
Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle



Abstract
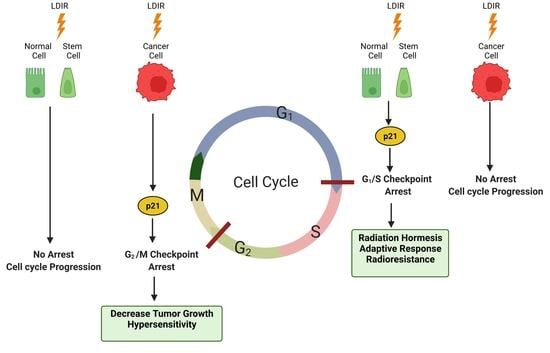
{kind=link}
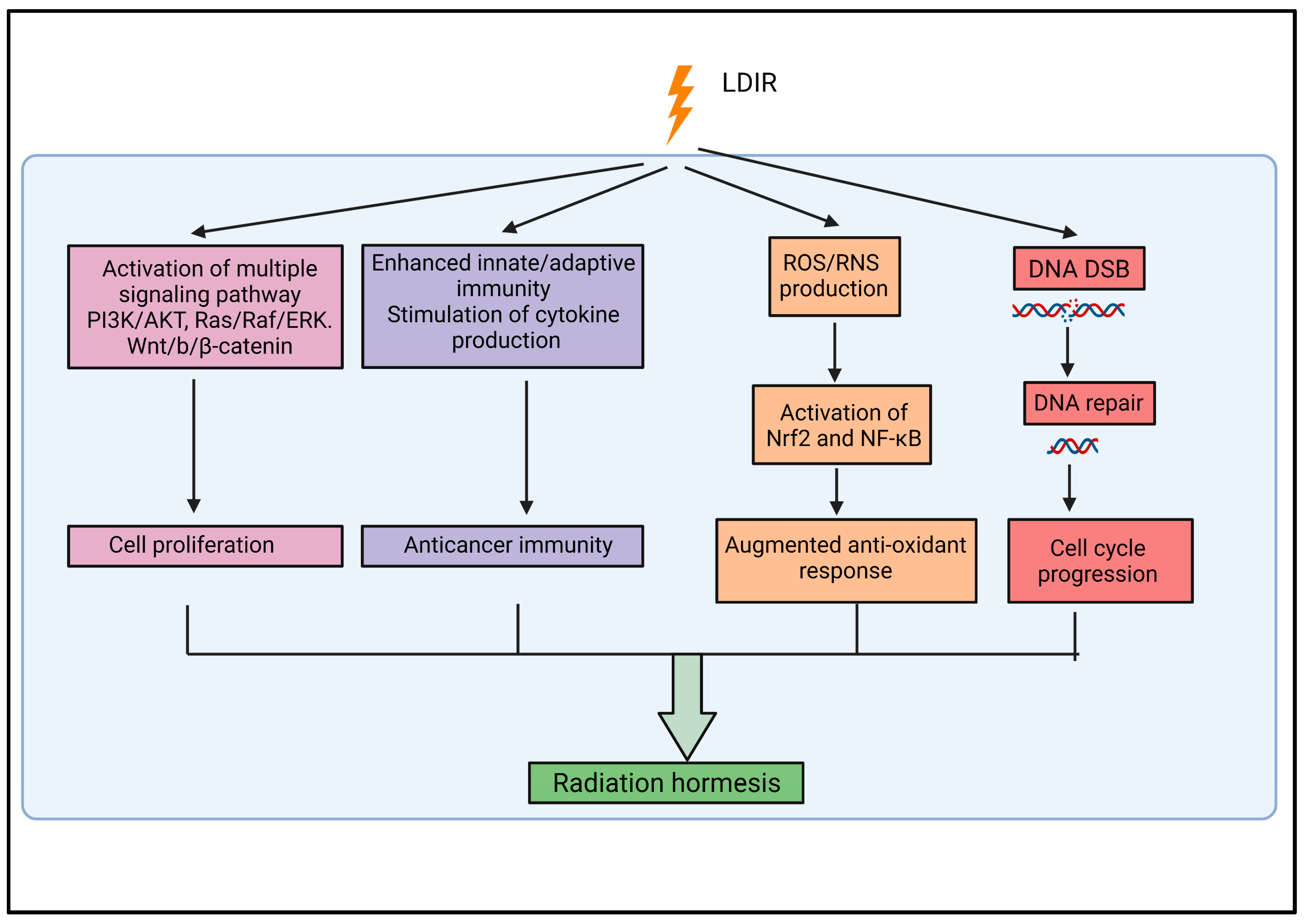{kind=link}
{kind=link}
1. Introduction
2. Molecular Mechanisms of Low-Dose Ionizing Radiation
2.1. Controversy over the Linear No-Threshold Model
2.2. Radiation Hormesis
2.3. Biological Responses Associated with Low-Dose Ionizing Radiation
3. Cell Cycle and Radiation
4. Effect of Low-Dose Ionizing Radiation on the Cell Cycle
4.1. Cancer Cells
4.2. Normal Cells
4.3. Stem Cells
5. LDIR Might Regulate Nucleocytoplasmic Localization of p21Waf1
6. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hall, E.J.; Giaccia, A. Radiobiology for the Radiologist; Elsevier Inc.: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Grubbe, E.H. The Origin and Birth of X-ray Therapy. Urol. Cutan. Rev. 1947, 51, 375–379. [Google Scholar]
- Shah, D.J.; Sachs, R.K.; Wilson, D.J. Radiation-Induced Cancer: A Modern View. Br. J. Radiol. 2012, 85, e1166–e1173. [Google Scholar] [CrossRef]
- Baskar, R.; Lee, K.A.; Yeo, R.; Yeoh, K.-W. Cancer and Radiation Therapy: Current Advances and Future Directions. Int. J. Med. Sci. 2012, 9, 193–199. [Google Scholar] [CrossRef]
- Mettler, F.A.; Thomadsen, B.R.; Bhargavan, M.; Gilley, D.B.; Gray, J.E.; Lipoti, J.A.; McCrohan, J.; Yoshizumi, T.T.; Mahesh, M. Medical Radiation Exposure in the U.S. in 2006: Preliminary Results. Health Phys. 2008, 95, 502–507. [Google Scholar] [CrossRef]
- Einstein, A.J. Medical Radiation Exposure to the U.S. Population: The Turning Tide. Radiology 2020, 295, 428–429. [Google Scholar] [CrossRef] [PubMed]
- Sokolov, M.; Neumann, R. Global Gene Expression Alterations as a Crucial Constituent of Human Cell Response to Low Doses of Ionizing Radiation Exposure. Int. J. Mol. Sci. 2015, 17, 55. [Google Scholar] [CrossRef] [PubMed]
- Jia, C.; Wang, Q.; Yao, X.; Yang, J. The Role of DNA Damage Induced by Low/High Dose Ionizing Radiation in Cell Carcinogenesis. Explor. Res. Hypothesis Med. 2021, 6, 177–184. [Google Scholar] [CrossRef]
- Feinendegen, L.E.; Pollycove, M.; Sondhaus, C.A. Responses to Low Doses of Ionizing Radiation in Biological Systems. Nonlinearity Biol. Toxicol. Med. 2004, 2, 154014204905074. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, M. Dose–Effect Relationship and Estimation of the Carcinogenic Effects of Low Doses of Ionizing Radiation: The Joint Report of the Académie Des Sciences (Paris) and of the Académie Nationale de Médecine. Int. J. Radiat. Oncol. 2005, 63, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Tubiana, M.; Feinendegen, L.E.; Yang, C.; Kaminski, J.M. The Linear No-Threshold Relationship Is Inconsistent with Radiation Biologic and Experimental Data. Radiology 2009, 251, 13–22. [Google Scholar] [CrossRef]
- Azzam, E.I.; de Toledo, S.M.; Raaphorst, G.P.; Mitchel, R.E. Low-Dose Ionizing Radiation Decreases the Frequency of Neoplastic Transformation to a Level below the Spontaneous Rate in C3H 10T1/2 Cells. Radiat. Res. 1996, 146, 369–373. [Google Scholar] [CrossRef]
- Li, W.; Wang, G.; Cui, J.; Xue, L.; Cai, L. Low-Dose Radiation (LDR) Induces Hematopoietic Hormesis: LDR-Induced Mobilization of Hematopoietic Progenitor Cells into Peripheral Blood Circulation. Exp. Hematol. 2004, 32, 1088–1096. [Google Scholar] [CrossRef]
- Liang, X.; So, Y.H.; Cui, J.; Ma, K.; Xu, X.; Zhao, Y.; Cai, L.; Li, W. The Low-Dose Ionizing Radiation Stimulates Cell Proliferation via Activation of the MAPK/ERK Pathway in Rat Cultured Mesenchymal Stem Cells. J. Radiat. Res. 2011, 52, 380–386. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Xu, Y.; Li, W.; Ma, K.; Cai, L.; Wang, G. Low-Dose Radiation Does Not Induce Proliferation in Tumor Cells in Vitro and in Vivo. Radiat. Res. 2008, 170, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Little, J.B. Delayed Initiation of DNA Synthesis in Irradiated Human Diploid Cells. Nature 1968, 218, 1064–1065. [Google Scholar] [CrossRef] [PubMed]
- De Toledo, S.M.; Azzam, E.I.; Keng, P.; Laffrenier, S.; Little, J.B. Regulation by Ionizing Radiation of CDC2, Cyclin A, Cyclin B, Thymidine Kinase, Topoisomerase IIalpha, and RAD51 Expression in Normal Human Diploid Fibroblasts Is Dependent on P53/P21Waf1. Cell Growth Differ. Mol. Biol. J. Am. Assoc. Cancer Res. 1998, 9, 887–896. [Google Scholar]
- Zhang, J.; de Toledo, S.M.; Pandey, B.N.; Guo, G.; Pain, D.; Li, H.; Azzam, E.I. Role of the Translationally Controlled Tumor Protein in DNA Damage Sensing and Repair. Proc. Natl. Acad. Sci. USA 2012, 109, E926–E933. [Google Scholar] [CrossRef]
- Wilson, G.D. Radiation and the Cell Cycle, Revisited. Cancer Metastasis Rev. 2004, 23, 209–225. [Google Scholar] [CrossRef]
- Lonati, L.; Barbieri, S.; Guardamagna, I.; Ottolenghi, A.; Baiocco, G. Radiation-Induced Cell Cycle Perturbations: A Computational Tool Validated with Flow-Cytometry Data. Sci. Rep. 2021, 11, 925. [Google Scholar] [CrossRef]
- Iliakis, G.; Wang, Y.; Guan, J.; Wang, H. DNA Damage Checkpoint Control in Cells Exposed to Ionizing Radiation. Oncogene 2003, 22, 5834–5847. [Google Scholar] [CrossRef]
- Chen, H.; Goodus, M.T.; de Toledo, S.M.; Azzam, E.I.; Levison, S.W.; Souayah, N. Ionizing Radiation Perturbs Cell Cycle Progression of Neural Precursors in the Subventricular Zone Without Affecting Their Long-Term Self-Renewal. ASN Neuro 2015, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Kim, J.K.; Nam, S.Y.; Yang, K.H.; Jeong, M.; Kim, H.S.; Kim, C.S.; Jin, Y.-W.; Kim, J. Low-Dose Radiation Stimulates the Proliferation of Normal Human Lung Fibroblasts via a Transient Activation of Raf and Akt. Mol. Cells 2007, 24, 424–430. [Google Scholar] [PubMed]
- Yang, G.; Li, W.; Jiang, H.; Liang, X.; Zhao, Y.; Yu, D.; Zhou, L.; Wang, G.; Tian, H.; Han, F.; et al. Low-Dose Radiation May Be a Novel Approach to Enhance the Effectiveness of Cancer Therapeutics. Int. J. Cancer 2016, 139, 2157–2168. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, G.; Bodycote, J.; Wolff, S. Adaptive Response of Human Lymphocytes to Low Concentrations of Radioactive Thymidine. Science 1984, 223, 594–597. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.M.; Fan, M.; Nantajit, D.; Cao, N.; Li, J.J. Cyclin D1 in Low-Dose Radiation-Induced Adaptive Resistance. Oncogene 2008, 27, 6738–6748. [Google Scholar] [CrossRef]
- Hendrikse, S.; Hunter, A.J.; Keraan, M.; Blekkenhorst, G.H. Effects of Low Dose Irradiation on TK6 and U937 Cells: Induction of P53 and Its Role in Cell-Cycle Delay and the Adaptive Response. Int. J. Radiat. Biol. 2000, 76, 11–21. [Google Scholar] [CrossRef]
- Li, S.-J.; Liang, X.-Y.; Li, H.-J.; Li, W.; Zhou, L.; Chen, H.-Q.; Ye, S.-G.; Yu, D.-H.; Cui, J.-W. Low-Dose Irradiation Promotes Proliferation of the Human Breast Cancer MDA-MB-231 Cells through Accumulation of Mutant P53. Int. J. Oncol. 2017, 50, 290–296. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sekihara, K.; Saitoh, K.; Yang, H.; Kawashima, H.; Kazuno, S.; Kikkawa, M.; Arai, H.; Miida, T.; Hayashi, N.; Sasai, K.; et al. Low-Dose Ionizing Radiation Exposure Represses the Cell Cycle and Protein Synthesis Pathways in in Vitro Human Primary Keratinocytes and U937 Cell Lines. PLoS ONE 2018, 13, e0199117. [Google Scholar] [CrossRef]
- Azzam, E.I.; de Toledo, S.M.; Waker, A.J.; Little, J.B. High and Low Fluences of Alpha-Particles Induce a G1 Checkpoint in Human Diploid Fibroblasts. Cancer Res. 2000, 60, 2623–2631. [Google Scholar]
- Lindell, B.; Sowby, D. The 1958 UNSCEAR Report. J. Radiol. Prot. 2008, 28, 277–282. [Google Scholar] [CrossRef]
- Zielinski, J.; Garner, M.; Band, P.; Krewski, D.; Shilnikova, N.; Jiang, H.; Ashmore, P.; Sont, W.; Fair, M.; Letourneau, E.; et al. Health Outcomes of Low-Dose Ionizing Radiation Exposure among Medical Workers: A Cohort Study of the Canadian National Dose Registry of Radiation Workers. Int. J. Occup. Med. Environ. Health 2009, 22, 149–156. [Google Scholar] [CrossRef]
- Hazelton, W.D.; Moolgavkar, S.H.; Curtis, S.B.; Zielinski, J.M.; Patrick Ashmore, J.; Krewski, D. Biologically Based Analysis of Lung Cancer Incidence in a Large Canadian Occupational Cohort with Low-Dose Ionizing Radiation Exposure, and Comparison with Japanese Atomic Bomb Survivors. J. Toxicol. Environ. Health Part A 2006, 69, 1013–1038. [Google Scholar] [CrossRef] [PubMed]
- Zablotska, L.B.; Little, M.P.; Cornett, R.J. Potential Increased Risk of Ischemic Heart Disease Mortality With Significant Dose Fractionation in the Canadian Fluoroscopy Cohort Study. Am. J. Epidemiol. 2014, 179, 120–131. [Google Scholar] [CrossRef] [PubMed]
- Sanders, C.L. (Ed.) Radiation Hormesis and the Linear-No-Threshold Assumption; Springer: Berlin/Heidelberg, Geramny, 2010; ISBN 978-3-642-03719-1. [Google Scholar]
- Golden, R.; Bus, J.; Calabrese, E. An Examination of the Linear No-Threshold Hypothesis of Cancer Risk Assessment: Introduction to a Series of Reviews Documenting the Lack of Biological Plausibility of LNT. Chem. Biol. Interact. 2019, 301, 2–5. [Google Scholar] [CrossRef]
- Luckey, T.D. Radiation Hormesis: The Good, the Bad, and the Ugly. Dose-Response 2006, 4, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Lehrer, S.; Rosenzweig, K.E. Lung Cancer Hormesis in High Impact States Where Nuclear Testing Occurred. Clin. Lung Cancer 2015, 16, 152–155. [Google Scholar] [CrossRef]
- Dobrzyński, L.; Fornalski, K.W.; Feinendegen, L.E. Cancer Mortality Among People Living in Areas With Various Levels of Natural Background Radiation. Dose-Response 2015, 13, 155932581559239. [Google Scholar] [CrossRef]
- Tharmalingam, S.; Sreetharan, S.; Brooks, A.L.; Boreham, D.R. Re-Evaluation of the Linear No-Threshold (LNT) Model Using New Paradigms and Modern Molecular Studies. Chem. Biol. Interact. 2019, 301, 54–67. [Google Scholar] [CrossRef]
- Kant, K.; Chauhan, R.P.; Sharma, G.S.; Chakarvarti, S.K. Hormesis in Humans Exposed to Low-Level Ionising Radiation. Int. J. Low Radiat. 2003, 1, 76. [Google Scholar] [CrossRef]
- Mitchel, R.E.J. Low Doses of Radiation Are Protective in Vitro and in Vivo: Evolutionary Origins. Dose-Response 2006, 4, 75–90. [Google Scholar] [CrossRef]
- Kim, C.S.; Kim, J.-M.; Nam, S.Y.; Yang, K.H.; Jeong, M.; Kim, H.S.; Lim, Y.-K.; Kim, C.S.; Jin, Y.-W.; Kim, J. Low-Dose of Ionizing Radiation Enhances Cell Proliferation via Transient ERK1/2 and P38 Activation in Normal Human Lung Fibroblasts. J. Radiat. Res. 2007, 48, 407–415. [Google Scholar] [CrossRef]
- Wei, L.-C.; Ding, Y.-X.; Liu, Y.-H.; Duan, L.; Bai, Y.; Shi, M.; Chen, L.-W. Low-Dose Radiation Stimulates Wnt/β-Catenin Signaling, Neural Stem Cell Proliferation and Neurogenesis of the Mouse Hippocampus in Vitro and in Vivo. Curr. Alzheimer Res. 2012, 9, 278–289. [Google Scholar] [CrossRef]
- Rothkamm, K.; Lobrich, M. Evidence for a Lack of DNA Double-Strand Break Repair in Human Cells Exposed to Very Low x-Ray Doses. Proc. Natl. Acad. Sci. USA 2003, 100, 5057–5062. [Google Scholar] [CrossRef]
- Scott, B.R. Low-Dose Radiation-Induced Protective Process and Implications for Risk Assessment, Cancer Prevention, and Cancer Therapy. Dose-Response 2007, 5, 131–149. [Google Scholar] [CrossRef]
- Pandey, R.; Shankar, B.S.; Sharma, D.; Sainis, K.B. Low Dose Radiation Induced Immunomodulation: Effect on Macrophages and CD8 + T Cells. Int. J. Radiat. Biol. 2005, 81, 801–812. [Google Scholar] [CrossRef]
- Shigematsu, A.; Adachi, Y.; Koike-Kiriyama, N.; Suzuki, Y.; Iwasaki, M.; Koike, Y.; Nakano, K.; Mukaide, H.; Imamura, M.; Ikehara, S. Effects of Low-Dose Irradiation on Enhancement of Immunity by Dendritic Cells. J. Radiat. Res. 2007, 48, 51–55. [Google Scholar] [CrossRef]
- Yang, G.; Kong, Q.; Wang, G.; Jin, H.; Zhou, L.; Yu, D.; Niu, C.; Han, W.; Li, W.; Cui, J. Low-Dose Ionizing Radiation Induces Direct Activation of Natural Killer Cells and Provides a Novel Approach for Adoptive Cellular Immunotherapy. Cancer Biother. Radiopharm. 2014, 29, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, B.; Dai, Z.; Ren, S.; Bai, M.; Wang, Z.; Li, Z.; Lin, S.; Wang, Z.; Huang, N.; et al. Low-Dose Splenic Radiation Inhibits Liver Tumor Development of Rats through Functional Changes in CD4+CD25+Treg Cells. Int. J. Biochem. Cell Biol. 2014, 55, 98–108. [Google Scholar] [CrossRef]
- Lau, Y.S.; Chew, M.T.; Alqahtani, A.; Jones, B.; Hill, M.A.; Nisbet, A.; Bradley, D.A. Low Dose Ionising Radiation-Induced Hormesis: Therapeutic Implications to Human Health. Appl. Sci. 2021, 11, 8909. [Google Scholar] [CrossRef]
- Tang, F.R.; Loke, W.K. Molecular Mechanisms of Low Dose Ionizing Radiation-Induced Hormesis, Adaptive Responses, Radioresistance, Bystander Effects, and Genomic Instability. Int. J. Radiat. Biol. 2015, 91, 13–27. [Google Scholar] [CrossRef] [PubMed]
- Guéguen, Y.; Bontemps, A.; Ebrahimian, T.G. Adaptive Responses to Low Doses of Radiation or Chemicals: Their Cellular and Molecular Mechanisms. Cell. Mol. Life Sci. 2019, 76, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Saini, D.; Shelke, S.; Mani Vannan, A.; Toprani, S.; Jain, V.; Das, B.; Seshadri, M. Transcription Profile of DNA Damage Response Genes at G0 Lymphocytes Exposed to Gamma Radiation. Mol. Cell. Biochem. 2012, 364, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Portess, D.I.; Bauer, G.; Hill, M.A.; O’Neill, P. Low-Dose Irradiation of Nontransformed Cells Stimulates the Selective Removal of Precancerous Cells via Intercellular Induction of Apoptosis. Cancer Res. 2007, 67, 1246–1253. [Google Scholar] [CrossRef]
- Ermakov, A.V.; Konkova, M.S.; Kostyuk, S.V.; Egolina, N.A.; Efremova, L.V.; Veiko, N.N. Oxidative Stress as a Significant Factor for Development of an Adaptive Response in Irradiated and Nonirradiated Human Lymphocytes after Inducing the Bystander Effect by Low-Dose X-Radiation. Mutat. Res./Fundam. Mol. Mech. Mutagenesis 2009, 669, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Abdelrazzak, A.B.; Stevens, D.L.; Bauer, G.; O’Neill, P.; Hill, M.A. The Role of Radiation Quality in the Stimulation of Intercellular Induction of Apoptosis in Transformed Cells at Very Low Doses. Radiat. Res. 2011, 176, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.N.; Canter, B.S.; Rajon, D.; Bäck, T.A.; Fritton, J.C.; Azzam, E.I.; Howell, R.W. Dose-Dependent Growth Delay of Breast Cancer Xenografts in the Bone Marrow of Mice Treated with 223Ra: The Role of Bystander Effects and Their Potential for Therapy. J. Nucl. Med. 2020, 61, 89–95. [Google Scholar] [CrossRef]
- Joiner, M.C.; Marples, B.; Lambin, P.; Short, S.C.; Turesson, I. Low-Dose Hypersensitivity: Current Status and Possible Mechanisms. Int. J. Radiat. Oncol. 2001, 49, 379–389. [Google Scholar] [CrossRef]
- Calkins, J. The T-N-PR Model of Radiation Response. J. Theor. Biol. 1973, 39, 609–622. [Google Scholar] [CrossRef]
- Marples, B. Is Low-Dose Hyper-Radiosensitivity a Measure of G2-Phase Cell Radiosensitivity? Cancer Metastasis Rev. 2004, 23, 197–207. [Google Scholar] [CrossRef]
- Marples, B.; Wouters, B.G.; Collis, S.J.; Chalmers, A.J.; Joiner, M.C. Low-Dose Hyper-Radiosensitivity: A Consequence of Ineffective Cell Cycle Arrest of Radiation-Damaged G2 -Phase Cells. Radiat. Res. 2004, 161, 247–255. [Google Scholar] [CrossRef]
- Shimura, T. Targeting the AKT/Cyclin D1 Pathway to Overcome Intrinsic and Acquired Radioresistance of Tumors for Effective Radiotherapy. Int. J. Radiat. Biol. 2017, 93, 381–385. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-Y.; Lee, C.-J.; Choi, J.-H.; Kim, J.-H.; Kim, J.-W.; Kim, J.-Y.; Nam, J.-S. The JAK2/STAT3/CCND2 Axis Promotes Colorectal Cancer Stem Cell Persistence and Radioresistance. J. Exp. Clin. Cancer Res. 2019, 38, 399. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Fertil, B.; Foray, N. Very Low-Dose Hyper-Radiosensitivity: Impact for Radiotherapy of Micrometastases. Cancer Radiother. J. Soc. Fr. Radiother. Oncol. 2007, 11, 260–265. [Google Scholar] [CrossRef]
- Amundson, S.A.; Lee, R.A.; Koch-Paiz, C.A.; Bittner, M.L.; Meltzer, P.; Trent, J.M.; Fornace, A.J. Differential Responses of Stress Genes to Low Dose-Rate Gamma Irradiation. Mol. Cancer Res. MCR 2003, 1, 445–452. [Google Scholar] [PubMed]
- Short, C.; Kelly, J.; Mayes, C.R.; Woodcock, M.; Joiner, M.C. Low-Dose Hypersensitivity after Fractionated Low-Dose Irradiation in Vitro. Int. J. Radiat. Biol. 2001, 77, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Tewari, S.; Khan, K.; Husain, N.; Rastogi, M.; Mishra, S.P.; Srivastav, A.K. Peripheral Blood Lymphocytes as In Vitro Model to Evaluate Genomic Instability Caused by Low Dose Radiation. Asian Pac. J. Cancer Prev. 2016, 17, 1773–1777. [Google Scholar] [CrossRef] [PubMed]
- Shimura, T.; Sasatani, M.; Kamiya, K.; Kawai, H.; Inaba, Y.; Kunugita, N. Mitochondrial Reactive Oxygen Species Perturb AKT/Cyclin D1 Cell Cycle Signaling via Oxidative Inactivation of PP2A in Lowdose Irradiated Human Fibroblasts. Oncotarget 2016, 7, 3559–3570. [Google Scholar] [CrossRef]
- Alexandrou, A.T.; Li, J.J. Cell Cycle Regulators Guide Mitochondrial Activity in Radiation-Induced Adaptive Response. Antioxid. Redox Signal. 2014, 20, 1463–1480. [Google Scholar] [CrossRef]
- Shimura, N.; Kojima, S. The Lowest Radiation Dose Having Molecular Changes in the Living Body. Dose-Response 2018, 16, 1–17. [Google Scholar] [CrossRef]
- Harashima, H.; Dissmeyer, N.; Schnittger, A. Cell Cycle Control across the Eukaryotic Kingdom. Trends Cell Biol. 2013, 23, 345–356. [Google Scholar] [CrossRef]
- Barnum, K.J.; O’Connell, M.J. Cell Cycle Regulation by Checkpoints. In Cell Cycle Control; Humana Press: New York, NY, USA, 2014; pp. 29–40. [Google Scholar]
- Sia, J.; Szmyd, R.; Hau, E.; Gee, H.E. Molecular Mechanisms of Radiation-Induced Cancer Cell Death: A Primer. Front. Cell Dev. Biol. 2020, 8, 41. [Google Scholar] [CrossRef]
- Niida, H.; Nakanishi, M. DNA Damage Checkpoints in Mammals. Mutagenesis 2006, 21, 3–9. [Google Scholar] [CrossRef]
- Maier, P.; Hartmann, L.; Wenz, F.; Herskind, C. Cellular Pathways in Response to Ionizing Radiation and Their Targetability for Tumor Radiosensitization. Int. J. Mol. Sci. 2016, 17, 102. [Google Scholar] [CrossRef]
- Nagasawa, H.; Li, C.Y.; Maki, C.G.; Imrich, A.C.; Little, J.B. Relationship between Radiation-Induced G1 Phase Arrest and P53 Function in Human Tumor Cells. Cancer Res. 1995, 55, 1842–1846. [Google Scholar] [PubMed]
- El-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y. WAF1/CIP1 Is Induced in P53-Mediated G1 Arrest and Apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar] [PubMed]
- Fei, P.; El-Deiry, W.S. P53 and Radiation Responses. Oncogene 2003, 22, 5774–5783. [Google Scholar] [CrossRef] [PubMed]
- Kastan, M.B.; Radin, A.I.; Kuerbitz, S.J.; Onyekwere, O.; Wolkow, C.A.; Civin, C.I.; Stone, K.D.; Woo, T.; Ravindranath, Y.; Craig, R.W. Levels of P53 Protein Increase with Maturation in Human Hematopoietic Cells. Cancer Res. 1991, 51, 4279–4286. [Google Scholar] [PubMed]
- Kastan, M.B.; Onyekwere, O.; Sidransky, D.; Vogelstein, B.; Craig, R.W. Participation of P53 Protein in the Cellular Response to DNA Damage. Cancer Res. 1991, 51, 6304–6311. [Google Scholar] [CrossRef] [PubMed]
- Domogauer, J.D.; de Toledo, S.M.; Howell, R.W.; Azzam, E.I. Acquired Radioresistance in Cancer Associated Fibroblasts Is Concomitant with Enhanced Antioxidant Potential and DNA Repair Capacity. Cell Commun. Signal. 2021, 19, 30. [Google Scholar] [CrossRef]
- Wang, B. Analyzing Cell Cycle Checkpoints in Response to Ionizing Radiation in Mammalian Cells. In Cell Cycle Control; Humana Press: New York, NY, USA, 2014; pp. 313–320. [Google Scholar]
- Huang, R.-X.; Zhou, P.-K. DNA Damage Response Signaling Pathways and Targets for Radiotherapy Sensitization in Cancer. Signal Transduct. Target. Ther. 2020, 5, 60. [Google Scholar] [CrossRef] [PubMed]
- Fornace, A.J.; Amundson, S.A.; Do, K.T.; Meltzer, P.; Trent, J.; Bittner, M. Stress-Gene Induction by Low-Dose Gamma Irradiation. Mil. Med. 2002, 167, 13. [Google Scholar]
- Daino, K.; Ichimura, S.; Nenoi, M. Early Induction of CDKN1A (P21) and GADD45 MRNA by a Low Dose of Ionizing Radiation Is Due to Their Dose-Dependent Post-Transcriptional Regulation. Radiat. Res. 2002, 157, 478–482. [Google Scholar] [CrossRef]
- Xiong, Y.; Hannon, G.J.; Zhang, H.; Casso, D.; Kobayashi, R.; Beach, D. P21 Is a Universal Inhibitor of Cyclin Kinases. Nature 1993, 366, 701–704. [Google Scholar] [CrossRef]
- El-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a Potential Mediator of P53 Tumor Suppression. Cell 1993, 75, 817–825. [Google Scholar] [CrossRef]
- Wang, X.W.; Zhan, Q.; Coursen, J.D.; Khan, M.A.; Kontny, H.U.; Yu, L.; Hollander, M.C.; O’Connor, P.M.; Fornace, A.J.; Harris, C.C. GADD45 Induction of a G2/M Cell Cycle Checkpoint. Proc. Natl. Acad. Sci. USA 1999, 96, 3706–3711. [Google Scholar] [CrossRef]
- Abou-El-Ardat, K. Response to Low-Dose X-Irradiation Is P53-Dependent in a Papillary Thyroid Carcinoma Model System. Int. J. Oncol. 2011, 39, 1429–1441. [Google Scholar] [CrossRef] [PubMed]
- Li, S.-J.; Liang, X.-Y.; Li, H.-J.; Yang, G.-Z.; Li, W.; Li, Z.; Zhou, L.; Wen, X.; Yu, D.-H.; Cui, J.-W. Low-Dose Irradiation Inhibits Proliferation of the P53null Type Human Prostate Cancer Cells through the ATM/P21 Pathway. Int. J. Mol. Med. 2017, 41, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Rainey, M.D.; Black, E.J.; Zachos, G.; Gillespie, D.A.F. Chk2 Is Required for Optimal Mitotic Delay in Response to Irradiation-Induced DNA Damage Incurred in G2 Phase. Oncogene 2008, 27, 896–906. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, C.Y.; Nagasawa, H.; Dahlberg, W.K.; Little, J.B. Diminished Capacity for P53 in Mediating a Radiation-Induced G1 Arrest in Established Human Tumor Cell Lines. Oncogene 1995, 11, 1885–1892. [Google Scholar] [PubMed]
- Marples, B.; Wouters, B.G.; Joiner, M.C. An Association between the Radiation-Induced Arrest of G2-Phase Cells and Low-Dose Hyper-Radiosensitivity: A Plausible Underlying Mechanism? Radiat. Res. 2003, 160, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Hou, D.; Ma, Y.; Feng, L.; Li, W.; Liu, W.; Qiao, J.; Jia, X.; Li, K. Effect of 0.1 Gy X-Ray Irradiation on Gene Expression Profiles in Normal Human Lymphoblastoid Cells. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi = Zhonghua Laodong Weisheng Zhiyebing Zazhi = Chin. J. Ind. Hyg. Occup. Dis. 2013, 31, 749–752. [Google Scholar]
- Chin, L.; Pomerantz, J.; DePinho, R.A. The INK4a/ARF Tumor Suppressor: One Gene–Two Products–Two Pathways. Trends Biochem. Sci. 1998, 23, 291–296. [Google Scholar] [CrossRef]
- Sokolov, M.; Neumann, R. Effects of Low Doses of Ionizing Radiation Exposures on Stress-Responsive Gene Expression in Human Embryonic Stem Cells. Int. J. Mol. Sci. 2014, 15, 588–604. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. P21 in Cancer: Intricate Networks and Multiple Activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Yunis, R.; Albrecht, H.; Kalanetra, K.M.; WU, S.; Rocke, D.M. Genomic Characterization of a Three-Dimensional Skin Model Following Exposure to Ionizing Radiation. J. Radiat. Res. 2012, 53, 860–875. [Google Scholar] [CrossRef] [PubMed]
- Sergeeva, V.A.; Ershova, E.S.; Veiko, N.N.; Malinovskaya, E.M.; Kalyanov, A.A.; Kameneva, L.V.; Stukalov, S.V.; Dolgikh, O.A.; Konkova, M.S.; Ermakov, A.V.; et al. Low-Dose Ionizing Radiation Affects Mesenchymal Stem Cells via Extracellular Oxidized Cell-Free DNA: A Possible Mediator of Bystander Effect and Adaptive Response. Oxidat. Med. Cell. Longev. 2017, 2017, 9515809. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Cazzalini, O.; Scovassi, A.I.; Savio, M.; Stivala, L.A.; Prosperi, E. Multiple Roles of the Cell Cycle Inhibitor P21(CDKN1A) in the DNA Damage Response. Mutat. Res. 2010, 704, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Dotto, G.P. P21(WAF1/Cip1): More than a Break to the Cell Cycle? Biochim. Biophys. Acta 2000, 1471, M43–M56. [Google Scholar] [PubMed]
- Gartel, A.L.; Tyner, A.L. The Role of the Cyclin-Dependent Kinase Inhibitor P21 in Apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Romanov, V.S.; Rudolph, K.L. P21 Shapes Cancer Evolution. Nat. Cell Biol. 2016, 18, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Romanov, V.S.; Pospelov, V.A.; Pospelova, T.V. Cyclin-Dependent Kinase Inhibitor P21(Waf1): Contemporary View on Its Role in Senescence and Oncogenesis. Biochemistry 2012, 77, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Alt, J.R.; Gladden, A.B.; Diehl, J.A. P21Cip1 Promotes Cyclin D1 Nuclear Accumulation via Direct Inhibition of Nuclear Export. J. Biol. Chem. 2002, 277, 8517–8523. [Google Scholar] [CrossRef]
- Wang, Y.A.; Elson, A.; Leder, P. Loss of P21 Increases Sensitivity to Ionizing Radiation and Delays the Onset of Lymphoma in Atm-Deficient Mice. Proc. Natl. Acad. Sci. USA 1997, 94, 14590–14595. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, H.; Lindqvist, A. Bystander Communication and Cell Cycle Decisions after DNA Damage. Front. Genet. 2015, 6, 63. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, C.-M.C.; Luxton, G.; Orton, C.G. Pulsed Reduced Dose Rate Radiation Therapy Is Likely to Become the Treatment Modality of Choice for Recurrent Cancers. Med. Phys. 2011, 38, 4909–4911. [Google Scholar] [CrossRef]
- Zhang, P.; Wang, B.; Chen, X.; Cvetkovic, D.; Chen, L.; Lang, J.; Ma, C.-M. Local Tumor Control and Normal Tissue Toxicity of Pulsed Low-Dose Rate Radiotherapy for Recurrent Lung Cancer. Dose-Response 2015, 13, 155932581558850. [Google Scholar] [CrossRef]
- Todorovic, V.; Prevc, A.; Zakelj, M.N.; Savarin, M.; Bucek, S.; Groselj, B.; Strojan, P.; Cemazar, M.; Sersa, G. Pulsed Low Dose-Rate Irradiation Response in Isogenic HNSCC Cell Lines with Different Radiosensitivity. Radiol. Oncol. 2020, 54, 168–179. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, M.G.M.; Wang, Y. Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle. Cells 2022, 11, 356. https://doi.org/10.3390/cells11030356
Khan MGM, Wang Y. Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle. Cells. 2022; 11(3):356. https://doi.org/10.3390/cells11030356
Chicago/Turabian StyleKhan, Md Gulam Musawwir, and Yi Wang. 2022. "Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle" Cells 11, no. 3: 356. https://doi.org/10.3390/cells11030356
APA StyleKhan, M. G. M., & Wang, Y. (2022). Advances in the Current Understanding of How Low-Dose Radiation Affects the Cell Cycle. Cells, 11(3), 356. https://doi.org/10.3390/cells11030356