
Interconnections between Inflammageing and Immunosenescence during Ageing


Abstract
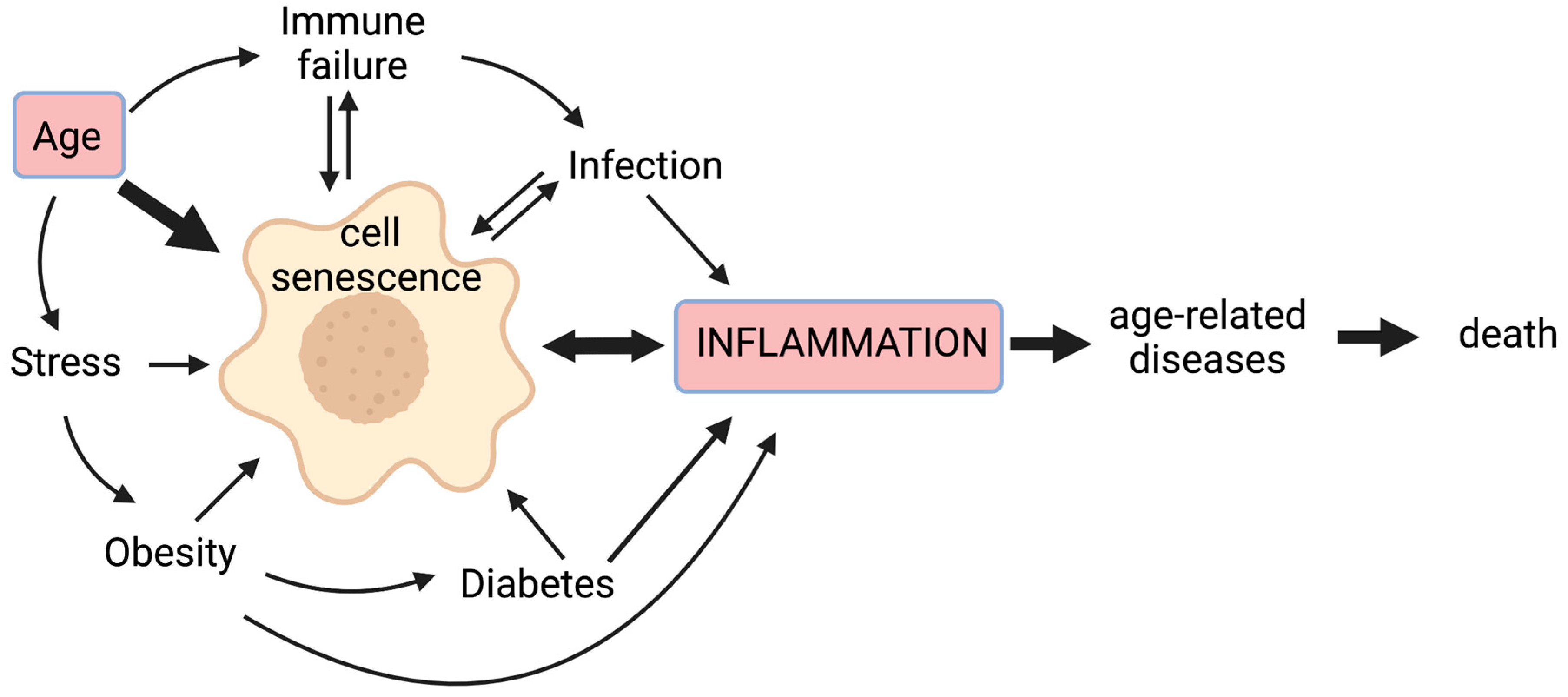
1. Introduction
1.1. Senescent Cells Contribute to a State of Chronic Low-Grade Inflammation
1.2. Inflammageing Depends on a Complex Interplay of Pro- and Anti-Inflammatory Mediators
1.2.1. Elevation of Clusters of Pro-Inflammatory Markers with Age

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IL-1β | IL-6 | CRP | SAA | ICAM | PAI-1 | Fibrinogen | Population Characteristics | Data Processing | Reference |
|---|---|---|---|---|---|---|---|---|---|
| No cluster reported | PCA1—Correlated with age, mortality, morbidity, and age-related diseases | NI | NI | NI | NI | 1010 participants (428 men, 582 women), aged 21–96 y.o. | Cluster analysis | [75] | |
| Absence of correlation with age | Positively correlated with age, mortality, morbidity, and some age-related disease | Positively correlated with age, mortality, morbidity, and some age-related disease | General population—InCHIANTI cohort | Individual associations | |||||
| Non-predictive of 5-year mortality | Significant independent predictor of 5- and 10-year mortality | Significant independent predictor of 5- and 10-year mortality | NI | NI | NI | NI | 7043 participants (2995 men, 4048 women), aged 65–102 y.o. | Fully adjusted correlation with 10-year mortality | [83] |
| Best predictor of 10-year mortality in combination with sTNFRI | General population—includes CHS and InCHIANTI cohorts | ||||||||
| IL-1/TGF—No adverse outcome reported | “Up”-regulation—Positively correlated with worsened mobility and frailty risk | NI | NI | NI | NI | 967 participants (416 men, 551 women), aged 65 y.o. and older | Cluster analysis | [76] | |
| General population—InCHIANTI cohort | |||||||||
| NI | CRP related—Negatively correlated with grip strength; Positively correlated with 400-m walk time | NI | NI | CRP related—Negatively correlated with grip strength; Positively correlated with 400-m walk time | NI | 1269 participants, aged 70–79 y.o. | Cluster analysis | [77] | |
| Highest R2 of the CRP-related component for knee and grip strength | Highest R2 of the CRP-related component for walking speed and 400-m walk time | Highest R2 of the CRP-related component for the HPPB score | General non-frail population | Individual associations | |||||
| NI | Positively associated with 4-year mortality | NI | NI | NI | NI | 285 participants (67 men, 218 women), aged 90 y.o. and older with a 4-year follow-up | Correlation with 4-year mortality | [93] | |
| Removed association with mortality | Predictor of 4-year mortality alone or in combination with IL-1RA | General population | Fully adjusted | ||||||
| NI | NI | Systemic inflammation | NI | Local inflammation-endothelial dysfunction | NI | Systemic inflammation | 320 participants (236 men, 84 women), aged 58–74 y.o. with a 1-year follow-up | Cluster analysis | [78] |
| Independent predictor of 1-year recurrent coronary events | No association with 1-year recurrent adverse cardiac events | No association with 1-year recurrent adverse cardiac events | Acute coronary syndrome patients | Individual associations | |||||
| Absence of correlation with age | Positively correlated with age | Positively correlated with age | 1327 participants (586 men, 741 women), aged 20–102 y.o. | Correlation with age | [80] | ||||
| NR | Greatly reduced the size of the regression coefficient for age | Removed the effect of age in men only | Removed the effect of age | General population—Includes InCHIANTI cohort | Adjustment for cardiovascular risk factors and morbidities | ||||
| NI | Proinflammation—Strong association with 4-year death rate | No cluster reported but is individually positively correlated with TNF-alpha, CRP, IL-6 and SAA | NI | NI | 580 women aged 31–85 y.o. with a 4.7-year follow-up | Cluster analysis | [79] | ||
| Clinically referred for coronary angiography | |||||||||
| No significant association with physical performance | Negatively correlated with global physical performance and hand-grip strength | Negatively correlated with global physical performance and hand-grip strength | NI | NI | NI | NI | 1020 participants (483 men, 537 women), aged 65–102 y.o. | Correlation with physical performance | [81] |
1.2.2. Do Anti-Inflammatory Factors Contribute to Inflammageing?
1.2.3. Inflammation Can Drive Morbidity in Older Adults—Thromboembolitic Events as an Example
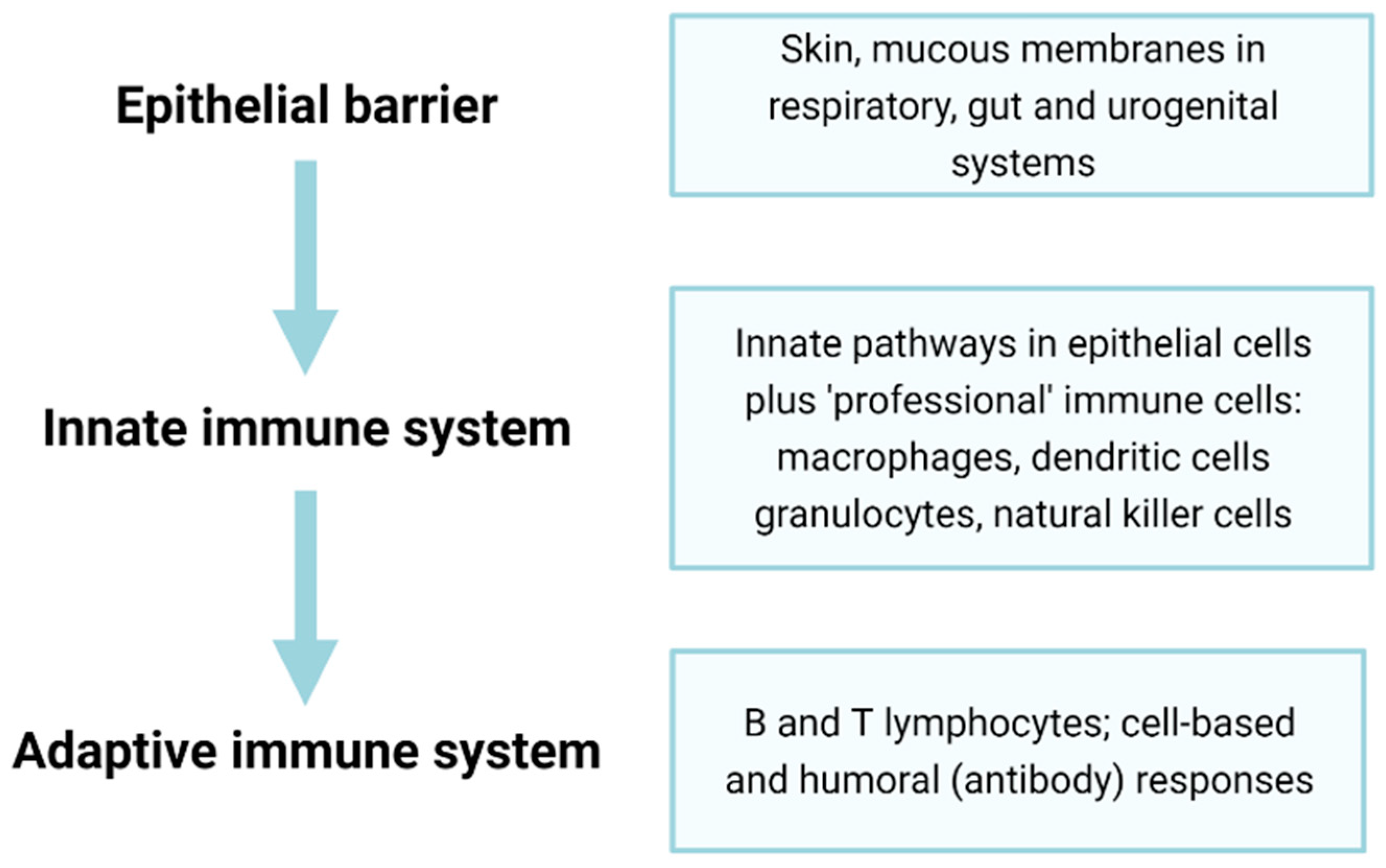
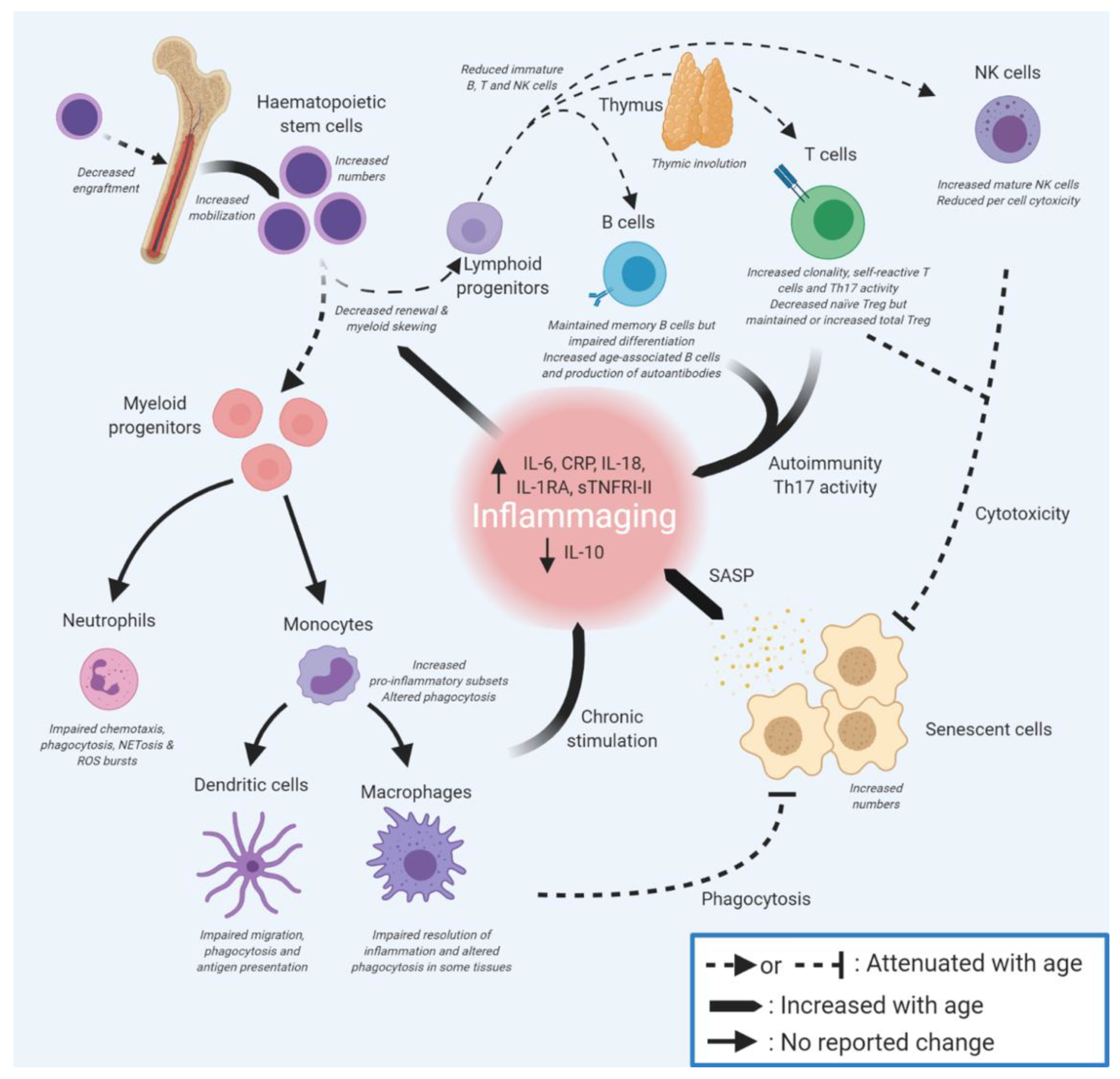
2. Immunosenescence Affects Many Components of the Immune System
3. Dysregulation of Innate Immunity on Ageing
3.1. Ageing of Haematopoietic Stem Cells Leads to Myeloid Skewing
3.2. Ageing in Innate Immune Effector Cells
3.2.1. Dendritic Cells
3.2.2. Granulocytes
3.2.3. Natural Killer Cells
3.2.4. Macrophages
3.3. The Inflammasome Drives Pyroptosis and Further Inflammation
3.4. Epigenetic Changes Provide Innate Immune Cells with Memory
4. Dysregulation of Adaptive Immunity during Ageing
4.1. Decreased B and T Cell Responses with Ageing
4.1.1. B Cells
4.1.2. T Cells
5. Strategies to Attenuate Inflammageing and Immunosenescence and Their Effects
5.1. Physical Exercise
5.2. Diet
5.3. Immune Rejuvenation
5.4. Interventions to Remove Senescent Cells
5.5. Molecular Modifiers of Senescence and Inflammation
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-Aging. An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef]
- Krabbe, K.S.; Pedersen, M.; Bruunsgaard, H. Inflammatory Mediators in the Elderly. Exp. Gerontol. 2004, 39, 687–699. [Google Scholar] [CrossRef]
- Arai, Y.; Martin-Ruiz, C.M.; Takayama, M.; Abe, Y.; Takebayashi, T.; Koyasu, S.; Suematsu, M.; Hirose, N.; von Zglinicki, T. Inflammation, But Not Telomere Length, Predicts Successful Ageing at Extreme Old Age: A Longitudinal Study of Semi-Supercentenarians. EBioMedicine 2015, 2, 1549–1558. [Google Scholar] [CrossRef] [PubMed]
- Bruunsgaard, H. The Clinical Impact of Systemic Low-Level Inflammation in Elderly Populations. With Special Reference to Cardiovascular Disease, Dementia and Mortality. Dan. Med. Bull. 2006, 53, 285–309. [Google Scholar]
- Franceschi, C.; Garagnani, P.; Vitale, G.; Capri, M.; Salvioli, S. Inflammaging and ‘Garb-Aging’. Trends Endocrinol. Metab. 2017, 28, 199–212. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The Hallmarks of Aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, N.; Karlseder, J. Complex Interactions between the DNA-Damage Response and Mammalian Telomeres. Nat. Struct. Mol. Biol. 2015, 22, 859–866. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a Break: Cellular Senescence as a DNA-Damage Response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The Limited In Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Brandl, A.; Meyer, M.; Bechmann, V.; Nerlich, M.; Angele, P. Oxidative Stress Induces Senescence in Human Mesenchymal Stem Cells. Exp. Cell Res. 2011, 317, 1541–1547. [Google Scholar] [CrossRef]
- Chen, J.-H.; Ozanne, S.E.; Hales, C.N. Methods of Cellular Senescence Induction Using Oxidative Stress. Methods Mol. Biol. 2007, 371, 179–189. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic Ras Provokes Premature Cell Senescence Associated with Accumulation of P53 and P16INK4a. Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Toussaint, O.; Dumont, P.; Remacle, J.; Dierick, J.-F.; Pascal, T.; Frippiat, C.; Magalhaes, J.P.; Zdanov, S.; Chainiaux, F. Stress-Induced Premature Senescence or Stress-Induced Senescence-like Phenotype: One in Vivo Reality, Two Possible Definitions? ScientificWorldJournal 2002, 2, 230–247. [Google Scholar] [CrossRef] [PubMed]
- Victorelli, S.; Passos, J.F. Telomeres and Cell Senescence—Size Matters Not. EBioMedicine 2017, 21, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Herranz, N.; Gallage, S.; Mellone, M.; Wuestefeld, T.; Klotz, S.; Hanley, C.J.; Raguz, S.; Acosta, J.C.; Innes, A.J.; Banito, A.; et al. MTOR Regulates MAPKAPK2 Translation to Control the Senescence-Associated Secretory Phenotype. Nat. Cell Biol. 2015, 17, 1205–1217. [Google Scholar] [CrossRef]
- Astle, M.V.; Hannan, K.M.; Ng, P.Y.; Lee, R.S.; George, A.J.; Hsu, A.K.; Haupt, Y.; Hannan, R.D.; Pearson, R.B. AKT Induces Senescence in Human Cells via MTORC1 and P53 in the Absence of DNA Damage: Implications for Targeting MTOR during Malignancy. Oncogene 2012, 31, 1949–1962. [Google Scholar] [CrossRef]
- Castilho, R.M.; Squarize, C.H.; Chodosh, L.A.; Williams, B.O.; Gutkind, J.S. MTOR Mediates Wnt-Induced Epidermal Stem Cell Exhaustion and Aging. Cell Stem Cell 2009, 5, 279–289. [Google Scholar] [CrossRef]
- Walters, H.E.; Deneka-Hannemann, S.; Cox, L.S. Reversal of Phenotypes of Cellular Senescence by Pan-MTOR Inhibition. Aging (Albany NY) 2016, 8, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Raj, K. DNA Methylation-Based Biomarkers and the Epigenetic Clock Theory of Ageing. Nat. Rev. Genet. 2018, 19, 371. [Google Scholar] [CrossRef]
- Horvath, S. DNA Methylation Age of Human Tissues and Cell Types. Genome Biol. 2013, 14, 3156. [Google Scholar] [CrossRef]
- Dou, Z.; Ghosh, K.; Vizioli, M.G.; Zhu, J.; Sen, P.; Wangensteen, K.J.; Simithy, J.; Lan, Y.; Lin, Y.; Zhou, Z.; et al. Cytoplasmic Chromatin Triggers Inflammation in Senescence and Cancer. Nature 2017, 550, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four Faces of Cellular Senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef]
- Malaquin, N.; Martinez, A.; Rodier, F. Keeping the Senescence Secretome under Control: Molecular Reins on the Senescence-Associated Secretory Phenotype. Exp. Gerontol. 2016, 82, 39–49. [Google Scholar] [CrossRef]
- Sagiv, A.; Krizhanovsky, V. Immunosurveillance of Senescent Cells: The Bright Side of the Senescence Program. Biogerontology 2013, 14, 617–628. [Google Scholar] [CrossRef]
- van Deursen, J.M. The Role of Senescent Cells in Ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Antonangeli, F.; Zingoni, A.; Soriani, A.; Santoni, A. Senescent Cells: Living or Dying Is a Matter of NK Cells. J. Leukoc. Biol. 2019, 105, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Burton, D.G.A.; Stolzing, A. Cellular Senescence: Immunosurveillance and Future Immunotherapy. Ageing Res. Rev. 2018, 43, 17–25. [Google Scholar] [CrossRef]
- Sagiv, A.; Biran, A.; Yon, M.; Simon, J.; Lowe, S.W.; Krizhanovsky, V. Granule Exocytosis Mediates Immune Surveillance of Senescent Cells. Oncogene 2013, 32, 1971–1977. [Google Scholar] [CrossRef]
- Kale, A.; Sharma, A.; Stolzing, A.; Desprez, P.-Y.; Campisi, J. Role of Immune Cells in the Removal of Deleterious Senescent Cells. Immun. Ageing 2020, 17, 16. [Google Scholar] [CrossRef]
- Storer, M.; Mas, A.; Robert-Moreno, A.; Pecoraro, M.; Ortells, M.C.; Di Giacomo, V.; Yosef, R.; Pilpel, N.; Krizhanovsky, V.; Sharpe, J.; et al. Senescence Is a Developmental Mechanism That Contributes to Embryonic Growth and Patterning. Cell 2013, 155, 1119–1130. [Google Scholar] [CrossRef]
- Walters, H.E.; Yun, M.H. Rising from the Ashes: Cellular Senescence in Regeneration. Curr. Opin. Genet. Dev. 2020, 64, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Davaapil, H.; Brockes, J.P.; Yun, M.H. Conserved and Novel Functions of Programmed Cellular Senescence during Vertebrate Development. Development 2017, 144, 106–114. [Google Scholar] [CrossRef] [PubMed]
- Mosteiro, L.; Pantoja, C.; Alcazar, N.; Marión, R.M.; Chondronasiou, D.; Rovira, M.; Fernandez-Marcos, P.J.; Muñoz-Martin, M.; Blanco-Aparicio, C.; Pastor, J.; et al. Tissue Damage and Senescence Provide Critical Signals for Cellular Reprogramming in Vivo. Science 2016, 354, aaf4445. [Google Scholar] [CrossRef]
- Yun, M.H.; Davaapil, H.; Brockes, J.P. Recurrent Turnover of Senescent Cells during Regeneration of a Complex Structure. eLife 2015, 4, e05505. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Espín, D.; Cañamero, M.; Maraver, A.; Gómez-López, G.; Contreras, J.; Murillo-Cuesta, S.; Rodríguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Campisi, J. Cellular Senescence as a Tumor-Suppressor Mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Ovadya, Y.; Landsberger, T.; Leins, H.; Vadai, E.; Gal, H.; Biran, A.; Yosef, R.; Sagiv, A.; Agrawal, A.; Shapira, A.; et al. Impaired Immune Surveillance Accelerates Accumulation of Senescent Cells and Aging. Nat. Commun. 2018, 9, 5435. [Google Scholar] [CrossRef]
- Muñoz, D.P.; Yannone, S.M.; Daemen, A.; Sun, Y.; Vakar-Lopez, F.; Kawahara, M.; Freund, A.M.; Rodier, F.; Wu, J.D.; Desprez, P.-Y.; et al. Targetable Mechanisms Driving Immunoevasion of Persistent Senescent Cells Link Chemotherapy-Resistant Cancer to Aging. JCI Insight 2019, 5, 124716. [Google Scholar] [CrossRef]
- Yousefzadeh, M.J.; Flores, R.R.; Zhu, Y.; Schmiechen, Z.C.; Brooks, R.W.; Trussoni, C.E.; Cui, Y.; Angelini, L.; Lee, K.-A.; McGowan, S.J.; et al. An Aged Immune System Drives Senescence and Ageing of Solid Organs. Nature 2021, 594, 100–105. [Google Scholar] [CrossRef]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Freund, A.; Orjalo, A.V.; Desprez, P.-Y.; Campisi, J. Inflammatory Networks during Cellular Senescence: Causes and Consequences. Trends Mol. Med. 2010, 16, 238–246. [Google Scholar] [CrossRef]
- Basisty, N.; Kale, A.; Jeon, O.H.; Kuehnemann, C.; Payne, T.; Rao, C.; Holtz, A.; Shah, S.; Sharma, V.; Ferrucci, L.; et al. A Proteomic Atlas of Senescence-Associated Secretomes for Aging Biomarker Development. PLOS Biol. 2020, 18, e3000599. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.; Scuoppo, C.; Wang, X.; Fang, X.; Balgley, B.; Bolden, J.E.; Premsrirut, P.; Luo, W.; Chicas, A.; Lee, C.S.; et al. Control of the Senescence-Associated Secretory Phenotype by NF-ΚB Promotes Senescence and Enhances Chemosensitivity. Genes Dev. 2011, 25, 2125–2136. [Google Scholar] [CrossRef] [PubMed]
- Meyer, P.; Maity, P.; Burkovski, A.; Schwab, J.; Müssel, C.; Singh, K.; Ferreira, F.F.; Krug, L.; Maier, H.J.; Wlaschek, M.; et al. A Model of the Onset of the Senescence Associated Secretory Phenotype after DNA Damage Induced Senescence. PLOS Comput. Biol. 2017, 13, e1005741. [Google Scholar] [CrossRef]
- Salminen, A.; Kauppinen, A.; Kaarniranta, K. Emerging Role of NF-ΚB Signaling in the Induction of Senescence-Associated Secretory Phenotype (SASP). Cell. Signal. 2012, 24, 835–845. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.-W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 Mediates a Switch between Two Distinct Secretomes during Senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Neri, F.; Basisty, N.; Desprez, P.-Y.; Campisi, J.; Schilling, B. Quantitative Proteomic Analysis of the Senescence-Associated Secretory Phenotype by Data-Independent Acquisition. Curr. Protoc. 2021, 1, e32. [Google Scholar] [CrossRef] [PubMed]
- Nelson, G.; Wordsworth, J.; Wang, C.; Jurk, D.; Lawless, C.; Martin-Ruiz, C.; von Zglinicki, T. A Senescent Cell Bystander Effect: Senescence-Induced Senescence. Aging Cell 2012, 11, 345–349. [Google Scholar] [CrossRef]
- Jeon, O.H.; David, N.; Campisi, J.; Elisseeff, J.H. Senescent Cells and Osteoarthritis: A Painful Connection. J. Clin. Investig. 2018, 128, 1229–1237. [Google Scholar] [CrossRef]
- Xu, M.; Bradley, E.W.; Weivoda, M.M.; Hwang, S.M.; Pirtskhalava, T.; Decklever, T.; Curran, G.L.; Ogrodnik, M.; Jurk, D.; Johnson, K.O.; et al. Transplanted Senescent Cells Induce an Osteoarthritis-Like Condition in Mice. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 780–785. [Google Scholar] [CrossRef]
- Jeon, O.H.; Kim, C.; Laberge, R.-M.; Demaria, M.; Rathod, S.; Vasserot, A.P.; Chung, J.W.; Kim, D.H.; Poon, Y.; David, N.; et al. Local Clearance of Senescent Cells Attenuates the Development of Post-Traumatic Osteoarthritis and Creates a pro-Regenerative Environment. Nat. Med. 2017, 23, 775–781. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics Improve Physical Function and Increase Lifespan in Old Age. Nat. Med. 2018, 24, 1246. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Liu, Z.; Chen, V.P.; Wang, L.; Inman, C.L.; Zhou, Y.; Guo, C.; Tchkonia, T.; Rowe, D.W.; Kuchel, G.A.; et al. Transplanting Cells from Old but Not Young Donors Causes Physical Dysfunction in Older Recipients. Aging Cell 2020, 19, e13106. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic Inflammation Induces Telomere Dysfunction and Accelerates Ageing in Mice. Nat. Commun. 2014, 5, 4172. [Google Scholar] [CrossRef] [PubMed]
- Ryder, J.R.; Northrop, E.; Rudser, K.D.; Kelly, A.S.; Gao, Z.; Khoury, P.R.; Kimball, T.R.; Dolan, L.M.; Urbina, E.M. Accelerated Early Vascular Aging Among Adolescents With Obesity and/or Type 2 Diabetes Mellitus. J. Am. Heart Assoc. 2020, 9, e014891. [Google Scholar] [CrossRef]
- Teissier, T.; Boulanger, É. The Receptor for Advanced Glycation End-Products (RAGE) Is an Important Pattern Recognition Receptor (PRR) for Inflammaging. Biogerontology 2019, 20, 279–301. [Google Scholar] [CrossRef]
- Teissier, T.; Quersin, V.; Gnemmi, V.; Daroux, M.; Howsam, M.; Delguste, F.; Lemoine, C.; Fradin, C.; Schmidt, A.-M.; Cauffiez, C.; et al. Knockout of Receptor for Advanced Glycation End-Products Attenuates Age-Related Renal Lesions. Aging Cell 2019, 18, e12850. [Google Scholar] [CrossRef]
- Davalos, A.R.; Kawahara, M.; Malhotra, G.K.; Schaum, N.; Huang, J.; Ved, U.; Beausejour, C.M.; Coppe, J.-P.; Rodier, F.; Campisi, J. P53-Dependent Release of Alarmin HMGB1 Is a Central Mediator of Senescent Phenotypes. J. Cell Biol. 2013, 201, 613–629. [Google Scholar] [CrossRef]
- Chuprin, A.; Gal, H.; Biron-Shental, T.; Biran, A.; Amiel, A.; Rozenblatt, S.; Krizhanovsky, V. Cell Fusion Induced by ERVWE1 or Measles Virus Causes Cellular Senescence. Genes Dev. 2013, 27, 2356–2366. [Google Scholar] [CrossRef] [PubMed]
- Kohli, J.; Veenstra, I.; Demaria, M. The Struggle of a Good Friend Getting Old: Cellular Senescence in Viral Responses and Therapy. EMBO Rep. 2021, 22, e52243. [Google Scholar] [CrossRef]
- Camell, C.D.; Yousefzadeh, M.J.; Zhu, Y.; Langhi Prata, L.G.P.; Huggins, M.A.; Pierson, M.; Zhang, L.; O’Kelly, R.D.; Pirtskhalava, T.; Xun, P.; et al. Senolytics Reduce Coronavirus-Related Mortality in Old Mice. Science 2021, 373, eabe4832. [Google Scholar] [CrossRef]
- Cox, L.S.; Lord, J.M. Targeting Aging Cells Improves Survival. Science 2021, 373, 281–282. [Google Scholar] [CrossRef]
- Ibler, A.E.M.; ElGhazaly, M.; Naylor, K.L.; Bulgakova, N.A.; El-Khamisy, S.F.; Humphreys, D. Typhoid Toxin Exhausts the RPA Response to DNA Replication Stress Driving Senescence and Salmonella Infection. Nat. Commun. 2019, 10, 4040. [Google Scholar] [CrossRef] [PubMed]
- Corona, G.; Pizzocaro, A.; Vena, W.; Rastrelli, G.; Semeraro, F.; Isidori, A.M.; Pivonello, R.; Salonia, A.; Sforza, A.; Maggi, M. Diabetes Is Most Important Cause for Mortality in COVID-19 Hospitalized Patients: Systematic Review and Meta-Analysis. Rev. Endocr. Metab. Disord. 2021, 22, 275–296. [Google Scholar] [CrossRef]
- Poly, T.N.; Islam, M.M.; Yang, H.C.; Lin, M.C.; Jian, W.-S.; Hsu, M.-H.; Jack Li, Y.-C. Obesity and Mortality Among Patients Diagnosed With COVID-19: A Systematic Review and Meta-Analysis. Front. Med. 2021, 8, 620044. [Google Scholar] [CrossRef] [PubMed]
- Aghili, S.M.M.; Ebrahimpur, M.; Arjmand, B.; Shadman, Z.; Pejman Sani, M.; Qorbani, M.; Larijani, B.; Payab, M. Obesity in COVID-19 Era, Implications for Mechanisms, Comorbidities, and Prognosis: A Review and Meta-Analysis. Int. J. Obes. 2021, 45, 998–1016. [Google Scholar] [CrossRef]
- Lee, S.; Yu, Y.; Trimpert, J.; Benthani, F.; Mairhofer, M.; Richter-Pechanska, P.; Wyler, E.; Belenki, D.; Kaltenbrunner, S.; Pammer, M.; et al. Virus-Induced Senescence Is a Driver and Therapeutic Target in COVID-19. Nature 2021, 599, 283–289. [Google Scholar] [CrossRef]
- Ershler, W.B.; Keller, E.T. Age-Associated Increased Interleukin-6 Gene Expression, Late-Life Diseases, and Frailty. Annu. Rev. Med. 2000, 51, 245–270. [Google Scholar] [CrossRef]
- Minciullo, P.L.; Catalano, A.; Mandraffino, G.; Casciaro, M.; Crucitti, A.; Maltese, G.; Morabito, N.; Lasco, A.; Gangemi, S.; Basile, G. Inflammaging and Anti-Inflammaging: The Role of Cytokines in Extreme Longevity. Arch. Immunol. Ther. Exp. 2016, 64, 111–126. [Google Scholar] [CrossRef]
- Fagiolo, U.; Cossarizza, A.; Scala, E.; Fanales-Belasio, E.; Ortolani, C.; Cozzi, E.; Monti, D.; Franceschi, C.; Paganelli, R. Increased Cytokine Production in Mononuclear Cells of Healthy Elderly People. Eur. J. Immunol. 1993, 23, 2375–2378. [Google Scholar] [CrossRef]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-Dependent Alterations of Monocyte Subsets and Monocyte-Related Chemokine Pathways in Healthy Adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Gerli, R.; Monti, D.; Bistoni, O.; Mazzone, A.M.; Peri, G.; Cossarizza, A.; Di Gioacchino, M.; Cesarotti, M.E.; Doni, A.; Mantovani, A.; et al. Chemokines, STNF-Rs and SCD30 Serum Levels in Healthy Aged People and Centenarians. Mech. Ageing Dev. 2000, 121, 37–46. [Google Scholar] [CrossRef]
- Mariani, E.; Cattini, L.; Neri, S.; Malavolta, M.; Mocchegiani, E.; Ravaglia, G.; Facchini, A. Simultaneous Evaluation of Circulating Chemokine and Cytokine Profiles in Elderly Subjects by Multiplex Technology: Relationship with Zinc Status. Biogerontology 2006, 7, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Morrisette-Thomas, V.; Cohen, A.A.; Fülöp, T.; Riesco, É.; Legault, V.; Li, Q.; Milot, E.; Dusseault-Bélanger, F.; Ferrucci, L. Inflamm-Aging Does Not Simply Reflect Increases in pro-Inflammatory Markers. Mech. Ageing Dev. 2014, 139, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Bandeen-Roche, K.; Walston, J.D.; Huang, Y.; Semba, R.D.; Ferrucci, L. Measuring Systemic Inflammatory Regulation in Older Adults: Evidence and Utility. Rejuvenation Res. 2009, 12, 403–410. [Google Scholar] [CrossRef]
- Hsu, F.-C.; Kritchevsky, S.B.; Liu, Y.; Kanaya, A.; Newman, A.B.; Perry, S.E.; Visser, M.; Pahor, M.; Harris, T.B.; Nicklas, B.J.; et al. Association between Inflammatory Components and Physical Function in the Health, Aging, and Body Composition Study: A Principal Component Analysis Approach. J. Gerontol. A Biol. Sci. Med. Sci. 2009, 64, 581–589. [Google Scholar] [CrossRef]
- Tziakas, D.N.; Chalikias, G.K.; Kaski, J.C.; Kekes, A.; Hatzinikolaou, E.I.; Stakos, D.A.; Tentes, I.K.; Kortsaris, A.X.; Hatseras, D.I. Inflammatory and Anti-Inflammatory Variable Clusters and Risk Prediction in Acute Coronary Syndrome Patients: A Factor Analysis Approach. Atherosclerosis 2007, 193, 196–203. [Google Scholar] [CrossRef]
- Kip, K.E.; Marroquin, O.C.; Shaw, L.J.; Arant, C.B.; Wessel, T.R.; Olson, M.B.; Johnson, B.D.; Mulukutla, S.; Sopko, G.; Merz, C.N.B.; et al. Global Inflammation Predicts Cardiovascular Risk in Women: A Report from the Women’s Ischemia Syndrome Evaluation (WISE) Study. Am. Heart J. 2005, 150, 900–906. [Google Scholar] [CrossRef]
- Ferrucci, L.; Corsi, A.; Lauretani, F.; Bandinelli, S.; Bartali, B.; Taub, D.D.; Guralnik, J.M.; Longo, D.L. The Origins of Age-Related Proinflammatory State. Blood 2005, 105, 2294–2299. [Google Scholar] [CrossRef]
- Cesari, M.; Penninx, B.W.J.H.; Pahor, M.; Lauretani, F.; Corsi, A.M.; Rhys Williams, G.; Guralnik, J.M.; Ferrucci, L. Inflammatory Markers and Physical Performance in Older Persons: The InCHIANTI Study. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 242–248. [Google Scholar] [CrossRef]
- Collerton, J.; Martin-Ruiz, C.; Davies, K.; Hilkens, C.M.; Isaacs, J.; Kolenda, C.; Parker, C.; Dunn, M.; Catt, M.; Jagger, C.; et al. Frailty and the Role of Inflammation, Immunosenescence and Cellular Ageing in the Very Old: Cross-Sectional Findings from the Newcastle 85+ Study. Mech. Ageing Dev. 2012, 133, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Varadhan, R.; Yao, W.; Matteini, A.; Beamer, B.A.; Xue, Q.-L.; Yang, H.; Manwani, B.; Reiner, A.; Jenny, N.; Parekh, N.; et al. Simple Biologically Informed Inflammatory Index of Two Serum Cytokines Predicts 10 Year All-Cause Mortality in Older Adults. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 165–173. [Google Scholar] [CrossRef]
- Weinhold, B.; Rüther, U. Interleukin-6-Dependent and -Independent Regulation of the Human C-Reactive Protein Gene. Biochem. J. 1997, 327, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lu, L.; Xie, Y.; Chen, X.; Tian, L.; Liang, Y.; Li, H.; Zhang, J.; Liu, Y.; Yu, X. Interleukin-6 Knockout Inhibits Senescence of Bone Mesenchymal Stem Cells in High-Fat Diet-Induced Bone Loss. Front. Endocrinol. 2020, 11, 622950. [Google Scholar] [CrossRef] [PubMed]
- Cardoso, A.L.; Fernandes, A.; Aguilar-Pimentel, J.A.; de Angelis, M.H.; Guedes, J.R.; Brito, M.A.; Ortolano, S.; Pani, G.; Athanasopoulou, S.; Gonos, E.S.; et al. Towards Frailty Biomarkers: Candidates from Genes and Pathways Regulated in Aging and Age-Related Diseases. Ageing Res. Rev. 2018, 47, 214–277. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Castellino, F.J.; Ploplis, V.A. Plasminogen Activator Inhibitor-1 (PAI-1) Is Cardioprotective in Mice by Maintaining Microvascular Integrity and Cardiac Architecture. Blood 2010, 115, 2038–2047. [Google Scholar] [CrossRef] [PubMed]
- Miles, L.A.; Parmer, R.J. PAI-1: Cardiac Friend or Foe? Blood 2010, 115, 1862–1863. [Google Scholar] [CrossRef]
- Eren, M.; Boe, A.E.; Klyachko, E.A.; Vaughan, D.E. Role of Plasminogen Activator Inhibitor-1 in Senescence and Aging. Semin. Thromb. Hemost. 2014, 40, 645–651. [Google Scholar] [CrossRef]
- Eren, M.; Boe, A.E.; Murphy, S.B.; Place, A.T.; Nagpal, V.; Morales-Nebreda, L.; Urich, D.; Quaggin, S.E.; Budinger, G.R.S.; Mutlu, G.M.; et al. PAI-1–Regulated Extracellular Proteolysis Governs Senescence and Survival in Klotho Mice. Proc. Natl. Acad. Sci. USA 2014, 111, 7090–7095. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Rai, R.; Park, K.E.; Eren, M.; Miyata, T.; Wilsbacher, L.D.; Vaughan, D.E. A Small Molecule Inhibitor of PAI-1 Protects against Doxorubicin-Induced Cellular Senescence. Oncotarget 2016, 7, 72443–72457. [Google Scholar] [CrossRef] [PubMed]
- Mailliez, A.; Guilbaud, A.; Puisieux, F.; Dauchet, L.; Boulanger, É. Circulating Biomarkers Characterizing Physical Frailty: CRP, Hemoglobin, Albumin, 25OHD and Free Testosterone as Best Biomarkers. Results of a Meta-Analysis. Exp. Gerontol. 2020, 139, 111014. [Google Scholar] [CrossRef]
- Jylhä, M.; Paavilainen, P.; Lehtimäki, T.; Goebeler, S.; Karhunen, P.J.; Hervonen, A.; Hurme, M. Interleukin-1 Receptor Antagonist, Interleukin-6, and C-Reactive Protein as Predictors of Mortality in Nonagenarians: The Vitality 90+ Study. J. Gerontol. A Biol. Sci. Med. Sci. 2007, 62, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Suh, Y.; Atzmon, G.; Cho, M.-O.; Hwang, D.; Liu, B.; Leahy, D.J.; Barzilai, N.; Cohen, P. Functionally Significant Insulin-like Growth Factor I Receptor Mutations in Centenarians. Proc. Natl. Acad. Sci. USA 2008, 105, 3438–3442. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S.; Pirazzini, C.; Bacalini, M.G.; Gentilini, D.; Di Blasio, A.M.; Delledonne, M.; Mari, D.; Arosio, B.; Monti, D.; Passarino, G.; et al. Decreased Epigenetic Age of PBMCs from Italian Semi-Supercentenarians and Their Offspring. Aging (Albany NY) 2015, 7, 1159–1170. [Google Scholar] [CrossRef]
- Atzmon, G.; Schechter, C.; Greiner, W.; Davidson, D.; Rennert, G.; Barzilai, N. Clinical Phenotype of Families with Longevity. J. Am. Geriatr. Soc. 2004, 52, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Atzmon, G.; Barzilai, N.; Hollowell, J.G.; Surks, M.I.; Gabriely, I. Extreme Longevity Is Associated with Increased Serum Thyrotropin. J. Clin. Endocrinol. Metab. 2009, 94, 1251–1254. [Google Scholar] [CrossRef]
- Del Giudice, M.; Gangestad, S.W. Rethinking IL-6 and CRP: Why They Are More than Inflammatory Biomarkers, and Why It Matters. Brain Behav. Immun. 2018, 70, 61–75. [Google Scholar] [CrossRef]
- Weyh, C.; Krüger, K.; Strasser, B. Physical Activity and Diet Shape the Immune System during Aging. Nutrients 2020, 12, 622. [Google Scholar] [CrossRef]
- Brandt, C.; Pedersen, B.K. The Role of Exercise-Induced Myokines in Muscle Homeostasis and the Defense against Chronic Diseases. J. Biomed. Biotechnol. 2010, 2010, 520258. [Google Scholar] [CrossRef]
- Steensberg, A.; Fischer, C.P.; Keller, C.; Møller, K.; Pedersen, B.K. IL-6 Enhances Plasma IL-1ra, IL-10, and Cortisol in Humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E433–E437. [Google Scholar] [CrossRef]
- Starkie, R.; Ostrowski, S.R.; Jauffred, S.; Febbraio, M.; Pedersen, B.K. Exercise and IL-6 Infusion Inhibit Endotoxin-Induced TNF-Alpha Production in Humans. FASEB J. 2003, 17, 884–886. [Google Scholar] [CrossRef] [PubMed]
- Ubaida-Mohien, C.; Lyashkov, A.; Gonzalez-Freire, M.; Tharakan, R.; Shardell, M.; Moaddel, R.; Semba, R.D.; Chia, C.W.; Gorospe, M.; Sen, R.; et al. Discovery Proteomics in Aging Human Skeletal Muscle Finds Change in Spliceosome, Immunity, Proteostasis and Mitochondria. eLife 2019, 8, e49874. [Google Scholar] [CrossRef]
- Dinarello, C.A. Interleukin 1 and Interleukin 18 as Mediators of Inflammation and the Aging Process. Am. J. Clin. Nutr. 2006, 83, 447S–455S. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Novick, D.; Kim, S.; Kaplanski, G. Interleukin-18 and IL-18 Binding Protein. Front. Immunol. 2013, 4, 289. [Google Scholar] [CrossRef]
- Pu, Z.; Che, Y.; Zhang, W.; Sun, H.; Meng, T.; Xie, H.; Cao, L.; Hao, H. Dual Roles of IL-18 in Colitis through Regulation of the Function and Quantity of Goblet Cells. Int. J. Mol. Med. 2019, 43, 2291–2302. [Google Scholar] [CrossRef]
- Macia, L.; Tan, J.; Vieira, A.T.; Leach, K.; Stanley, D.; Luong, S.; Maruya, M.; Ian McKenzie, C.; Hijikata, A.; Wong, C.; et al. Metabolite-Sensing Receptors GPR43 and GPR109A Facilitate Dietary Fibre-Induced Gut Homeostasis through Regulation of the Inflammasome. Nat. Commun. 2015, 6, 6734. [Google Scholar] [CrossRef] [PubMed]
- Eisenmesser, E.Z.; Gottschlich, A.; Redzic, J.S.; Paukovich, N.; Nix, J.C.; Azam, T.; Zhang, L.; Zhao, R.; Kieft, J.S.; The, E.; et al. Interleukin-37 Monomer Is the Active Form for Reducing Innate Immunity. Proc. Natl. Acad. Sci. USA 2019, 116, 5514–5522. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Xie, B.; Wu, G.; Hu, J.; Wang, D.; Cai, X.; Li, J. Interleukin-37: The Effect of Anti-Inflammatory Response in Human Coronary Artery Endothelial Cells. Available online: https://www.hindawi.com/journals/mi/2019/2650590/ (accessed on 16 June 2020).
- Henry, C.J.; Casás-Selves, M.; Kim, J.; Zaberezhnyy, V.; Aghili, L.; Daniel, A.E.; Jimenez, L.; Azam, T.; McNamee, E.N.; Clambey, E.T.; et al. Aging-Associated Inflammation Promotes Selection for Adaptive Oncogenic Events in B Cell Progenitors. J. Clin. Investig. 2015, 125, 4666–4680. [Google Scholar] [CrossRef]
- Henry, C.J.; Eisenmesser, E.; Dinarello, C.A.; DeGregori, J. Interleukin-37 Improves Aging-Associated Declines in Adaptive Immunity Leading to Enhanced Suppression of Ph+ Leukemia. J. Immunol. 2019, 202, 65.8. [Google Scholar]
- Sapinsley, Z.J.; Ballak, D.B.; Brunt, V.E.; Zigler, M.C.; Gioscia-Ryan, R.A.; Richey, J.J.; Dinarello, C.A.; Seals, D.R. Administration of Anti-Inflammatory Interleukin-37 Ameliorates Age-Related Vascular, Metabolic and Physical Dysfunction in Mice. FASEB J. 2017, 31, 1079. [Google Scholar] [CrossRef]
- Ballak, D.B.; Brunt, V.E.; Sapinsley, Z.J.; Ziemba, B.P.; Richey, J.J.; Zigler, M.C.; Johnson, L.C.; Gioscia-Ryan, R.A.; Culp-Hill, R.; Eisenmesser, E.Z.; et al. Short-Term Interleukin-37 Treatment Improves Vascular Endothelial Function, Endurance Exercise Capacity, and Whole-Body Glucose Metabolism in Old Mice. Aging Cell 2020, 19, e13074. [Google Scholar] [CrossRef]
- Sanjabi, S.; Zenewicz, L.A.; Kamanaka, M.; Flavell, R.A. Anti- and Pro-Inflammatory Roles of TGF-β, IL-10, and IL-22 In Immunity and Autoimmunity. Curr. Opin. Pharm. 2009, 9, 447–453. [Google Scholar] [CrossRef]
- Takahashi, T.; Ellingson, M.K.; Wong, P.; Israelow, B.; Lucas, C.; Klein, J.; Silva, J.; Mao, T.; Oh, J.E.; Tokuyama, M.; et al. Sex Differences in Immune Responses That Underlie COVID-19 Disease Outcomes. Nature 2020, 588, 315–320. [Google Scholar] [CrossRef]
- Yu, B.; Qi, Y.; Li, R.; Shi, Q.; Satpathy, A.T.; Chang, H.Y. B Cell-Specific XIST Complex Enforces X-Inactivation and Restrains Atypical B Cells. Cell 2021, 184, 1790–1803. [Google Scholar] [CrossRef]
- Girard, S.; Kadhim, H.; Larouche, A.; Roy, M.; Gobeil, F.; Sébire, G. Pro-Inflammatory Disequilibrium of the IL-1 Beta/IL-1ra Ratio in an Experimental Model of Perinatal Brain Damages Induced by Lipopolysaccharide and Hypoxia-Ischemia. Cytokine 2008, 43, 54–62. [Google Scholar] [CrossRef]
- Arend, W.P. The Balance between IL-1 and IL-1Ra in Disease. Cytokine Growth Factor Rev. 2002, 13, 323–340. [Google Scholar] [CrossRef]
- Esmon, C.T. The Interactions between Inflammation and Coagulation. Br. J. Haematol. 2005, 131, 417–430. [Google Scholar] [CrossRef]
- Branchford, B.R.; Carpenter, S.L. The Role of Inflammation in Venous Thromboembolism. Front. Pediatr. 2018, 6, 142. [Google Scholar] [CrossRef]
- Engbers, M.J.; Vlieg, A.V.H.; Rosendaal, F.R. Venous Thrombosis in the Elderly: Incidence, Risk Factors and Risk Groups. J. Thromb. Haemost. 2010, 8, 2105–2112. [Google Scholar] [CrossRef]
- Cohen, H.J.; Harris, T.; Pieper, C.F. Coagulation and Activation of Inflammatory Pathways in the Development of Functional Decline and Mortality in the Elderly. Am. J. Med. 2003, 114, 180–187. [Google Scholar] [CrossRef]
- Ferrucci, L.; Harris, T.B.; Guralnik, J.M.; Tracy, R.P.; Corti, M.C.; Cohen, H.J.; Penninx, B.; Pahor, M.; Wallace, R.; Havlik, R.J. Serum IL-6 Level and the Development of Disability in Older Persons. J. Am. Geriatr. Soc. 1999, 47, 639–646. [Google Scholar] [CrossRef]
- Grobler, C.; Maphumulo, S.C.; Grobbelaar, L.M.; Bredenkamp, J.C.; Laubscher, G.J.; Lourens, P.J.; Steenkamp, J.; Kell, D.B.; Pretorius, E. Covid-19: The Rollercoaster of Fibrin(Ogen), D-Dimer, Von Willebrand Factor, P-Selectin and Their Interactions with Endothelial Cells, Platelets and Erythrocytes. Int. J. Mol. Sci. 2020, 21, 5168. [Google Scholar] [CrossRef]
- Coppola, R.; Mari, D.; Lattuada, A.; Franceschi, C. Von Willebrand Factor in Italian Centenarians. Haematologica 2003, 88, 39–43. [Google Scholar]
- Mari, D.; Mannucci, P.M.; Coppola, R.; Bottasso, B.; Bauer, K.A.; Rosenberg, R.D. Hypercoagulability in Centenarians: The Paradox of Successful Aging. Blood 1995, 85, 3144–3149. [Google Scholar] [CrossRef]
- Mari, D.; Coppola, R.; Provenzano, R. Hemostasis Factors and Aging. Exp. Gerontol. 2008, 43, 66–73. [Google Scholar] [CrossRef]
- Sanada, F.; Taniyama, Y.; Muratsu, J.; Otsu, R.; Shimizu, H.; Rakugi, H.; Morishita, R. Source of Chronic Inflammation in Aging. Front. Cardiovasc. Med. 2018, 5. [Google Scholar] [CrossRef]
- Zhang, H.; Puleston, D.J.; Simon, A.K. Autophagy and Immune Senescence. Trends Mol. Med. 2016, 22, 671–686. [Google Scholar] [CrossRef]
- Wijsman, C.A.; Maier, A.B.; de Craen, A.J.M.; van den Biggelaar, A.H.J.; Westendorp, R.G.J. An Unopposed Proinflammatory Response Is Beneficial for Survival in the Oldest Old. Results of the Leiden 85-Plus Study. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66A, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Panda, A.; Arjona, A.; Sapey, E.; Bai, F.; Fikrig, E.; Montgomery, R.R.; Lord, J.M.; Shaw, A.C. Human Innate Immunosenescence: Causes and Consequences for Immunity in Old Age. Trends Immunol. 2009, 30, 325–333. [Google Scholar] [CrossRef]
- Ogata, K.; An, E.; Shioi, Y.; Nakamura, K.; Luo, S.; Yokose, N.; Minami, S.; Dan, K. Association between Natural Killer Cell Activity and Infection in Immunologically Normal Elderly People. Clin. Exp. Immunol. 2001, 124, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Hazeldine, J.; Lord, J.M. The Impact of Ageing on Natural Killer Cell Function and Potential Consequences for Health in Older Adults. Ageing Res. Rev. 2013, 12, 1069–1078. [Google Scholar] [CrossRef]
- Zimmermann, P.; Curtis, N. Why Is COVID-19 Less Severe in Children? A Review of the Proposed Mechanisms Underlying the Age-Related Difference in Severity of SARS-CoV-2 Infections. Arch. Dis. Child. 2021, 106, 429–439. [Google Scholar] [CrossRef] [PubMed]
- Fábián, T.K.; Hermann, P.; Beck, A.; Fejérdy, P.; Fábián, G. Salivary Defense Proteins: Their Network and Role in Innate and Acquired Oral Immunity. Int. J. Mol. Sci. 2012, 13, 4295–4320. [Google Scholar] [CrossRef]
- Mun, J.J.; Tam, C.; Evans, D.J.; Fleiszig, S.M.J. Modulation of Epithelial Immunity by Mucosal Fluid. Sci. Rep. 2011, 1, 8. [Google Scholar] [CrossRef] [PubMed]
- Voynow, J.A.; Rubin, B.K. Mucins, Mucus, and Sputum. Chest 2009, 135, 505–512. [Google Scholar] [CrossRef]
- Zhang, Y.-W.; Bi, L.-T.; Hou, S.-P.; Zhao, X.-L.; Song, Y.-L.; Ma, T.-H. Reduced Lung Water Transport Rate Associated with Downregulation of Aquaporin-1 and Aquaporin-5 in Aged Mice. Clin. Exp. Pharm. Physiol. 2009, 36, 734–738. [Google Scholar] [CrossRef]
- Ho, J.C.; Chan, K.N.; Hu, W.H.; Lam, W.K.; Zheng, L.; Tipoe, G.L.; Sun, J.; Leung, R.; Tsang, K.W. The Effect of Aging on Nasal Mucociliary Clearance, Beat Frequency, and Ultrastructure of Respiratory Cilia. Am. J. Respir. Crit. Care Med. 2001, 163, 983–988. [Google Scholar] [CrossRef]
- Grudzinska, F.S.; Brodlie, M.; Scholefield, B.R.; Jackson, T.; Scott, A.; Thickett, D.R.; Sapey, E. Neutrophils in Community-Acquired Pneumonia: Parallels in Dysfunction at the Extremes of Age. Thorax 2020, 75, 164–171. [Google Scholar] [CrossRef]
- Sapey, E.; Patel, J.M.; Greenwood, H.L.; Walton, G.M.; Hazeldine, J.; Sadhra, C.; Parekh, D.; Dancer, R.C.A.; Nightingale, P.; Lord, J.M.; et al. Pulmonary Infections in the Elderly Lead to Impaired Neutrophil Targeting, Which Is Improved by Simvastatin. Am. J. Respir. Crit. Care Med. 2017, 196, 1325–1336. [Google Scholar] [CrossRef]
- Thevaranjan, N.; Puchta, A.; Schulz, C.; Naidoo, A.; Szamosi, J.C.; Verschoor, C.P.; Loukov, D.; Schenck, L.P.; Jury, J.; Foley, K.P.; et al. Age-Associated Microbial Dysbiosis Promotes Intestinal Permeability, Systemic Inflammation, and Macrophage Dysfunction. Cell Host Microbe 2017, 21, 455–466. [Google Scholar] [CrossRef] [PubMed]
- Hollander, D.; Kaunitz, J.D. The “Leaky Gut”: Tight Junctions but Loose Associations? Dig. Dis. Sci. 2020, 65, 1277–1287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-J.; Guo, D.-W.; Tian, L.; Shang, D.-S.; Zhao, W.-D.; Li, B.; Fang, W.-G.; Zhu, L.; Chen, Y.-H. Peripheral T Cells Derived from Alzheimer’s Disease Patients Overexpress CXCR2 Contributing to Its Transendothelial Migration, Which Is Microglial TNF-α-Dependent. Neurobiol. Aging 2010, 31, 175–188. [Google Scholar] [CrossRef] [PubMed]
- Marques, F.; Sousa, J.C.; Sousa, N.; Palha, J.A. Blood–Brain-Barriers in Aging and in Alzheimer’s Disease. Mol. Neurodegener. 2013, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Pellicanò, M.; Larbi, A.; Goldeck, D.; Colonna-Romano, G.; Buffa, S.; Bulati, M.; Rubino, G.; Iemolo, F.; Candore, G.; Caruso, C.; et al. Immune Profiling of Alzheimer Patients. J. Neuroimmunol. 2012, 242, 52–59. [Google Scholar] [CrossRef]
- Shang, D.S.; Yang, Y.M.; Zhang, H.; Tian, L.; Jiang, J.S.; Dong, Y.B.; Zhang, K.; Li, B.; Zhao, W.D.; Fang, W.G.; et al. Intracerebral GM-CSF Contributes to Transendothelial Monocyte Migration in APP/PS1 Alzheimer’s Disease Mice. J. Cereb. Blood Flow Metab. 2016, 36, 1978–1991. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Dong, G.; Xiao, W.; Xiao, E.; Miao, F.; Syverson, A.; Missaghian, N.; Vafa, R.; Cabrera-Ortega, A.A.; Rossa, C.; et al. Effect of Aging on Periodontal Inflammation, Microbial Colonization, and Disease Susceptibility. J. Dent. Res. 2016, 95, 460–466. [Google Scholar] [CrossRef] [PubMed]
- Nadim, R.; Tang, J.; Dilmohamed, A.; Yuan, S.; Wu, C.; Bakre, A.T.; Partridge, M.; Ni, J.; Copeland, J.R.; Anstey, K.J.; et al. Influence of Periodontal Disease on Risk of Dementia: A Systematic Literature Review and a Meta-Analysis. Eur. J. Epidemiol. 2020, 35, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Watts, A.; Crimmins, E.M.; Gatz, M. Inflammation as a Potential Mediator for the Association between Periodontal Disease and Alzheimer’s Disease. Neuropsychiatr. Dis. Treat. 2008, 4, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Müller, L.; Fülöp, T.; Pawelec, G. Immunosenescence in Vertebrates and Invertebrates. Immun. Ageing 2013, 10, 12. [Google Scholar] [CrossRef]
- Kaufmann, S.H.E.; Dorhoi, A. Molecular Determinants in Phagocyte-Bacteria Interactions. Immunity 2016, 44, 476–491. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; Siracusa, M.C.; Yap, G.S.; Gause, W.C. Innate Cell Communication Kick-Starts Pathogen-Specific Immunity. Nat. Immunol. 2016, 17, 356–363. [Google Scholar] [CrossRef]
- Franchi, L.; Warner, N.; Viani, K.; Nuñez, G. Function of Nod-like Receptors in Microbial Recognition and Host Defense. Immunol. Rev. 2009, 227, 106–128. [Google Scholar] [CrossRef] [PubMed]
- Kato, H.; Takahasi, K.; Fujita, T. RIG-I-like Receptors: Cytoplasmic Sensors for Non-Self RNA. Immunol. Rev. 2011, 243, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Ingle, H.; Prasad, D.V.R.; Kumar, H. Recognition of Bacterial Infection by Innate Immune Sensors. Crit. Rev. Microbiol. 2013, 39, 229–246. [Google Scholar] [CrossRef] [PubMed]
- Plato, A.; Willment, J.A.; Brown, G.D. C-Type Lectin-Like Receptors of the Dectin-1 Cluster: Ligands and Signaling Pathways. Int. Rev. Immunol. 2013, 32, 134–156. [Google Scholar] [CrossRef]
- Roers, A.; Hiller, B.; Hornung, V. Recognition of Endogenous Nucleic Acids by the Innate Immune System. Immunity 2016, 44, 739–754. [Google Scholar] [CrossRef]
- Lin, L. RAGE on the Toll Road? Cell. Mol. Immunol. 2006, 3, 351–358. [Google Scholar]
- Cruickshanks, H.A.; McBryan, T.; Nelson, D.M.; VanderKraats, N.D.; Shah, P.P.; van Tuyn, J.; Singh Rai, T.; Brock, C.; Donahue, G.; Dunican, D.S.; et al. Senescent Cells Harbour Features of the Cancer Epigenome. Nat. Cell Biol. 2013, 15, 1495–1506. [Google Scholar] [CrossRef]
- Simon, M.; Meter, M.V.; Ablaeva, J.; Ke, Z.; Gonzalez, R.S.; Taguchi, T.; Cecco, M.D.; Leonova, K.I.; Kogan, V.; Helfand, S.L.; et al. LINE1 Derepression in Aged Wild Type and SIRT6 Deficient Mice Drives Inflammation. Cell Metab. 2019, 29, 871–885. [Google Scholar] [CrossRef]
- Dranoff, G. Cytokines in Cancer Pathogenesis and Cancer Therapy. Nat. Rev. Cancer 2004, 4, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Markiewski, M.M.; Lambris, J.D. The Role of Complement in Inflammatory Diseases from behind the Scenes into the Spotlight. Am. J. Pathol. 2007, 171, 715–727. [Google Scholar] [CrossRef]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human Bone Marrow Hematopoietic Stem Cells Are Increased in Frequency and Myeloid-Biased with Age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell Intrinsic Alterations Underlie Hematopoietic Stem Cell Aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef] [PubMed]
- Geiger, H.; de Haan, G.; Florian, M.C. The Ageing Haematopoietic Stem Cell Compartment. Nat. Rev. Immunol. 2013, 13, 376–389. [Google Scholar] [CrossRef]
- Jung, H.; Kim, D.O.; Byun, J.-E.; Kim, W.S.; Kim, M.J.; Song, H.Y.; Kim, Y.K.; Kang, D.-K.; Park, Y.-J.; Kim, T.-D.; et al. Thioredoxin-Interacting Protein Regulates Haematopoietic Stem Cell Ageing and Rejuvenation by Inhibiting P38 Kinase Activity. Nat. Commun. 2016, 7, 13674. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Moon, H.-B.; Spangrude, G.J. Major Age-Related Changes of Mouse Hematopoietic Stem/Progenitor Cells. Ann. N. Y. Acad. Sci. 2003, 996, 195–208. [Google Scholar] [CrossRef]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of Aging on the Homing and Engraftment of Murine Hematopoietic Stem and Progenitor Cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef] [PubMed]
- Xing, Z.; Ryan, M.A.; Daria, D.; Nattamai, K.J.; Van Zant, G.; Wang, L.; Zheng, Y.; Geiger, H. Increased Hematopoietic Stem Cell Mobilization in Aged Mice. Blood 2006, 108, 2190–2197. [Google Scholar] [CrossRef] [PubMed]
- de Haan, G.; Lazare, S.S. Aging of Hematopoietic Stem Cells. Blood 2018, 131, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Yoon, S.R.; Choi, I.; Jung, H. Causes and Mechanisms of Hematopoietic Stem Cell Aging. Int. J. Mol. Sci. 2019, 20, 1272. [Google Scholar] [CrossRef]
- Mejia-Ramirez, E.; Florian, M.C. Understanding Intrinsic Hematopoietic Stem Cell Aging. Haematologica 2020, 105, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Ergen, A.V.; Boles, N.C.; Goodell, M.A. Rantes/Ccl5 Influences Hematopoietic Stem Cell Subtypes and Causes Myeloid Skewing. Blood 2012, 119, 2500–2509. [Google Scholar] [CrossRef]
- He, H.; Xu, P.; Zhang, X.; Liao, M.; Dong, Q.; Cong, T.; Tang, B.; Yang, X.; Ye, M.; Chang, Y.-J.; et al. Aging-Induced IL27Ra Signaling Impairs Hematopoietic Stem Cells. Blood 2020, 136, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-C.; Yu, K.-R. Impact of Mesenchymal Stem Cell Senescence on Inflammaging. BMB Rep. 2020, 53, 65–73. [Google Scholar] [CrossRef]
- Esplin, B.L.; Shimazu, T.; Welner, R.S.; Garrett, K.P.; Nie, L.; Zhang, Q.; Humphrey, M.B.; Yang, Q.; Borghesi, L.A.; Kincade, P.W. Chronic Exposure to a TLR Ligand Injures Hematopoietic Stem Cells. J. Immunol. 2011, 186, 5367–5375. [Google Scholar] [CrossRef]
- Kapsenberg, M.L. Dendritic-Cell Control of Pathogen-Driven T-Cell Polarization. Nat. Rev. Immunol. 2003, 3, 984–993. [Google Scholar] [CrossRef]
- Behrens, G.; Li, M.; Smith, C.M.; Belz, G.T.; Mintern, J.; Carbone, F.R.; Heath, W.R. Helper T Cells, Dendritic Cells and CTL Immunity. Immunol. Cell Biol. 2004, 82, 84–90. [Google Scholar] [CrossRef]
- Agrawal, A.; Agrawal, S.; Cao, J.-N.; Su, H.; Osann, K.; Gupta, S. Altered Innate Immune Functioning of Dendritic Cells in Elderly Humans: A Role of Phosphoinositide 3-Kinase-Signaling Pathway. J. Immunol. 2007, 178, 6912–6922. [Google Scholar] [CrossRef]
- Agrawal, A.; Tay, J.; Ton, S.; Agrawal, S.; Gupta, S. Increased Reactivity of Dendritic Cells from Aged Subjects to Self-Antigen, the Human DNA. J. Immunol. 2009, 182, 1138–1145. [Google Scholar] [CrossRef]
- Agrawal, S.; Gollapudi, S.; Gupta, S.; Agrawal, A. Dendritic Cells from the Elderly Display an Intrinsic Defect in the Production of IL-10 in Response to Lithium Chloride. Exp. Gerontol. 2013, 48, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Goldstein, D.R. Impact of Aging on Antigen Presentation Cell Function of Dendritic Cells. Curr. Opin. Immunol. 2013, 25, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Kornete, M.; Piccirillo, C.A. Functional Crosstalk between Dendritic Cells and Foxp3+ Regulatory T Cells in the Maintenance of Immune Tolerance. Front. Immunol. 2012, 3, 165. [Google Scholar] [CrossRef]
- Aydar, Y.; Balogh, P.; Tew, J.G.; Szakal, A.K. Age-Related Depression of FDC Accessory Functions and CD21 Ligand-Mediated Repair of Co-Stimulation. Eur. J. Immunol. 2002, 32, 2817–2826. [Google Scholar] [CrossRef]
- Agrawal, A.; Agrawal, S.; Gupta, S. Role of Dendritic Cells in Inflammation and Loss of Tolerance in the Elderly. Front. Immunol. 2017, 8, 896. [Google Scholar] [CrossRef]
- Orsini, G.; Legitimo, A.; Failli, A.; Massei, F.; Biver, P.; Consolini, R. Enumeration of Human Peripheral Blood Dendritic Cells throughout the Life. Int. Immunol. 2012, 24, 347–356. [Google Scholar] [CrossRef]
- Garbe, K.; Bratke, K.; Wagner, S.; Virchow, J.C.; Lommatzsch, M. Plasmacytoid Dendritic Cells and Their Toll-like Receptor 9 Expression Selectively Decrease with Age. Hum. Immunol. 2012, 73, 493–497. [Google Scholar] [CrossRef]
- Jing, Y.; Shaheen, E.; Drake, R.R.; Chen, N.; Gravenstein, S.; Deng, Y. Aging Is Associated with a Numerical and Functional Decline in Plasmacytoid Dendritic Cells, Whereas Myeloid Dendritic Cells Are Relatively Unaltered in Human Peripheral Blood. Hum. Immunol. 2009, 70, 777–784. [Google Scholar] [CrossRef]
- Chatta, G.S.; Andrews, R.G.; Rodger, E.; Schrag, M.; Hammond, W.P.; Dale, D.C. Hematopoietic Progenitors and Aging: Alterations in Granulocytic Precursors and Responsiveness to Recombinant Human G-CSF, GM-CSF, and IL-3. J. Gerontol. 1993, 48, M207–M212. [Google Scholar] [CrossRef]
- Wenisch, C.; Patruta, S.; Daxböck, F.; Krause, R.; Hörl, W. Effect of Age on Human Neutrophil Function. J. Leukoc. Biol. 2000, 67, 40–45. [Google Scholar] [CrossRef]
- Niwa, Y.; Kasama, T.; Miyachi, Y.; Kanoh, T. Neutrophil Chemotaxis, Phagocytosis and Parameters of Reactive Oxygen Species in Human Aging: Cross-Sectional and Longitudinal Studies. Life Sci. 1989, 44, 1655–1664. [Google Scholar] [CrossRef]
- Simell, B.; Vuorela, A.; Ekström, N.; Palmu, A.; Reunanen, A.; Meri, S.; Käyhty, H.; Väkeväinen, M. Aging Reduces the Functionality of Anti-Pneumococcal Antibodies and the Killing of Streptococcus Pneumoniae by Neutrophil Phagocytosis. Vaccine 2011, 29, 1929–1934. [Google Scholar] [CrossRef]
- Nomellini, V.; Brubaker, A.L.; Mahbub, S.; Palmer, J.L.; Gomez, C.R.; Kovacs, E.J. Dysregulation of Neutrophil CXCR2 and Pulmonary Endothelial Icam-1 Promotes Age-Related Pulmonary Inflammation. Aging Dis. 2012, 3, 234–247. [Google Scholar]
- Gomez, C.R.; Hirano, S.; Cutro, B.T.; Birjandi, S.; Baila, H.; Nomellini, V.; Kovacs, E.J. Advanced Age Exacerbates the Pulmonary Inflammatory Response after Lipopolysaccharide Exposure. Crit. Care Med. 2007, 35, 246–251. [Google Scholar] [CrossRef]
- Naccache, P.H.; Lefebvre, J.S. A Straight Neutrophil Path to Healthy Aging? Blood 2014, 123, 154–156. [Google Scholar] [CrossRef]
- Sapey, E.; Greenwood, H.; Walton, G.; Mann, E.; Love, A.; Aaronson, N.; Insall, R.H.; Stockley, R.A.; Lord, J.M. Phosphoinositide 3-Kinase Inhibition Restores Neutrophil Accuracy in the Elderly: Toward Targeted Treatments for Immunosenescence. Blood 2014, 123, 239–248. [Google Scholar] [CrossRef]
- Barkaway, A.; Rolas, L.; Joulia, R.; Bodkin, J.; Lenn, T.; Owen-Woods, C.; Reglero-Real, N.; Stein, M.; Vázquez-Martínez, L.; Girbl, T.; et al. Age-Related Changes in the Local Milieu of Inflamed Tissues Cause Aberrant Neutrophil Trafficking and Subsequent Remote Organ Damage. Immunity 2021, 54, 1494–1510. [Google Scholar] [CrossRef]
- Sauce, D.; Dong, Y.; Campillo-Gimenez, L.; Casulli, S.; Bayard, C.; Autran, B.; Boddaert, J.; Appay, V.; Elbim, C. Reduced Oxidative Burst by Primed Neutrophils in the Elderly Individuals Is Associated With Increased Levels of the CD16bright/CD62Ldim Immunosuppressive Subset. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 163–172. [Google Scholar] [CrossRef]
- Ogawa, K.; Suzuki, K.; Okutsu, M.; Yamazaki, K.; Shinkai, S. The Association of Elevated Reactive Oxygen Species Levels from Neutrophils with Low-Grade Inflammation in the Elderly. Immun. Ageing 2008, 5, 13. [Google Scholar] [CrossRef]
- Braga, P.C.; Sala, M.T.; Sasso, M.D.; Mancini, L.; Sandrini, M.C.; Annoni, G. Influence of Age on Oxidative Bursts (Chemiluminescence) of Polymorphonuclear Neutrophil Leukocytes. Gerontology 1998, 44, 192–197. [Google Scholar] [CrossRef]
- Kovalenko, E.I.; Boyko, A.A.; Semenkov, V.F.; Lutsenko, G.V.; Grechikhina, M.V.; Kanevskiy, L.M.; Azhikina, T.L.; Telford, W.G.; Sapozhnikov, A.M. ROS Production, Intracellular HSP70 Levels and Their Relationship in Human Neutrophils: Effects of Age. Oncotarget 2014, 5, 11800–11812. [Google Scholar] [CrossRef]
- Hazeldine, J.; Harris, P.; Chapple, I.L.; Grant, M.; Greenwood, H.; Livesey, A.; Sapey, E.; Lord, J.M. Impaired Neutrophil Extracellular Trap Formation: A Novel Defect in the Innate Immune System of Aged Individuals. Aging Cell 2014, 13, 690–698. [Google Scholar] [CrossRef]
- Brubaker, A.L.; Rendon, J.L.; Ramirez, L.; Choudhry, M.A.; Kovacs, E.J. Reduced Neutrophil Chemotaxis and Infiltration Contributes to Delayed Resolution of Cutaneous Wound Infection with Advanced Age. J. Immunol. 2013, 190, 1746–1757. [Google Scholar] [CrossRef]
- Butcher, S.; Chahel, H.; Lord, J.M. Ageing and the Neutrophil: No Appetite for Killing? Immunology 2000, 100, 411–416. [Google Scholar] [CrossRef]
- Wessels, I.; Jansen, J.; Rink, L.; Uciechowski, P. Immunosenescence of Polymorphonuclear Neutrophils. ScientificWorldJournal 2010, 10, 145–160. [Google Scholar] [CrossRef]
- De Martinis, M.; Modesti, M.; Ginaldi, L. Phenotypic and Functional Changes of Circulating Monocytes and Polymorphonuclear Leucocytes from Elderly Persons. Immunol. Cell Biol. 2004, 82, 415–420. [Google Scholar] [CrossRef]
- Tortorella, C.; Polignano, A.; Piazzolla, G.; Serrone, M.; Jirillo, E.; Antonaci, S. Lipopolysaccharide-, Granulocyte-Monocyte Colony Stimulating Factor and Pentoxifylline-Mediated Effects on Formyl-Methionyl-Leucine-Phenylalanine-Stimulated Neutrophil Respiratory Burst in the Elderly. Microbios 1996, 85, 189–198. [Google Scholar]
- Uciechowski, P.; Rink, L. Neutrophil Granulocyte Functions in the Elderly. In Handbook on Immunosenescence: Basic Understanding and Clinical Applications; Fulop, T., Franceschi, C., Hirokawa, K., Pawelec, G., Eds.; Springer: Dordrecht, The Netherlands, 2009; pp. 439–455. ISBN 978-1-4020-9063-9. [Google Scholar]
- Lipschitz, D.A.; Udupa, K.B.; Boxer, L.A. The Role of Calcium in the Age-Related Decline of Neutrophil Function. Blood 1988, 71, 659–665. [Google Scholar] [CrossRef]
- Tanji-Matsuba, K.; van Eeden, S.F.; Saito, Y.; Okazawa, M.; Klut, M.E.; Hayashi, S.; Hogg, J.C. Functional Changes in Aging Polymorphonuclear Leukocytes. Circulation 1998, 97, 91–98. [Google Scholar] [CrossRef]
- Rao, K.M. Age-Related Decline in Ligand-Induced Actin Polymerization in Human Leukocytes and Platelets. J. Gerontol. 1986, 41, 561–566. [Google Scholar] [CrossRef]
- Ito, T.K.; Yokoyama, M.; Yoshida, Y.; Nojima, A.; Kassai, H.; Oishi, K.; Okada, S.; Kinoshita, D.; Kobayashi, Y.; Fruttiger, M.; et al. A Crucial Role for CDC42 in Senescence-Associated Inflammation and Atherosclerosis. PLoS ONE 2014, 9, e102186. [Google Scholar] [CrossRef]
- Uciechowski, P.; Rink, L. Basophil, Eosinophil, and Neutrophil Functions in the Elderly. In Immunology of Aging; Massoud, A., Rezaei, N., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 47–63. ISBN 978-3-642-39495-9. [Google Scholar]
- van Beek, A.A.; Fransen, F.; Meijer, B.; de Vos, P.; Knol, E.F.; Savelkoul, H.F.J. Aged Mice Display Altered Numbers and Phenotype of Basophils, and Bone Marrow-Derived Basophil Activation, with a Limited Role for Aging-Associated Microbiota. Immun Ageing 2018, 15, 32. [Google Scholar] [CrossRef]
- He, Z.; Allers, C.; Sugimoto, C.; Ahmed, N.; Fujioka, H.; Kim, W.-K.; Didier, E.S.; Kuroda, M.J. Rapid Turnover and High Production Rate of Myeloid Cells in Adult Rhesus Macaques with Compensations during Aging. J. Immunol. 2018, 200, 4059–4067. [Google Scholar] [CrossRef]
- Song, C.; Vandewoude, M.; Stevens, W.; De Clerck, L.; Van der Planken, M.; Whelan, A.; Anisman, H.; Dossche, A.; Maes, M. Alterations in Immune Functions during Normal Aging and Alzheimer’s Disease. Psychiatry Res. 1999, 85, 71–80. [Google Scholar] [CrossRef]
- Schwarzenbach, H.R.; Nakagawa, T.; Conroy, M.C.; de Weck, A.L. Skin Reactivity, Basophil Degranulation and IgE Levels in Ageing. Clin. Allergy 1982, 12, 465–473. [Google Scholar] [CrossRef]
- Mathur, S.K.; Schwantes, E.A.; Jarjour, N.N.; Busse, W.W. Age-Related Changes in Eosinophil Function in Human Subjects. Chest 2008, 133, 412–419. [Google Scholar] [CrossRef]
- Abernathy-Close, L.; Dieterle, M.G.; Vendrov, K.C.; Bergin, I.L.; Rao, K.; Young, V.B. Aging Dampens the Intestinal Innate Immune Response during Severe Clostridioides Difficile Infection and Is Associated with Altered Cytokine Levels and Granulocyte Mobilization. Infect. Immun. 2020, 88, e00960-19. [Google Scholar] [CrossRef]
- Yagi, T.; Sato, A.; Hayakawa, H.; Ide, K. Failure of Aged Rats to Accumulate Eosinophils in Allergic Inflammation of the Airway. J. Allergy Clin. Immunol. 1997, 99, 38–47. [Google Scholar] [CrossRef]
- Karasuyama, H.; Miyake, K.; Yoshikawa, S.; Kawano, Y.; Yamanishi, Y. How Do Basophils Contribute to Th2 Cell Differentiation and Allergic Responses? Int. Immunol. 2018, 30, 391–396. [Google Scholar] [CrossRef]
- Faunce, D.E.; Palmer, J.L.; Paskowicz, K.K.; Witte, P.L.; Kovacs, E.J. CD1d-Restricted NKT Cells Contribute to the Age-Associated Decline of T Cell Immunity. J. Immunol. 2005, 175, 3102–3109. [Google Scholar] [CrossRef]
- Inui, T.; Nakagawa, R.; Ohkura, S.; Habu, Y.; Koike, Y.; Motoki, K.; Kuranaga, N.; Fukasawa, M.; Shinomiya, N.; Seki, S. Age-Associated Augmentation of the Synthetic Ligand- Mediated Function of Mouse NK1.1 Ag(+) T Cells: Their Cytokine Production and Hepatotoxicity in Vivo and in Vitro. J. Immunol. 2002, 169, 6127–6132. [Google Scholar] [CrossRef]
- Borrego, F.; Alonso, M.C.; Galiani, M.D.; Carracedo, J.; Ramirez, R.; Ostos, B.; Peña, J.; Solana, R. NK Phenotypic Markers and IL2 Response in NK Cells from Elderly People. Exp. Gerontol. 1999, 34, 253–265. [Google Scholar] [CrossRef]
- Facchini, A.; Mariani, E.; Mariani, A.R.; Papa, S.; Vitale, M.; Manzoli, F.A. Increased Number of Circulating Leu 11+ (CD 16) Large Granular Lymphocytes and Decreased NK Activity during Human Ageing. Clin. Exp. Immunol. 1987, 68, 340–347. [Google Scholar]
- Vitale, M.; Zamai, L.; Neri, L.M.; Galanzi, A.; Facchini, A.; Rana, R.; Cataldi, A.; Papa, S. The Impairment of Natural Killer Function in the Healthy Aged Is Due to a Postbinding Deficient Mechanism. Cell. Immunol. 1992, 145, 1–10. [Google Scholar] [CrossRef]
- DelaRosa, O.; Tarazona, R.; Casado, J.G.; Alonso, C.; Ostos, B.; Peña, J.; Solana, R. Valpha24+ NKT Cells Are Decreased in Elderly Humans. Exp. Gerontol. 2002, 37, 213–217. [Google Scholar] [CrossRef]
- Peralbo, E.; DelaRosa, O.; Gayoso, I.; Pita, M.L.; Tarazona, R.; Solana, R. Decreased Frequency and Proliferative Response of Invariant Valpha24Vbeta11 Natural Killer T (INKT) Cells in Healthy Elderly. Biogerontology 2006, 7, 483–492. [Google Scholar] [CrossRef]
- Campos, C.; Pera, A.; Sanchez-Correa, B.; Alonso, C.; Lopez-Fernandez, I.; Morgado, S.; Tarazona, R.; Solana, R. Effect of Age and CMV on NK Cell Subpopulations. Exp. Gerontol. 2014, 54, 130–137. [Google Scholar] [CrossRef]
- Chidrawar, S.M.; Khan, N.; Chan, Y.L.T.; Nayak, L.; Moss, P.A. Ageing Is Associated with a Decline in Peripheral Blood CD56bright NK Cells. Immun. Ageing 2006, 3, 10. [Google Scholar] [CrossRef]
- Solana, R.; Campos, C.; Pera, A.; Tarazona, R. Shaping of NK Cell Subsets by Aging. Curr. Opin. Immunol. 2014, 29, 56–61. [Google Scholar] [CrossRef]
- Almeida-Oliveira, A.; Smith-Carvalho, M.; Porto, L.C.; Cardoso-Oliveira, J.; dos Santos Ribeiro, A.; Falcão, R.R.; Abdelhay, E.; Bouzas, L.F.; Thuler, L.C.S.; Ornellas, M.H.; et al. Age-Related Changes in Natural Killer Cell Receptors from Childhood through Old Age. Hum. Immunol. 2011, 72, 319–329. [Google Scholar] [CrossRef]
- Le Garff-Tavernier, M.; Béziat, V.; Decocq, J.; Siguret, V.; Gandjbakhch, F.; Pautas, E.; Debré, P.; Merle-Beral, H.; Vieillard, V. Human NK Cells Display Major Phenotypic and Functional Changes over the Life Span. Aging Cell 2010, 9, 527–535. [Google Scholar] [CrossRef]
- Camous, X.; Pera, A.; Solana, R.; Larbi, A. NK Cells in Healthy Aging and Age-Associated Diseases. Available online: https://www.hindawi.com/journals/bmri/2012/195956/ (accessed on 28 November 2018).
- Solana, R.; Tarazona, R.; Gayoso, I.; Lesur, O.; Dupuis, G.; Fulop, T. Innate Immunosenescence: Effect of Aging on Cells and Receptors of the Innate Immune System in Humans. Semin. Immunol. 2012, 24, 331–341. [Google Scholar] [CrossRef]
- Hazeldine, J.; Hampson, P.; Lord, J.M. Reduced Release and Binding of Perforin at the Immunological Synapse Underlies the Age-Related Decline in Natural Killer Cell Cytotoxicity. Aging Cell 2012, 11, 751–759. [Google Scholar] [CrossRef]
- Haynes, L.; Linton, P.-J.; Eaton, S.M.; Tonkonogy, S.L.; Swain, S.L. Interleukin 2, but Not Other Common γ Chain–Binding Cytokines, Can Reverse the Defect in Generation of Cd4 Effector T Cells from Naive T Cells of Aged Mice. J. Exp. Med. 1999, 190, 1013–1024. [Google Scholar] [CrossRef]
- Myśliwska, J.; Bryl, E.; Foerster, J.; Myśliwski, A. Increase of Interleukin 6 and Decrease of Interleukin 2 Production during the Ageing Process Are Influenced by the Health Status. Mech. Ageing Dev. 1998, 100, 313–328. [Google Scholar] [CrossRef]
- Whisler, R.L.; Beiqing, L.; Chen, M. Age-Related Decreases in IL-2 Production by Human T Cells Are Associated with Impaired Activation of Nuclear Transcriptional Factors AP-1 and NF-AT. Cell. Immunol. 1996, 169, 185–195. [Google Scholar] [CrossRef]
- Gounder, S.S.; Abdullah, B.J.J.; Radzuanb, N.E.I.B.M.; Zain, F.D.B.M.; Sait, N.B.M.; Chua, C.; Subramani, B. Effect of Aging on NK Cell Population and Their Proliferation at Ex Vivo Culture Condition. Anal. Cell Pathol. 2018, 2018, 7871814. [Google Scholar] [CrossRef]
- Albright, J.M.; Dunn, R.C.; Shults, J.A.; Boe, D.M.; Afshar, M.; Kovacs, E.J. Advanced Age Alters Monocyte and Macrophage Responses. Antioxid. Redox Signal. 2016, 25, 805–815. [Google Scholar] [CrossRef]
- Ortega-Gómez, A.; Perretti, M.; Soehnlein, O. Resolution of Inflammation: An Integrated View. EMBO Mol. Med. 2013, 5, 661–674. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Abdulnour, R.-E.E.; Walker, K.H.; Engstrom, B.D.; Levy, B.D. Specialized Proresolving Mediators in Innate and Adaptive Immune Responses in Airway Diseases. Physiol. Rev. 2018, 98, 1335–1370. [Google Scholar] [CrossRef]
- Duffney, P.F.; Falsetta, M.L.; Rackow, A.R.; Thatcher, T.H.; Phipps, R.P.; Sime, P.J. Key Roles for Lipid Mediators in the Adaptive Immune Response. J. Clin. Investig. 2018, 128, 2724–2731. [Google Scholar] [CrossRef]
- Krishnamoorthy, N.; Burkett, P.R.; Dalli, J.; Abdulnour, R.-E.E.; Colas, R.; Ramon, S.; Phipps, R.P.; Petasis, N.A.; Kuchroo, V.K.; Serhan, C.N.; et al. Cutting Edge: Maresin-1 Engages Regulatory T Cells to Limit Type 2 Innate Lymphoid Cell Activation and Promote Resolution of Lung Inflammation. J. Immunol. 2015, 194, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Chiurchiù, V.; Leuti, A.; Dalli, J.; Jacobsson, A.; Battistini, L.; Maccarrone, M.; Serhan, C.N. Proresolving Lipid Mediators Resolvin D1, Resolvin D2, and Maresin 1 Are Critical in Modulating T Cell Responses. Sci. Transl. Med. 2016, 8, 353ra111. [Google Scholar] [CrossRef] [PubMed]
- Rea, I.M.; Gibson, D.S.; McGilligan, V.; McNerlan, S.E.; Alexander, H.D.; Ross, O.A. Age and Age-Related Diseases: Role of Inflammation Triggers and Cytokines. Front. Immunol. 2018, 9, 586. [Google Scholar] [CrossRef]
- Duvall, M.G.; Levy, B.D. DHA- and EPA-Derived Resolvins, Protectins, and Maresins in Airway Inflammation. Eur. J. Pharm. 2016, 785, 144–155. [Google Scholar] [CrossRef] [PubMed]
- Moro, K.; Nagahashi, M.; Ramanathan, R.; Takabe, K.; Wakai, T. Resolvins and Omega Three Polyunsaturated Fatty Acids: Clinical Implications in Inflammatory Diseases and Cancer. World J. Clin. Cases 2016, 4, 155–164. [Google Scholar] [CrossRef]
- Serhan, C.N.; Petasis, N.A. Resolvins and Protectins in Inflammation-Resolution. Chem. Rev. 2011, 111, 5922–5943. [Google Scholar] [CrossRef] [PubMed]
- Börgeson, E.; McGillicuddy, F.C.; Harford, K.A.; Corrigan, N.; Higgins, D.F.; Maderna, P.; Roche, H.M.; Godson, C. Lipoxin A4 Attenuates Adipose Inflammation. FASEB J. 2012, 26, 4287–4294. [Google Scholar] [CrossRef]
- Gangemi, S.; Pescara, L.; D’Urbano, E.; Basile, G.; Nicita-Mauro, V.; Davì, G.; Romano, M. Aging Is Characterized by a Profound Reduction in Anti-Inflammatory Lipoxin A4 Levels. Exp. Gerontol. 2005, 40, 612–614. [Google Scholar] [CrossRef]
- Doyle, R.; Sadlier, D.M.; Godson, C. Pro-Resolving Lipid Mediators: Agents of Anti-Ageing? Semin. Immunol. 2018, 40, 36–48. [Google Scholar] [CrossRef]
- Arnardottir, H.H.; Dalli, J.; Colas, R.A.; Shinohara, M.; Serhan, C.N. Aging Delays Resolution of Acute Inflammation in Mice: Reprogramming the Host Response with Novel Nano-Proresolving Medicines. J. Immunol. 2014, 193, 4235–4244. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid Storm in Infection and Inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed]
- Serhan, C.N. Pro-Resolving Lipid Mediators Are Leads for Resolution Physiology. Nature 2014, 510, 92–101. [Google Scholar] [CrossRef]
- Dalli, J.; Serhan, C.N. Pro-Resolving Mediators in Regulating and Conferring Macrophage Function. Front. Immunol. 2017, 8, 1400. [Google Scholar] [CrossRef]
- Chen, D.; Chao, D.L.; Rocha, L.; Kolar, M.; Huu, V.A.N.; Krawczyk, M.; Dasyani, M.; Wang, T.; Jafari, M.; Jabari, M.; et al. The Lipid Elongation Enzyme ELOVL2 Is a Molecular Regulator of Aging in the Retina. Aging Cell 2020, 19, e13100. [Google Scholar] [CrossRef] [PubMed]
- Bacalini, M.G.; Deelen, J.; Pirazzini, C.; De Cecco, M.; Giuliani, C.; Lanzarini, C.; Ravaioli, F.; Marasco, E.; van Heemst, D.; Suchiman, H.E.D.; et al. Systemic Age-Associated DNA Hypermethylation of ELOVL2 Gene: In Vivo and In Vitro Evidences of a Cell Replication Process. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 1015–1023. [Google Scholar] [CrossRef]
- Garagnani, P.; Bacalini, M.G.; Pirazzini, C.; Gori, D.; Giuliani, C.; Mari, D.; Di Blasio, A.M.; Gentilini, D.; Vitale, G.; Collino, S.; et al. Methylation of ELOVL2 Gene as a New Epigenetic Marker of Age. Aging Cell 2012, 11, 1132–1134. [Google Scholar] [CrossRef] [PubMed]
- Slieker, R.C.; Relton, C.L.; Gaunt, T.R.; Slagboom, P.E.; Heijmans, B.T. Age-Related DNA Methylation Changes Are Tissue-Specific with ELOVL2 Promoter Methylation as Exception. Epigenetics Chromatin 2018, 11, 25. [Google Scholar] [CrossRef]
- Ginhoux, F.; Guilliams, M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity 2016, 44, 439–449. [Google Scholar] [CrossRef]
- Nayar, S.; Dasgupta, P.; Galustian, C. Extending the Lifespan and Efficacies of Immune Cells Used in Adoptive Transfer for Cancer Immunotherapies-A Review. Oncoimmunology 2015, 4, e1002720. [Google Scholar] [CrossRef]
- Sathe, P.; Shortman, K. The Steady-State Development of Splenic Dendritic Cells. Mucosal Immunol. 2008, 1, 425–431. [Google Scholar] [CrossRef]
- Dimri, G.P.; Lee, X.; Basile, G.; Acosta, M.; Scott, G.; Roskelley, C.; Medrano, E.E.; Linskens, M.; Rubelj, I.; Pereira-Smith, O. A Biomarker That Identifies Senescent Human Cells in Culture and in Aging Skin in Vivo. Proc. Natl. Acad. Sci. USA 1995, 92, 9363–9367. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Cruz, D.; Asamoah, N.; Buxbaum, A.; Sohar, I.; Lobel, P.; Maxfield, F.R. Activation of Microglia Acidifies Lysosomes and Leads to Degradation of Alzheimer Amyloid Fibrils. Mol. Biol. Cell 2007, 18, 1490–1496. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, A.; Capetillo-Zarate, E.; Cruz, D.; Gouras, G.K.; Maxfield, F.R. Degradation of Alzheimer’s Amyloid Fibrils by Microglia Requires Delivery of ClC-7 to Lysosomes. Mol. Biol. Cell 2011, 22, 1664–1676. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, I.; Ohmoto, E.; Aoyama, S.; Takizawa, M.; Oda, Y.; Nonaka, K.; Nakada, H.; Yorimitsu, S.; Kimura, I. Monocyte Chemiluminescence and Macrophage Precursors in the Aged. Acta Med. Okayama 1985, 39, 447–451. [Google Scholar] [CrossRef]
- Hearps, A.C.; Martin, G.E.; Angelovich, T.A.; Cheng, W.-J.; Maisa, A.; Landay, A.L.; Jaworowski, A.; Crowe, S.M. Aging Is Associated with Chronic Innate Immune Activation and Dysregulation of Monocyte Phenotype and Function. Aging Cell 2012, 11, 867–875. [Google Scholar] [CrossRef]
- Nyugen, J.; Agrawal, S.; Gollapudi, S.; Gupta, S. Impaired Functions of Peripheral Blood Monocyte Subpopulations in Aged Humans. J. Clin. Immunol. 2010, 30, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Linehan, E.; Dombrowski, Y.; Snoddy, R.; Fallon, P.G.; Kissenpfennig, A.; Fitzgerald, D.C. Aging Impairs Peritoneal but Not Bone Marrow-Derived Macrophage Phagocytosis. Aging Cell 2014, 13, 699–708. [Google Scholar] [CrossRef]
- Takahashi, R.; Totsuka, S.; Ishigami, A.; Kobayashi, Y.; Nagata, K. Attenuated Phagocytosis of Secondary Necrotic Neutrophils by Macrophages in Aged and SMP30 Knockout Mice. Geriatr. Gerontol. Int. 2016, 16, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Kohut, M.L.; Senchina, D.S.; Madden, K.S.; Martin, A.E.; Felten, D.L.; Moynihan, J.A. Age Effects on Macrophage Function Vary by Tissue Site, Nature of Stimulant, and Exercise Behavior. Exp. Gerontol. 2004, 39, 1347–1360. [Google Scholar] [CrossRef]
- Gomez, C.R.; Karavitis, J.; Palmer, J.L.; Faunce, D.E.; Ramirez, L.; Nomellini, V.; Kovacs, E.J. Interleukin-6 Contributes to Age-Related Alteration of Cytokine Production by Macrophages. Mediat. Inflamm. 2010, 2010, 475139. [Google Scholar] [CrossRef]
- Liang, S.; Domon, H.; Hosur, K.B.; Wang, M.; Hajishengallis, G. Age-Related Alterations in Innate Immune Receptor Expression and Ability of Macrophages to Respond to Pathogen Challenge in Vitro. Mech. Ageing Dev. 2009, 130, 538–546. [Google Scholar] [CrossRef]
- Chelvarajan, R.L.; Liu, Y.; Popa, D.; Getchell, M.L.; Getchell, T.V.; Stromberg, A.J.; Bondada, S. Molecular Basis of Age-Associated Cytokine Dysregulation in LPS-Stimulated Macrophages. J. Leukoc. Biol. 2006, 79, 1314–1327. [Google Scholar] [CrossRef]
- Boehmer, E.D.; Goral, J.; Faunce, D.E.; Kovacs, E.J. Age-Dependent Decrease in Toll-like Receptor 4-Mediated Proinflammatory Cytokine Production and Mitogen-Activated Protein Kinase Expression. J. Leukoc. Biol. 2004, 75, 342–349. [Google Scholar] [CrossRef]
- Boehmer, E.D.; Meehan, M.J.; Cutro, B.T.; Kovacs, E.J. Aging Negatively Skews Macrophage TLR2- and TLR4-Mediated pro-Inflammatory Responses without Affecting the IL-2-Stimulated Pathway. Mech. Ageing Dev. 2005, 126, 1305–1313. [Google Scholar] [CrossRef] [PubMed]
- Boyd, A.R.; Shivshankar, P.; Jiang, S.; Berton, M.T.; Orihuela, C.J. Age-Related Defects in TLR2 Signaling Diminish the Cytokine Response by Alveolar Macrophages during Murine Pneumococcal Pneumonia. Exp. Gerontol. 2012, 47, 507–518. [Google Scholar] [CrossRef]
- Vande Walle, L.; Lamkanfi, M. Pyroptosis. Curr. Biol. 2016, 26, R568–R572. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by Inflammatory Caspases Determines Pyroptotic Cell Death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Muñoz-Planillo, R.; Núñez, G. Sensing and Reacting to Microbes through the Inflammasomes. Nat. Immunol. 2012, 13, 325–332. [Google Scholar] [CrossRef]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The Inflammasome: A Caspase-1-Activation Platform That Regulates Immune Responses and Disease Pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 Cleaves Gasdermin D for Non-Canonical Inflammasome Signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef]
- He, W.; Wan, H.; Hu, L.; Chen, P.; Wang, X.; Huang, Z.; Yang, Z.-H.; Zhong, C.-Q.; Han, J. Gasdermin D Is an Executor of Pyroptosis and Required for Interleukin-1β Secretion. Cell Res. 2015, 25, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of Action, Role in Disease, and Therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty Acid–Induced NLRP3-ASC Inflammasome Activation Interferes with Insulin Signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 Inflammasome Instigates Obesity-Induced Inflammation and Insulin Resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Niepmann, S.; Knolle, P.A.; Hornung, V. Aging-Associated TNF Production Primes Inflammasome Activation and NLRP3-Related Metabolic Disturbances. J. Immunol. 2016, 197, 2900–2908. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A Role for Mitochondria in NLRP3 Inflammasome Activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Shimada, K.; Crother, T.R.; Karlin, J.; Dagvadorj, J.; Chiba, N.; Chen, S.; Ramanujan, V.K.; Wolf, A.J.; Vergnes, L.; Ojcius, D.M.; et al. Oxidized Mitochondrial DNA Activates the NLRP3 Inflammasome during Apoptosis. Immunity 2012, 36, 401–414. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Grant, R.W.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Kanneganti, T.-D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 Inflammasome Promotes Age-Related Thymic Demise and Immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.-L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal Hematopoiesis Associated with TET2 Deficiency Accelerates Atherosclerosis Development in Mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef] [PubMed]
- Mejias, N.H.; Martinez, C.C.; Stephens, M.E.; de Rivero Vaccari, J.P. Contribution of the Inflammasome to Inflammaging. J. Inflamm. 2018, 15, 23. [Google Scholar] [CrossRef] [PubMed]
- Franklin, B.S.; Bossaller, L.; De Nardo, D.; Ratter, J.M.; Stutz, A.; Engels, G.; Brenker, C.; Nordhoff, M.; Mirandola, S.R.; Al-Amoudi, A.; et al. The Adaptor ASC Has Extracellular and “prionoid” Activities That Propagate Inflammation. Nat. Immunol. 2014, 15, 727–737. [Google Scholar] [CrossRef] [PubMed]
- Kleinnijenhuis, J.; Quintin, J.; Preijers, F.; Joosten, L.A.B.; Ifrim, D.C.; Saeed, S.; Jacobs, C.; van Loenhout, J.; de Jong, D.; Stunnenberg, H.G.; et al. Bacille Calmette-Guerin Induces NOD2-Dependent Nonspecific Protection from Reinfection via Epigenetic Reprogramming of Monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, 17537–17542. [Google Scholar] [CrossRef] [PubMed]
- Netea, M.G.; van der Meer, J.W.M. Trained Immunity: An Ancient Way of Remembering. Cell Host Microbe 2017, 21, 297–300. [Google Scholar] [CrossRef]
- Netea, M.G.; Joosten, L.A.B.; Latz, E.; Mills, K.H.G.; Natoli, G.; Stunnenberg, H.G.; O’Neill, L.A.J.; Xavier, R.J. Trained Immunity: A Program of Innate Immune Memory in Health and Disease. Science 2016, 352, aaf1098. [Google Scholar] [CrossRef]
- Zhong, C.; Yang, X.; Feng, Y.; Yu, J. Trained Immunity: An Underlying Driver of Inflammatory Atherosclerosis. Front. Immunol. 2020, 11. [Google Scholar] [CrossRef]
- Zhang, Q.; Cao, X. Epigenetic Regulation of the Innate Immune Response to Infection. Nat. Rev. Immunol. 2019, 19, 417–432. [Google Scholar] [CrossRef]
- Wen, H.; Dou, Y.; Hogaboam, C.M.; Kunkel, S.L. Epigenetic Regulation of Dendritic Cell-Derived Interleukin-12 Facilitates Immunosuppression after a Severe Innate Immune Response. Blood 2008, 111, 1797–1804. [Google Scholar] [CrossRef]
- He, X.; Xu, J.; Li, G.; Li, M.; Li, L.; Pei, Z.; Zhang, L.; Hu, X. NLRP3-Dependent Microglial Training Impaired the Clearance of Amyloid-Beta and Aggravated the Cognitive Decline in Alzheimer’s Disease. Cell Death Dis. 2020, 11, 849. [Google Scholar] [CrossRef]
- Vineis, P.; Robinson, O.; Chadeau-Hyam, M.; Dehghan, A.; Mudway, I.; Dagnino, S. What Is New in the Exposome? Environ. Int. 2020, 143, 105887. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; Buchanan, S.; Selman, C.; Stenvinkel, P. Allostatic Load and Ageing: Linking the Microbiome and Nutrition with Age-Related Health. Biochem. Soc. Trans. 2019, 47, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Shiels, P.G.; McGuinness, D.; Eriksson, M.; Kooman, J.P.; Stenvinkel, P. The Role of Epigenetics in Renal Ageing. Nat. Rev. Nephrol. 2017, 13, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent Epigenetic Differences Associated with Prenatal Exposure to Famine in Humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef]
- Tsurumi, A.; Li, W. Global Heterochromatin Loss. Epigenetics 2012, 7, 680–688. [Google Scholar] [CrossRef]
- Villeponteau, B. The Heterochromatin Loss Model of Aging. Exp. Gerontol. 1997, 32, 383–394. [Google Scholar] [CrossRef]
- Djeghloul, D.; Kuranda, K.; Kuzniak, I.; Barbieri, D.; Naguibneva, I.; Choisy, C.; Bories, J.-C.; Dosquet, C.; Pla, M.; Vanneaux, V.; et al. Age-Associated Decrease of the Histone Methyltransferase SUV39H1 in HSC Perturbs Heterochromatin and B Lymphoid Differentiation. Stem Cell Rep. 2016, 6, 970–984. [Google Scholar] [CrossRef]
- Sidler, C.; Woycicki, R.; Li, D.; Wang, B.; Kovalchuk, I.; Kovalchuk, O. A Role for SUV39H1-Mediated H3K9 Trimethylation in the Control of Genome Stability and Senescence in WI38 Human Diploid Lung Fibroblasts. Aging (Albany NY) 2014, 6, 545–563. [Google Scholar] [CrossRef]
- Jintaridth, P.; Mutirangura, A. Distinctive Patterns of Age-Dependent Hypomethylation in Interspersed Repetitive Sequences. Physiol. Genom. 2010, 41, 194–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Wilson, R.; Heiss, J.; Breitling, L.P.; Saum, K.-U.; Schöttker, B.; Holleczek, B.; Waldenberger, M.; Peters, A.; Brenner, H. DNA Methylation Signatures in Peripheral Blood Strongly Predict All-Cause Mortality. Nat. Commun. 2017, 8, 14617. [Google Scholar] [CrossRef]
- Gowers, I.R.; Walters, K.; Kiss-Toth, E.; Read, R.C.; Duff, G.W.; Wilson, A.G. Age-Related Loss of CpG Methylation in the Tumour Necrosis Factor Promoter. Cytokine 2011, 56, 792–797. [Google Scholar] [CrossRef]
- Steegenga, W.T.; Boekschoten, M.V.; Lute, C.; Hooiveld, G.J.; de Groot, P.J.; Morris, T.J.; Teschendorff, A.E.; Butcher, L.M.; Beck, S.; Müller, M. Genome-Wide Age-Related Changes in DNA Methylation and Gene Expression in Human PBMCs. Age 2014, 36, 9648. [Google Scholar] [CrossRef]
- Bradburn, S.; McPhee, J.; Bagley, L.; Carroll, M.; Slevin, M.; Al-Shanti, N.; Barnouin, Y.; Hogrel, J.-Y.; Pääsuke, M.; Gapeyeva, H.; et al. Dysregulation of C-X-C Motif Ligand 10 during Aging and Association with Cognitive Performance. Neurobiol. Aging 2018, 63, 54–64. [Google Scholar] [CrossRef]
- Khalil, H.; Tazi, M.; Caution, K.; Ahmed, A.; Kanneganti, A.; Assani, K.; Kopp, B.; Marsh, C.; Dakhlallah, D.; Amer, A.O. Aging Is Associated with Hypermethylation of Autophagy Genes in Macrophages. Epigenetics 2016, 11, 381–388. [Google Scholar] [CrossRef]
- Stranks, A.J.; Hansen, A.L.; Panse, I.; Mortensen, M.; Ferguson, D.J.P.; Puleston, D.J.; Shenderov, K.; Watson, A.S.; Veldhoen, M.; Phadwal, K.; et al. Autophagy Controls Acquisition of Aging Features in Macrophages. J. Innate Immun. 2015, 7, 375–391. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of Adaptive Immunity by the Innate Immune System. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Kondo, M. T and B Lymphocyte Differentiation from Hematopoietic Stem Cell. Semin. Immunol. 2008, 20, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Andersen, M.H.; Schrama, D.; thor Straten, P.; Becker, J.C. Cytotoxic T Cells. J. Investig. Dermatol. 2006, 126, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, S.; Prommersberger, S.; Pfeiffer, I.A.; Schuler-Thurner, B.; Schuler, G.; Dörrie, J.; Schaft, N. Concurrent Interaction of DCs with CD4(+) and CD8(+) T Cells Improves Secondary CTL Expansion: It Takes Three to Tango. Eur. J. Immunol. 2014, 44, 3543–3559. [Google Scholar] [CrossRef]
- Zhang, N.; Bevan, M.J. CD8+ T Cells: Foot Soldiers of the Immune System. Immunity 2011, 35, 161. [Google Scholar] [CrossRef]
- LeBien, T.W.; Tedder, T.F. B Lymphocytes: How They Develop and Function. Blood 2008, 112, 1570–1580. [Google Scholar] [CrossRef]
- Takahama, Y. Journey through the Thymus: Stromal Guides for T-Cell Development and Selection. Nat. Rev. Immunol. 2006, 6, 127–135. [Google Scholar] [CrossRef]
- Bayly-Jones, C.; Bubeck, D.; Dunstone, M.A. The Mystery behind Membrane Insertion: A Review of the Complement Membrane Attack Complex. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372, 0160221. [Google Scholar] [CrossRef] [PubMed]
- Frank, M.M.; Joiner, K.; Hammer, C. The Function of Antibody and Complement in the Lysis of Bacteria. Rev. Infect. Dis. 1987, 9 (Suppl. 5), S537–S545. [Google Scholar] [CrossRef] [PubMed]
- Radbruch, A.; Muehlinghaus, G.; Luger, E.O.; Inamine, A.; Smith, K.G.C.; Dörner, T.; Hiepe, F. Competence and Competition: The Challenge of Becoming a Long-Lived Plasma Cell. Nat. Rev. Immunol. 2006, 6, 741–750. [Google Scholar] [CrossRef]
- Cancro, M.P.; Hao, Y.; Scholz, J.L.; Riley, R.L.; Frasca, D.; Dunn-Walters, D.K.; Blomberg, B.B. B Cells and Aging: Molecules and Mechanisms. Trends Immunol. 2009, 30, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Koohy, H.; Bolland, D.J.; Matheson, L.S.; Schoenfelder, S.; Stellato, C.; Dimond, A.; Várnai, C.; Chovanec, P.; Chessa, T.; Denizot, J.; et al. Genome Organization and Chromatin Analysis Identify Transcriptional Downregulation of Insulin-like Growth Factor Signaling as a Hallmark of Aging in Developing B Cells. Genome Biol. 2018, 19, 126. [Google Scholar] [CrossRef]
- Stephan, R.P.; Lill-Elghanian, D.A.; Witte, P.L. Development of B Cells in Aged Mice: Decline in the Ability of pro-B Cells to Respond to IL-7 but Not to Other Growth Factors. J. Immunol. 1997, 158, 1598–1609. [Google Scholar] [PubMed]
- Labrie, J.E.; Sah, A.P.; Allman, D.M.; Cancro, M.P.; Gerstein, R.M. Bone Marrow Microenvironmental Changes Underlie Reduced RAG-Mediated Recombination and B Cell Generation in Aged Mice. J. Exp. Med. 2004, 200, 411–423. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. The Generation of Memory B Cells Is Maintained, but the Antibody Response Is Not, in the Elderly after Repeated Influenza Immunizations. Vaccine 2016, 34, 2834–2840. [Google Scholar] [CrossRef]
- Hao, Y.; O’Neill, P.; Naradikian, M.S.; Scholz, J.L.; Cancro, M.P. A B-Cell Subset Uniquely Responsive to Innate Stimuli Accumulates in Aged Mice. Blood 2011, 118, 1294–1304. [Google Scholar] [CrossRef]
- Rubtsov, A.V.; Rubtsova, K.; Fischer, A.; Meehan, R.T.; Gillis, J.Z.; Kappler, J.W.; Marrack, P. Toll-like Receptor 7 (TLR7)–Driven Accumulation of a Novel CD11c+ B-Cell Population Is Important for the Development of Autoimmunity. Blood 2011, 118, 1305–1315. [Google Scholar] [CrossRef]
- Ma, S.; Wang, C.; Mao, X.; Hao, Y. B Cell Dysfunction Associated With Aging and Autoimmune Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Naismith, E.; Pangrazzi, L.; Grasse, M.; Keller, M.; Miggitsch, C.; Weinberger, B.; Trieb, K.; Grubeck-Loebenstein, B. Peripheral Antibody Concentrations Are Associated with Highly Differentiated T Cells and Inflammatory Processes in the Human Bone Marrow. Immun. Ageing 2019, 16, 21. [Google Scholar] [CrossRef]
- Salminen, A. Activation of Immunosuppressive Network in the Aging Process. Ageing Res. Rev. 2020, 57, 100998. [Google Scholar] [CrossRef] [PubMed]
- Anthony, R.M.; Rutitzky, L.I.; Urban, J.F.; Stadecker, M.J.; Gause, W.C. Protective Immune Mechanisms in Helminth Infection. Nat. Rev. Immunol. 2007, 7, 975–987. [Google Scholar] [CrossRef] [PubMed]
- Sallusto, F.; Geginat, J.; Lanzavecchia, A. Central Memory and Effector Memory T Cell Subsets: Function, Generation, and Maintenance. Annu. Rev. Immunol. 2004, 22, 745–763. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and Function of MTOR Signalling in T Cell Fate Decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef]
- Mannick, J.B.; Del Giudice, G.; Lattanzi, M.; Valiante, N.M.; Praestgaard, J.; Huang, B.; Lonetto, M.A.; Maecker, H.T.; Kovarik, J.; Carson, S.; et al. MTOR Inhibition Improves Immune Function in the Elderly. Sci. Transl. Med. 2014, 6, 268ra179. [Google Scholar] [CrossRef]
- Mannick, J.B.; Morris, M.; Hockey, H.-U.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 Inhibition Enhances Immune Function and Reduces Infections in the Elderly. Sci. Transl. Med. 2018, 10, aaq1564. [Google Scholar] [CrossRef]
- Mannick, J.B.; Teo, G.; Bernardo, P.; Quinn, D.; Russell, K.; Klickstein, L.; Marshall, W.; Shergill, S. Targeting the Biology of Ageing with MTOR Inhibitors to Improve Immune Function in Older Adults: Phase 2b and Phase 3 Randomised Trials. Lancet Healthy Longev 2021, 2, e250–e262. [Google Scholar] [CrossRef]
- Walters, H.E.; Cox, L.S. MTORC Inhibitors as Broad-Spectrum Therapeutics for Age-Related Diseases. Int. J. Mol. Sci. 2018, 19, 2325. [Google Scholar] [CrossRef]
- Min, H.; Montecino-Rodriguez, E.; Dorshkind, K. Reduction in the Developmental Potential of Intrathymic T Cell Progenitors with Age. J. Immunol. 2004, 173, 245–250. [Google Scholar] [CrossRef]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-Associated Characteristics of Murine Hematopoietic Stem Cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef]
- Sun, L.; Brown, R.; Chen, S.; Zhuge, Q.; Su, D.-M. Aging Induced Decline in T-Lymphopoiesis Is Primarily Dependent on Status of Progenitor Niches in the Bone Marrow and Thymus. Aging (Albany NY) 2012, 4, 606–619. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, A.L.; Seach, N.; Reiseger, J.J.; Lowen, T.E.; Hammett, M.V.; Scott, H.S.; Boyd, R.L. Reduced Thymic Aire Expression and Abnormal NF-ΚB2 Signaling in a Model of Systemic Autoimmunity. J. Immunol. 2009, 182, 2690–2699. [Google Scholar] [CrossRef] [PubMed]
- Latchney, S.E.; Calvi, L.M. The Aging Hematopoietic Stem Cell Niche: Phenotypic and Functional Changes and Mechanisms That Contribute to Hematopoietic Aging. Semin. Hematol. 2017, 54, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Emmrich, S.; Tolibzoda Zakusilo, F.; Trapp, A.; Zhou, X.; Zhang, Q.; Irving, E.M.; Drage, M.G.; Zhang, Z.; Gladyshev, V.N.; Seluanov, A.; et al. Ectopic Cervical Thymi and No Thymic Involution until Midlife in Naked Mole Rats. Aging Cell 2021, 20, e13477. [Google Scholar] [CrossRef]
- Thomas, R.; Wang, W.; Su, D.-M. Contributions of Age-Related Thymic Involution to Immunosenescence and Inflammaging. Immun Ageing 2020, 17, 2. [Google Scholar] [CrossRef] [PubMed]
- Hale, J.S.; Boursalian, T.E.; Turk, G.L.; Fink, P.J. Thymic Output in Aged Mice. Proc. Natl. Acad. Sci. USA 2006, 103, 8447–8452. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Lee, W.-W.; Weyand, C.M. Aging and T-Cell Diversity. Exp. Gerontol. 2007, 42, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Swain, S.; Clise-Dwyer, K.; Haynes, L. Homeostasis and the Age-Associated Defect of CD4 T Cells. Semin. Immunol. 2005, 17, 370–377. [Google Scholar] [CrossRef]
- Merani, S.; Pawelec, G.; Kuchel, G.A.; McElhaney, J.E. Impact of Aging and Cytomegalovirus on Immunological Response to Influenza Vaccination and Infection. Front. Immunol. 2017, 8, 784. [Google Scholar] [CrossRef]
- Nguyen, T.H.O.; Sant, S.; Bird, N.L.; Grant, E.J.; Clemens, E.B.; Koutsakos, M.; Valkenburg, S.A.; Gras, S.; Lappas, M.; Jaworowski, A.; et al. Perturbed CD8+ T Cell Immunity across Universal Influenza Epitopes in the Elderly. J. Leukoc. Biol. 2018, 103, 321–339. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Immunosenescence and Cancer. Biogerontology 2017, 18, 717–721. [Google Scholar] [CrossRef] [PubMed]
- den Braber, I.; Mugwagwa, T.; Vrisekoop, N.; Westera, L.; Mögling, R.; de Boer, A.B.; Willems, N.; Schrijver, E.H.R.; Spierenburg, G.; Gaiser, K.; et al. Maintenance of Peripheral Naive T Cells Is Sustained by Thymus Output in Mice but Not Humans. Immunity 2012, 36, 288–297. [Google Scholar] [CrossRef] [PubMed]
- van der Geest, K.S.M.; Abdulahad, W.H.; Tete, S.M.; Lorencetti, P.G.; Horst, G.; Bos, N.A.; Kroesen, B.-J.; Brouwer, E.; Boots, A.M.H. Aging Disturbs the Balance between Effector and Regulatory CD4+ T Cells. Exp. Gerontol. 2014, 60, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, F.; Zhang, B.; Yan, P.; Rowan, B.G.; Abdel-Mageed, A.B.; Steele, C.; Jazwinski, S.M.; Moroz, K.; Norton, E.B.; et al. CD4+ T Helper 17 Cell Response of Aged Mice Promotes Prostate Cancer Cell Migration and Invasion. Prostate 2020, 80, 764–776. [Google Scholar] [CrossRef]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Kroemer, G.; Zitvogel, L. CD4+ T Cells at the Center of Inflammaging. Cell Metab. 2020, 32, 4–5. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.-A.; Lee, J.; Park, J.-S.; Jhun, J.-Y.; Moon, Y.-M.; Cho, M.-L.; Kim, H.-Y. Increased Th17 Differentiation in Aged Mice Is Significantly Associated with High IL-1β Level and Low IL-2 Expression. Exp. Gerontol. 2014, 49, 55–62. [Google Scholar] [CrossRef]
- Kimura, A.; Kishimoto, T. IL-6: Regulator of Treg/Th17 Balance. Eur. J. Immunol. 2010, 40, 1830–1835. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Ding, S.; Wang, Y.M.; Xu, X.; Shen, Z.; Fu, R.; Liu, M.; Hu, C.; Zhang, C.; Cao, Q.; et al. Age-Associated Alteration in Th17 Cell Response Is Related to Endothelial Cell Senescence and Atherosclerotic Cerebral Infarction. Am. J. Transl. Res. 2017, 9, 5160–5168. [Google Scholar] [PubMed]
- Coder, B.D.; Wang, H.; Ruan, L.; Su, D.-M. Thymic Involution Perturbs Negative Selection Leading to Autoreactive T Cells That Induce Chronic Inflammation. J. Immunol. 2015, 194, 5825–5837. [Google Scholar] [CrossRef]
- Xia, J.; Wang, H.; Guo, J.; Zhang, Z.; Coder, B.; Su, D.-M. Age-Related Disruption of Steady-State Thymic Medulla Provokes Autoimmune Phenotype via Perturbing Negative Selection. Aging Dis. 2012, 3, 248–259. [Google Scholar]
- Coder, B.; Su, D.-M. Thymic Involution beyond T-Cell Insufficiency. Oncotarget 2015, 6, 21777–21778. [Google Scholar] [CrossRef] [PubMed]
- Gallegos, A.M.; Bevan, M.J. Central Tolerance: Good but Imperfect. Immunol. Rev. 2006, 209, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.; Wang, W.; Thomas, R.; Su, D.-M. Capacity of TTreg Generation Is Not Impaired in the Atrophied Thymus. PLoS Biol. 2017, 15, e2003352. [Google Scholar] [CrossRef]
- Kalia, V.; Sarkar, S. Regulation of Effector and Memory CD8 T Cell Differentiation by IL-2—A Balancing Act. Front. Immunol. 2018, 9, 2987. [Google Scholar] [CrossRef]
- Goronzy, J.J.; Fang, F.; Cavanagh, M.M.; Qi, Q.; Weyand, C.M. Naive T Cell Maintenance and Function in Human Aging. J. Immunol. 2015, 194, 4073–4080. [Google Scholar] [CrossRef]
- Martínez-Zamudio, R.I.; Dewald, H.K.; Vasilopoulos, T.; Gittens-Williams, L.; Fitzgerald-Bocarsly, P.; Herbig, U. Senescence-Associated β-Galactosidase Reveals the Abundance of Senescent CD8+ T Cells in Aging Humans. Aging Cell 2021, 20, e13344. [Google Scholar] [CrossRef] [PubMed]
- Perez-Lanzon, M.; Zitvogel, L.; Kroemer, G. Failure of Immunosurveillance Accelerates Aging. Oncoimmunology 2019, 8, 1575117. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Borsa, M.; Simon, A.K. Hallmarks and Detection Techniques of Cellular Senescence and Cellular Ageing in Immune Cells. Aging Cell 2021, 20, e13316. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Q.; Wagner, W.M.; Voehringer, D.; Wikby, A.; Klatt, T.; Walter, S.; Müller, C.A.; Pircher, H.; Pawelec, G. Age-Associated Accumulation of CMV-Specific CD8+ T Cells Expressing the Inhibitory Killer Cell Lectin-like Receptor G1 (KLRG1). Exp. Gerontol. 2003, 38, 911–920. [Google Scholar] [CrossRef]
- Voehringer, D.; Koschella, M.; Pircher, H. Lack of Proliferative Capacity of Human Effector and Memory T Cells Expressing Killer Cell Lectinlike Receptor G1 (KLRG1). Blood 2002, 100, 3698–3702. [Google Scholar] [CrossRef] [PubMed]
- Desdín-Micó, G.; Soto-Heredero, G.; Aranda, J.F.; Oller, J.; Carrasco, E.; Gabandé-Rodríguez, E.; Blanco, E.M.; Alfranca, A.; Cussó, L.; Desco, M.; et al. T Cells with Dysfunctional Mitochondria Induce Multimorbidity and Premature Senescence. Science 2020, 368, 1371–1376. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Meryk, A.; Naismith, E.; Koziel, R.; Lair, J.; Krismer, M.; Trieb, K.; Grubeck-Loebenstein, B. “Inflamm-aging” Influences Immune Cell Survival Factors in Human Bone Marrow. Eur. J. Immunol. 2017, 47, 481–492. [Google Scholar] [CrossRef]
- Moreau, J.-F.; Pradeu, T.; Grignolio, A.; Nardini, C.; Castiglione, F.; Tieri, P.; Capri, M.; Salvioli, S.; Taupin, J.-L.; Garagnani, P.; et al. The Emerging Role of ECM Crosslinking in T Cell Mobility as a Hallmark of Immunosenescence in Humans. Ageing Res. Rev. 2017, 35, 322–335. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.I.; De Maeyer, R.P.H.; Covre, L.P.; Nehar-Belaid, D.; Lanna, A.; Ward, S.; Marches, R.; Chambers, E.S.; Gomes, D.C.O.; Riddell, N.E.; et al. Sestrins Induce Natural Killer Function in Senescent-like CD8+ T Cells. Nat. Immunol. 2020, 21, 684–694. [Google Scholar] [CrossRef]
- Quinn, K.M.; Linterman, M.A. Senescence Blurs the Line between Innate and Adaptive Immune Cells. Immunol. Cell Biol. 2020, 98, 431–433. [Google Scholar] [CrossRef]
- Nilsson, M.I.; Bourgeois, J.M.; Nederveen, J.P.; Leite, M.R.; Hettinga, B.P.; Bujak, A.L.; May, L.; Lin, E.; Crozier, M.; Rusiecki, D.R.; et al. Lifelong Aerobic Exercise Protects against Inflammaging and Cancer. PLoS ONE 2019, 14, e0210863. [Google Scholar] [CrossRef] [PubMed]
- Lavin, K.M.; Perkins, R.K.; Jemiolo, B.; Raue, U.; Trappe, S.W.; Trappe, T.A. Effects of Aging and Lifelong Aerobic Exercise on Basal and Exercise-Induced Inflammation. J. Appl. Physiol. 2019, 128, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Duggal, N.A.; Pollock, R.D.; Lazarus, N.R.; Harridge, S.; Lord, J.M. Major Features of Immunesenescence, Including Reduced Thymic Output, Are Ameliorated by High Levels of Physical Activity in Adulthood. Aging Cell 2018, 17, 12750. [Google Scholar] [CrossRef]
- Beyer, I.; Mets, T.; Bautmans, I. Chronic Low-Grade Inflammation and Age-Related Sarcopenia. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 12. [Google Scholar] [CrossRef]
- Tartibian, B.; Hajizadeh Maleki, B.; Kanaley, J.; Sadeghi, K. Long-Term Aerobic Exercise and Omega-3 Supplementation Modulate Osteoporosis through Inflammatory Mechanisms in Post-Menopausal Women: A Randomized, Repeated Measures Study. Nutr. Metab. (Lond.) 2011, 8, 71. [Google Scholar] [CrossRef]
- Nicklas, B.J.; Hsu, F.-C.; Brinkley, T.J.; Church, T.; Goodpaster, B.H.; Kritchevsky, S.B.; Pahor, M. Exercise Training and Plasma C-Reactive Protein and Interleukin-6 in Elderly People. J. Am. Geriatr. Soc. 2008, 56, 2045–2052. [Google Scholar] [CrossRef] [PubMed]
- Bautmans, I.; Njemini, R.; Vasseur, S.; Chabert, H.; Moens, L.; Demanet, C.; Mets, T. Biochemical Changes in Response to Intensive Resistance Exercise Training in the Elderly. Gerontology 2005, 51, 253–265. [Google Scholar] [CrossRef]
- Bautmans, I.; Salimans, L.; Njemini, R.; Beyer, I.; Lieten, S.; Liberman, K. The Effects of Exercise Interventions on the Inflammatory Profile of Older Adults: A Systematic Review of the Recent Literature. Exp. Gerontol. 2021, 146, 111236. [Google Scholar] [CrossRef] [PubMed]
- Girven, M.; Dugdale, H.F.; Owens, D.J.; Hughes, D.C.; Stewart, C.E.; Sharples, A.P. L-Glutamine Improves Skeletal Muscle Cell Differentiation and Prevents Myotube Atrophy After Cytokine (TNF-α) Stress Via Reduced P38 MAPK Signal Transduction. J. Cell. Physiol. 2016, 231, 2720–2732. [Google Scholar] [CrossRef]
- Dalle, S.; Rossmeislova, L.; Koppo, K. The Role of Inflammation in Age-Related Sarcopenia. Front. Physiol. 2017, 8, 1045. [Google Scholar] [CrossRef]
- Minuzzi, L.G.; Rama, L.; Chupel, M.U.; Rosado, F.; Dos Santos, J.V.; Simpson, R.; Martinho, A.; Paiva, A.; Teixeira, A.M. Effects of Lifelong Training on Senescence and Mobilization of T Lymphocytes in Response to Acute Exercise. Exerc. Immunol. Rev. 2018, 24, 72–84. [Google Scholar] [PubMed]
- Bartlett, D.B.; Slentz, C.A.; Willis, L.H.; Hoselton, A.; Huebner, J.L.; Kraus, V.B.; Moss, J.; Muehlbauer, M.J.; Spielmann, G.; Muoio, D.M.; et al. Rejuvenation of Neutrophil Functions in Association With Reduced Diabetes Risk Following Ten Weeks of Low-Volume High Intensity Interval Walking in Older Adults With Prediabetes—A Pilot Study. Front. Immunol. 2020, 11, 729. [Google Scholar] [CrossRef]
- Spielmann, G.; McFarlin, B.K.; O’Connor, D.P.; Smith, P.J.W.; Pircher, H.; Simpson, R.J. Aerobic Fitness Is Associated with Lower Proportions of Senescent Blood T-Cells in Man. Brain Behav. Immun. 2011, 25, 1521–1529. [Google Scholar] [CrossRef] [PubMed]
- Mathot, E.; Liberman, K.; Cao Dinh, H.; Njemini, R.; Bautmans, I. Systematic Review on the Effects of Physical Exercise on Cellular Immunosenescence-Related Markers—An Update. Exp. Gerontol. 2021, 149, 111318. [Google Scholar] [CrossRef] [PubMed]
- Chan, R.S.M.; Yu, B.W.M.; Leung, J.; Lee, J.S.W.; Auyeung, T.W.; Kwok, T.; Woo, J. How Dietary Patterns Are Related to Inflammaging and Mortality in Community-Dwelling Older Chinese Adults in Hong Kong—A Prospective Analysis. J. Nutr. Health Aging 2019, 23, 181–194. [Google Scholar] [CrossRef]
- Alonso-Pedrero, L.; Ojeda-Rodríguez, A.; Martínez-González, M.A.; Zalba, G.; Bes-Rastrollo, M.; Marti, A. Ultra-Processed Food Consumption and the Risk of Short Telomeres in an Elderly Population of the Seguimiento Universidad de Navarra (SUN) Project. Am. J. Clin. Nutr. 2020, 111, 1259–1266. [Google Scholar] [CrossRef] [PubMed]
- Ojeda-Rodríguez, A.; Zazpe, I.; Alonso-Pedrero, L.; Zalba, G.; Guillen-Grima, F.; Martinez-Gonzalez, M.A.; Marti, A. Association between Diet Quality Indexes and the Risk of Short Telomeres in an Elderly Population of the SUN Project. Clin. Nutr. 2020, 39, 2487–2494. [Google Scholar] [CrossRef]
- Urpi-Sarda, M.; Casas, R.; Sacanella, E.; Corella, D.; Andrés-Lacueva, C.; Llorach, R.; Garrabou, G.; Cardellach, F.; Sala-Vila, A.; Ros, E.; et al. The 3-Year Effect of the Mediterranean Diet Intervention on Inflammatory Biomarkers Related to Cardiovascular Disease. Biomedicines 2021, 9, 862. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, K.; Aggarwal, B.B.; Singh, R.B.; Buttar, H.S.; Wilson, D.; De Meester, F. Food Antioxidants and Their Anti-Inflammatory Properties: A Potential Role in Cardiovascular Diseases and Cancer Prevention. Diseases 2016, 4, 28. [Google Scholar] [CrossRef]
- Rodriguez, A.J.; Mousa, A.; Ebeling, P.R.; Scott, D.; de Courten, B. Effects of Vitamin D Supplementation on Inflammatory Markers in Heart Failure: A Systematic Review and Meta-Analysis of Randomized Controlled Trials. Sci. Rep. 2018, 8, 1169. [Google Scholar] [CrossRef]
- Grübler, M.R.; Zittermann, A.; Verheyen, N.D.; Trummer, C.; Theiler-Schwetz, V.; Keppel, M.H.; Malle, O.; Richtig, G.; Gängler, S.; Bischoff-Ferrari, H.; et al. Randomized Trial of Vitamin D versus Placebo Supplementation on Markers of Systemic Inflammation in Hypertensive Patients. Nutr. Metab Cardiovasc. Dis. 2021, 31, 3202–3209. [Google Scholar] [CrossRef]
- Gonçalves de Carvalho, C.M.R.; Ribeiro, S.M.L. Aging, Low-Grade Systemic Inflammation and Vitamin D: A Mini-Review. Eur. J. Clin. Nutr. 2017, 71, 434–440. [Google Scholar] [CrossRef]
- Adelani, I.B.; Ogadi, E.O.; Onuzulu, C.; Rotimi, O.A.; Maduagwu, E.N.; Rotimi, S.O. Dietary Vitamin D Ameliorates Hepatic Oxidative Stress and Inflammatory Effects of Diethylnitrosamine in Rats. Heliyon 2020, 6, e04842. [Google Scholar] [CrossRef] [PubMed]
- L Bishop, E.; Ismailova, A.; Dimeloe, S.; Hewison, M.; White, J.H. Vitamin D and Immune Regulation: Antibacterial, Antiviral, Anti-Inflammatory. JBMR Plus 2021, 5, e10405. [Google Scholar] [CrossRef]
- Chang, S.H.; Chung, Y.; Dong, C. Vitamin D Suppresses Th17 Cytokine Production by Inducing C/EBP Homologous Protein (CHOP) Expression. J. Biol. Chem. 2010, 285, 38751–38755. [Google Scholar] [CrossRef]
- De Spiegeleer, A.; Beckwée, D.; Bautmans, I.; Petrovic, M.; Sarcopenia Guidelines Development group of the Belgian Society of Gerontology and Geriatrics (BSGG). Pharmacological Interventions to Improve Muscle Mass, Muscle Strength and Physical Performance in Older People: An Umbrella Review of Systematic Reviews and Meta-Analyses. Drugs Aging 2018, 35, 719–734. [Google Scholar] [CrossRef] [PubMed]
- Fontana, L.; Partridge, L.; Longo, V.D. Dietary Restriction, Growth Factors and Aging: From Yeast to Humans. Science 2010, 328, 321–326. [Google Scholar] [CrossRef]
- Kim, D.H.; Bang, E.; Jung, H.J.; Noh, S.G.; Yu, B.P.; Choi, Y.J.; Chung, H.Y. Anti-Aging Effects of Calorie Restriction (CR) and CR Mimetics Based on the Senoinflammation Concept. Nutrients 2020, 12, 422. [Google Scholar] [CrossRef]
- Chung, H.Y.; Kim, H.J.; Kim, J.W.; Yu, B.P. The Inflammation Hypothesis of Aging. Ann. N. Y. Acad. Sci. 2001, 928, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Zhang, H.; Powell, T.J.; Lipina, E.; Sims, S.; Panse, I.; Watson, A.S.; Cerundolo, V.; Townsend, A.R.; Klenerman, P.; et al. Autophagy Is a Critical Regulator of Memory CD8+ T Cell Formation. eLife 2014, 3, e03706. [Google Scholar] [CrossRef]
- Mattison, J.A.; Roth, G.S.; Beasley, T.M.; Tilmont, E.M.; Handy, A.M.; Herbert, R.L.; Longo, D.L.; Allison, D.B.; Young, J.E.; Bryant, M.; et al. Impact of Caloric Restriction on Health and Survival in Rhesus Monkeys from the NIA Study. Nature 2012, 489, 318–321. [Google Scholar] [CrossRef] [PubMed]
- Mattison, J.A.; Colman, R.J.; Beasley, T.M.; Allison, D.B.; Kemnitz, J.W.; Roth, G.S.; Ingram, D.K.; Weindruch, R.; de Cabo, R.; Anderson, R.M. Caloric Restriction Improves Health and Survival of Rhesus Monkeys. Nat. Commun. 2017, 8, 14063. [Google Scholar] [CrossRef]
- Colman, R.J.; Anderson, R.M.; Johnson, S.C.; Kastman, E.K.; Kosmatka, K.J.; Beasley, T.M.; Allison, D.B.; Cruzen, C.; Simmons, H.A.; Kemnitz, J.W.; et al. Caloric Restriction Delays Disease Onset and Mortality in Rhesus Monkeys. Science 2009, 325, 201–204. [Google Scholar] [CrossRef]
- Meydani, S.N.; Das, S.K.; Pieper, C.F.; Lewis, M.R.; Klein, S.; Dixit, V.D.; Gupta, A.K.; Villareal, D.T.; Bhapkar, M.; Huang, M.; et al. Long-Term Moderate Calorie Restriction Inhibits Inflammation without Impairing Cell-Mediated Immunity: A Randomized Controlled Trial in Non-Obese Humans. Aging (Albany NY) 2016, 8, 1416–1426. [Google Scholar] [CrossRef]
- Cox, L.S.; Mattison, J.A. Increasing Longevity through Caloric Restriction or Rapamycin Feeding in Mammals: Common Mechanisms for Common Outcomes? Aging Cell 2009, 8, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Lührs, H.; Gerke, T.; Müller, J.G.; Melcher, R.; Schauber, J.; Boxberge, F.; Scheppach, W.; Menzel, T. Butyrate Inhibits NF-KappaB Activation in Lamina Propria Macrophages of Patients with Ulcerative Colitis. Scand. J. Gastroenterol. 2002, 37, 458–466. [Google Scholar] [CrossRef] [PubMed]
- Parada Venegas, D.; De la Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10. [Google Scholar] [CrossRef]
- Pirozzi, C.; Francisco, V.; Guida, F.D.; Gómez, R.; Lago, F.; Pino, J.; Meli, R.; Gualillo, O. Butyrate Modulates Inflammation in Chondrocytes via GPR43 Receptor. Cell. Physiol. Biochem. 2018, 51, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Inan, M.S.; Rasoulpour, R.J.; Yin, L.; Hubbard, A.K.; Rosenberg, D.W.; Giardina, C. The Luminal Short-Chain Fatty Acid Butyrate Modulates NF-KappaB Activity in a Human Colonic Epithelial Cell Line. Gastroenterology 2000, 118, 724–734. [Google Scholar] [CrossRef]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium Butyrate Inhibits the NF-Kappa B Signaling Pathway and Histone Deacetylation, and Attenuates Experimental Colitis in an IL-10 Independent Manner. Int. Immunopharmacol. 2017, 51, 47–56. [Google Scholar] [CrossRef]
- Shintouo, C.M.; Mets, T.; Beckwee, D.; Bautmans, I.; Ghogomu, S.M.; Souopgui, J.; Leemans, L.; Meriki, H.D.; Njemini, R. Is Inflammageing Influenced by the Microbiota in the Aged Gut? A Systematic Review. Exp. Gerontol. 2020, 141, 111079. [Google Scholar] [CrossRef]
- Asnicar, F.; Berry, S.E.; Valdes, A.M.; Nguyen, L.H.; Piccinno, G.; Drew, D.A.; Leeming, E.; Gibson, R.; Le Roy, C.; Khatib, H.A.; et al. Microbiome Connections with Host Metabolism and Habitual Diet from 1,098 Deeply Phenotyped Individuals. Nat. Med. 2021, 27, 321–332. [Google Scholar] [CrossRef]
- Fahy, G.M.; Brooke, R.T.; Watson, J.P.; Good, Z.; Vasanawala, S.S.; Maecker, H.; Leipold, M.D.; Lin, D.T.S.; Kobor, M.S.; Horvath, S. Reversal of Epigenetic Aging and Immunosenescent Trends in Humans. Aging Cell 2019, 18, e13028. [Google Scholar] [CrossRef]
- Kim, M.-J.; Miller, C.M.; Shadrach, J.L.; Wagers, A.J.; Serwold, T. Young, Proliferative Thymic Epithelial Cells Engraft and Function in Aging Thymuses. J. Immunol. 2015, 194, 4784–4795. [Google Scholar] [CrossRef]
- Bredenkamp, N.; Ulyanchenko, S.; O’Neill, K.E.; Manley, N.R.; Vaidya, H.J.; Blackburn, C.C. An Organized and Functional Thymus Generated from FOXN1-Reprogrammed Fibroblasts. Nat. Cell Biol. 2014, 16, 902–908. [Google Scholar] [CrossRef] [PubMed]
- Henson, S.M.; Snelgrove, R.; Hussell, T.; Wells, D.J.; Aspinall, R. An IL-7 Fusion Protein That Shows Increased Thymopoietic Ability. J. Immunol. 2005, 175, 4112–4118. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Guo, J.; Brown, R.; Amagai, T.; Zhao, Y.; Su, D.-M. Declining Expression of a Single Epithelial Cell-Autonomous Gene Accelerates Age-Related Thymic Involution. Aging Cell 2010, 9, 347–357. [Google Scholar] [CrossRef]
- Bredenkamp, N.; Nowell, C.S.; Blackburn, C.C. Regeneration of the Aged Thymus by a Single Transcription Factor. Development 2014, 141, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Keren, Z.; Naor, S.; Nussbaum, S.; Golan, K.; Itkin, T.; Sasaki, Y.; Schmidt-Supprian, M.; Lapidot, T.; Melamed, D. B-Cell Depletion Reactivates B Lymphopoiesis in the BM and Rejuvenates the B Lineage in Aging. Blood 2011, 117, 3104–3112. [Google Scholar] [CrossRef]
- Avivi, I.; Zisman-Rozen, S.; Naor, S.; Dai, I.; Benhamou, D.; Shahaf, G.; Tabibian-Keissar, H.; Rosenthal, N.; Rakovsky, A.; Hanna, A.; et al. Depletion of B Cells Rejuvenates the Peripheral B-Cell Compartment but Is Insufficient to Restore Immune Competence in Aging. Aging Cell 2019, 18, e12959. [Google Scholar] [CrossRef]
- Wahlestedt, M.; Erlandsson, E.; Kristiansen, T.; Lu, R.; Brakebusch, C.; Weissman, I.L.; Yuan, J.; Martin-Gonzalez, J.; Bryder, D. Clonal Reversal of Ageing-Associated Stem Cell Lineage Bias via a Pluripotent Intermediate. Nat. Commun. 2017, 8, 14533. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef]
- Borgoni, S.; Kudryashova, K.S.; Burka, K.; de Magalhães, J.P. Targeting Immune Dysfunction in Aging. Ageing Res. Rev. 2021, 70, 101410. [Google Scholar] [CrossRef] [PubMed]
- Cox, L.S.; Bellantuono, I.; Lord, J.M.; Sapey, E.; Mannick, J.B.; Partridge, L.; Gordon, A.L.; Steves, C.J.; Witham, M.D. Tackling Immunosenescence to Improve COVID-19 Outcomes and Vaccine Response in Older Adults. Lancet Healthy Longev. 2020, 1, e55–e57. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T Cells Reverse Senescence-Associated Pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef]
- Song, P.; An, J.; Zou, M.-H. Immune Clearance of Senescent Cells to Combat Ageing and Chronic Diseases. Cells 2020, 9, 671. [Google Scholar] [CrossRef]
- Baar, M.P.; Brandt, R.M.C.; Putavet, D.A.; Klein, J.D.D.; Derks, K.W.J.; Bourgeois, B.R.M.; Stryeck, S.; Rijksen, Y.; van Willigenburg, H.; Feijtel, D.A.; et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 2017, 169, 132–147. [Google Scholar] [CrossRef]
- Muñoz-Espín, D.; Rovira, M.; Galiana, I.; Giménez, C.; Lozano-Torres, B.; Paez-Ribes, M.; Llanos, S.; Chaib, S.; Muñoz-Martín, M.; Ucero, A.C.; et al. A Versatile Drug Delivery System Targeting Senescent Cells. EMBO Mol. Med. 2018, 10, 9355. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.-M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of Senescent Cells by ABT263 Rejuvenates Aged Hematopoietic Stem Cells in Mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin Is a Senotherapeutic That Extends Health and Lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin Fed Late in Life Extends Lifespan in Genetically Heterogeneous Mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR Regulates the Pro-Tumorigenic Senescence-Associated Secretory Phenotype by Promoting IL1A Translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Rolt, A.; Nair, A.; Cox, L.S. Optimisation of a Screening Platform for Determining IL-6 Inflammatory Signalling in the Senescence-Associated Secretory Phenotype (SASP). Biogerontology 2019, 20, 359–371. [Google Scholar] [CrossRef]
- Davis, T.; Haughton, M.F.; Jones, C.J.; Kipling, D. Prevention of Accelerated Cell Aging in the Werner Syndrome. Ann. N. Y. Acad. Sci. 2006, 1067, 243–247. [Google Scholar] [CrossRef]
- Alimbetov, D.; Davis, T.; Brook, A.J.C.; Cox, L.S.; Faragher, R.G.A.; Nurgozhin, T.; Zhumadilov, Z.; Kipling, D. Suppression of the Senescence-Associated Secretory Phenotype (SASP) in Human Fibroblasts Using Small Molecule Inhibitors of P38 MAP Kinase and MK2. Biogerontology 2016, 17, 305–315. [Google Scholar] [CrossRef]
- Correia-Melo, C.; Birch, J.; Fielder, E.; Rahmatika, D.; Taylor, J.; Chapman, J.; Lagnado, A.; Carroll, B.M.; Miwa, S.; Richardson, G.; et al. Rapamycin Improves Healthspan but Not Inflammaging in Nfκb1−/− Mice. Aging Cell 2019, 18, e12882. [Google Scholar] [CrossRef]
- Zhang, X.-J.; Qin, J.-J.; Cheng, X.; Shen, L.; Zhao, Y.-C.; Yuan, Y.; Lei, F.; Chen, M.-M.; Yang, H.; Bai, L.; et al. In-Hospital Use of Statins Is Associated with a Reduced Risk of Mortality among Individuals with COVID-19. Cell Metab 2020, 32, 176–187. [Google Scholar] [CrossRef]
- Alsaleh, G.; Panse, I.; Swadling, L.; Zhang, H.; Richter, F.C.; Meyer, A.; Lord, J.; Barnes, E.; Klenerman, P.; Green, C.; et al. Autophagy in T Cells from Aged Donors Is Maintained by Spermidine and Correlates with Function and Vaccine Responses. Elife 2020, 9, e57950. [Google Scholar] [CrossRef]
- Zhang, H.; Alsaleh, G.; Feltham, J.; Sun, Y.; Napolitano, G.; Riffelmacher, T.; Charles, P.; Frau, L.; Hublitz, P.; Yu, Z.; et al. Polyamines Control EIF5A Hypusination, TFEB Translation, and Autophagy to Reverse B Cell Senescence. Mol. Cell 2019, 76, 110–125. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin Inhibits the Senescence-Associated Secretory Phenotype by Interfering with IKK/NF-ΚB Activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Bannister, C.A.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.P.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can People with Type 2 Diabetes Live Longer than Those without? A Comparison of Mortality in People Initiated with Metformin or Sulphonylurea Monotherapy and Matched, Non-Diabetic Controls. Diabetes Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef]
- Glossmann, H.H.; Lutz, O.M.D. Metformin and Aging: A Review. Gerontology 2019, 65, 581–590. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef]
- Menegazzo, L.; Scattolini, V.; Cappellari, R.; Bonora, B.M.; Albiero, M.; Bortolozzi, M.; Romanato, F.; Ceolotto, G.; Vigili de Kreutzeberg, S.; Avogaro, A.; et al. The Antidiabetic Drug Metformin Blunts NETosis in Vitro and Reduces Circulating NETosis Biomarkers in Vivo. Acta Diabetol. 2018, 55, 593–601. [Google Scholar] [CrossRef]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin Improves Healthspan and Lifespan in Mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef]
- He, M.; Chiang, H.-H.; Luo, H.; Zheng, Z.; Qiao, Q.; Wang, L.; Tan, M.; Ohkubo, R.; Mu, W.-C.; Zhao, S.; et al. An Acetylation Switch of the NLRP3 Inflammasome Regulates Aging-Associated Chronic Inflammation and Insulin Resistance. Cell Metab. 2020, 31, 580–591. [Google Scholar] [CrossRef]
- Luo, H.; Mu, W.-C.; Karki, R.; Chiang, H.-H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945. [Google Scholar] [CrossRef]
- Marín-Aguilar, F.; Castejón-Vega, B.; Alcocer-Gómez, E.; Lendines-Cordero, D.; Cooper, M.A.; de la Cruz, P.; Andújar-Pulido, E.; Pérez-Alegre, M.; Muntané, J.; Pérez-Pulido, A.J.; et al. NLRP3 Inflammasome Inhibition by MCC950 in Aged Mice Improves Health via Enhanced Autophagy and PPARα Activity. J. Gerontol. A Biol. Sci. Med. Sci. 2020, 75, 1457–1464. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Mitchell, S.J.; Bernier, M.; Aon, M.A.; Cortassa, S.; Kim, E.Y.; Fang, E.F.; Palacios, H.H.; Ali, A.; Navas-Enamorado, I.; Di Francesco, A.; et al. Nicotinamide Improves Aspects of Healthspan, but Not Lifespan, in Mice. Cell Metab. 2018, 27, 667–676. [Google Scholar] [CrossRef]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, But Not Resveratrol or Simvastatin, Extends Life Span of Genetically Heterogeneous Mice. J. Gerontol. Ser. A 2011, 66A, 191–201. [Google Scholar] [CrossRef]
- Schmidt, C. GSK/Sirtris Compounds Dogged by Assay Artifacts. Nat. Biotechnol. 2010, 28, 185–186. [Google Scholar] [CrossRef]
- Kitada, M.; Ogura, Y.; Monno, I.; Koya, D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front. Endocrinol. 2019, 10, 187. [Google Scholar] [CrossRef]
- Aman, Y.; Qiu, Y.; Tao, J.; Fang, E.F. Therapeutic Potential of Boosting NAD+ in Aging and Age-Related Diseases. Transl. Med. Aging 2018, 2, 30–37. [Google Scholar] [CrossRef]
- Martens, C.R.; Denman, B.A.; Mazzo, M.R.; Armstrong, M.L.; Reisdorph, N.; McQueen, M.B.; Chonchol, M.; Seals, D.R. Chronic Nicotinamide Riboside Supplementation Is Well-Tolerated and Elevates NAD + in Healthy Middle-Aged and Older Adults. Nat. Commun. 2018, 9, 1286. [Google Scholar] [CrossRef]
- Foster, A.D.; Sivarapatna, A.; Gress, R.E. The Aging Immune System and Its Relationship with Cancer. Aging Health 2011, 7, 707–718. [Google Scholar] [CrossRef]
- Sayed, N.; Huang, Y.; Nguyen, K.; Krejciova-Rajaniemi, Z.; Grawe, A.P.; Gao, T.; Tibshirani, R.; Hastie, T.; Alpert, A.; Cui, L.; et al. An Inflammatory Aging Clock (IAge) Based on Deep Learning Tracks Multimorbidity, Immunosenescence, Frailty and Cardiovascular Aging. Nat. Aging 2021, 1, 598–615. [Google Scholar] [CrossRef]





| Pro- and Anti-Inflammatory Factors | Evidence | |||
|---|---|---|---|---|
| IL-18 | TNF-α | Population Characteristics | Data Processing | Reference |
| PCA1—Correlated with age, mortality, morbidity and age-related diseases | 1010 participants (428 men, 582 women), aged 21–96 y | Cluster analysis | [75] | |
| Positively correlated with age | Absence of correlation with age, mortality, and morbidity. Positively correlated with some age-related diseases | General population—InCHIANTI cohort | Individual associations | |
| Significant independent predictor of 5- and 10-year mortality | Non-predictive of 5-year mortality | 7043 participants (2995 men, 4048 women), aged 65–102 y | Fully adjusted correlation with 10-year mortality | [83] |
| General population—includes CHS and InCHIANTI cohorts | ||||
| No cluster reported | “Down”-regulation—Correlated with frailty outcomes | 967 participants (416 men, 551 women), aged 65 y and older | Cluster analysis | [76] |
| Individually correlated with worsened mobility | General population—InCHIANTI cohort | |||
| NI | TNF-α related—Negatively correlated with knee strength, HPPB score, and grip strength; Positively correlated with 400-m walk time | 1269 participants, aged 70–79 y | Cluster analysis | [77] |
| Highest R2 of the TNF-α-related component for the HPPB score and knee strength | General non-frail population | Individual associations | ||
| NI | NI | 285 participants (67 men, 218 women), aged 90 y and older with a 4-year follow-up | Correlation with 4-year mortality | [93] |
| General population | Fully adjusted | |||
| Local inflammation-endothelial dysfunction | NI | 320 participants (236 men, 84 women), aged 58–74 y with a 1-year follow-up | Cluster analysis | [78] |
| Independent predictor of 1-year recurrent coronary events | Acute coronary syndrome patients | Individual associations | ||
| Positively correlated with age | Absence of correlation with age | 1327 participants (586 men, 741 women), aged 20–102 y | Correlation with age | [80] |
| Reduced the size of the regression coefficient for age | NR | General population—Includes InCHIANTI cohort | Adjustment for cardiovascular risk factors and morbidities | |
| Proinflammation and anti-inflammation—Absence of association with 4-year death rate | 580 women aged 31–85 y with a 4.7-year follow-up | Cluster analysis | [79] | |
| Clinically referred for coronary angiography | ||||
| NI | Negatively correlated with walking and standing balance performance | 1020 participants (483 men, 537 women), aged 65–102 y | Correlation with physical performance | [81] |
| IL-1RA | IL-2sR | IL-6sR | sTNFRI | sTNFRII | IL-10 | TGF-β | HDL Cholesterol | Population Characteristics | Data Processing | Reference |
|---|---|---|---|---|---|---|---|---|---|---|
| PCA1—Correlated with age, mortality, morbidity, and age-related diseases | NI | No cluster reported | PCA1—Correlated with age, mortality, morbidity, and age-related diseases | No cluster reported | No cluster reported | NI | 1010 participants (428 men, 582 women), aged 21–96 y.o | Cluster analysis | [75] | |
| Positively correlated with age | Absence of correlation with age | Positively correlated with age, mortality, morbidity and some age-related disease | Positively correlated with age | Negatively correlated with age | Absence of correlation with age | General population—InCHIANTI cohort | Individual associations | |||
| Significant independent predictor of 5- and 10-year mortality | NI | Non-predictive of 5-year mortality | Significant independent predictor of 5- and 10-year mortality | Non-predictive of 5-year mortality | Non-predictive of 5-year mortality | NI | NI | 7043 participants (2995 men, 4048 women), aged 65–102 y.o. | Fully adjusted correlation with 10-year mortality | [83] |
| Best predictor of 10-year mortality in combination with IL-6 | General population—Includes CHS and InCHIANTI cohorts | |||||||||
| “Up”-regulation—Positively correlated with worsened mobility and frailty risk | NI | NI | NI | NI | NI | IL-1/TGF—No adverse outcome reported | NI | 967 participants (416 men, 551 women), aged 65 y.o. and older | Cluster analysis | [76] |
| General population—InCHIANTI cohort | ||||||||||
| NI | TNF-α related—Negatively correlated with knee strength, HPPB score and grip strength; positively correlated with 400-m walk time | NI | NI | NI | 1269 participants, aged 70–79 y.o. | Cluster analysis | [77] | |||
| Highest R2 of the TNF-α-related component for walking speed | Highest R2 of the TNF-α-related component for grip strength | Highest R2 of the TNF-α-related component for the HPPB score and 400-m walk time | General non-frail population | Individual associations | ||||||
| Positively associated with 4-year mortality | NI | NI | NI | NI | NI | NI | NI | 285 participants (67 men, 218 women), aged 90 y.o. and older with a 4-year follow-up | Correlation with 4-year mortality | [93] |
| Strong independent predictor of 4-year mortality alone or in combination with CRP | General population | Fully adjusted | ||||||||
| NI | NI | NI | NI | NI | Protective or anti-inflammation—negative predictor of adverse cardiac events | NI | Protective or anti-inflammation—negative predictor of adverse cardiac events | 320 participants (236 men, 84 women), aged 58–74 y.o. with a 1-year follow-up | Cluster analysis | [78] |
| Significant negative predictor of 1-year adverse cardiac events | Significant negative predictor of 1-year adverse cardiac events | Acute coronary syndrome patients | Individual associations | |||||||
| Positively correlated with age | NI | Positively correlated with age in women only | NI | NI | NI | Absence of correlation with age | NI | 1327 participants (586 men, 741 women), aged 20–102 y.o. | Correlation with age | [80] |
| Removed the effect of age | Increased the size of the regression coefficient for age | NR | General population—includes InCHIANTI cohort | Adjustment for cardiovascular risk factors and morbidities | ||||||
| NI | NI | NI | NI | NI | NI | Immunosuppressive—absence of association with 4-year death rate | NI | 580 women aged 31–85 y.o. with a 4.7-year follow-up | Cluster analysis | [79] |
| Clinically referred for coronary angiography | ||||||||||
| Negatively correlated with global physical performance but not with hand-grip strength | NI | No significant association with physical performance | NI | NI | No significant association with physical performance | NI | NI | 1020 participants (483 men, 537 women), aged 65–102 y.o. | Correlation with physical performance | [81] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Teissier, T.; Boulanger, E.; Cox, L.S. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells 2022, 11, 359. https://doi.org/10.3390/cells11030359
Teissier T, Boulanger E, Cox LS. Interconnections between Inflammageing and Immunosenescence during Ageing. Cells. 2022; 11(3):359. https://doi.org/10.3390/cells11030359
Chicago/Turabian StyleTeissier, Thibault, Eric Boulanger, and Lynne S. Cox. 2022. "Interconnections between Inflammageing and Immunosenescence during Ageing" Cells 11, no. 3: 359. https://doi.org/10.3390/cells11030359
APA StyleTeissier, T., Boulanger, E., & Cox, L. S. (2022). Interconnections between Inflammageing and Immunosenescence during Ageing. Cells, 11(3), 359. https://doi.org/10.3390/cells11030359