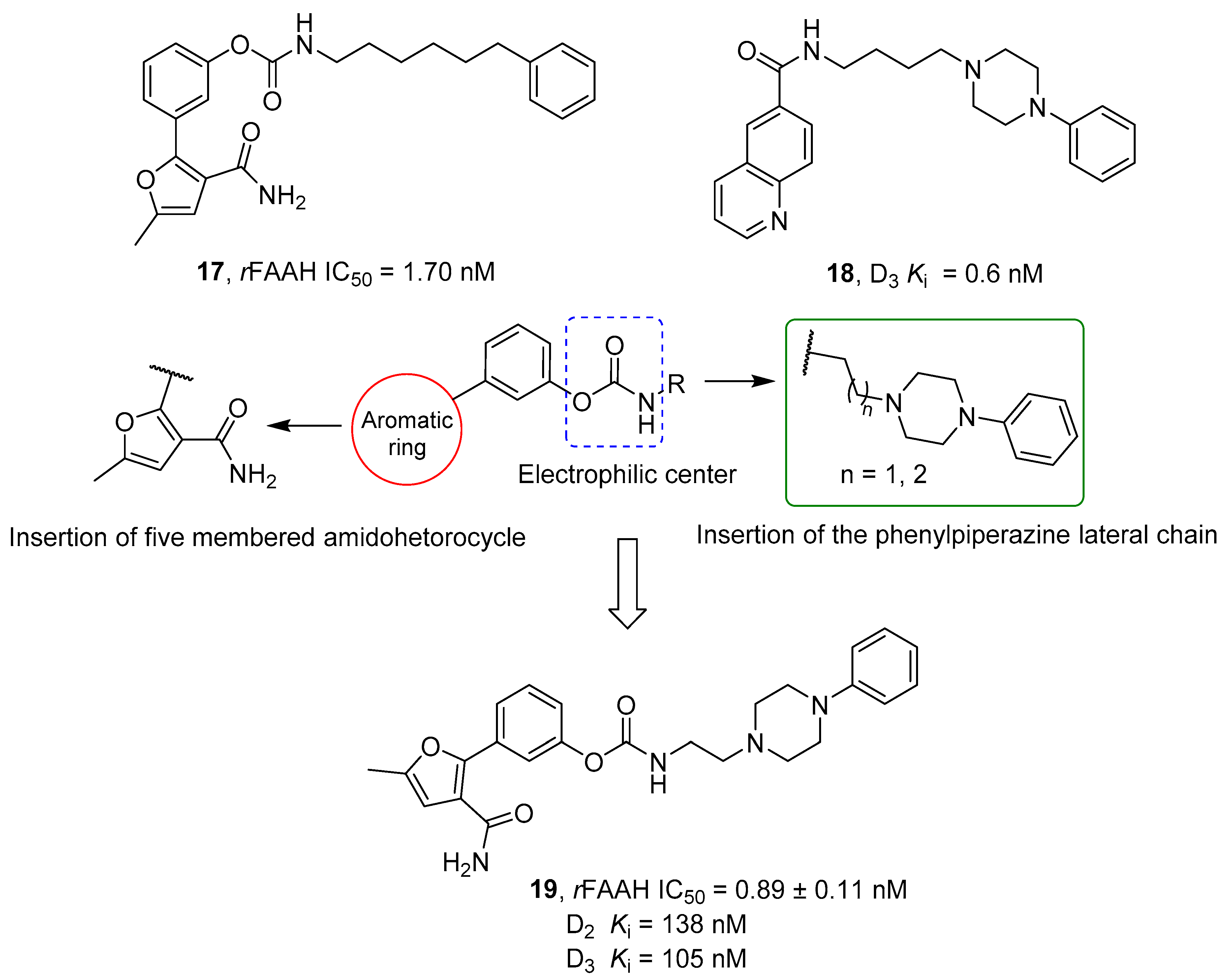
| Cpds | n | R | rFAAH IC50 (nM) | hFAAH IC50 (nM) | D3 EC50 (nM) | D2 EC50 (nM) | CB1 EC50 (nM) |
| 16a | 2 |  | 0.3 | 1.6 | 6.5 | >1000 | 0.9 |
| 16b | 1 |  | 0.1 | 1.3 | 3.9 | 240.0 | 0.3 |
| 16c | 2 |  | 22.0 | 6.1 | 1.3 | 209.0 | 420.0 |
 | |||||
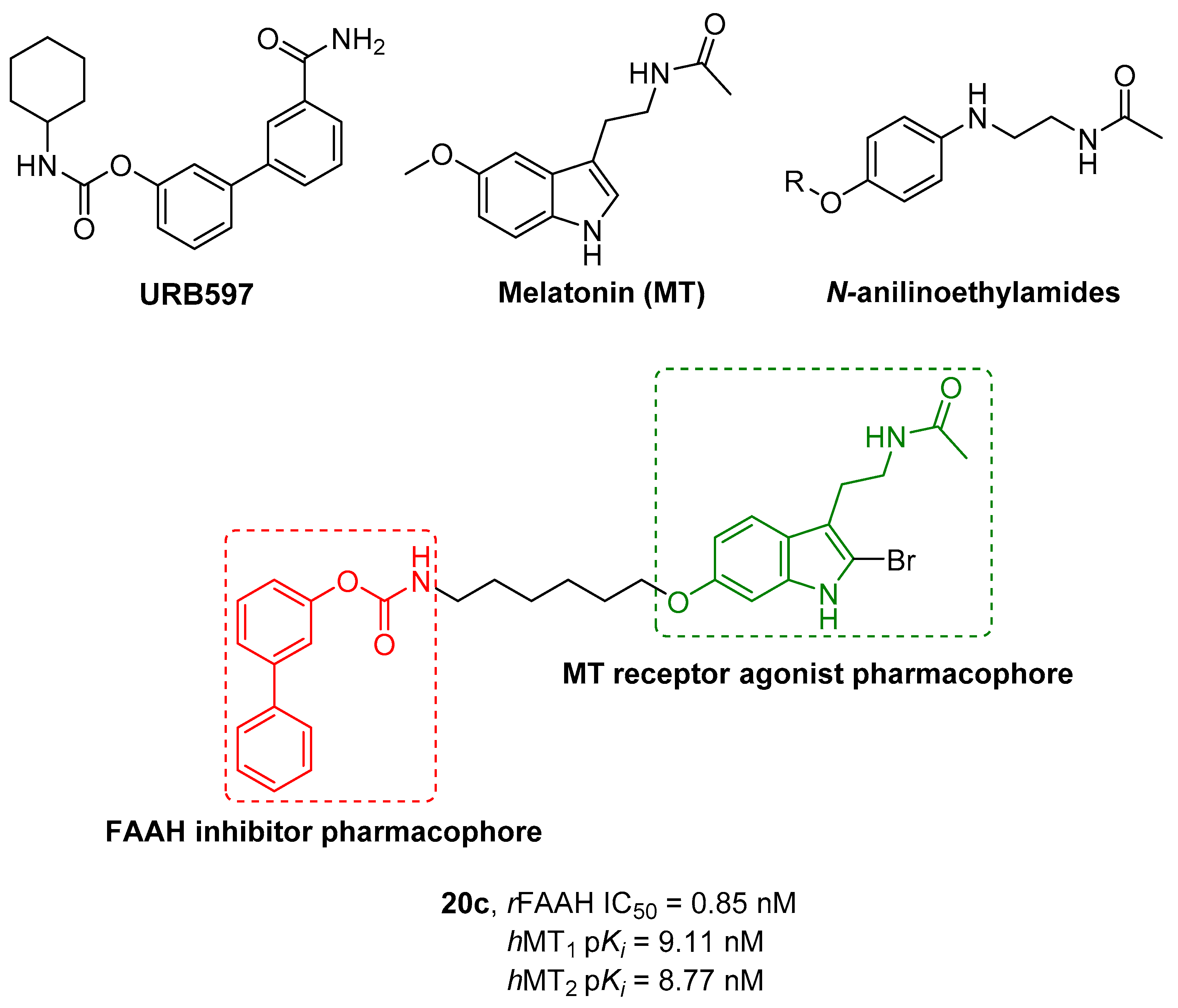
|---|---|---|---|---|---|
| Cpds | R1 | R2 | hMT1 pKi ± SD | hMT2 pKi ± SD | rFAAH IC50 (nM) ± SD |
| 20a | -CONH2 |  | 7.41 ± 0.03 | 7.81 ± 0.05 | 0.43 ± 0.01 |
| 20b | -H |  | 8.22 ± 0.01 | 8.34 ± 0.09 | 2.38 ± 0.16 |
| 20c | -H |  | 9.11 ± 0.10 | 8.77 ± 0.03 | 0.85 ± 0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Papa, A.; Pasquini, S.; Contri, C.; Gemma, S.; Campiani, G.; Butini, S.; Varani, K.; Vincenzi, F. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells 2022, 11, 471. https://doi.org/10.3390/cells11030471
Papa A, Pasquini S, Contri C, Gemma S, Campiani G, Butini S, Varani K, Vincenzi F. Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells. 2022; 11(3):471. https://doi.org/10.3390/cells11030471
Chicago/Turabian StylePapa, Alessandro, Silvia Pasquini, Chiara Contri, Sandra Gemma, Giuseppe Campiani, Stefania Butini, Katia Varani, and Fabrizio Vincenzi. 2022. "Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition" Cells 11, no. 3: 471. https://doi.org/10.3390/cells11030471
APA StylePapa, A., Pasquini, S., Contri, C., Gemma, S., Campiani, G., Butini, S., Varani, K., & Vincenzi, F. (2022). Polypharmacological Approaches for CNS Diseases: Focus on Endocannabinoid Degradation Inhibition. Cells, 11(3), 471. https://doi.org/10.3390/cells11030471