
Mast Cell–Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth

 ,
, 

 , , , and
, , , and
Abstract
1. Introduction
2. The Multifaceted Role of Mast Cells in Solid Tumors
3. Signaling Pathways and Chemotactic Molecules Involved in Mast Cell-Migration to Solid Tumors
3.1. Tumor-Derived Cytokines and Growth Factors That Promote MC Migration to Tumors
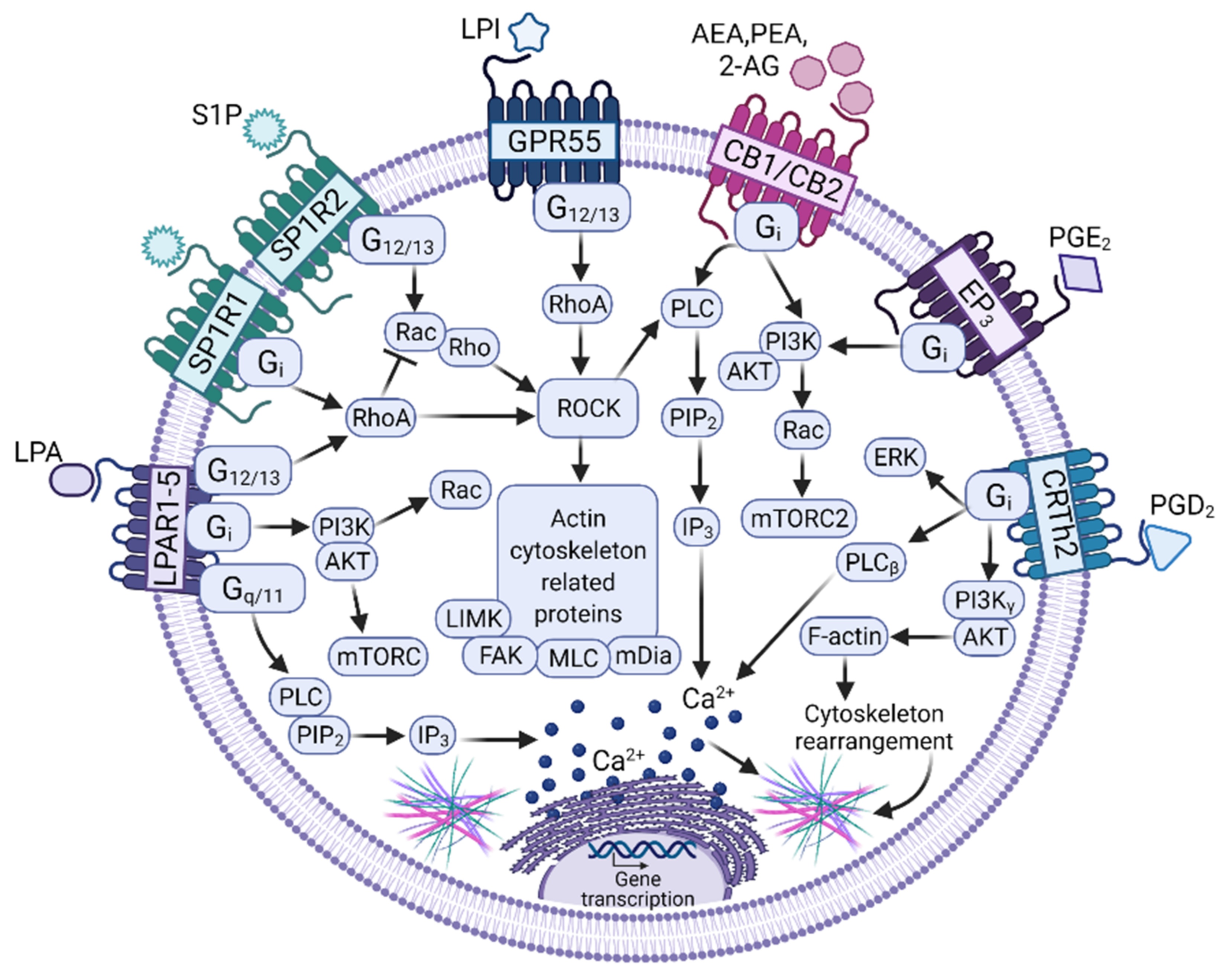
3.2. Bioactive Lipids as Key Molecules Involved in MC–Tumor Interactions
4. Influence of TME Conditions on MC Physiology: Can We Talk about Mast Cell Polarization?
4.1. Hypoxia and MCs: A Dangerous Connection in Cancer?
5. Tumor-Derived Molecules That Activate MCs
5.1. Tumor-Derived DAMPs That Activate Toll-like and RAGE Receptors in MCs
5.2. Tumor-Derived Mediators That Activate RTKs in MCs
5.3. Tumor-Derived TGF-β Activate MCs
5.4. Potential Role of Tumor-Derived Adenosine and Neuropeptides in MC Activation
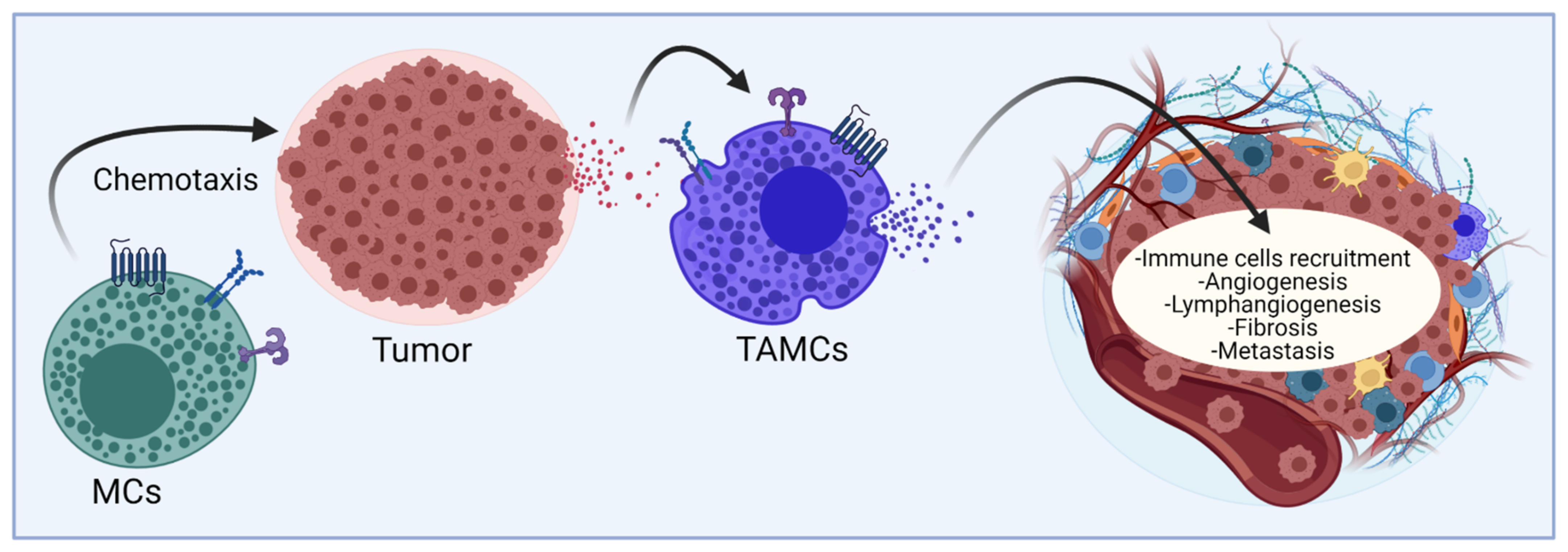
6. MC-Derived Mediators That Promote the Recruitment of Other Immune Cells to the TME
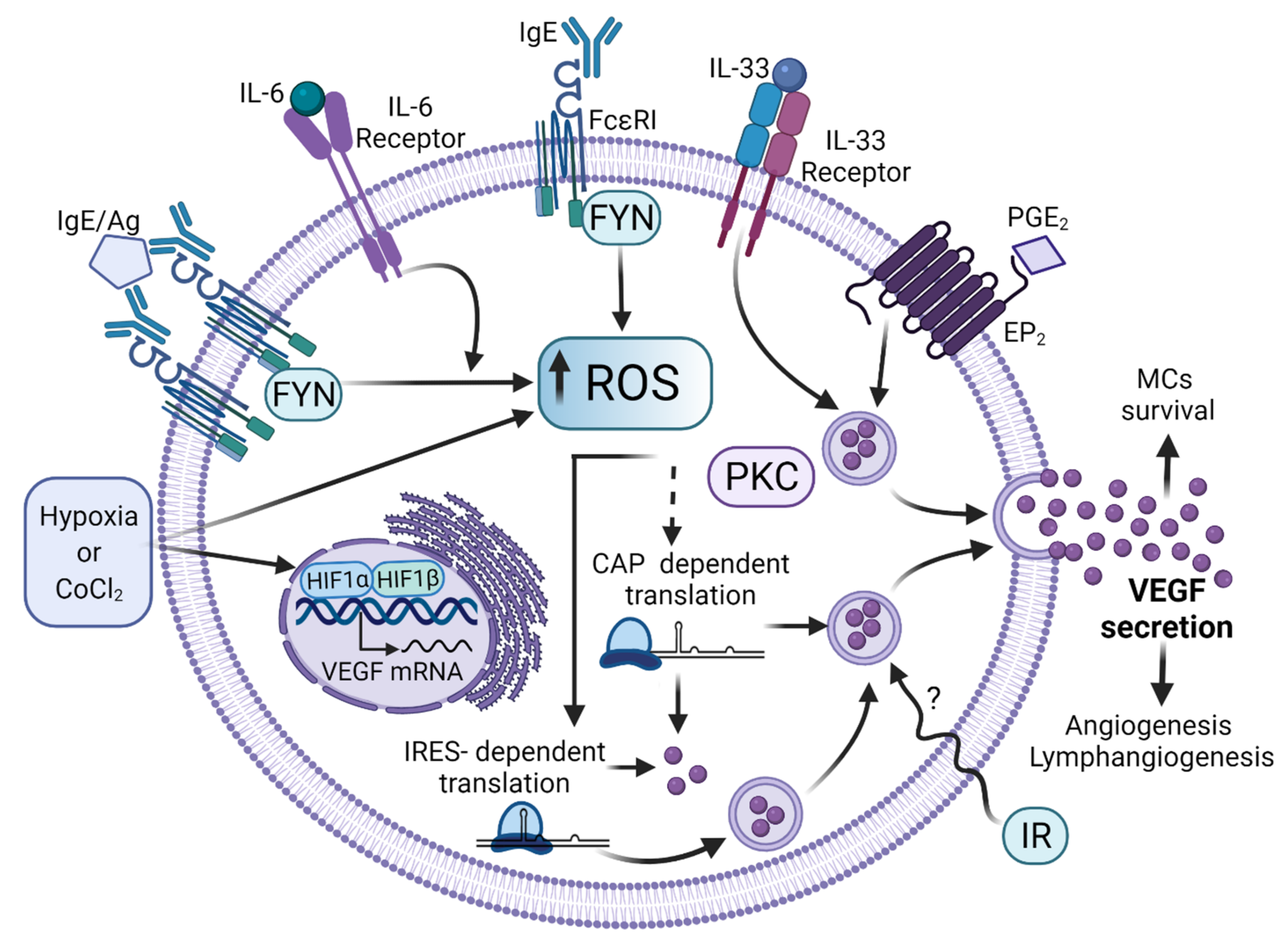
7. MC-Derived Mediators That Participate in Angiogenesis: The Case of the Production of VEGF
8. MC-Derived Mediators and Signaling Pathways That Regulate Lymphangiogenesis
9. MC-Derived Mediators That Contribute to the Fibrotic Process
10. MC-Derived Mediators as Promoters of Metastatic Processes
11. Targeting Tumor-Associated MCs to Design Novel Cancer Therapies
12. Tumor-Associated MCs: Soldiers on the Front Lines or Instigators of the Flames of Cancer?
Author Contributions
Funding
Conflicts of Interest
References
- Abbott, M.; Ustoyev, Y. Cancer and the Immune System: The History and Background of Immunotherapy. Semin. Oncol. Nurs. 2019, 35, 150923. [Google Scholar] [CrossRef] [PubMed]
- Candeias, S.M.; Gaipl, U.S. The Immune System in Cancer Prevention, Development and Therapy. Anticancer Agents Med. Chem. 2016, 16, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Coley, W.B. The Treatment of Inoperable Sarcoma by Bacterial Toxins (the Mixed Toxins of the Streptococcus erysipelas and the Bacillus prodigiosus). Proc. R. Soc. Med. 1910, 3, 1–48. Available online: http://www.ncbi.nlm.nih.gov/pubmed/19974799 (accessed on 2 October 2021). [CrossRef] [PubMed]
- Ehrlich, P. Über den jetzigen Stand der Chemotherapie. Ber. Dtsch. Chem. Ges. 1909, 42, 17–47. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Muenst, S.; Läubli, H.; Soysal, S.D.; Zippelius, A.; Tzankov, A.; Hoeller, S. The immune system and cancer evasion strategies: Therapeutic concepts. J. Intern. Med. 2016, 279, 541–562. [Google Scholar] [CrossRef]
- Espinosa-Riquer, Z.P.; Segura-Villalobos, D.; Ramírez-Moreno, I.G.; Pérez Rodríguez, M.J.; Lamas, M.; Gonzalez-Espinosa, C. Signal Transduction Pathways Activated by Innate Immunity in Mast Cells: Translating Sensing of Changes into Specific Responses. Cells 2020, 9, 2411. [Google Scholar] [CrossRef]
- Mukai, K.; Tsai, M.; Starkl, P.; Marichal, T.; Galli, S.J. IgE and mast cells in host defense against parasites and venoms. Semin. Immunopathol. 2016, 38, 581–603. [Google Scholar] [CrossRef]
- Matsuguchi, T. Mast cells as critical effectors of host immune defense against Gram-negative bacteria. Curr. Med. Chem. 2012, 19, 1432–1442. [Google Scholar] [CrossRef]
- Marshall, J.S.; Portales-Cervantes, L.; Leong, E. Mast Cell Responses to Viruses and Pathogen Products. Int. J. Mol. Sci. 2019, 20, 4241. [Google Scholar] [CrossRef]
- Varricchi, G.; Galdiero, M.R.; Loffredo, S.; Marone, G.; Iannone, R.; Marone, G.; Granata, F. Are mast cells MASTers in cancer? Front. Immunol. 2017, 8, 424. [Google Scholar] [CrossRef] [PubMed]
- Marichal, T.; Tsai, M.; Galli, S.J. Mast cells: Potential positive and negative roles in tumor biology. Cancer Immunol. Res. 2013, 1, 269–279. [Google Scholar] [CrossRef] [PubMed]
- Carlini, M.J.; Dalurzo, M.C.L.; Lastiri, J.M.; Smith, D.E.; Vasallo, B.C.; Puricelli, L.I.; Lauría de Cidre, L.S. Mast cell phenotypes and microvessels in non-small cell lung cancer and its prognostic significance. Hum. Pathol. 2010, 41, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Aoki, M.; Pawankar, R.; Niimi, Y.; Kawana, S. Mast cells in basal cell carcinoma express VEGF, IL-8 and RANTES. Int. Arch. Allergy Immunol. 2003, 130, 216–223. [Google Scholar] [CrossRef]
- Amini, R.-M.; Aaltonen, K.; Nevanlinna, H.; Carvalho, R.; Salonen, L.; Heikkilä, P.; Blomqvist, C. Mast cells and eosinophils in invasive breast carcinoma. BMC Cancer 2007, 7, 165. [Google Scholar] [CrossRef] [PubMed]
- Ranieri, G.; Ammendola, M.; Patruno, R.; Celano, G.; Zito, F.A.; Montemurro, S.; Rella, A.; Di Lecce, V.; Gadaleta, C.D.; Battista De Sarro, G.; et al. Tryptase-positive mast cells correlate with angiogenesis in early breast cancer patients. Int. J. Oncol. 2009, 35, 115–120. [Google Scholar] [CrossRef]
- Mangia, A.; Malfettone, A.; Rossi, R.; Paradiso, A.; Ranieri, G.; Simone, G.; Resta, L. Tissue remodelling in breast cancer: Human mast cell tryptase as an initiator of myofibroblast differentiation. Histopathology 2011, 58, 1096–1106. [Google Scholar] [CrossRef]
- Johnson, C.; Huynh, V.; Hargrove, L.; Kennedy, L.; Graf-Eaton, A.; Owens, J.; Trzeciakowski, J.P.; Hodges, K.; DeMorrow, S.; Han, Y.; et al. Inhibition of Mast Cell-Derived Histamine Decreases Human Cholangiocarcinoma Growth and Differentiation via c-Kit/Stem Cell Factor-Dependent Signaling. Am. J. Pathol. 2016, 186, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.-Y.; Fan, Y.; Luo, H.-S.; Shen, Z.-X.; Guo, Y.; Zhao, L.-J. Prognostic significance of cell infiltrations of immunosurveillance in colorectal cancer. World J. Gastroenterol. 2005, 11, 1210–1214. [Google Scholar] [CrossRef]
- Acikalin, M.F.; Oner, U.; Topçu, I.; Yaşar, B.; Kiper, H.; Colak, E. Tumour angiogenesis and mast cell density in the prognostic assessment of colorectal carcinomas. Dig. Liver Dis. 2005, 37, 162–169. [Google Scholar] [CrossRef]
- Malfettone, A.; Silvestris, N.; Saponaro, C.; Ranieri, G.; Russo, A.; Caruso, S.; Popescu, O.; Simone, G.; Paradiso, A.; Mangia, A. High density of tryptase-positive mast cells in human colorectal cancer: A poor prognostic factor related to protease-activated receptor 2 expression. J. Cell. Mol. Med. 2013, 17, 1025–1037. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, S.; Ichikawa, Y.; Nakagawa, K.; Kumamoto, T.; Mori, R.; Matsuyama, R.; Takeda, K.; Ota, M.; Tanaka, K.; Tamura, T.; et al. High infiltration of mast cells positive to tryptase predicts worse outcome following resection of colorectal liver metastases. BMC Cancer 2015, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guidolin, D.; Marinaccio, C.; Tortorella, C.; Annese, T.; Ruggieri, S.; Finato, N.; Crivellato, E.; Ribatti, D. Non-random spatial relationships between mast cells and microvessels in human endometrial carcinoma. Clin. Exp. Med. 2017, 17, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Ammendola, M.; Sacco, R.; Zuccalà, V.; Luposella, M.; Patruno, R.; Gadaleta, P.; Zizzo, N.; Gadaleta, C.D.; De Sarro, G.; Sammarco, G.; et al. Mast Cells Density Positive to Tryptase Correlate with Microvascular Density in both Primary Gastric Cancer Tissue and Loco-Regional Lymph Node Metastases from Patients That Have Undergone Radical Surgery. Int. J. Mol. Sci. 2016, 17, 1905. Available online: https://pubmed.ncbi.nlm.nih.gov/27854307/ (accessed on 1 October 2021). [CrossRef]
- Liu, X.; Jin, H.; Zhang, G.; Lin, X.; Chen, C.; Sun, J.; Zhang, Y.; Zhang, Q.; Yu, J. Intratumor IL-17-positive mast cells are the major source of the IL-17 that is predictive of survival in gastric cancer patients. PLoS ONE 2014, 9, e106834. [Google Scholar] [CrossRef]
- Lv, Y.; Zhao, Y.; Wang, X.; Chen, N.; Mao, F.; Teng, Y.; Wang, T.; Peng, L.; Zhang, J.; Cheng, P.; et al. Increased intratumoral mast cells foster immune suppression and gastric cancer progression through TNF-α-PD-L1 pathway. J. Immunother. Cancer 2019, 7, 54. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Finato, N.; Crivellato, E.; Marzullo, A.; Mangieri, D.; Nico, B.; Vacca, A.; Beltrami, C.A. Neovascularization and mast cells with tryptase activity increase simultaneously with pathologic progression in human endometrial cancer. Am. J. Obstet. Gynecol. 2005, 193, 1961–1965. [Google Scholar] [CrossRef]
- Jiang, L.; Hua, Y.; Shen, Q.; Ding, S.; Jiang, W.; Zhang, W.; Zhu, X. Role of mast cells in gynecological neoplasms. Front. Biosci. Landmark Ed. 2013, 18, 773–781. [Google Scholar] [CrossRef][Green Version]
- Ju, M.-J.; Qiu, S.-J.; Gao, Q.; Fan, J.; Cai, M.-Y.; Li, Y.-W.; Tang, Z.-Y. Combination of peritumoral mast cells and T-regulatory cells predicts prognosis of hepatocellular carcinoma. Cancer Sci. 2009, 100, 1267–1274. [Google Scholar] [CrossRef]
- Tu, J.-F.; Pan, H.-Y.; Ying, X.-H.; Lou, J.; Ji, J.-S.; Zou, H. Mast Cells Comprise the Major of Interleukin 17-Producing Cells and Predict a Poor Prognosis in Hepatocellular Carcinoma. Medicine 2016, 95, e3220. [Google Scholar] [CrossRef]
- Banat, G.-A.; Tretyn, A.; Pullamsetti, S.S.; Wilhelm, J.; Weigert, A.; Olesch, C.; Ebel, K.; Stiewe, T.; Grimminger, F.; Seeger, W.; et al. Immune and Inflammatory Cell Composition of Human Lung Cancer Stroma. PLoS ONE 2015, 10, e0139073. [Google Scholar] [CrossRef] [PubMed]
- Shikotra, A.; Ohri, C.M.; Green, R.H.; Waller, D.A.; Bradding, P. Mast cell phenotype, TNFα expression and degranulation status in non-small cell lung cancer. Sci. Rep. 2016, 6, 38352. [Google Scholar] [CrossRef] [PubMed]
- Öhrvik, H.; Grujic, M.; Waern, I.; Gustafson, A.-M.; Ernst, N.; Roers, A.; Hartmann, K.; Pejler, G. Mast cells promote melanoma colonization of lungs. Oncotarget 2016, 7, 68990–69001. [Google Scholar] [CrossRef] [PubMed]
- Siiskonen, H.; Poukka, M.; Bykachev, A.; Tyynelä-Korhonen, K.; Sironen, R.; Pasonen-Seppänen, S.; Harvima, I.T. Low numbers of tryptase+ and chymase+ mast cells associated with reduced survival and advanced tumor stage in melanoma. Melanoma Res. 2015, 25, 479–485. [Google Scholar] [CrossRef]
- Dantas, R.C.M.; de Souza, R.O.; Valverde, L.D.F.; Vidal, M.T.A.; Sales, C.B.S.; Sousa, L.P.; Dos Santos, J.N.; Ramos, E.A.G.; Gurgel Rocha, C.A. Evaluation of Mast Cell Density in the Tumor Microenvironment in Oral Epithelial Dysplasia and Oral Squamous Cell Carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2017, 25, e83–e88. [Google Scholar] [CrossRef]
- Ma, Y.; Hwang, R.F.; Logsdon, C.D.; Ullrich, S.E. Dynamic mast cell-stromal cell interactions promote growth of pancreatic cancer. Cancer Res. 2013, 73, 3927–3937. [Google Scholar] [CrossRef]
- Chang, D.Z.; Ma, Y.; Ji, B.; Wang, H.; Deng, D.; Liu, Y.; Abbruzzese, J.L.; Liu, Y.; Logsdon, C.D.; Hwu, P. Mast cells in tumor microenvironment promotes the in vivo growth of pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2011, 17, 7015–7023. [Google Scholar] [CrossRef]
- Pittoni, P.; Tripodo, C.; Piconese, S.; Mauri, G.; Parenza, M.; Rigoni, A.; Sangaletti, S.; Colombo, M.P. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011, 71, 5987–5997. [Google Scholar] [CrossRef]
- Johansson, A.; Rudolfsson, S.; Hammarsten, P.; Halin, S.; Pietras, K.; Jones, J.; Stattin, P.; Egevad, L.; Granfors, T.; Wikström, P.; et al. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am. J. Pathol. 2010, 177, 1031–1041. [Google Scholar] [CrossRef]
- Li, L.; Dang, Q.; Xie, H.; Yang, Z.; He, D.; Liang, L.; Song, W.; Yeh, S.; Chang, C. Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget 2015, 6, 14179–14190. [Google Scholar] [CrossRef]
- Pittoni, P.; Colombo, M.P. The dark side of mast cell-targeted therapy in prostate cancer. Cancer Res. 2012, 72, 831–835. [Google Scholar] [CrossRef]
- Hempel, H.A.; Cuka, N.S.; Kulac, I.; Barber, J.R.; Cornish, T.C.; Platz, E.A.; De Marzo, A.M.; Sfanos, K.S. Low Intratumoral Mast Cells Are Associated With a Higher Risk of Prostate Cancer Recurrence. Prostate 2017, 77, 412–424. [Google Scholar] [CrossRef]
- Fleischmann, A.; Schlomm, T.; Köllermann, J.; Sekulic, N.; Huland, H.; Mirlacher, M.; Sauter, G.; Simon, R.; Erbersdobler, A. Immunological microenvironment in prostate cancer: High mast cell densities are associated with favorable tumor characteristics and good prognosis. Prostate 2009, 69, 976–981. [Google Scholar] [CrossRef]
- Chen, Y.; Li, C.; Xie, H.; Fan, Y.; Yang, Z.; Ma, J.; He, D.; Li, L. Infiltrating mast cells promote renal cell carcinoma angiogenesis by modulating PI3K→AKT→GSK3β→AM signaling. Oncogene 2017, 36, 2879–2888. [Google Scholar] [CrossRef]
- Watanabe, S.; Miyata, Y.; Matsuo, T.; Mochizuki, Y.; Nishikido, M.; Hayashi, T.; Sakai, H. High density of tryptase-positive mast cells in patients with renal cell carcinoma on hemodialysis: Correlation with expression of stem cell factor and protease activated receptor-2. Hum. Pathol. 2012, 43, 888–897. [Google Scholar] [CrossRef]
- Cherdantseva, T.M.; Bobrov, I.P.; Avdalyan, A.M.; Klimachev, V.V.; Kazartsev, A.V.; Kryuchkova, N.G.; Klimachev, I.V.; Myadelets, M.N.; Lepilov, A.V.; Lushnikova, E.L.; et al. Mast Cells in Renal Cancer: Clinical Morphological Correlations and Prognosis. Bull. Exp. Biol. Med. 2017, 163, 801–804. [Google Scholar] [CrossRef]
- Fu, H.; Zhu, Y.; Wang, Y.; Liu, Z.; Zhang, J.; Wang, Z.; Xie, H.; Dai, B.; Xu, J.; Ye, D. Tumor Infiltrating Mast Cells (TIMs) Confers a Marked Survival Advantage in Nonmetastatic Clear-Cell Renal Cell Carcinoma. Ann. Surg. Oncol. 2017, 24, 1435–1442. [Google Scholar] [CrossRef]
- Melillo, R.M.; Guarino, V.; Avilla, E.; Galdiero, M.R.; Liotti, F.; Prevete, N.; Rossi, F.W.; Basolo, F.; Ugolini, C.; de Paulis, A.; et al. Mast cells have a protumorigenic role in human thyroid cancer. Oncogene 2010, 29, 6203–6215. [Google Scholar] [CrossRef] [PubMed]
- Grimbaldeston, M.A.; Chen, C.-C.; Piliponsky, A.M.; Tsai, M.; Tam, S.-Y.; Galli, S.J. Mast cell-deficient W-sash c-kit mutant Kit W-sh/W-sh mice as a model for investigating mast cell biology in vivo. Am. J. Pathol. 2005, 167, 835–848. [Google Scholar] [CrossRef]
- Zhou, J.S.; Xing, W.; Friend, D.S.; Austen, K.F.; Katz, H.R. Mast cell deficiency in Kit(W-sh) mice does not impair antibody-mediated arthritis. J. Exp. Med. 2007, 204, 2797–2802. [Google Scholar] [CrossRef]
- Jachetti, E.; D’Incà, F.; Danelli, L.; Magris, R.; Dal Secco, C.; Vit, F.; Cancila, V.; Tripodo, C.; Scapini, P.; Colombo, M.P.; et al. Frontline Science: Mast cells regulate neutrophil homeostasis by influencing macrophage clearance activity. J. Leukoc. Biol. 2019, 105, 633–644. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Gray, D.H.D.; Jones, T.; Hallgren, J.; Kuo, F.C.; Chaletzky, B.; Gurish, M.; Mathis, D.; Benoist, C.; Lee, D.M. Genetic inversion in mast cell-deficient (Wsh) mice interrupts corin and manifests as hematopoietic and cardiac aberrancy. Am. J. Pathol. 2008, 173, 1693–1701. [Google Scholar] [CrossRef] [PubMed]
- Oldford, S.A.; Haidl, I.D.; Howatt, M.A.; Leiva, C.A.; Johnston, B.; Marshall, J.S. A critical role for mast cells and mast cell-derived IL-6 in TLR2-mediated inhibition of tumor growth. J. Immunol. 2010, 185, 7067–7076. [Google Scholar] [CrossRef] [PubMed]
- Jiménez-Andrade, G.Y.; Ibarra-Sánchez, A.; González, D.; Lamas, M.; González-Espinosa, C. Immunoglobulin E induces VEGF production in mast cells and potentiates their pro-tumorigenic actions through a Fyn kinase-dependent mechanism. J. Hematol. Oncol. 2013, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, S.; Wölbing, F.; Kempf, W.E.; Skabytska, Y.; Köberle, M.; Volz, T.; Sinnberg, T.; Amaral, T.; Möckel, S.; Yazdi, A.; et al. Targeting tumor-resident mast cells for effective anti-melanoma immune responses. JCI Insight 2019, 4, 1–15. [Google Scholar] [CrossRef]
- Sinnamon, M.J.; Carter, K.J.; Sims, L.P.; Lafleur, B.; Fingleton, B.; Matrisian, L.M. A protective role of mast cells in intestinal tumorigenesis. Carcinogenesis 2008, 29, 880–886. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Wasiuk, A.; Dalton, D.K.; Schpero, W.L.; Stan, R.V.; Conejo-Garcia, J.R.; Noelle, R.J. Mast cells impair the development of protective anti-tumor immunity. Cancer Immunol. Immunother. 2012, 61, 2273–2282. [Google Scholar] [CrossRef]
- He, L.; Zhu, Z.; Chen, S.; Wang, Y.; Gu, H. Mammary tumor growth and metastasis are reduced in c-Kit mutant Sash mice. Cancer Med. 2016, 5, 1292–1297. [Google Scholar] [CrossRef]
- Majorini, M.T.; Cancila, V.; Rigoni, A.; Botti, L.; Dugo, M.; Triulzi, T.; De Cecco, L.; Fontanella, E.; Jachetti, E.; Tagliabue, E.; et al. Infiltrating Mast Cell-Mediated Stimulation of Estrogen Receptor Activity in Breast Cancer Cells Promotes the Luminal Phenotype. Cancer Res. 2020, 80, 2311–2324. Available online: https://pubmed.ncbi.nlm.nih.gov/32179512/ (accessed on 1 October 2021). [CrossRef]
- Eissmann, M.F.; Dijkstra, C.; Jarnicki, A.; Phesse, T.; Brunnberg, J.; Poh, A.R.; Etemadi, N.; Tsantikos, E.; Thiem, S.; Huntington, N.D.; et al. IL-33-mediated mast cell activation promotes gastric cancer through macrophage mobilization. Nat. Commun. 2019, 10, 2735. [Google Scholar] [CrossRef] [PubMed]
- Bergot, A.-S.; Ford, N.; Leggatt, G.R.; Wells, J.W.; Frazer, I.H.; Grimbaldeston, M.A. HPV16-E7 expression in squamous epithelium creates a local immune suppressive environment via CCL2- and CCL5- mediated recruitment of mast cells. PLoS Pathog. 2014, 10, e1004466. [Google Scholar] [CrossRef]
- Giannou, A.D.; Marazioti, A.; Spella, M.; Kanellakis, N.I.; Apostolopoulou, H.; Psallidas, I.; Prijovich, Z.M.; Vreka, M.; Zazara, D.E.; Lilis, I.; et al. Mast cells mediate malignant pleural effusion formation. J. Clin. Investig. 2015, 125, 2317–2334. [Google Scholar] [CrossRef]
- Lilis, I.; Ntaliarda, G.; Papaleonidopoulos, V.; Giotopoulou, G.A.; Oplopoiou, M.; Marazioti, A.; Spella, M.; Marwitz, S.; Goldmann, T.; Bravou, V.; et al. Interleukin-1β provided by KIT-competent mast cells is required for KRAS-mutant lung adenocarcinoma. Oncoimmunology 2019, 8, e1593802. [Google Scholar] [CrossRef]
- Mcculloch, E.A.; Siminovitch, L.; Till, J.E. Spleen-Colony Formation in Anemic Mice of Genotype WW. Science 1964, 144, 844–846. [Google Scholar] [CrossRef]
- Kitamura, Y.; Go, S.; Hatanaka, K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood 1978, 52, 447–452. [Google Scholar] [CrossRef]
- Nocka, K.; Tan, J.C.; Chiu, E.; Chu, T.Y.; Ray, P.; Traktman, P.; Besmer, P. Molecular bases of dominant negative and loss of function mutations at the murine c-kit/white spotting locus: W37, Wv, W41 and W. EMBO J. 1990, 9, 1805–1813. [Google Scholar] [CrossRef] [PubMed]
- Starkey, J.R.; Crowle, P.K.; Taubenberger, S. Mast-cell-deficient W/Wv mice exhibit a decreased rate of tumor angiogenesis. Int. J. Cancer 1988, 42, 48–52. [Google Scholar] [CrossRef]
- Siebenhaar, F.; Metz, M.; Maurer, M. Mast cells protect from skin tumor development and limit tumor growth during cutaneous de novo carcinogenesis in a Kit-dependent mouse model. Exp. Dermatol. 2014, 23, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Raymond, W.W.; Bergers, G.; Laig-Webster, M.; Behrendtsen, O.; Werb, Z.; Caughey, G.H.; Hanahan, D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999, 13, 1382–1397. [Google Scholar] [CrossRef]
- Wedemeyer, J.; Galli, S.J. Decreased susceptibility of mast cell-deficient Kit(W)/Kit(W-v) mice to the development of 1, 2-dimethylhydrazine-induced intestinal tumors. Lab. Investig. 2005, 85, 388–396. [Google Scholar] [CrossRef]
- Burtin, C.; Ponvert, C.; Fray, A.; Scheinmann, P.; Lespinats, G.; Loridon, B.; Canu, P.; Paupe, J. Inverse correlation between tumor incidence and tissue histamine levels in W/WV, WV/+, and +/+ mice. J. Natl. Cancer Inst. 1985, 74, 671–674. [Google Scholar]
- Feyerabend, T.B.; Weiser, A.; Tietz, A.; Stassen, M.; Harris, N.; Kopf, M.; Radermacher, P.; Möller, P.; Benoist, C.; Mathis, D.; et al. Cre-mediated cell ablation contests mast cell contribution in models of antibody- and T cell-mediated autoimmunity. Immunity 2011, 35, 832–844. [Google Scholar] [CrossRef] [PubMed]
- Stieglitz, D.; Lamm, S.; Braig, S.; Feuerer, L.; Kuphal, S.; Dietrich, P.; Arndt, S.; Echtenacher, B.; Hellerbrand, C.; Karrer, S.; et al. BMP6-induced modulation of the tumor micro-milieu. Oncogene 2019, 38, 609–621. [Google Scholar] [CrossRef]
- Antsiferova, M.; Martin, C.; Huber, M.; Feyerabend, T.B.; Förster, A.; Hartmann, K.; Rodewald, H.-R.; Hohl, D.; Werner, S. Mast cells are dispensable for normal and activin-promoted wound healing and skin carcinogenesis. J. Immunol. 2013, 191, 6147–6155. [Google Scholar] [CrossRef]
- Scholten, J.; Hartmann, K.; Gerbaulet, A.; Krieg, T.; Müller, W.; Testa, G.; Roers, A. Mast cell-specific Cre/loxP-mediated recombination in vivo. Transgenic Res. 2008, 17, 307–315. [Google Scholar] [CrossRef]
- Dudeck, A.; Dudeck, J.; Scholten, J.; Petzold, A.; Surianarayanan, S.; Köhler, A.; Peschke, K.; Vöhringer, D.; Waskow, C.; Krieg, T.; et al. Mast cells are key promoters of contact allergy that mediate the adjuvant effects of haptens. Immunity 2011, 34, 973–984. [Google Scholar] [CrossRef]
- Ghouse, S.M.; Polikarpova, A.; Muhandes, L.; Dudeck, J.; Tantcheva-Poór, I.; Hartmann, K.; Lesche, M.; Dahl, A.; Eming, S.; Müller, W.; et al. Although Abundant in Tumor Tissue, Mast Cells Have No Effect on Immunological Micro-milieu or Growth of HPV-Induced or Transplanted Tumors. Cell Rep. 2018, 22, 27–35. [Google Scholar] [CrossRef]
- Lilla, J.N.; Chen, C.-C.; Mukai, K.; BenBarak, M.J.; Franco, C.B.; Kalesnikoff, J.; Yu, M.; Tsai, M.; Piliponsky, A.M.; Galli, S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011, 118, 6930–6938. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Kitamura, Y. Genetically mast-cell-deficient W/Wv and Sl/Sld mice. Their value for the analysis of the roles of mast cells in biologic responses in vivo. Am. J. Pathol. 1987, 127, 191–198. [Google Scholar] [PubMed]
- Kitamura, Y.; Go, S. Decreased production of mast cells in S1/S1d anemic mice. Blood 1979, 53, 492–497. Available online: http://www.ncbi.nlm.nih.gov/pubmed/367470 (accessed on 4 January 2022). [CrossRef] [PubMed]
- Arguello, F.; Furlanetto, R.W.; Baggs, R.B.; Graves, B.T.; Harwell, S.E.; Cohen, H.J.; Frantz, C.N. Incidence and distribution of experimental metastases in mutant mice with defective organ microenvironments (genotypes Sl/Sld and W/Wv). Cancer Res. 1992, 52, 2304–2309. [Google Scholar] [PubMed]
- Niwa, Y.; Kasugai, T.; Ohno, K.; Morimoto, M.; Yamazaki, M.; Dohmae, K.; Nishimune, Y.; Kondo, K.; Kitamura, Y. Anemia and mast cell depletion in mutant rats that are homozygous at “white spotting (Ws)” locus. Blood 1991, 78, 1936–1941. [Google Scholar] [CrossRef]
- Tsujimura, T.; Hirota, S.; Nomura, S.; Niwa, Y.; Yamazaki, M.; Tono, T.; Morii, E.; Kim, H.M.; Kondo, K.; Nishimune, Y. Characterization of Ws mutant allele of rats: A 12-base deletion in tyrosine kinase domain of c-kit gene. Blood 1991, 78, 1942–1946. [Google Scholar] [CrossRef]
- Onoue, H.; Maeyama, K.; Nomura, S.; Kasugai, T.; Tei, H.; Kim, H.M.; Watanabe, T.; Kitamura, Y. Absence of immature mast cells in the skin of Ws/Ws rats with a small deletion at tyrosine kinase domain of the c-kit gene. Am. J. Pathol. 1993, 142, 1001–1007. [Google Scholar]
- Maffini, M.V.; Soto, A.M.; Sonnenschein, C.; Papadopoulos, N.; Theoharides, T.C. Lack of c-kit receptor promotes mammary tumors in N-nitrosomethylurea-treated Ws/Ws rats. Cancer Cell Int. 2008, 8, 5. [Google Scholar] [CrossRef]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Yu, Y.R.; Ho, P.C. Sculpting tumor microenvironment with immune system: From immunometabolism to immunoediting. Clin. Exp. Immunol. 2019, 197, 153–160. [Google Scholar] [CrossRef]
- Ávila-Rodríguez, D.; Segura-Villalobos, D.L.; Ibarra-Sánchez, A.; González-Espinosa, C.; Macías-Silva, M. TGF-β y células cebadas: Reguladores del desarrollo del tumor. TIP Rev. Espec. Cienc. Químico-Biológicas 2020, 23, 1–11. [Google Scholar] [CrossRef]
- Costa, N.L.; Oton-Leite, A.F.; Cheim-Júnior, A.P.; Alencar, R.D.C.G.; Bittar, G.O.J.; Silva, T.A.; Batista, A.C. Density and migration of mast cells in lip squamous cell carcinoma and actinic cheilitis. Histol. Histopathol. 2009, 24, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Coum, A.; Marinescu, V.D.; Põlajeva, J.; Smits, A.; Nelander, S.; Uhrbom, L.; Westermark, B.; Forsberg-Nilsson, K.; Pontén, F.; et al. Glioma-derived plasminogen activator inhibitor-1 (PAI-1) regulates the recruitment of LRP1 positive mast cells. Oncotarget 2015, 6, 23647–23661. [Google Scholar] [CrossRef]
- Zhong, B.; Li, Y.; Liu, X.; Wang, D. Association of mast cell infiltration with gastric cancer progression. Oncol. Lett. 2018, 15, 755–764. [Google Scholar] [CrossRef]
- Somasundaram, R.; Connelly, T.; Choi, R.; Choi, H.; Samarkina, A.; Li, L.; Gregorio, E.; Chen, Y.; Thakur, R.; Abdel-Mohsen, M.; et al. Tumor-infiltrating mast cells are associated with resistance to anti-PD-1 therapy. Nat. Commun. 2021, 12, 346. [Google Scholar] [CrossRef]
- Põlajeva, J.; Sjösten, A.M.; Lager, N.; Kastemar, M.; Waern, I.; Alafuzoff, I.; Smits, A.; Westermark, B.; Pejler, G.; Uhrbom, L.; et al. Mast cell accumulation in glioblastoma with a potential role for stem cell factor and chemokine CXCL12. PLoS ONE 2011, 6, e25222. [Google Scholar] [CrossRef]
- Halova, I.; Draberova, L.; Draber, P. Mast cell chemotaxis-chemoattractants and signaling pathways. Front. Immunol. 2012, 3, 119. [Google Scholar] [CrossRef]
- Kwok, E.; Everingham, S.; Zhang, S.; Greer, P.A.; Allingham, J.S.; Craig, A.W.B. FES kinase promotes mast cell recruitment to mammary tumors via the stem cell factor/KIT receptor signaling axis. Mol. Cancer Res. 2012, 10, 881–891. [Google Scholar] [CrossRef][Green Version]
- Huang, B.; Lei, Z.; Zhang, G.-M.; Li, D.; Song, C.; Li, B.; Liu, Y.; Yuan, Y.; Unkeless, J.; Xiong, H.; et al. SCF-mediated mast cell infiltration and activation exacerbate the inflammation and immunosuppression in tumor microenvironment. Blood 2008, 112, 1269–1279. [Google Scholar] [CrossRef]
- Yu, Y.; Blokhuis, B.; Derks, Y.; Kumari, S.; Garssen, J.; Redegeld, F. Human mast cells promote colon cancer growth via bidirectional crosstalk: Studies in 2D and 3D coculture models. Oncoimmunology 2018, 7, e1504729. [Google Scholar] [CrossRef]
- Draberova, L.; Draberova, H.; Potuckova, L.; Halova, I.; Bambouskova, M.; Mohandas, N.; Draber, P. Cytoskeletal Protein 4.1R Is a Positive Regulator of the FcεRI Signaling and Chemotaxis in Mast Cells. Front. Immunol. 2019, 10, 3068. [Google Scholar] [CrossRef]
- Baines, A.J.; Lu, H.C.; Bennett, P.M. The Protein 4.1 family: Hub proteins in animals for organizing membrane proteins. Biochim. Biophys. Acta-Biomembr. 2014, 1838, 605–619. [Google Scholar] [CrossRef]
- Fischer, M.; Juremalm, M.; Olsson, N.; Backlin, C.; Sundström, C.; Nilsson, K.; Enblad, G.; Nilsson, G. Expression of CCL5/RANTES by Hodgkin and Reed-Sternberg cells and its possible role in the recruitment of mast cells into lymphomatous tissue. Int. J. cancer 2003, 107, 197–201. [Google Scholar] [CrossRef]
- Zhu, X.-Q.; Lv, J.-Q.; Lin, Y.; Xiang, M.; Gao, B.-H.; Shi, Y.-F. Expression of chemokines CCL5 and CCL11 by smooth muscle tumor cells of the uterus and its possible role in the recruitment of mast cells. Gynecol. Oncol. 2007, 105, 650–656. [Google Scholar] [CrossRef]
- Gruber, B.L.; Marchese, M.J.; Kew, R. Angiogenic factors stimulate mast-cell migration. Blood 1995, 86, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- Ronca, R.; Tamma, R.; Coltrini, D.; Ruggieri, S.; Presta, M.; Ribatti, D. Fibroblast growth factor modulates mast cell recruitment in a murine model of prostate cancer. Oncotarget 2017, 8, 82583–82592. [Google Scholar] [CrossRef] [PubMed]
- Komi, D.E.A.; Redegeld, F.A. Role of Mast Cells in Shaping the Tumor Microenvironment. Clin. Rev. Allergy Immunol. 2020, 58, 313–325. [Google Scholar] [CrossRef]
- Oldford, S.A.; Marshall, J.S. Mast cells as targets for immunotherapy of solid tumors. Mol. Immunol. 2015, 63, 113–124. [Google Scholar] [CrossRef]
- Evans, J.F.; Hutchinson, J.H. Seeing the future of bioactive lipid drug targets. Nat. Chem. Biol. 2010, 6, 476–479. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Maccarrone, M. Bioactive lipids as modulators of immunity, inflammation and emotions. Curr. Opin. Pharmacol. 2016, 29, 54–62. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Leuti, A.; Maccarrone, M. Bioactive Lipids and Chronic Inflammation: Managing the Fire Within. Front. Immunol. 2018, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Taiyab, A.; Hussain, A.; Alajmi, M.F.; Islam, A.; Hassan, M.I. Targeting the Sphingosine Kinase/Sphingosine-1-Phosphate Signaling Axis in Drug Discovery for Cancer Therapy. Cancers 2021, 13, 1898. [Google Scholar] [CrossRef] [PubMed]
- Takabe, K.; Spiegel, S. Export of sphingosine-1-phosphate and cancer progression. J. Lipid Res. 2014, 55, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Rivera, J. An emerging role for the lipid mediator sphingosine-1-phosphate in mast cell effector function and allergic disease. Adv. Exp. Med. Biol. 2011, 716, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Jolly, P.S.; Bektas, M.; Olivera, A.; Gonzalez-Espinosa, C.; Proia, R.L.; Rivera, J.; Milstien, S.; Spiegel, S. Transactivation of Sphingosine-1-Phosphate Receptors by FcεRI Triggering Is Required for Normal Mast Cell Degranulation and Chemotaxis. J. Exp. Med. 2004, 199, 959–970. [Google Scholar] [CrossRef] [PubMed]
- Oskeritzian, C.A.; Price, M.M.; Hait, N.C.; Kapitonov, D.; Falanga, Y.T.; Morales, J.K.; Ryan, J.J.; Milstien, S.; Spiegel, S. Essential roles of sphingosine-1-phosphate receptor 2 in human mast cell activation, anaphylaxis, and pulmonary edema. J. Exp. Med. 2010, 207, 465–474. [Google Scholar] [CrossRef]
- Kihara, Y.; Maceyka, M.; Spiegel, S.; Chun, J. Lysophospholipid receptor nomenclature review: IUPHAR Review 8. Br. J. Pharmacol. 2014, 171, 3575–3594. [Google Scholar] [CrossRef]
- Olivera, A.; Rivera, J. Sphingolipids and the Balancing of Immune Cell Function: Lessons from the Mast Cell. J. Immunol. 2005, 174, 1153–1158. [Google Scholar] [CrossRef]
- Piñeiro, R.; Maffucci, T.; Falasca, M. The putative cannabinoid receptor GPR55 defines a novel autocrine loop in cancer cell proliferation. Oncogene 2011, 30, 142–152. [Google Scholar] [CrossRef]
- Zhou, X.; Guo, X.; Song, Y.; Zhu, C.; Zou, W. The LPI/GPR55 axis enhances human breast cancer cell migration via HBXIP and p-MLC signaling. Acta Pharmacol. Sin. 2018, 39, 459–471. [Google Scholar] [CrossRef]
- Kargl, J.; Andersen, L.; Hasenöhrl, C.; Feuersinger, D.; Stančić, A.; Fauland, A.; Magnes, C.; El-Heliebi, A.; Lax, S.; Uranitsch, S.; et al. GPR55 promotes migration and adhesion of colon cancer cells indicating a role in metastasis. Br. J. Pharmacol. 2016, 173, 142–154. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, N.A.; Yang, J.; Trauger, S.A.; Nakayama, H.; Huang, L.; Strunk, D.; Moses, M.A.; Klagsbrun, M.; Bischoff, J.; Graier, W.F. The GPR 55 agonist, L-α-lysophosphatidylinositol, mediates ovarian carcinoma cell-induced angiogenesis. Br. J. Pharmacol. 2015, 172, 4107–4118. [Google Scholar] [CrossRef] [PubMed]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef]
- Cantarella, G.; Scollo, M.; Lempereur, L.; Saccani-Jotti, G.; Basile, F.; Bernardini, R. Endocannabinoids inhibit release of nerve growth factor by inflammation-activated mast cells. Biochem. Pharmacol. 2011, 82, 380–388. [Google Scholar] [CrossRef]
- Cruz, S.L.; Sánchez-Miranda, E.; Castillo-Arellano, J.I.; Cervantes-Villagrana, R.D.; Ibarra-Sánchez, A.; González-Espinosa, C. Anandamide inhibits FcεRI-dependent degranulation and cytokine synthesis in mast cells through CB2 and GPR55 receptor activation. Possible involvement of CB2-GPR55 heteromers. Int. Immunopharmacol. 2018, 64, 298–307. [Google Scholar] [CrossRef]
- Aoki, J.; Inoue, A.; Okudaira, S. Two pathways for lysophosphatidic acid production. Biochim. Biophys. Acta 2008, 1781, 513–518. [Google Scholar] [CrossRef]
- Stracke, M.L.; Krutzsch, H.C.; Unsworth, E.J.; Arestad, A.; Cioce, V.; Schiffmann, E.; Liotta, L.A. Identification, purification, and partial sequence analysis of autotaxin, a novel motility-stimulating protein. J. Biol. Chem. 1992, 267, 2524–2529. Available online: http://www.ncbi.nlm.nih.gov/pubmed/1733949 (accessed on 2 September 2021). [CrossRef]
- Liu, S.; Murph, M.; Panupinthu, N.; Mills, G.B. ATX-LPA receptor axis in inflammation and cancer. Cell Cycle 2009, 8, 3695–3701. [Google Scholar] [CrossRef]
- Stassar, M.J.; Devitt, G.; Brosius, M.; Rinnab, L.; Prang, J.; Schradin, T.; Simon, J.; Petersen, S.; Kopp-Schneider, A.; Zöller, M. Identification of human renal cell carcinoma associated genes by suppression subtractive hybridization. Br. J. Cancer 2001, 85, 1372–1382. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kawagoe, H.; Stracke, M.L.; Nakamura, H.; Sano, K. Expression and transcriptional regulation of the PD-Ialpha/autotaxin gene in neuroblastoma. Cancer Res. 1997, 57, 2516–2521. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9192834 (accessed on 2 September 2021).
- Lee, S.C.; Dacheux, M.A.; Norman, D.D.; Balázs, L.; Torres, R.M.; Augelli-Szafran, C.E.; Tigyi, G.J. Regulation of Tumor Immunity by Lysophosphatidic Acid. Cancers 2020, 12, 1202. [Google Scholar] [CrossRef] [PubMed]
- Balijepalli, P.; Sitton, C.C.; Meier, K.E. Lysophosphatidic Acid Signaling in Cancer Cells: What Makes LPA So Special? Cells 2021, 10, 2059. [Google Scholar] [CrossRef]
- Lundequist, A.; Boyce, J.A. LPA5 is abundantly expressed by human mast cells and important for lysophosphatidic acid induced MIP-1β release. PLoS ONE 2011, 6, e18192. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.A.; Boyce, J.A. IL-4 regulates MEK expression required for lysophosphatidic acid-mediated chemokine generation by human mast cells. J. Immunol. 2005, 175, 5430–5438. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, T.; Ohata, H.; Momose, K.; Honda, K. Lysophosphatidic acid induces histamine release from mast cells and skin fragments. Pharmacology 2005, 75, 13–20. [Google Scholar] [CrossRef]
- Shlyonsky, V.; Naeije, R.; Mies, F. Possible role of lysophosphatidic acid in rat model of hypoxic pulmonary vascular remodeling. Pulm. Circ. 2014, 4, 471–481. Available online: http://www.ncbi.nlm.nih.gov/pubmed/25621161 (accessed on 2 September 2021). [CrossRef]
- Rask, K.; Zhu, Y.; Wang, W.; Hedin, L.; Sundfeldt, K. Ovarian epithelial cancer: A role for PGE2-synthesis and signalling in malignant transformation and progression. Mol. Cancer 2006, 5, 62. [Google Scholar] [CrossRef]
- Sales, K.J.; Katz, A.A.; Davis, M.; Hinz, S.; Soeters, R.P.; Hofmeyr, M.D.; Millar, R.P.; Jabbour, H.N. Cyclooxygenase-2 expression and prostaglandin E(2) synthesis are up-regulated in carcinomas of the cervix: A possible autocrine/paracrine regulation of neoplastic cell function via EP2/EP4 receptors. J. Clin. Endocrinol. Metab. 2001, 86, 2243–2249. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Lee, Y.; Kim, M.S.; Kang, Y.J.; Park, Y.-J.; Jung, H.; Kim, T.-D.; Lee, H.G.; Choi, I.; Yoon, S.R. Prostaglandin E2 Secreted by Thyroid Cancer Cells Contributes to Immune Escape Through the Suppression of Natural Killer (NK) Cell Cytotoxicity and NK Cell Differentiation. Front. Immunol. 2018, 9, 1859. [Google Scholar] [CrossRef]
- Zang, S.; Ni, M.; Lian, Y.; Zhang, Y.; Liu, J.; Huang, A. Expression of microsomal prostaglandin E2 synthase-1 and its role in human hepatocellular carcinoma. Hum. Pathol. 2013, 44, 1681–1687. [Google Scholar] [CrossRef]
- Finetti, F.; Travelli, C.; Ercoli, J.; Colombo, G.; Buoso, E.; Trabalzini, L. Prostaglandin E2 and Cancer: Insight into Tumor Progression and Immunity. Biology 2020, 9, 434. [Google Scholar] [CrossRef]
- Gomi, K.; Zhu, F.G.; Marshall, J.S. Prostaglandin E2 selectively enhances the IgE-mediated production of IL-6 and granulocyte-macrophage colony-stimulating factor by mast cells through an EP1/EP3-dependent mechanism. J. Immunol. 2000, 165, 6545–6552. [Google Scholar] [CrossRef]
- Kunikata, T.; Yamane, H.; Segi, E.; Matsuoka, T.; Sugimoto, Y.; Tanaka, S.; Tanaka, H.; Nagai, H.; Ichikawa, A.; Narumiya, S. Suppression of allergic inflammation by the prostaglandin E receptor subtype EP3. Nat. Immunol. 2005, 6, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Shirata, N.; Taketomi, Y.; Tsuchiya, S.; Segi-Nishida, E.; Inazumi, T.; Kabashima, K.; Tanaka, S.; Murakami, M.; Narumiya, S.; et al. Prostaglandin E2-EP3 signaling induces inflammatory swelling by mast cell activation. J. Immunol. 2014, 192, 1130–1137. [Google Scholar] [CrossRef] [PubMed]
- Kuehn, H.S.; Jung, M.-Y.; Beaven, M.A.; Metcalfe, D.D.; Gilfillan, A.M. Prostaglandin E2 activates and utilizes mTORC2 as a central signaling locus for the regulation of mast cell chemotaxis and mediator release. J. Biol. Chem. 2011, 286, 391–402. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.R.; Do Amaral, N.S.; Marchi, F.A.; Silva, F.I.D.B.; Da Costa, A.A.B.A.; Carvalho, K.C.; Baiocchi, G.; Soares, F.A.; De Brot, L.; Rocha, R.M. Prostaglandin D2 expression is prognostic in high-grade serous ovarian cancer. Oncol. Rep. 2019, 41, 2254–2264. [Google Scholar] [CrossRef]
- Zhang, B.; Bie, Q.; Wu, P.; Zhang, J.; You, B.; Shi, H.; Qian, H.; Xu, W. PGD2/PTGDR2 Signaling Restricts the Self-Renewal and Tumorigenesis of Gastric Cancer. Stem Cells 2018, 36, 990–1003. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Aritake, K.; Matsumoto, S.; Kamauchi, S.; Nakagawa, T.; Hori, M.; Momotani, E.; Urade, Y.; Ozaki, H. Prostagladin D2 is a mast cell-derived antiangiogenic factor in lung carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 19802–19807. [Google Scholar] [CrossRef]
- Boehme, S.A.; Franz-Bacon, K.; Chen, E.P.; Ly, T.W.; Kawakami, Y.; Bacon, K.B. Murine bone marrow-derived mast cells express chemoattractant receptor-homologous molecule expressed on T-helper class 2 cells (CRTh2). Int. Immunol. 2009, 21, 621–632. [Google Scholar] [CrossRef][Green Version]
- Nakamura, T.; Fujiwara, Y.; Yamada, R.; Fujii, W.; Hamabata, T.; Lee, M.Y.; Maeda, S.; Aritake, K.; Roers, A.; Sessa, W.C.; et al. Mast cell-derived prostaglandin D2 attenuates anaphylactic reactions in mice. J. Allergy Clin. Immunol. 2017, 140, 630–632.e9. [Google Scholar] [CrossRef]
- Samson, M.-T.; Small-Howard, A.; Shimoda, L.M.N.; Koblan-Huberson, M.; Stokes, A.J.; Turner, H. Differential roles of CB1 and CB2 cannabinoid receptors in mast cells. J. Immunol. 2003, 170, 4953–4962. [Google Scholar] [CrossRef]
- Sugawara, K.; Bíró, T.; Tsuruta, D.; Tóth, B.I.; Kromminga, A.; Zákány, N.; Zimmer, A.; Funk, W.; Gibbs, B.F.; Zimmer, A.; et al. Endocannabinoids limit excessive mast cell maturation and activation in human skin. J. Allergy Clin. Immunol. 2012, 129, 726–738.e8. [Google Scholar] [CrossRef]
- Vannacci, A.; Giannini, L.; Passani, M.B.; Di Felice, A.; Pierpaoli, S.; Zagli, G.; Fantappiè, O.; Mazzanti, R.; Masini, E.; Mannaioni, P.F. The endocannabinoid 2-arachidonylglycerol decreases the immunological activation of Guinea pig mast cells: Involvement of nitric oxide and eicosanoids. J. Pharmacol. Exp. Ther. 2004, 311, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Espinosa-Riquer, Z.P.; Ibarra-Sánchez, A.; Vibhushan, S.; Bratti, M.; Charles, N.; Blank, U.; Rodríguez-Manzo, G.; González-Espinosa, C. TLR4 Receptor Induces 2-AG-Dependent Tolerance to Lipopolysaccharide and Trafficking of CB2 Receptor in Mast Cells. J. Immunol. 2019, 202, 2360–2371. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Crupi, R.; Impellizzeri, D.; Campolo, M.; Marino, A.; Esposito, E.; Cuzzocrea, S. Administration of palmitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain. Behav. Immun. 2012, 26, 1310–1321. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F. Immunological Genome Project Consortium Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Plum, T.; Wang, X.; Rettel, M.; Krijgsveld, J.; Feyerabend, T.B.; Rodewald, H.R. Human Mast Cell Proteome Reveals Unique Lineage, Putative Functions, and Structural Basis for Cell Ablation. Immunity 2020, 52, 404–416.e5. [Google Scholar] [CrossRef]
- Frossi, B.; Mion, F.; Sibilano, R.; Danelli, L.; Pucillo, C.E.M. Is it time for a new classification of mast cells? What do we know about mast cell heterogeneity? Immunol. Rev. 2018, 282, 35–46. [Google Scholar] [CrossRef]
- Presta, I.; Donato, A.; Zaffino, P.; Spadea, M.F.; Mancuso, T.; Malara, N.; Chiefari, E.; Donato, G. Does a polarization state exist for mast cells in cancer? Med. Hypotheses 2019, 131, 109281. [Google Scholar] [CrossRef]
- Zhao, Y.-B.; Yang, S.-H.; Shen, J.; Deng, K.; Li, Q.; Wang, Y.; Cui, W.; Ye, H. Interaction between regulatory T cells and mast cells via IL-9 and TGF-β production. Oncol. Lett. 2020, 20, 360. [Google Scholar] [CrossRef]
- Varricchi, G.; de Paulis, A.; Marone, G.; Galli, S.J. Future Needs in Mast Cell Biology. Int. J. Mol. Sci. 2019, 20, 4397. [Google Scholar] [CrossRef]
- Colgan, S.P.; Furuta, G.T.; Taylor, C.T. Hypoxia and Innate Immunity: Keeping Up with the HIFsters. Annu. Rev. Immunol. 2020, 38, 341–363. [Google Scholar] [CrossRef] [PubMed]
- Palazon, A.; Goldrath, A.W.; Nizet, V.; Johnson, R.S. HIF transcription factors, inflammation, and immunity. Immunity 2014, 41, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Saxena, K.; Jolly, M.K. Acute vs. Chronic vs. cyclic hypoxia: Their differential dynamics, molecular mechanisms, and effects on tumor progression. Biomolecules 2019, 9, 339. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Zhao, Q.; Zhang, Y.; Liu, Z.; Zheng, Z.; Liu, S.; Meng, L.; Xin, Y.; Jiang, X. Targeting hypoxia in the tumor microenvironment: A potential strategy to improve cancer immunotherapy. J. Exp. Clin. Cancer Res. 2021, 40, 24. [Google Scholar] [CrossRef] [PubMed]
- Gulliksson, M.; Carvalho, R.F.S.; Ullerås, E.; Nilsson, G. Mast cell survival and mediator secretion in response to hypoxia. PLoS ONE 2010, 5, e12360. [Google Scholar] [CrossRef]
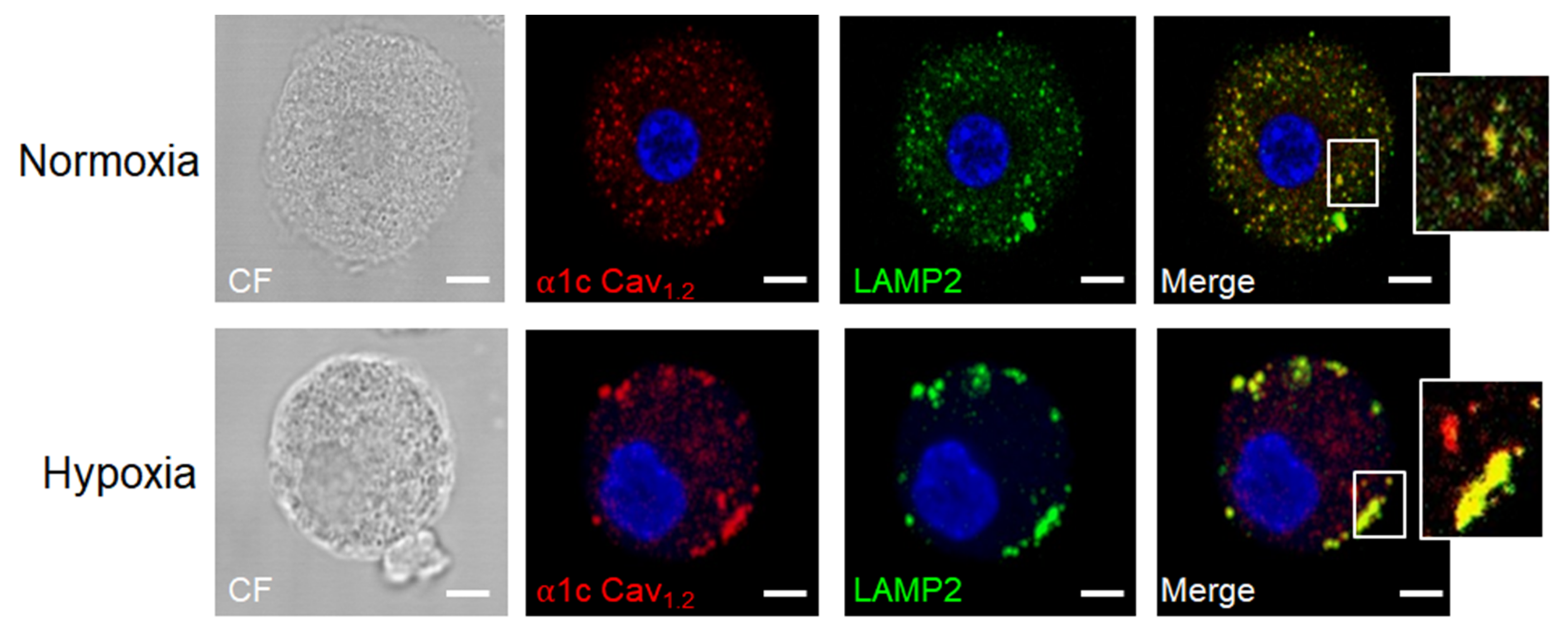
- Ramírez-Moreno, I.G.; Ibarra-Sánchez, A.; Castillo-Arellano, J.I.; Blank, U.; González-Espinosa, C. Mast Cells Localize in Hypoxic Zones of Tumors and Secrete CCL-2 under Hypoxia through Activation of L-Type Calcium Channels. J. Immunol. 2020, 204, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Grützkau, A.; Smorodchenko, A.; Lippert, U.; Kirchhof, L.; Artuc, M.; Henz, B.M. LAMP-1 and LAMP-2, but not LAMP-3, are reliable markers for activation-induced secretion of human mast cells. Cytometry A 2004, 61, 62–68. [Google Scholar] [CrossRef]
- Knol, E.F.; Mul, F.P.J.; Jansen, H.; Calafat, J.; Roos, D. Monitoring human basophil activation via CD63 monoclonal antibody 435. J. Allergy Clin. Immunol. 1991, 88, 328–338. [Google Scholar] [CrossRef]
- Matsumoto, S.; Yasui, H.; Mitchell, J.B.; Krishna, M.C. Imaging cycling tumor hypoxia. Cancer Res. 2010, 70, 10019–10023. [Google Scholar] [CrossRef]
- Larsen, K.M.; Minaya, M.K.; Vaish, V.; Peña, M.M.O. The Role of IL-33/ST2 Pathway in Tumorigenesis. Int. J. Mol. Sci. 2018, 19, 2676. [Google Scholar] [CrossRef]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Haskó, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef]
- Luo, Y.; Chihara, Y.; Fujimoto, K.; Sasahira, T.; Kuwada, M.; Fujiwara, R.; Fujii, K.; Ohmori, H.; Kuniyasu, H. High mobility group box 1 released from necrotic cells enhances regrowth and metastasis of cancer cells that have survived chemotherapy. Eur. J. Cancer 2013, 49, 741–751. [Google Scholar] [CrossRef] [PubMed]
- Ghavami, S.; Rashedi, I.; Dattilo, B.M.; Eshraghi, M.; Chazin, W.J.; Hashemi, M.; Wesselborg, S.; Kerkhoff, C.; Los, M. S100A8/A9 at low concentration promotes tumor cell growth via RAGE ligation and MAP kinase-dependent pathway. J. Leukoc. Biol. 2008, 83, 1484–1492. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef] [PubMed]
- Madera-Salcedo, I.K.; Cruz, S.L.; Gonzalez-Espinosa, C. Morphine prevents lipopolysaccharide-induced TNF secretion in mast cells blocking IκB kinase activation and SNAP-23 phosphorylation: Correlation with the formation of a β-arrestin/TRAF6 complex. J. Immunol. 2013, 191, 3400–3409. [Google Scholar] [CrossRef]
- Martín-Ávila, A.; Medina-Tamayo, J.; Ibarra-Sánchez, A.; Vázquez-Victorio, G.; Castillo-Arellano, J.I.; Hernández-Mondragón, A.C.; Rivera, J.; Madera-Salcedo, I.K.; Blank, U.; Macías-Silva, M.; et al. Protein Tyrosine Kinase Fyn Regulates TLR4-Elicited Responses on Mast Cells Controlling the Function of a PP2A-PKCα/β Signaling Node Leading to TNF Secretion. J. Immunol. 2016, 196, 5075–5088. [Google Scholar] [CrossRef]
- Guzmán-Mejía, F.; López-Rubalcava, C.; González-Espinosa, C. Stimulation of nAchRα7 Receptor Inhibits TNF Synthesis and Secretion in Response to LPS Treatment of Mast Cells by Targeting ERK1/2 and TACE Activation. J. Neuroimmune Pharmacol. 2018, 13, 39–52. [Google Scholar] [CrossRef]
- Supajatura, V.; Ushio, H.; Nakao, A.; Akira, S.; Okumura, K.; Ra, C.; Ogawa, H. Differential responses of mast cell Toll-like receptors 2 and 4 in allergy and innate immunity. J. Clin. Investig. 2002, 109, 1351–1359. [Google Scholar] [CrossRef]
- Rojas, A.; Delgado-López, F.; Perez-Castro, R.; Gonzalez, I.; Romero, J.; Rojas, I.; Araya, P.; Añazco, C.; Morales, E.; Llanos, J. HMGB1 enhances the protumoral activities of M2 macrophages by a RAGE-dependent mechanism. Tumour Biol. 2016, 37, 3321–3329. [Google Scholar] [CrossRef]
- Witsch, E.; Sela, M.; Yarden, Y. Roles for growth factors in cancer progression. Physiology 2010, 25, 85–101. [Google Scholar] [CrossRef]
- Hundley, T.R.; Gilfillan, A.M.; Tkaczyk, C.; Andrade, M.V.; Metcalfe, D.D.; Beaven, M.A. Kit and FcepsilonRI mediate unique and convergent signals for release of inflammatory mediators from human mast cells. Blood 2004, 104, 2410–2417. [Google Scholar] [CrossRef]
- Kim, M.-S.; Kuehn, H.S.; Metcalfe, D.D.; Gilfillan, A.M. Activation and function of the mTORC1 pathway in mast cells. J. Immunol. 2008, 180, 4586–4595. [Google Scholar] [CrossRef]
- Oliveira, S.H.P.; Lukacs, N.W. Stem cell factor: A hemopoietic cytokine with important targets in asthma. Curr. Drug Targets. Inflamm. Allergy 2003, 2, 313–318. [Google Scholar] [CrossRef]
- Drube, S.; Heink, S.; Walter, S.; Löhn, T.; Grusser, M.; Gerbaulet, A.; Berod, L.; Schons, J.; Dudeck, A.; Freitag, J.; et al. The receptor tyrosine kinase c-Kit controls IL-33 receptor signaling in mast cells. Blood 2010, 115, 3899–3906. [Google Scholar] [CrossRef]
- Batlle, E.; Massagué, J. Transforming Growth Factor-β Signaling in Immunity and Cancer. Immunity 2019, 50, 924–940. [Google Scholar] [CrossRef]
- Olsson, N.; Piek, E.; Sundström, M.; ten Dijke, P.; Nilsson, G. Transforming growth factor-beta-mediated mast cell migration depends on mitogen-activated protein kinase activity. Cell. Signal. 2001, 13, 483–490. [Google Scholar] [CrossRef]
- Gruber, B.L.; Marchese, M.J.; Kew, R.R. Transforming growth factor-beta 1 mediates mast cell chemotaxis. J. Immunol. 1994, 152, 5860–5867. Available online: http://www.ncbi.nlm.nih.gov/pubmed/7515916 (accessed on 3 January 2022). [PubMed]
- Edlund, S.; Landström, M.; Heldin, C.-H.; Aspenström, P. Transforming growth factor-beta-induced mobilization of actin cytoskeleton requires signaling by small GTPases Cdc42 and RhoA. Mol. Biol. Cell 2002, 13, 902–914. [Google Scholar] [CrossRef]
- Ramírez-Valadez, K.A.; Vázquez-Victorio, G.; Macías-Silva, M.; González-Espinosa, C. Fyn kinase mediates cortical actin ring depolymerization required for mast cell migration in response to TGF-β in mice. Eur. J. Immunol. 2017, 47, 1305–1316. [Google Scholar] [CrossRef]
- Norrby, K. Mast cells and angiogenesis. Apmis 2002, 110, 355–371. [Google Scholar] [CrossRef]
- Zhao, W.; Gomez, G.; Yu, S.-H.; Ryan, J.J.; Schwartz, L.B. TGF-beta1 attenuates mediator release and de novo Kit expression by human skin mast cells through a Smad-dependent pathway. J. Immunol. 2008, 181, 7263–7272. [Google Scholar] [CrossRef] [PubMed]
- Ndaw, V.S.; Abebayehu, D.; Spence, A.J.; Paez, P.A.; Kolawole, E.M.; Taruselli, M.T.; Caslin, H.L.; Chumanevich, A.P.; Paranjape, A.; Baker, B.; et al. TGF-β1 Suppresses IL-33-Induced Mast Cell Function. J. Immunol. 2017, 199, 866–873. [Google Scholar] [CrossRef]
- Miller, H.R.; Wright, S.H.; Knight, P.A.; Thornton, E.M. A novel function for transforming growth factor-beta1: Upregulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific beta-chymase, mouse mast cell protease-1. Blood 1999, 93, 3473–3486. [Google Scholar] [CrossRef]
- Funaba, M.; Ikeda, T.; Murakami, M.; Ogawa, K.; Nishino, Y.; Tsuchida, K.; Sugino, H.; Abe, M. Involvement of p38 MAP kinase and Smad3 in TGF-beta-mediated mast cell functions. Cell. Signal. 2006, 18, 2154–2161. [Google Scholar] [CrossRef] [PubMed]
- Funaba, M.; Ikeda, T.; Murakami, M.; Ogawa, K.; Nishino, Y.; Tsuchida, K.; Sugino, H.; Abe, M. Transcriptional regulation of mouse mast cell protease-7 by TGF-beta. Biochim. Biophys. Acta 2006, 1759, 166–170. [Google Scholar] [CrossRef]
- Funaba, M.; Ikeda, T.; Murakami, M.; Ogawa, K.; Abe, M. Up-regulation of mouse mast cell protease-6 gene by transforming growth factor-beta and activin in mast cell progenitors. Cell. Signal. 2005, 17, 121–128. [Google Scholar] [CrossRef]
- Krstic, J.; Santibanez, J.F. Transforming growth factor-beta and matrix metalloproteinases: Functional interactions in tumor stroma-infiltrating myeloid cells. Sci. World J. 2014, 2014, 521754. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Nucleotide- and nucleoside-converting ectoenzymes: Important modulators of purinergic signalling cascade. Biochim. Biophys. Acta 2008, 1783, 673–694. [Google Scholar] [CrossRef]
- Blay, J.; White, T.D.; Hoskin, D.W. The extracellular fluid of solid carcinomas contains immunosuppressive concentrations of adenosine. Cancer Res. 1997, 57, 2602–2605. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9205063 (accessed on 5 January 2022).
- De Andrade Mello, P.; Coutinho-Silva, R.; Savio, L.E.B. Multifaceted Effects of Extracellular Adenosine Triphosphate and Adenosine in the Tumor-Host Interaction and Therapeutic Perspectives. Front. Immunol. 2017, 8, 1526. [Google Scholar] [CrossRef]
- Gorzalczany, Y.; Sagi-Eisenberg, R. Role of mast cell-derived adenosine in cancer. Int. J. Mol. Sci. 2019, 20, 2603. [Google Scholar] [CrossRef] [PubMed]
- Rudich, N.; Ravid, K.; Sagi-Eisenberg, R. Mast cell adenosine receptors function: A focus on the a3 adenosine receptor and inflammation. Front. Immunol. 2012, 3, 134. [Google Scholar] [CrossRef]
- Gorzalczany, Y.; Akiva, E.; Klein, O.; Merimsky, O.; Sagi-Eisenberg, R. Mast cells are directly activated by contact with cancer cells by a mechanism involving autocrine formation of adenosine and autocrine/paracrine signaling of the adenosine A3 receptor. Cancer Lett. 2017, 397, 23–32. [Google Scholar] [CrossRef]
- Gorzalczany, Y.; Merimsky, O.; Sagi-Eisenberg, R. Mast Cells Are Directly Activated by Cancer Cell-Derived Extracellular Vesicles by a CD73- and Adenosine-Dependent Mechanism. Transl. Oncol. 2019, 12, 1549–1556. [Google Scholar] [CrossRef]
- Mancino, M.; Ametller, E.; Gascón, P.; Almendro, V. The neuronal influence on tumor progression. Biochim. Biophys. Acta 2011, 1816, 105–118. [Google Scholar] [CrossRef]
- Faulkner, S.; Jobling, P.; March, B.; Jiang, C.C.; Hondermarck, H. Tumor Neurobiology and the War of Nerves in Cancer. Cancer Discov. 2019, 9, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Rozengurt, E. Neuropeptides as growth factors for normal and cancerous cells. Trends Endocrinol. Metab. 2002, 13, 128–134. [Google Scholar] [CrossRef]
- Tan, X.; Sivakumar, S.; Bednarsch, J.; Wiltberger, G.; Kather, J.N.; Niehues, J.; de Vos-Geelen, J.; Valkenburg-van Iersel, L.; Kintsler, S.; Roeth, A.; et al. Nerve fibers in the tumor microenvironment in neurotropic cancer—Pancreatic cancer and cholangiocarcinoma. Oncogene 2021, 40, 899–908. [Google Scholar] [CrossRef]
- Schonkeren, S.L.; Thijssen, M.S.; Vaes, N.; Boesmans, W.; Melotte, V. The emerging role of nerves and glia in colorectal cancer. Cancers 2021, 13, 152. [Google Scholar] [CrossRef]
- Wang, N.; Wang, J.; Zhang, Y.; Hu, S.; Zhang, T.; Wu, Y.; Sun, X.; Zhang, T.; Yang, S.; He, L. Substance P-induced lung inflammation in mice is mast cell dependent. Clin. Exp. Allergy 2021, 52, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Danelli, L.; Frossi, B.; Gri, G.; Mion, F.; Guarnotta, C.; Bongiovanni, L.; Tripodo, C.; Mariuzzi, L.; Marzinotto, S.; Rigoni, A.; et al. Mast cells boost myeloid-derived suppressor cell activity and contribute to the development of tumor-favoring microenvironment. Cancer Immunol. Res. 2015, 3, 85–95. [Google Scholar] [CrossRef]
- Yang, Z.; Zhang, B.; Li, D.; Lv, M.; Huang, C.; Shen, G.-X.; Huang, B. Mast cells mobilize myeloid-derived suppressor cells and Treg cells in tumor microenvironment via IL-17 pathway in murine hepatocarcinoma model. PLoS ONE 2010, 5, e8922. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Feng, Q.; Zheng, P.; Yang, L.; Zhu, D.; Chang, W.; Ji, M.; He, G.; Xu, J. Low tumor infiltrating mast cell density confers prognostic benefit and reflects immunoactivation in colorectal cancer. Int. J. Cancer 2018, 143, 2271–2280. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.W.; Keshavarzian, A.; Gounaris, E.; Melson, J.E.; Cheon, E.C.; Blatner, N.R.; Chen, Z.E.; Tsai, F.-N.; Lee, G.; Ryu, H.; et al. PI3K/AKT signaling is essential for communication between tissue-infiltrating mast cells, macrophages, and epithelial cells in colitis-induced cancer. Clin. Cancer Res. 2013, 19, 2342–2354. [Google Scholar] [CrossRef] [PubMed]
- Drobits, B.; Holcmann, M.; Amberg, N.; Swiecki, M.; Grundtner, R.; Hammer, M.; Colonna, M.; Sibilia, M. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Investig. 2012, 122, 575–585. [Google Scholar] [CrossRef]
- Al-Ostoot, F.H.; Salah, S.; Khamees, H.A.; Khanum, S.A. Tumor angiogenesis: Current challenges and therapeutic opportunities. Cancer Treat. Res. Commun. 2021, 28, 100422. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Kessler, D.A.; Langer, R.S.; Pless, N.A.; Folkman, J. Mast cells and tumor angiogenesis. Int. J. cancer 1976, 18, 703–709. [Google Scholar] [CrossRef]
- Grützkau, A.; Krüger-Krasagakes, S.; Baumeister, H.; Schwarz, C.; Kögel, H.; Welker, P.; Lippert, U.; Henz, B.M.; Möller, A. Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: Implications for the biological significance of VEGF206. Mol. Biol. Cell 1998, 9, 875–884. [Google Scholar] [CrossRef]
- Boesiger, J.; Tsai, M.; Maurer, M.; Yamaguchi, M.; Brown, L.F.; Claffey, K.P.; Dvorak, H.F.; Galli, S.J. Mast cells can secrete vascular permeability factor/vascular endothelial cell growth factor and exhibit enhanced release after immunoglobulin E-dependent upregulation of fc epsilon receptor I expression. J. Exp. Med. 1998, 188, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Kalesnikoff, J.; Huber, M.; Lam, V.; Damen, J.E.; Zhang, J.; Siraganian, R.P.; Krystal, G. Monomeric IgE stimulates signaling pathways in mast cells that lead to cytokine production and cell survival. Immunity 2001, 14, 801–811. [Google Scholar] [CrossRef]
- Sly, L.M.; Kalesnikoff, J.; Lam, V.; Wong, D.; Song, C.; Omeis, S.; Chan, K.; Lee, C.W.K.; Siraganian, R.P.; Rivera, J.; et al. IgE-Induced Mast Cell Survival Requires the Prolonged Generation of Reactive Oxygen Species. J. Immunol. 2008, 181, 3850–3860. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.; Mohammed, Z.; Deppen, J.; Gomez, G. Interleukin-6 potentiates FcεRI-induced PGD2 biosynthesis and induces VEGF from human in situ-matured skin mast cells. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 1069–1078. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.X.; Kaieda, S.; Ameri, S.; Fishgal, N.; Dwyer, D.; Dellinger, A.; Kepley, C.L.; Gurish, M.F.; Nigrovic, P.A. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc. Natl. Acad. Sci. USA 2014, 111, 10281–10286. [Google Scholar] [CrossRef]
- Cristinziano, L.; Poto, R.; Criscuolo, G.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Loffredo, S.; de Paulis, A.; Marone, G.; Spadaro, G.; et al. Il-33 and superantigenic activation of human lung mast cells induce the release of angiogenic and lymphangiogenic factors. Cells 2021, 10, 145. [Google Scholar] [CrossRef]
- Abdel-Majid, R.M.; Marshall, J.S. Prostaglandin E 2 Induces Degranulation-Independent Production of Vascular Endothelial Growth Factor by Human Mast Cells. J. Immunol. 2004, 172, 1227–1236. [Google Scholar] [CrossRef]
- Marcella, S.; Petraroli, A.; Braile, M.; Parente, R.; Ferrara, A.L.; Galdiero, M.R.; Modestino, L.; Cristinziano, L.; Rossi, F.W.; Varricchi, G.; et al. Vascular endothelial growth factors and angiopoietins as new players in mastocytosis. Clin. Exp. Med. 2021, 21, 415–427. [Google Scholar] [CrossRef] [PubMed]
- Heissig, B.; Rafii, S.; Akiyama, H.; Ohki, Y.; Sato, Y.; Rafael, T.; Zhu, Z.; Hicklin, D.J.; Okumura, K.; Ogawa, H.; et al. Low-dose irradiation promotes tissue revascularization through VEGF release from mast cells and MMP-9-mediated progenitor cell mobilization. J. Exp. Med. 2005, 202, 739–750. [Google Scholar] [CrossRef]
- García-Román, J.; Ibarra-Sánchez, A.; Lamas, M.; González Espinosa, C. VEGF secretion during hypoxia depends on free radicals-induced Fyn kinase activity in mast cells. Biochem. Biophys. Res. Commun. 2010, 401, 262–267. [Google Scholar] [CrossRef]
- Ma, Q.; Dieterich, L.C.; Ikenberg, K.; Bachmann, S.B.; Mangana, J.; Proulx, S.T.; Amann, V.C.; Levesque, M.P.; Dummer, R.; Baluk, P.; et al. Unexpected contribution of lymphatic vessels to promotion of distant metastatic tumor spread. Sci. Adv. 2018, 4, eaat4758. [Google Scholar] [CrossRef]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular Mechanisms and Future Promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef]
- Mäkinen, T.; Veikkola, T.; Mustjoki, S.; Karpanen, T.; Catimel, B.; Nice, E.C.; Wise, L.; Mercer, A.; Kowalski, H.; Kerjaschki, D.; et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001, 20, 4762–4773. [Google Scholar] [CrossRef] [PubMed]
- Detoraki, A.; Staiano, R.I.; Granata, F.; Giannattasio, G.; Prevete, N.; de Paulis, A.; Ribatti, D.; Genovese, A.; Triggiani, M.; Marone, G. Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J. Allergy Clin. Immunol. 2009, 123, 1142–1149.e5. [Google Scholar] [CrossRef]
- Belhabib, I.; Zaghdoudi, S.; Lac, C.; Bousquet, C.; Jean, C. Extracellular Matrices and Cancer-Associated Fibroblasts: Targets for Cancer Diagnosis and Therapy? Cancers 2021, 13, 3466. [Google Scholar] [CrossRef]
- Piersma, B.; Hayward, M.K.; Weaver, V.M. Fibrosis and cancer: A strained relationship. Biochim. Biophys. Acta. Rev. Cancer 2020, 1873, 188356. [Google Scholar] [CrossRef]
- Gilkes, D.M.; Semenza, G.L.; Wirtz, D. Hypoxia and the extracellular matrix: Drivers of tumour metastasis. Nat. Rev. Cancer 2014, 14, 430–439. [Google Scholar] [CrossRef]
- Biffi, G.; Tuveson, D.A. Diversity and Biology of Cancer-Associated Fibroblasts. Physiol. Rev. 2021, 101, 147–176. [Google Scholar] [CrossRef]
- Samoszuk, M.; Kanakubo, E.; Chan, J.K. Degranulating mast cells in fibrotic regions of human tumors and evidence that mast cell heparin interferes with the growth of tumor cells through a mechanism involving fibroblasts. BMC Cancer 2005, 5, 121. [Google Scholar] [CrossRef]
- Yuan, H.; Hsiao, Y.-H.; Zhang, Y.; Wang, J.; Yin, C.; Shen, R.; Su, Y. Destructive impact of T-lymphocytes, NK and Mast cells on basal cell layers: Implications for tumor invasion. BMC Cancer 2013, 13, 258. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hartmann, K.; Eckes, B.; Krieg, T. Mast cells enhance contraction of three-dimensional collagen lattices by fibroblasts by cell-cell interaction: Role of stem cell factor/c-kit. Immunology 2000, 99, 435–439. [Google Scholar] [CrossRef]
- Dabbous, M.K.; North, S.M.; Haney, L.; Tipton, D.A.; Nicolson, G.L. Effects of mast cell-macrophage interactions on the production of collagenolytic enzymes by metastatic tumor cells and tumor-derived and stromal fibroblasts. Clin. Exp. Metastasis 1995, 13, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Korpal, M.; Kang, Y. Targeting the transforming growth factor-beta signalling pathway in metastatic cancer. Eur. J. Cancer 2010, 46, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, S.; Yokote, T.; Hiraoka, N.; Nishiwaki, U.; Hanafusa, T.; Nishimura, Y.; Tsuji, M. Role of mast cells in fibrosis of classical Hodgkin lymphoma. Int. J. Immunopathol. Pharmacol. 2016, 29, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Argentieri, R.L.; Farrell, F.X.; Bracht, M.; Sheng, H.; Whitaker, B.; Beck, H.; Tsui, P.; Cochlin, K.; Evanoff, H.L.; et al. Hyper-responsiveness of IPF/UIP fibroblasts: Interplay between TGFbeta1, IL-13 and CCL2. Int. J. Biochem. Cell Biol. 2008, 40, 2174–2182. [Google Scholar] [CrossRef]
- Gruber, B.L.; Kew, R.R.; Jelaska, A.; Marchese, M.J.; Garlick, J.; Ren, S.; Schwartz, L.B.; Korn, J.H. Human mast cells activate fibroblasts: Tryptase is a fibrogenic factor stimulating collagen messenger ribonucleic acid synthesis and fibroblast chemotaxis. J. Immunol. 1997, 158, 2310–2317. Available online: http://www.ncbi.nlm.nih.gov/pubmed/9036979 (accessed on 4 January 2022).
- Akers, I.A.; Parsons, M.; Hill, M.R.; Hollenberg, M.D.; Sanjar, S.; Laurent, G.J.; McAnulty, R.J. Mast cell tryptase stimulates human lung fibroblast proliferation via protease-activated receptor-2. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L193–L201. [Google Scholar] [CrossRef]
- Tchougounova, E.; Lundequist, A.; Fajardo, I.; Winberg, J.-O.; Abrink, M.; Pejler, G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J. Biol. Chem. 2005, 280, 9291–9296. [Google Scholar] [CrossRef]
- Iddamalgoda, A.; Le, Q.T.; Ito, K.; Tanaka, K.; Kojima, H.; Kido, H. Mast cell tryptase and photoaging: Possible involvement in the degradation of extra cellular matrix and basement membrane proteins. Arch. Dermatol. Res. 2008, 300, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Peinado, H.; Zhang, H.; Matei, I.R.; Costa-Silva, B.; Hoshino, A.; Rodrigues, G.; Psaila, B.; Kaplan, R.N.; Bromberg, J.F.; Kang, Y.; et al. Pre-metastatic niches: Organ-specific homes for metastases. Nat. Rev. Cancer 2017, 17, 302–317. Available online: https://pubmed.ncbi.nlm.nih.gov/28303905/ (accessed on 1 October 2021). [CrossRef]
- Paget, S. The distribution of secondary growths in cancer of the breast. Lancet 1889, 133, 571–573. Available online: http://www.ncbi.nlm.nih.gov/pubmed/2673568 (accessed on 1 October 2021). [CrossRef]
- Chin, A.R.; Wang, S.E. Cancer Tills the Premetastatic Field: Mechanistic Basis and Clinical Implications. Clin. Cancer Res. 2016, 22, 3725–3733. [Google Scholar] [CrossRef]
- Kaplan, R.N.; Riba, R.D.; Zacharoulis, S.; Bramley, A.H.; Vincent, L.; Costa, C.; MacDonald, D.D.; Jin, D.K.; Shido, K.; Kerns, S.A.; et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 2005, 438, 820–827. Available online: https://pubmed.ncbi.nlm.nih.gov/16341007/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Aponte-López, A.; Muñoz-Cruz, S. Mast Cells in the Tumor Microenvironment. Adv. Exp. Med. Biol. 2020, 1273, 159–173. Available online: https://pubmed.ncbi.nlm.nih.gov/33119881/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Hu, G.; Wang, S.; Cheng, P. Tumor-infiltrating tryptase+ mast cells predict unfavorable clinical outcome in solid tumors. Int. J. Cancer 2018, 142, 813–821. Available online: https://pubmed.ncbi.nlm.nih.gov/29023696/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Sang, J.; Yi, D.; Tang, X.; Zhang, Y.; Huang, T. The associations between mast cell infiltration, clinical features and molecular types of invasive breast cancer. Oncotarget 2016, 7, 81661–81669. Available online: https://pubmed.ncbi.nlm.nih.gov/27835573/ (accessed on 1 October 2021). [CrossRef]
- Fragomeni, S.M.; Sciallis, A.; Jeruss, J.S. Molecular Subtypes and Local-Regional Control of Breast Cancer. Surg. Oncol. Clin. N. Am. 2018, 27, 95–120. [Google Scholar] [CrossRef] [PubMed]
- Yano, H.; Kinuta, M.; Tateishi, H.; Nakano, Y.; Matsui, S.; Monden, T.; Okamura, J.; Sakai, M.; Okamoto, S. Mast cell infiltration around gastric cancer cells correlates with tumor angiogenesis and metastasis. Gastric Cancer 1999, 2, 26–32. Available online: https://pubmed.ncbi.nlm.nih.gov/11957067/ (accessed on 1 October 2021). [CrossRef]
- Cabanillas-Saez, A.; Schalper, J.A.; Nicovani, S.M.; Rudolph, M.I. Characterization of mast cells according to their content of tryptase and chymase in normal and neoplastic human uterine cervix. Int. J. Gynecol. Cancer 2002, 12, 92–98. Available online: https://pubmed.ncbi.nlm.nih.gov/11860542/ (accessed on 1 October 2021). [CrossRef]
- Diaconu, N.-C.; Rummukainen, J.; Naukkarinen, A.; Mättö, M.; Harvima, R.J.; Pelkonen, J.; Harvima, I.T. Mast cell chymase is present in uterine cervical carcinoma and it detaches viable and growing cervical squamous carcinoma cells from substratum in vitro. Arch. Dermatol. Res. 2011, 303, 499–512. Available online: https://pubmed.ncbi.nlm.nih.gov/21274549/ (accessed on 1 October 2021). [CrossRef]
- Jiang, Y.; Wu, Y.; Hardie, W.J.; Zhou, X. Mast cell chymase affects the proliferation and metastasis of lung carcinoma cells in vitro. Oncol. Lett. 2017, 14, 3193–3198. Available online: https://pubmed.ncbi.nlm.nih.gov/28927065/ (accessed on 1 October 2021). [CrossRef]
- Valiente, M.; Ahluwalia, M.S.; Boire, A.; Brastianos, P.K.; Goldberg, S.B.; Lee, E.Q.; Le Rhun, E.; Preusser, M.; Winkler, F.; Soffietti, R. The Evolving Landscape of Brain Metastasis. Trends Cancer 2018, 4, 176–196. Available online: https://pubmed.ncbi.nlm.nih.gov/29506669/ (accessed on 1 October 2021). [CrossRef]
- Roy, A.; Libard, S.; Weishaupt, H.; Gustavsson, I.; Uhrbom, L.; Hesselager, G.; Swartling, F.J.; Pontén, F.; Alafuzoff, I.; Tchougounova, E. Mast Cell Infiltration in Human Brain Metastases Modulates the Microenvironment and Contributes to the Metastatic Potential. Front. Oncol. 2017, 7, 115. Available online: https://pubmed.ncbi.nlm.nih.gov/28626727/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Skokos, D.; Goubran-Botros, H.; Roa, M.; Mécheri, S. Immunoregulatory properties of mast cell-derived exosomes. Mol. Immunol. 2002, 38, 1359–1362. Available online: https://pubmed.ncbi.nlm.nih.gov/12217408/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. Available online: https://pubmed.ncbi.nlm.nih.gov/17486113/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Skotland, T.; Hessvik, N.P.; Sandvig, K.; Llorente, A. Exosomal lipid composition and the role of ether lipids and phosphoinositides in exosome biology. J. Lipid Res. 2019, 60, 9–18. Available online: https://pubmed.ncbi.nlm.nih.gov/30076207/ (accessed on 1 October 2021). [CrossRef] [PubMed]
- Bland, C.L.; Byrne-Hoffman, C.N.; Fernandez, A.; Rellick, S.L.; Deng, W.; Klinke, D.J. Exosomes derived from B16F0 melanoma cells alter the transcriptome of cytotoxic T cells that impacts mitochondrial respiration. FEBS J. 2018, 285, 1033–1050. Available online: https://pubmed.ncbi.nlm.nih.gov/29399967/ (accessed on 1 October 2021). [CrossRef]
- Linton, S.S.; Abraham, T.; Liao, J.; Clawson, G.A.; Butler, P.J.; Fox, T.; Kester, M.; Matters, G.L. Tumor-promoting effects of pancreatic cancer cell exosomes on THP-1-derived macrophages. PLoS ONE 2018, 13, e0206759. Available online: https://pubmed.ncbi.nlm.nih.gov/30383833/ (accessed on 1 October 2021). [CrossRef]
- Xiao, H.; He, M.; Xie, G.; Liu, Y.; Zhao, Y.; Ye, X.; Li, X.; Zhang, M. The release of tryptase from mast cells promote tumor cell metastasis via exosomes. BMC Cancer 2019, 19, 1015. Available online: https://pubmed.ncbi.nlm.nih.gov/31664930/ (accessed on 1 October 2021). [CrossRef]
- Dubreuil, P.; Letard, S.; Ciufolini, M.; Gros, L.; Humbert, M.; Castéran, N.; Borge, L.; Hajem, B.; Lermet, A.; Sippl, W.; et al. Masitinib (AB1010), a potent and selective tyrosine kinase inhibitor targeting KIT. PLoS ONE 2009, 4, e7258. Available online: https://pubmed.ncbi.nlm.nih.gov/19789626/ (accessed on 2 December 2021). [CrossRef]
- Humbert, M.; Castéran, N.; Letard, S.; Hanssens, K.; Iovanna, J.; Finetti, P.; Bertucci, F.; Bader, T.; Mansfield, C.D.; Moussy, A.; et al. Masitinib combined with standard gemcitabine chemotherapy: In vitro and in vivo studies in human pancreatic tumour cell lines and ectopic mouse model. PLoS ONE 2010, 5, e9430. Available online: https://pubmed.ncbi.nlm.nih.gov/20209107/ (accessed on 2 December 2021). [CrossRef]
- Mitry, E.; Hammel, P.; Deplanque, G.; Mornex, F.; Levy, P.; Seitz, J.F.; Moussy, A.; Kinet, J.P.; Hermine, O.; Rougier, P.; et al. Safety and activity of masitinib in combination with gemcitabine in patients with advanced pancreatic cancer. Cancer Chemother. Pharmacol. 2010, 66, 395–403. Available online: https://pubmed.ncbi.nlm.nih.gov/20364428/ (accessed on 2 December 2021). [CrossRef] [PubMed]
- Deplanque, G.; Demarchi, M.; Hebbar, M.; Flynn, P.; Melichar, B.; Atkins, J.; Nowara, E.; Moyé, L.; Piquemal, D.; Ritter, D.; et al. A randomized, placebo-controlled phase III trial of masitinib plus gemcitabine in the treatment of advanced pancreatic cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 1194–1200. Available online: https://pubmed.ncbi.nlm.nih.gov/25858497/ (accessed on 2 December 2021). [CrossRef] [PubMed]
- Guo, X.; Zhai, L.; Xue, R.; Shi, J.; Zeng, Q.; Gao, C. Mast Cell Tryptase Contributes to Pancreatic Cancer Growth through Promoting Angiogenesis via Activation of Angiopoietin-1. Int. J. Mol. Sci. 2016, 17, 834. Available online: https://pubmed.ncbi.nlm.nih.gov/27240355/ (accessed on 2 December 2021). [CrossRef] [PubMed]
- Heuston, S.; Hyland, N.P. Chymase inhibition as a pharmacological target: A role in inflammatory and functional gastrointestinal disorders? Br. J. Pharmacol. 2012, 167, 732–740. Available online: https://pubmed.ncbi.nlm.nih.gov/22646261/ (accessed on 2 December 2021). [CrossRef] [PubMed]
- Yarla, N.S.; Pathuri, G.; Terzyan, S.; Zhang, Y.; Singh, A.; Scotti, M.T.; Madka, V.; Rao, C.V. Abstract LB074: Chymostatin, a cathepsin L inhibitor, inhibits lung cancer cell proliferation and COVID-19 M pro in vitro. In Proceedings of the COVID-19 and Cancer, American Association for Cancer Research Annual Meeting 2021, Philadelphia, PA, USA, 9–14 April 2021; p. LB074. [Google Scholar]
- Zhang, T.; Finn, D.F.; Barlow, J.W.; Walsh, J.J. Mast cell stabilisers. Eur. J. Pharmacol. 2016, 778, 158–168. Available online: https://pubmed.ncbi.nlm.nih.gov/26130122/ (accessed on 2 December 2021). [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Type | Mast Cell Activity/Function | Localization/Prognosis | References |
|---|---|---|---|
| Basal Cell Carcinoma (BCC) ** | High numbers of MCs were found in BCC patients. MCs secrete IL-8 and RANTES to recruit lymphocytes and VEGF to promote angiogenesis. | Peritumoral | [14] |
| Breast Cancer ** | MCs positive for estrogen receptor in the stroma of tumors are associated with low-grade tumors in BC patients. | Tumor stroma | [15] |
| MCs may play a role in primary breast cancer angiogenesis. Infiltrated MCs may contribute to stromal remodeling and the α-SMA+ myofibroblast differentiation, both in BC patients. | Bad prognosis | [16,17] | |
| Cholangiocarcinoma (ICC) ** | Intratumoral MCs secrete histamine to promote angiogenesis, EMT of cancer cells, and ECM degradation in a mouse model. Cholangiocytes secrete Stem Cell Factor (SCF) to recruit MCs into the tumor in vitro. | Intratumoral | [18] |
| Colorectal Cancer (CRC) ** | MCs secrete chymase to recruit macrophages, neutrophils, and other immune cells to improve host immunity against cancer in CRC patients. | Good prognosis | [19] |
| Perivascular MCs promote angiogenesis and tumor progression at earlier and advanced stages. Peritumoral MCs positive for PAR-2 are associated with advanced CRC and numbers of MCs serve as a prognostic marker in CRC patients. | Peritumoral/Bad prognosis | [20,21,22] | |
| Endometrial carcinoma | MCs have a preferential localization along blood vessels and sites of new vessel formation in human endometrial carcinoma samples. | Intratumoral/Bad prognosis | [23] |
| Gastric Cancer (GC) ** and Gastrointestinal Cancer (GIC) ** | MCs promote angiogenesis and metastasis of cancer cells in GC patients. | Peritumoral/Bad prognosis | [24] and references therein |
| In GC patients exist a positive correlation between MC numbers, IL-17 production and microvessel density, and numbers of neutrophils and regulatory Tregs. | Intratumoral/Bad prognosis | [25] | |
| MCs levels increase with tumor progression and predict reduced overall survival in GC patients. | [26] | ||
| Gynecologic Cancer ** | High density of MCs in human samples of pre-malignant lesions of the cervix and endometrial cancer. MCs secrete tryptase to promote angiogenesis and invasion. | Peritumoral | [27] |
| MCs are detected in ovarian cancer, uterine leiomyomas, vulva cancer, and the trophoblastic disease in women. | Peritumoral | [28] | |
| Hepatocarcinoma (HCC) ** | Peritumoral MC density positively correlates with the numbers of Tregs in HCC patients. | Peritumoral/Bad prognosis | [29] |
| MCs secrete IL-17 to induce angiogenesis and tumor progression in HCC patients. | Peritumoral/Bad prognosis | [30] | |
| Lung Cancer ** | Intratumoral MCs indicate bad prognosis in human lung adenocarcinomas and advanced tumors. | Intratumoral/Bad prognosis | [31] |
| Cytotoxic activity of TNF-α from MCs confers improved survival in NSCLC (non-small cell lung cancer) patients. | Intratumoral/Good prognosis | [32] | |
| Melanoma ** | Perivascular MCs secrete VEGF to promote angiogenesis, which correlates with malignancy and metastasis in a mouse model. | Peritumoral/Bad prognosis | [33] and references therein |
| Low density of MCs indicates bad prognosis in human samples. | Peritumoral/Bad prognosis | [34] | |
| Oral Squamous Cell Carcinoma | A higher MC density in human OSCC tumors is associated with a better prognosis. | Good prognosis | [35] |
| Pancreatic Cancer ** | MCs secrete tryptase and IL-13 to promote cancer cells proliferation and invasion in vitro. Peritumoral MCs inter-communicate with pancreatic cancer cells by contact and high MC density indicates bad prognosis in a mouse model. | Peritumoral/Bad prognosis | [36,37] |
| Prostate Cancer | MCs secrete MMP-9 to promote angiogenesis and invasion and secrete high levels of FGF-2 in mouse and rat models. | [38,39] | |
| Metastasis is promoted by MCs via the regulation of the lncRNA-HOTAIR-PRC2-Androgen receptor-MMP9 signaling complex in human and mouse models. | [40] | ||
| Peritumoral MCs are biomarkers at early stages of prostate tumor and indicate bad prognosis in humans. | Bad prognosis | [41] and references therein | |
| Intratumoral MCs are associated with a lower risk of prostate cancer recurrence, favorable tumor characteristics and good prognosis in PC patients. | Intratumoral/Good prognosis | [42,43] | |
| Renal Cell Carcinoma | Intratumoral MCs promote tumor angiogenesis and acceleration of tumor growth in human and mouse models. | Bad prognosis | [44] |
| MCs are associated with cell proliferation and recurrence in RCC patients. | Peritumoral/Bad prognosis | [45] | |
| The presence of intratumoral MCs in patients with RCC without metastasis after surgery functions as a predictive marker of survival and relapse. | Intratumoral/Good prognosis | [46,47] | |
| Thyroid carcinoma | The secretion of histamine, CXCL1 and CXCL10 by human MCs promotes cancer cells’ proliferation, survival, and metastasis, as well as angiogenesis in vitro. | Bad prognosis | [48] |
| Sash Mice (c-kit Wsh/Wsh) Mice bearing a large inversion (close to 70 kb) comprising the promoter of the c-kit gene (Wsh mutation). Constitutive lack of MCs, melanocytes, and interstitial cells of Cajal. Extramedullary myelopoiesis and systemic neutrophilia. Cardiac defects due to abnormal corin expression. Normal MCs numbers and neutrophils are reached after reconstitution with BMMCs from WT mice [49,50,51,52]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Melanoma | -Tumor generation: s.c. administration of 1 × 105 B16.F10 cells above the right and left flanks. -Results: TLR-2 receptor agonist (Pam3CSK4) inhibited the growth of melanoma tumors, and this effect was attributed to MCs since it was not observed in MC-deficient mice. Tumor growth inhibition was restored in c-kit Wsh/Wsh mice through local reconstitution with WT, but not with TLR2-deficient MCs [53]. |
| -Tumor generation: s.c. administration of 0.5 × 106 B16.F1 melanoma cells into the left ear pinna. -Results: Monomeric IgE (mIgE) increased melanoma tumor growth, peritumoral MCs numbers, and blood vessels in WT but not in c-kit Wsh/Wsh mice. Effects of mIgE on melanoma tumor growth were restored after reconstitution of sash mice with WT but not with Fyn −/− BMMCs [54]. | |
| -Tumor generation: i.d. administration of B16-OVA melanoma cells and adoptive transfer of tumor-specific OT-I and OT-II T cells (TCs) was performed, followed by 3 consecutive peritumoral injections of LPS or vehicle. -Results: LPS-activated melanoma-resident MCs secreted CXCL10, which recruited tumor-infiltrating effector T cells (TILs) and started the immune response against melanoma. c-kit Wsh/Wsh mice failed in inducing protective immune activation upon LPS exposure. Reconstitution of the skin of c-kit Wsh/Wsh mice with WT BMMCs completely restored LPS-induced melanoma immune control [55]. | |
| Prostate adenocarcinoma | -Tumor generation: s.c. administration of 2 × 106 cells from the T1525 and T23 cell lines. -Results: MCs promoted prostate tumor growth through MMP-9 production. Reconstitution of c-kit Wsh/Wsh with WT but not with MMP-9−/− MCs was enough to restore tumor growth [38]. |
| Intestinal tumor | -Utilized cancer model: Murine model of multiple intestinal neoplasia (Min, APCMin/+). -Results: Sash mice developed 50% more adenomas, and the tumors were 33% larger than in their WT littermates. Eosinophils were fewer in adenomas from Min–Sash mice than in WT mice [56]. |
| Pancreatic tumor | -Tumor generation: i.p. administration of 1 mg of tamoxifen in mice (pIns-mycERTAM; RIP7-bcl-xL), a murine model of Myc-induced β-cell carcinoma. -Results: Myc-induced pancreatic tumors were not observable on c-kit Wsh/Wsh mice. MCs contributed to the generation of blood vessels and the macroscopic expansion of Myc-induced pancreatic islet tumors [57]. |
| -Utilized cancer model: Spontaneous mouse model of pancreatic ductal adenocarcinoma (PDAC) K-rasG12V. -Results: PDAC tumor growth was inhibited in c-kit Wsh/Wsh mice, but it was observable in WT mice. Tumor development was recovered in sash mice reconstituted with WT BMMCs [37]. | |
| Bladder carcinoma | -Tumor induction: i.d. administration of 2.5 × 105 MB49 cells. -Results: Sash female mice showed enhanced survival to MB49 tumors in comparison to WT female mice. Reconstitution of Wsh female mice with BMMCs diminished protective anti-tumor immunity [58]. |
| Breast cancer | -Utilized cancer model: c-kit Wsh/Wsh mice were crossed with mammary tumor model mice with the MMTV-Polyoma Middle T antigen (PyMT). -Results: c-kit Wsh/Wsh mice showed delayed tumor growth, diminished tumor size, and reduced lung metastasis and angiogenesis compared to MC-proficient mice [59]. |
| -Utilized cancer model: Mammary tumor transgenic mouse strain MMTV-Polyoma middle T antigen (PyMT). -Results: MCs induced a luminal phenotype and modified the outcome of breast cancer tumors. The absence of MCs was associated with a lower grade of mammary tumors, with an evident reduction in metastasis dissemination [60]. | |
| Gastric cancer | -Utilized cancer model: c-kit Wsh/Wsh mice were crossed with the gp130F/F intestinal-type gastric cancer murine model. -Results: gp130F/F/c-kit Wsh/Wsh mice showed significantly smaller and fewer tumors than their gp130F/F littermates. MC deficiency diminished gp130F/F-driven tumor mass [61]. |
| Squamous cell carcinoma | -Utilized cancer model: c-kit Wsh/Wsh mice were crossed with E7 mice to obtain MC-deficient mice expressing the HPV16-E7 oncoprotein (E7.Kit Wsh/Wsh mice). -Results: Whereas E7 mice showed tumor generation and no rejection of tumor grafts, E7 Kit Wsh/Wsh mice showed increased rejection of skin grafts [62]. |
| Malignant pleural effusion | -Utilized cancer model: malignant pleural effusions (MPEs) induced by LLC and MC38 adenocarcinomas. -Results: MPEs were not observed in c-kit Wsh/Wsh mice, and the adoptive transfer of WT bone marrow, as well as MC reconstitution, restored MPEs in MC–deficient mice [63]. |
| Lung adenocarcinoma (LADC) | -Utilized cancer model: LADC induced in KRASG12D-transgenic mice. -Results: c-kit Wsh/Wsh (KRASG12D; c-kit Wsh/Wsh) showed protection from KRAS-driven alveolar carcinomas compared with KRASG12D mice. Tumor induction: i.d. injection of 106 LLC cells into the rear flank dermis. -Results: c-kit Wsh/Wsh mice displayed delayed primary tumor growth, as well as decreased spontaneous metastasis to the lungs compared with controls [64]. |
| Kit Wv/Kit Wv Mice bearing a point mutation at the white spotting (W) locus together with a point mutation in the tyrosine kinase domain (Wv) of the protein CD117 (c-Kit). Expressed a truncated and inactive form of CD117. Constitutive lack of melanocytes, interstitial cells of Cajal, and MCs. Sterile, presented macrocytic anemia, gastric ulcers, and low numbers of bone marrow cells and circulating neutrophils. The very low number of MCs observed in these mice could be restored after reconstitution with BMMCs from WT mice [65,66,67]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Melanoma | -Tumor generation: s.c. administration of B16-BL6 melanoma cells (l05 cells) in ear pinna. -Results: Kit W/Wv mice showed slower angiogenesis and a lower metastatic colony development rate than their WT littermates. Angiogenic response and incidence of hematogenous metastases were restored with WT bone-marrow reconstitution in Kit W/Wv mice [68]. |
| Skin carcinogenesis | -Tumor generation: Administration of the carcinogen 7,12 dimethylbenz[a]-anthracene and subsequent treatment with the tumor promoter 12-tetradecanoyl phorbol-13-acetate (PMA). -Results: Tumor development and growth were increased in Kit W/Kit Wv mice. Reconstitution by local adoptive transfer of MCs normalized tumor incidence and growth [69]. |
| Squamous cell carcinoma | -Utilized cancer model: Transgenic mouse model of HPV-associated squamous cell carcinoma. -Results: Premalignant angiogenesis was abolished in Kit W/Kit Wwv HPV16 transgenic mice [70]. |
| Intestinal tumor | -Tumor generation: Administration of 1,2 dimethylhydrazine (DMH). -Results: Compared to WT mice, Kit W/Kit W-v mice developed fewer tumors after treatment with DMH. Adoptive transfer of WT bone marrow cells to W/Wv mice increased the susceptibility to the development of DMH-induced tumors [71]. |
| Lewis Lung Carcinoma (LLC) | -Tumor generation: s.c. administration of 2 × 106 tumor cells in the right flank. -Results: Tumor incidence was higher in MC-deficient mice (Kit W/Wv and Kit Wv/+) than in WT animals [72]. |
| Fibrosarcoma | -Tumor generation: s.c. administration of 104 MC-B6-1 tumor cells in the right flank. -Results: Tumor incidence was higher in MC-deficient mice (W/WV and in WV/+) than in WT animals [72]. |
| Cre-mediated mast cell eradication (Cre-Master) Mice generated by placing Cre expression under the control of the Cpa3 gene promoter. Deletion of 28 nucleotides of the first exon of Cpa3 locus. Constitutive lack of MCs in peritoneum, skin, and intestine. An important reduction in spleen basophil function. Some MC-dependent reactions could be reconstituted with BMMCs from WT mice [73]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Melanoma | -Utilized cancer model: Spontaneous tumor melanoma Tg(GRM1)EPv. -Results: Median tumor onset was significantly earlier in GRM1/Cre-Master mice than in Tg(GRM1)EPv control mice [74]. |
| Skin carcinogenesis | -Utilized cancer model: Topical administration of 25 mg of 7, 12-dimethylbenz(a) anthracene (DMBA) on the back skin of 8- to 10-wk-old female mice, and one week later, 7.5 mg of the tumor promoter 12-tetradecanoyl phorbol-13-acetate (PMA) was applied weekly for 20 wk. -Results: Tumorigenesis-associated vascularization was not impaired in Cre Master mice [75]. |
| Lung adenocarcinoma (LADC) | -Utilized cancer model: Mice received 10 consecutive weekly i.p. injections of urethane, a carcinogen contained in tobacco (1 g/Kg). -Results: Cpa3.Cre mice were markedly protected from urethane-induced bronchial carcinomas in terms of tumor multiplicity, size, and cellular proliferation rate. Tumor generation: i.d. administration of 106 LLC cells into the rear flank dermis. -Results: Cpa3.Cre mice displayed delayed primary tumor growth and decreased spontaneous metastasis to the lungs compared with controls [64]. |
| Malignant pleural effusion | Utilized cancer model: Malignant pleural effusions (MPEs) induced by LLC and MC38 adenocarcinomas. -Results: MCs were required for MPEs since Cpa3Cre/+ mice were protected from adenocarcinoma-induced effusions [63]. |
| Mcpt5-Cre+ R-DTA Mice created by the crossing of transgenic mice expressing Cre recombinase under the control of the MC protease-5 (Mcpt-5) promoter and DTR-floxed mice. Inducible reduction in skin, stomach, and peritoneal MCs, which normally express Mcpt-5 [76,77]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Melanoma | -Tumor generation: i.v. or s.c. administration of 1 × 105 B16.F10 cells into the tail vein or hip region of male mice. -Results: Reduced melanoma colonization in lungs from Mcpt5-Cre+ R-DTA+ vs. Mcpt5-Cre- R-DTA+ mice was observed [33]. |
| -Tumor generation: i.d. administration of B16-OVA melanoma cells and adoptive transfer of tumor-specific OT-I and OT-II T cells (TILs) was performed, followed by 3 consecutive peritumoral injections of LPS or vehicle. -Results: Mcpt5-cre+ R-DTAfl/fl mice failed to initiate melanoma immune defense upon LPS exposure as WT mice did [55]. | |
| Squamous cell carcinoma | -Utilized cancer model: Transgenic mouse model of HPV-associated squamous cell carcinoma. -Results: Absence of MCs had no impact on HPV-Induced epithelial growth and neovascularization, no differences were observed between MC-proficient (R26DTA/DTA Cre-negative) and MC-deficient (R26DTA/DTA Mcpt5-Cre + R-DTA) K14-HPV16 transgenic mice [78]. |
| “Hello Kitty” Cpa3-Cre; Mcl-1fl/fl Tg (Cpa3-cre) 3Glli; B6;129-Mcl1tm3sjkJ Mice created by a cross between transgenic mice expressing Cre under the control of Cpa3 promoter and Mcl-1 floxed mice. Constitutive low numbers of MCs in lung, peritoneum, skin, and trachea. Reconstitution of skin due to passive anaphylactic reactions caused by intradermal injection of BMMCs from WT mice. Lower numbers of basophils [79]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Gastric cancer | -Utilized cancer model: Mice Cpa3-Cre; Mcl-1fl/fl were crossed with the mouse model gp130F/F of intestinal-type gastric cancer. -Results: MC-deficient gp130F/F;Cpa3-Cre; Mcl-1/fl/fl mice had significantly reduced tumor mass compared to their MC-proficient controls. Reduced angiogenic vessel density in the tumors of gp130F/F; Cpa3-Cre;Mcl1fl/fl mice [61]. |
| SI/SId Mice presenting a mutation at the Sl locus, codifying for Stem Cell Factor (SCF). Constitutive lack of melanocytes, germ cells, and interstitial cells of Cajal and MCs. Animals could not be reconstituted with BMMCs from WT mice, but MCs could be restored after injection of SCF [65,80,81]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Melanoma | -Utilized cancer model: Systemic intra-arterial injection of B16-G3.26 melanoma cells. -Results: Tumor metastasis in mutant SI/SId mice was evaluated. Mutant Sl/SId mice were found to have a markedly lower incidence of ovarian metastasis compared to normal congenic mice [82]. |
| Ws/Ws Rats presented a deletion on the white locus (c-Kit gene, Ws, white spotting mutation). As a consequence, four amino acids close to the tyrosine autophosphorylation site of CD117 were absent in the protein. Constitutive of MC deficiency and anemia [83,84,85]. | |
| Tumor analyzed | Experimental approaches and main findings |
| Breast cancer | -Tumor generation: Administration of 50 mg/kg of the chemical carcinogen N-nitrosomethylurea (NMU). -Results: Ws/Ws rats did not show signs of neoplasia after treatment, compared to WT rats [86]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Segura-Villalobos, D.; Ramírez-Moreno, I.G.; Martínez-Aguilar, M.; Ibarra-Sánchez, A.; Muñoz-Bello, J.O.; Anaya-Rubio, I.; Padilla, A.; Macías-Silva, M.; Lizano, M.; González-Espinosa, C. Mast Cell–Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth. Cells 2022, 11, 349. https://doi.org/10.3390/cells11030349
Segura-Villalobos D, Ramírez-Moreno IG, Martínez-Aguilar M, Ibarra-Sánchez A, Muñoz-Bello JO, Anaya-Rubio I, Padilla A, Macías-Silva M, Lizano M, González-Espinosa C. Mast Cell–Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth. Cells. 2022; 11(3):349. https://doi.org/10.3390/cells11030349
Chicago/Turabian StyleSegura-Villalobos, Deisy, Itzel G. Ramírez-Moreno, Magnolia Martínez-Aguilar, Alfredo Ibarra-Sánchez, J. Omar Muñoz-Bello, Isabel Anaya-Rubio, Alejandro Padilla, Marina Macías-Silva, Marcela Lizano, and Claudia González-Espinosa. 2022. "Mast Cell–Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth" Cells 11, no. 3: 349. https://doi.org/10.3390/cells11030349
APA StyleSegura-Villalobos, D., Ramírez-Moreno, I. G., Martínez-Aguilar, M., Ibarra-Sánchez, A., Muñoz-Bello, J. O., Anaya-Rubio, I., Padilla, A., Macías-Silva, M., Lizano, M., & González-Espinosa, C. (2022). Mast Cell–Tumor Interactions: Molecular Mechanisms of Recruitment, Intratumoral Communication and Potential Therapeutic Targets for Tumor Growth. Cells, 11(3), 349. https://doi.org/10.3390/cells11030349