Abstract
Epithelial cells that form the kidney proximal tubule (PT) rely on an intertwined ecosystem of vesicular membrane trafficking pathways to ensure the reabsorption of essential nutrients—a key requisite for homeostasis. The endolysosome stands at the crossroads of this sophisticated network, internalizing molecules through endocytosis, sorting receptors and nutrient transporters, maintaining cellular quality control via autophagy, and toggling the balance between PT differentiation and cell proliferation. Dysregulation of such endolysosome-guided trafficking pathways might thus lead to a generalized dysfunction of PT cells, often causing chronic kidney disease and life-threatening complications. In this review, we highlight the biological functions of endolysosome-residing proteins from the perspectives of understanding—and potentially reversing—the pathophysiology of rare inherited diseases affecting the kidney PT. Using cystinosis as a paradigm of endolysosome disease causing PT dysfunction, we discuss how the endolysosome governs the homeostasis of specialized epithelial cells. This review also provides a critical analysis of the molecular mechanisms through which defects in autophagy pathways can contribute to PT dysfunction, and proposes potential interventions for affected tissues. These insights might ultimately accelerate the discovery and development of new therapeutics, not only for cystinosis, but also for other currently intractable endolysosome-related diseases, eventually transforming our ability to regulate homeostasis and health.
1. Introduction
Epithelial cells that line the proximal tubule (PT) of the kidney reabsorb a large variety of filtered macromolecules and low-molecular mass-nutrients through a particularly well-developed endolysosome system and through membrane trafficking pathways. Ever since its discovery by Christian De Duve in the 1960s, the endolysosome has come to be known as a single-membrane-enclosed organelle devoted to the degradation of damaged cellular constituents, including aged and/or misfolded proteins, and pathogens [1]. Extracellular and intracellular materials can reach the endolysosome through endocytosis and autophagy, respectively [2]. Fusion events subsequently enable the endolysosomes to recycle cargoes and/or substrates engulfed by endocytic and/or autophagic vesicles.
Beyond degradation and the disposal of cellular waste [3], the endolysosomes can also steer the metabolic trajectory of cells in response to nutrient availability, growth factors, and stress signals, hence guiding nearly every aspect of metabolic function, ultimately coordinating cell- and organism-wide growth [4]. As a consequence, dysregulation of endolysosomes and autophagy pathways might thus pose a devastating threat to many different cell types, eventually culminating in neurodegeneration, metabolic disease, cancer, and pathologies associated with ageing [2].
Over the last two decades, studies of rare inherited diseases, in combination with advances in technology and high-throughput omics, have provided novel insights into fundamental principles governing the contribution of the endolysosome to the maintenance of homeostasis [5,6]. Through converging approaches, these paradigms have helped to understand the pathogenesis of common kidney disease entities in which imbalances in the endolysosome system have been implicated in PT dysfunction [7]. Furthermore, the integration of genome-wide association studies (GWAS) with quantitative trait analyses (eQTLs) have identified that the expression of an endolysosome-residing enzyme, that is the beta-mannosidase (MANBA), is lower in the kidneys of subjects with chronic kidney disease (CKD), and that the MANBA risk allele shows evidence of structural and functional defects in endolysosome and autophagy pathways in kidney tubule cells [8]. In addition, common variants within genes closely linked to endolysosome function (e.g., CUBN, DAB2, RAB4, and LRP2) have recently been associated with proteinuria and CKD risk [9,10], highlighting the fundamental role of the endolysosome system in kidney health and its contribution to kidney disease risk within the population.
In this review, we discuss the biological functions of the endolysosome, not only as a “disposal-garbage system” of the cell, but also as a hub for homeostasis and signaling pathways, and delve into the breakdown and removal of damaged and/or dysfunctional mitochondria through autophagic pathways. Taking cystinosis as a paradigm of endolysosome disease causing PT dysfunction, we discuss how the endolysosome and autophagy govern the physiology of kidney tubule cells. We describe the cellular pathways and molecular underpinnings through which the absence of the endolysosome-residing transporter cystinosin might wreak havoc on autophagy, ultimately leading to dysfunction of the kidney PT. In the concluding section, we highlight potential new, attractive targets for therapeutically tackling cellular adversities linked to cystinosin dysfunction, offering new targetable pathways for this life-threatening disease.
2. Role of the Endolysosome System and Autophagy in Kidney Proximal Tubule
2.1. Receptor-Mediated Endocytosis and the Endolysosome
The kidney PT constitutes a paradigm of effective communication between the environment and endomembrane compartments, guiding the reabsorption of vital nutrients [5,6]. PT cells use receptor-mediated endocytosis and endolysosome-guided transport systems to efficiently reabsorb albumin and low molecular weight proteins (LMWPs) from the ultrafiltrate, preventing the urinary waste of essential proteins under physiological conditions [5,6]. The retrieval of albumin and LMWPs occurs through the multiligand receptors LRP2/megalin and cubilin [11,12,13], and the cooperating protein amnionless (AMN; Figure 1). The binding of filtered ligands to, and the interactions between both endocytic receptors, induces their internalization into clathrin-coated vesicles, and subsequent transport towards the endolysosomal compartments [14,15,16]. An essential component in this trafficking pathway is the apical endosomal compartment, where the ligands opportunely dissociate from their endocytic receptors through a process that requires sustained vesicular acidification (Figure 1) by the electrogenic vacuolar H+-ATPase (v-ATPase) proton pump [17,18]. In the kidney PT, additional proteins appear to be involved in the maintenance of the endolysosomal acidification, such as the anion transporter chloride channel 5 and 7 (ClC5 and ClC7; [19]); the cystic fibrosis transmembrane conductance regulator (CFTR; [19]); and the cation transporters mucolipin 1 and two pore calcium channel 1 (TPC1) and TCP2, which mediate Ca2+ and Na+ release from the endolysosome [20]. Once dissociated from their ligands, the endocytic receptors efficiently traffic to subapical Rab11+ apical recycling endosomes and successively reach the apical membrane in a microtubule-dependent manner [16], sustaining new cycles of ligand binding and internalization (Figure 1). The generation and maintenance of the endolysosomal pH gradient sustains not only the progression of cargo-filled vesicles towards the endocytic route, but also the activation acid hydrolases within the degradative compartments [21]. Iterative rounds of cargo sorting, coupled with maturation of the early endosomes, result in the formation of late endosomes that fuse with the lysosomes to form endolysosomes (Figure 1), where their accompanying cargoes are eventually degraded [22].
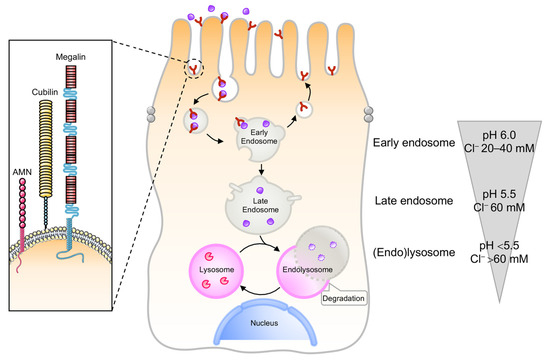
Figure 1.
The endolysosome system in the kidney PT. Epithelial cells lining the kidney PT have multifunctional endocytic receptors and a highly developed endolysosome system to take up plasma proteins that are filtered by the glomerulus. The endocytic pathway in PT cells requires coated pits and vesicles, followed by early endosomes that form recycling endosomes or mature to late endosomes that fuse with the lysosome to form the endolysosomes. The luminal pH drops from 7.4 in the tubule lumen to 6.0 in early endosomes, 5.5 in late endosomes, and below 5.0 in endolysosomes. Such vesicular acidification enables the dissociation between receptors and their ligands, the recycling of receptors back to the apical membrane, and the progression of ligands towards the endolysosomal compartments. In parallel, the Cl− concentrations drop from 110 mM in the extracellular space to 20–40 mM in early endosomes, 60 mM in late endosomes, and >80 mM in lysosomes, i.e., much higher than the 10–40 mM in the cytosol.
Accumulating evidence suggests that the endolysosome terminates autophagy—an evolutionary conserved pathway that degrades cellular components, such as defective organelles and misfolded proteins, to preserve homeostasis [1,23]. Furthermore, recent studies indicate that autophagy-mediated clearance pathways coordinate the renovation of cells and tissues during kidney development and differentiation, and are also involved in the prevention of genomic damage [24,25]. Therefore, its dysregulation might hasten not only PT dysfunction and kidney disease, but also other pathologies associated with kidney ageing [25,26].
Beyond its role in cellular destruction and quality control, the endolysosome system can steer the metabolic trajectories of cells in response to diverse microenvironmental cues in order to preserve homeostasis [2,4]. Crucial in this process is the (nutrient–dependent) recruitment of the evolutionarily conserved protein kinase called mTOR and its associated regulatory complex 1 (mTORC1) to the surface of the endolysosome through a multiprotein complex [27] comprising Rag guanosine triphosphatases (GTPases) [28,29], Ragulator [30,31], and vacuolar H+-adenosine triphosphatase ATPase (v-ATPase) [32]. In the presence of nutrients, the complex localizes on the surface of the endolysosome, where the growth-factor-directed activation of the endolysosome-bound GTPase Rheb [33] allosterically stimulates mTORC1 activity. Signaling from endolysosomes, mTORC1 initiates anabolic programs enhancing growth and proliferation, while suppressing catabolic autophagy and cellular quality control [2,34,35]. In addition, recent studies in rat kidney cells suggest that the reactivation of mTORC1, in combination with the precise regulation of phosphoinositide production, also coordinates autophagic lysosomal reformation (ALR)—an essential process that helps recycle a full complement of functional lysosomes from auto/endolysosomes during prolonged starvation [36]. Furthermore, the cytosolic face of the endolysosome drives the dynamic association of MiT/TFE family basic helix−loop−helix (bHLH) transcription factors, including TFEB, TFE3, TFEC, and MiTF, which that regulate endolysosome biogenesis [34], autophagy [35], and energy metabolism [37], as well as tethering factors that promote endolysosome fusion [2] or contact with other organelles to carry out specific metabolic programs [38]. Intriguingly, the association between mTOR, endolysosome, and the reabsorptive dysfunction in PT cells lacking Raptor [39]—the scaffold protein that docks mTOR kinase on the surface of the endolysosome—suggests potential interactions between nutrient sensing, endolysosome-directed mTORC1 signaling, and the maintenance of the kidney PT integrity.
2.2. Types of Autophagy
Three major routes for the delivery of autophagic cargos to endolysosomes have been reported: macroautophagy, chaperone-mediated autophagy, and microautophagy (Figure 2). Macroautophagy—the best-characterized form of autophagy—involves the sequestration of cellular material within a double-membrane vesicle, termed an autophagosome [40]. Induction factors and stress signals determine the choice of the autophagosome content that can proceed in a relatively nonselective manner, that is the bulk autophagy [41], or entail the tightly regulated disposal of individual cellular components [42]. For instance, mitophagy removes dysfunctional and/or damaged mitochondria; “ribophagy” for ribosomes; “pexophagy” for peroxisomes; “reticulophagy” specifically dismantles portions of the endoplasmic reticulum; ‘‘nucleophagy’’ parts of the nucleus; ‘‘aggrephagy’’selectively removes misfolded protein aggregates, ‘‘lipophagy’’ lipid droplets, and ‘‘xenophagy’’ specifically degrades intracellular bacteria that escape endosomes [42]. Irrespective of substrate specificity, selective autophagy relies on a set of cellular sensors that detect potentially dangerous cues and convert them into signals that are ultimately conveyed to the autophagic machinery [42].
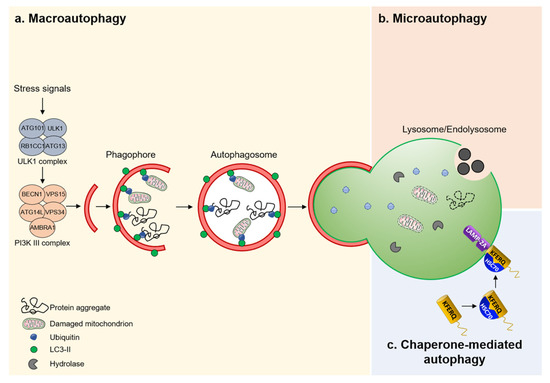
Figure 2.
Different mechanisms of autophagy. (a) Macroautophagy is a nonselective bulk process that removes protein aggregates and/or damaged mitochondria. (b) Microautophagy captures cytoplasmic constituents through direct invagination of endolysosome membranes. Resulting vesicles are subsequently released into the lumen of the endolysosome for degradation. (c) CMA identifies proteins containing a KFERQ pentapeptide-related motif by a chaperone complex HSC70 that transports them into the lumen of the endolysosome through a receptor/translocon containing LAMP2A.
Several distinct complexes containing autophagy-related proteins (ATGs) work with membrane trafficking components to regulate a well-oiled, multistep process that involves initiation, membrane nucleation and phagophore formation, phagophore elongation, cargo sequestration, expansion, autophagosome-lysosome fusion, and degradation. For example, a complex composed of serine/threonine protein kinases ULK1, ULK2, and other proteins stimulates the initiation of autophagy [43,44], while the class III phosphoinositide 3-kinase (PI3K) complex regulates the phagophore formation [45,46]. In addition, two ubiquitin-related systems, i.e., ATG12-ATG5-ATG16L and the microtubule-associated protein 1 light chain 3 (MAP1LC3, also known as LC3), govern the phagophore elongation and sealing of the autophagosome [47]. The autophagy-mediated turnover of damaged and/or dysfunctional mitochondria is required for protecting PT from a wide range of stimuli and insults, such as ischemia, acute kidney injury, sepsis, nutrient deprivation, exposure to toxins and/or pathogens, heat, radiation, hypoxia, and ureteral obstruction [20,48]. Conversely, the deletion of essential autophagy genes (e.g., Atg5 or Atg7; [20,49]) damages PT cells through defective mitochondrial clearance and increased reactive oxygen species (ROS), further substantiating the fundamental role of autophagy in the maintenance of kidney PT integrity and normal physiology. A fascinating interplay between ATG proteins and the membrane dynamics and the nutrient and/or energy-dependent signaling networks that induce autophagy has extensively been described in detail elsewhere [50].
Conversely, chaperone-mediated autophagy (CMA) operates as a protein-exclusive type of autophagy, whereby KFERQ-like motif-bearing proteins are recognized by the heat-shock cognate protein HSPA8/HSC70 and cross the surface of the endolysosome through the binding to lysosomal-associated membrane protein 2A (LAMP2A). This triggers the assembly of receptor/translocon containing LAMP2A that targets the degradation of CMA-flagged substrates by endolysosomes [51]. In contrast to CMA and macroautophagy, microautophagy enwraps, sequesters, and transports cytosolic components into the lumen of endolysosomes without the formation of autophagosomes [52]. The resulting breakdown products generated by the endolysosome-based degradation are eventually exported to the cytoplasm through dedicated nutrient transporters that span the membrane of the endolysosome, and are further utilized for energy or in other metabolic reactions [53]. These recent discoveries are now putting autophagy–endolysosome degradative systems under the spotlight, as they play a key role in safeguarding the homeostasis, integrity, and physiology of the kidney PT.
3. Cystinosis as a Paradigm of Endolysosome Disease Causing PT Dysfunction
The dysregulation of the endolysosome system causes a generalized dysfunction of PT cells, ultimately triggering losses of essential nutrients into the urine, thereby causing CKD [54] and life-threatening complications. Such PT dysfunction can stem from rare inherited disorders, owing to the malfunctioning of endolysosome-residing proteins, particularly in cystinosis [5,6].
Cystinosis—one of a family of approximately 70 rare inborn diseases of the metabolism known as lysosomal storage diseases [55]—is caused by inactivating mutations in the CTNS gene encoding the proton-driven transporter cystinosin [56], which exports cystine from the endolysosome (Figure 3a). Given that the low abundance of cystinosin in the lysosomal membrane is the rate-limiting step for cystine transport, its functional loss leads cystine to accumulate within the endolysosomes of tissues across the body, culminating in severe multiorgan dysfunctions that affect primarily the brain, eyes, liver, muscles, pancreas, and kidneys.
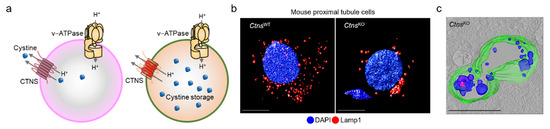
Figure 3.
Cystinosin: from endolysosome to disease. (a) Cystinosin works in tandem with v-ATPase to transport cystine from the endolysosome, and its absence triggers (b,c) the storage of cystine within enlarged endolysosomes in primary proximal tubule cells derived from mouse kidneys. Adapted from Festa et al. [62].
The renal Fanconi syndrome is often the first manifestation of cystinosis, usually presenting within the first year of life and characterized by the early and severe dysfunction of PT cells, highlighting the unique vulnerability of kidney cell types [57]. Infantile (MIM #219800) and juvenile (MIM #219900) forms of cystinosis represent a frequent cause of inherited PT dysfunction and renal Fanconi syndrome. In addition, children with cystinosis display early deposition of cystine crystals in the cornea, thereby causing photophobia and painful corneal erosions [58]. In their second to third decade of life, patients with cystinosis can also develop hypothyroidism, hypogonadism, diabetes, myopathy, and deterioration of fine vision and decline of the central nervous system [59,60,61].
The only available strategy to counteract cystine storage is the oral administration of cysteamine, which allows cystine to exit from the endolysosomes [63]. However, cysteamine treatment is hampered by side effects and poor tolerance, and it does not prevent or treat PT dysfunction [63]. Stem cells and gene therapy treatments, which rescued the eyes, kidneys, and thyroid in Ctns knockout (KO) mice, and are currently being tested in cystinosis patients, are limited by complexity and high costs [57,64]. Thus, there is an urgent need to identify safe and cost-effective therapeutics for patients with cystinosis. The advent of a growing number of animal and cell-based models that reproduce the human disease pathology has improved our understanding of disease mechanisms and the cellular pathways underlying PT dysfunction and renal Fanconi syndrome, ultimately accelerating the discovery and development of promising new therapeutic approaches. This progress and the recent discoveries are discussed in detail in the next sections.
4. Insights into Disease Pathways—The Role of Impaired Autophagy
Recent studies using a CtnsKO mouse model that recapitulates multiple features of cystinosis have suggested that the absence of cystinosin in PT cells leads cystine to accumulate within enlarged endolysosomes that move to the perinuclear region and exhibit structural, trafficking, and functional defects (Figure 3b,c). This presumably activates a signaling cascade that drives abnormal cell growth and apical dedifferentiation, ultimately leading to defective receptor-mediated endocytosis and urinary loss of LMW proteins in vivo [65,66]. The tight integration between endolysosome system, regulation of growth signaling pathways, and maintenance of PT differentiation suggests that endolysosome dysfunction driven by cystinosin loss might disrupt the homeostasis in cystinosis-affected PT cells. How, mechanistically, the absence of cystinosin wreaks havoc on cellular homeostasis has remained incompletely understood.
Accumulating evidence suggests that the endolysosome can capture and degrade aged and/or malfunctioning cellular constituents through macroautophagy/autophagy—an evolutionarily conserved, “self–eating” process through which potentially dangerous cytosolic entities are sequestered within autophagosomes and subsequently delivered to endolysosomes for degradation [1,67,68]. This homeostatic process is particularly active in PT cells, whose intense reabsorptive properties require the maintenance of the mitochondrial network [25,69]. Given the structural and functional defects of cystinosis-affected endolysosomes and considering that autophagy relies on catabolic properties of endolysosomes, the cystinosin loss-induced storage of cystine might compromise the degradation of autophagosomes in kidney PT cells. Using differentiated PT cell culture systems, which closely reproduce the key features of the in vivo disease phenotype [62,70] in combination with bona fide autophagy biosensors and assay technologies, Festa and colleagues revealed that primary cells derived from microdissected PT segments of CtnsKO (henceforward referred to as mPTCs) mice fail to dismantle Lc3b-flagged autophagosomes [62,71]. Evidence supporting incomplete autophagy flux in cystinosin-deficient PT cells include the following: (i) abnormally high numbers of autophagosomes under-normal growth conditions; (ii) failure to clear autophagic vesicles (AVs) formed after starvation-induced autophagy, mimicking bafilomycin (Bfn) A1 action; (iii) inability of BfnA1 to further elevate the Lc3-II and Sqstm1/p62 protein levels and the numbers of punctate Lc3b-flagged autophagosomes; (iv) and impaired degradation of the resting autophagosomes in CtnsKO mPTCs with a selective PI3K3/Vps34 inhibitor [62,71]. Similar autophagy defects (e.g., accumulation of AVs and their defective degradation, and increased p62 levels) have also been observed in several other LSDs (Table 1 [72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90]). Defects in autophagy–endolysosome degradative pathways, which are also encountered in ctns-deficient zebrafish, are reverted by exogenously expressing wild-type cystinosin in mutant cells [62,71]. Of note, treatment with the oral drug cysteamine, which efficiently depletes the storage of cystine within endolysosomes, does not restore the functioning of endolysosomes nor the catabolic autophagy in patient cells [91,92]. Thus, cystinosin—beyond its function in cystine transport—might act as an evolutionarily conserved, metabolic rheostat that regulates the response of endolysosomes to the arrival of endocytosed and autophagy cargoes, hence safeguarding the integrity and the physiological homeostasis of kidney tubule cells [62,71].

Table 1.
Autophagy pathways in lysosomal storage diseases.
Conversely, Napolitano and colleagues indicate that the macroautophagy/autophagy flux seems to be fully normal, despite the increased number of autophagosomes in mutant cells [93]. Thus, it is plausible that elevated numbers of autophagosome could stem from compensatory mechanisms due to defects in CMA. Indeed, studies in cultured cells (e.g., hepatocytes and T cells) have indicated a functional crosstalk between macroautophagy and CMA, whereby cells respond to the failure of one of these pathways by activating the other [94,95]. In line with this concept, studies on lysosomes purified from livers of starved CtnsKO mice unveil defects in the degradation of glyceraldehyde-3-posphate dehydrogenase (GADPH)—a well-established substrate for the CMA pathway [51]. These abnormalities are reflected by dislodgement of the lysosomal receptor Lamp2a required for CMA from its natural binding partner Lamp1 and co-localization with Rab11a-positive recycling endosomes. These trafficking defects appear to be specific for Lamp2a, as other Lamp proteins can normally reach the endolysosomes in cystinosis-affected fibroblasts [93]. The small GTPase Ras-related protein Rab-11A (RAB11), and the RAB7 effector Rab-interacting lysosomal protein (RILP), seem to be part of this trafficking machinery, as the correction of the lower levels observed for both proteins in patient cells is sufficient to repair LAMP2A mistargeting, and hence the CMA pathway in diseased cells [96]. Such an apparent discrepancy might be attributed to: (i) differential biochemistry of distinct cell types in the body; (ii) turnover rates of autophagy cargos and substrates; (iii) cell type and tissue/organ-dependent adaptative responses to counteract the primary storage defect, and (iv) whether the cells are renewing or terminally differentiated; and (v) differential threshold triggered by cystine storage to induce dysfunction in distinct cell types, and affected tissues and organs. However, in some studies, exogenous expression of the dynein subunit, e.g., DYNC1LI2 (dynein, cytoplasmic 1 light intermediate chain 2)—a key cytoskeletal motor protein involved intracellular transport of cargo, organelle trafficking, mitotic spindle assembly, and positioning—rescues the localization of the chaperone-mediated autophagy (CMA) receptor LAMP2A, CMA activity, and the cellular homeostasis in cystinosis-affected PT cells [97]. Regardless of the mechanisms involved, the concept that defects in endolysosomes and autophagy pathways might contribute to cystinosis pathogenesis is in line with recent studies that indicate an accumulation of autophagosomes engulfing damaged and/or dysfunction mitochondria, and increased formation of aggregate-prone SQSTM1/p62 inclusions in both kidney biopsies [98,99] and patient cells [100].
5. Autophagy, Mitochondria, and Epithelial Dysfunction in Cystinosis
The conjugation of defective endolysosome dynamics and impaired catabolic properties is strikingly similar to cellular alterations stemming from the accumulation of monoclonal light chains (κLCs) within the endolysosomes of PT cells, causing a similar epithelial dysfunction [7]. Furthermore, the uncontrolled increase in the endolysosomal PtdIns(4,5)P2 pool that arises from loss-of-function of the PtdIns(4,5)P2 5-phosphatase OCRL triggers endolysosome dysfunction and autophagosome accumulation of patients with Lowe syndrome [102,103]—another rare inherited disorders causing PT dysfunction and renal Fanconi syndrome. The storage of either cystine or κLCs or PtdIns(4,5)P2 might thus tamp down the homeostasis and transport functions of PT cells, emphasizing the crucial role of autophagy–endolysosome degradative systems in preserving the homeostasis and physiology of the kidney PT.
As a direct consequence of defective autophagy–endolysosome degradation systems, CtnsKO PT cells remarkably accumulate SQSTM1- and ubiquitin-forming aggregates with damaged and/or dysfunctional mitochondria within enlarged, non-degradative endolysosomes, ultimately overproducing mitochondrial-derived reactive oxygen species (ROS) [62,71]. Genetic (e.g., short hairpin RNA interference targeting Atg7) and pharmacological (e.g., inhibition of Beclin1/Vps-34 complex by using SAR-405 or Spautin-1) suppression of autophagy dampens the functioning of the mitochondrial network, inducing oxidative stress, while repressing the receptor-mediated endocytosis and transport properties of PT cells [62,71]. This evidence further reinforces the mechanistic connection between defective mitochondrial quality control, oxidative stress, and cellular dysfunction. Thus, the maintenance of degradative autophagy might serve as a bona fide (homeostasis-modifying) process that regulates the identity of the kidney tubule cells. How, mechanistically, defects in degradative autophagy disrupting the differentiation of PT cells have remained to be fully elucidated.
Recent insights have illuminated the biological functions of tight junction proteins in safeguarding the epithelial cell behavior and phenotype. In particular, tight junction adaptor protein 1 (Tjp1) represses the nuclear translocation of an Y box binding protein 3 (Ybx3) —a transcriptional factor that promotes cell proliferation while repressing PT differentiation during kidney development [104]. As oxidative stress damages tight junction integrity [105], the excessive mitochondrial ROS induced by cystinosin loss might trigger an abnormal activation of the tight junction associated Ybx3 (Y box binding protein) signaling, which would, in turn, lead to epithelial dysfunction in cystinosis PT cells. In line with this model, increased levels of mitochondrial ROS stimulate Gna12/Ga12-SRC-mediated phosphorylation of Tjp1 and its subsequent misrouting to enlarged, non-degradative endolysosomes. The disruption of tight junction integrity triggers the hyperactivation of tight junction-associated Ybx3 signaling, with increased proliferation (e.g., Ccnd1 and Pcna) and reduced apical differentiation (e.g., Lrp2), ultimately disabling receptor-mediated endocytosis and epithelial functions in CtnsKO cells [62,71] (Figure 4). Gain- and loss-of-function approaches targeting Gna12 or Tjp1 or Ybx3, or pharmacological interventions impeding activation of the Gna12-Src-directed signaling (e.g., with the mitochondrial-targeted antioxidant Mito-TEMPO or with the SRC inhibitor SU6656) restore epithelial functions in CtnsKO cells [62,71]. By regulating autophagy and the Tjp1-Yxb3 signaling, the crosstalk between cystinosin and the endolysosome system might thus dictate the balance between proliferation and differentiation of PT cells, and hence their role in homeostasis.
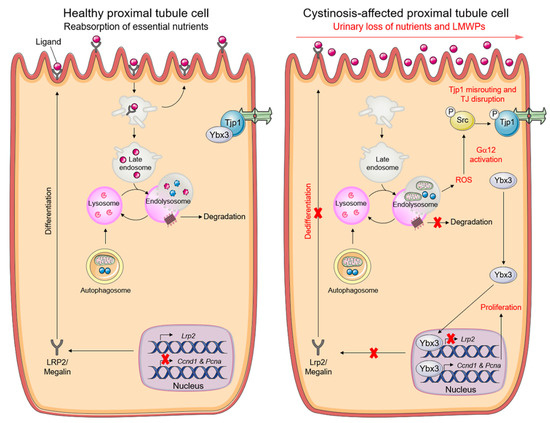
Figure 4.
Pathogenic cascade driving PT dysfunction in cystinosis cells. Graphical schematic illustrating that cystinosin-deficient PT cells accumulate dysfunctional mitochondria and reactive-oxygen species (ROS), triggering an abnormal activation of the tight junction—associated signalling that stimulates proliferation while suppressing apical dedifferentiation. Reprinted with permission from Ref. [6]. Copyright 2021 Springer Nature.
6. Pharmacological Modulation of Autophagy as a Targetable Pathway in Cystinosis
There are no curative treatments for cystinosis, and the current supportive care approaches have substantially decreased mortality and overall morbidity. For example, supplementation with water, bicarbonate, citrate, phosphate, salts, and vitamin D can rapidly attenuate the metabolic complications associated with renal Fanconi syndrome, and hence maintain an adequate body fluid and electrolyte homeostasis [6,57]. Beyond management care, patients with cystinosis can benefit from treatment with cysteamine [58]—an FDA-approved drug that depletes the endolysosomal cystine storage by cleaving cystine into free cysteine and cysteamine–cysteine mixed sulphide. These metabolites are subsequently exported from the endolysosome to cytoplasm through cationic amino acid transporter 2 (PQLC2), which spans the endolysosomal membrane [106]. Despite an improvement in patients’ quality of life, treatment is hampered by adverse effects, poor tolerance, and a strict dosing schedule, and it does not prevent or treat the renal Fanconi syndrome and kidney failure [58,63]. Therefore, there is an urgent need to yield promising new targetable interventions in the early course of cystinosis.
The molecular understanding of regulatory circuitries coupling endolysosome disease, autophagy, and epithelial dysfunction might thus guide the discovery and development of targeted therapeutics for cystinosis patients [57,64]. In this case, interventions that are aimed to target each step of the pathogenic cascade might mediate beneficial effects and potentially counteract the homeostatic perturbations imposed by cystinosin loss and the resulting cystine storage. For example, small molecule compounds that either activate CMA [107] or boost the excretion [108] of cystine-loaded endolysosomes might ameliorate clinical outcomes if they are used concomitantly with the cystine-depleting drug cysteamine [93,109] (Figure 5). Boosting CMA with small-molecule activators (e.g., QX77) increases the lifetime of the endocytic receptor megalin at the plasma membrane, ultimately improving the epithelial functions in human PT cells lacking cystinosin (e.g., CRISPR-Cas9-induced gene deletion) [101]. Consistent with these observations, combinatorial strategies using an mTORC1 inhibitor (e.g., everolimus) and cysteamine rescues the homeostasis and functioning of autophagy–endolysosome degradation systems in cystinosis patient-derived pluripotent stem cells (iPSCs) and kidney organoid models of the disease [110] (Figure 5).
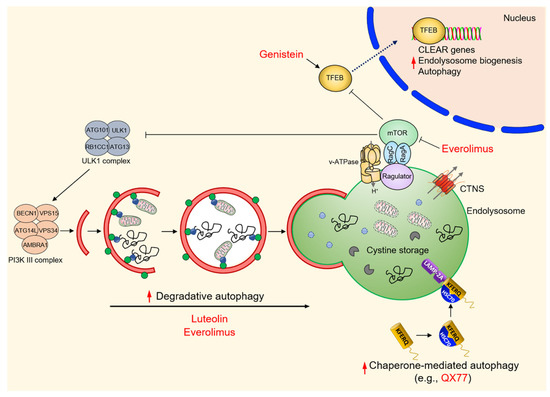
Figure 5.
Summary of emerging therapeutic strategies targeting autophagy pathways in cystinosis.
Despite normal mTORC1 activity in cystinotic iPSCs and their derived kidney organoids, the molecular mechanisms behind the beneficial effects of the combo treatment remain largely elusive. In this setting, a potential mediator could be the activation of TFEB—a master regulator that controls the expression of the genes involved in autophagy and endolysosome biogenesis [2,34,35] (Figure 5). Recent work showing that cystinosin might physically interact with many components of the v-ATPase–Ragulator–Rag complex [110,111], which regulates the mTORC1 lifetime and its activation at the surface of the endolysosome, and that the reconstitution of TFEB signaling stimulates the catabolic properties of endolysosomes and the completion of autophagy in conditionally immortalized PT epithelial cells (ciPTEC) derived from the urine of a cystinotic patient [92], further substantiate the concept. Indeed, the pharmacological induction (e.g., genistein) of nuclear translocation and the activation of TFEB-dependent transcriptional programs has recently been shown to empty cystine storage, to restore the functioning of endolysosomes and degradative autophagy, and to improve the processing of endocytosed cargos in cystinotic ciPTEC [92] (Figure 5).
Recently, drug discovery and repurposing strategies are gaining momentum as a default tool for providing affordable therapies in rare inherited diseases [112,113]. With the possibility to screen approved and investigational products, the process is well adapted to the curiosity-driven research culture in academia, hence mitigating the risk inherent in preclinical drug discovery. With this lag in mind, De Leo and colleagues recently identified small molecule drug candidates [100] that decrease the accumulation of the autophagy substrate p62/SQSTM1 and restore the autophagy–endolysosome degradative pathways, which are compromised in different models and cell systems of cystinosis [100]. Among several positive hits, luteolin—a natural flavonoid that is present in various fruits and vegetables—has emerged as the most interesting candidate. This compound has a good safety profile, owing to its similarity to genistein, and improves the endolysosome-mediated degradation of autophagy cargoes and substrates, including damaged and dysfunctional (ROS-overproducing) mitochondria [100] (Figure 5). In addition, treating cystinotic ciPTEC, mPTCs derived from mouse CtnsKO kidneys, and zebrafish models of cystinosis with luteolin not only repaired endolysosomes, autophagy degradation, and mitochondrial redox homeostasis and cellular distress, but also restored megalin expression at the plasma membrane, ultimately stimulating protein absorption and hence transport functions [100]. These findings extend previous observations demonstrating that structural and functional deformities of the kidney PT could be delayed in CtnsKO mice by administrating mitochondria-targeted ROS scavengers such as mitoquinone [114] or mito-TEMPO [62]. Thus, the modulation of autophagy–endolysosome degradative systems might offer a promising new therapeutic avenue not only for cystinosis, but also for other currently intractable diseases related to endolysosome storage.
7. Concluding Remarks
The maintenance of a healthy endolysosomal system is particularly crucial for preserving the homeostasis and physiology of kidney tubule cells, and loss-of-function mutations that impair the functioning of the endolysosome system can invariably lead to PT dysfunction and kidney disease. Rare inherited defects in an endolysosome-residing protein and the storage materials, as exemplified here by cystinosis, might disable autophagy and organelle quality control, triggering a level of mitochondrial distress that drives the dysfunction of the kidney PT. Further studies will be required for understanding whether other LSDs might have various degrees of PT dysfunction and kidney disease. In most cases, kidney disease manifestations might be overshadowed by more severe symptoms affecting the brain, underestimating the prevalence of kidney involvement in these disorders.
The mechanisms by which cystinosin deficiency wreaks havoc on homeostasis and function of the endolysosome system remain largely elusive. These defects could stem from defects in mannose-6-phosphate (M6P)-dependent trafficking [115] or megalin-directed reuptake of filtered lysosomal cathepsins [116] or endolysosome acidification [117]. Alternatively, the storage of cystine wrought by cystinosin loss might affect the folding of disulphide-bonded substrates for endoproteolytic attack or thiol-active catalytic sites of endolysosomal cathepsins, ultimately affecting processing and their lasting maturation [57,118].
As the endolysosome is the site for nutrient sensing and the activation of mTORC1 signaling—the master regulator that represses autophagy and endolysosome biogenesis—it will be important to evaluate whether cystinosin deficiency and cystine storage might contribute to hyperactive mTORC1. This might in turn inhibit endolysosome and autophagosome biogenesis, thus generating a vicious cycle that boosts metabolic dyshomeostasis and dysfunction in cystinosis cells. Although cystinosin could physically interact with many components of the v-ATPase–Ragulator–Rag complex [111] that regulates mTORC1 activity, the contribution of dysregulated nutrient sensing and mTOR signaling to disease pathogenesis remains an open question. The recent development of model organisms [62] and primary PT cell culture systems [65,70], which closely reproduce the key features of the disease phenotype, and mass spectrometry-based profiling of intact endolysosomes [119], presents an opportunity to address this critical point.
Decline in endolysosome function and mitochondrial autophagy are clear hallmarks of ageing, and correlate with metabolic dysfunction [3]. Indeed, the behavior of “aged” endolysosomes mimics the cellular phenotypes encountered in cystinosis and other LSD cells. We suspect that the dysregulation of adaptive response to mitochondrial distress might also contribute to maladaptation and disease in patients with cystinosis, and this will require further studies to understand the effects of CTNS mutations on organelle repair pathways, such as mitochondrial unfolded protein response (UPRmt) and mitochondrial biogenesis. The increasing power of organelle-specific purification and profiling via proteomic, lipidomic, and metabolomic-based approaches will be useful in filling these missing knowledge gaps. These questions are just examples of all the exciting work that lies ahead to comprehensively dissect the biological functions of cystinosin in the context of tissue homeostasis and disease.
A current challenge is to translate the knowledge gained from fundamental studies of endolysosome biology to the treatment of cystinosis and other endolysosome-related diseases. In this regard, the use of informative preclinical models, coupled with improved knowledge of disease signatures and the recent advances in multi-omics technologies, might accelerate the discovery and development of “first-in-class” therapeutics that can halt the progression of cystinosis, as well as other rare and more common diseases related to endolysosome dysfunction.
Author Contributions
L.R.R. and A.L. conceptualized the article. L.R.R., E.D.L. and A.L. wrote the article. D.N. and E.D.L. researched the data for the article and drew the figures under the supervision of L.R.R. and A.L. All of the authors contributed substantially to the discussion of the content and reviewed the article before submission. L.R.R. and A.L. edited the article. All authors have read and agreed to the published version of the manuscript.
Funding
A.L. is supported by Cystinosis Research Foundation (Irvine, CA, USA) and the Swiss National Centre of Competence in Research (NCCR) Kidney Control of Homeostasis (Kidney.CH). A.L. received financial support from the University Research Priority Program of the University of Zurich (URPP) ITINERARE—Innovative Therapies in Rare Diseases. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication. L.R. and E.D.L. are supported by Cystinosis Research Foundation (Irvine, CA, USA) and Italian Ministry of Health.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Levine, B.; Kroemer, G. Biological Functions of Autophagy Genes: A Disease Perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ballabio, A.; Bonifacino, J.S. Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat. Rev. Mol. Cell Biol. 2020, 21, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nat. Aging 2021, 1, 634–650. [Google Scholar] [CrossRef]
- Shin, H.R.; Zoncu, R. The Lysosome at the Intersection of Cellular Growth and Destruction. Dev. Cell 2020, 54, 226–238. [Google Scholar] [CrossRef]
- van der Wijst, J.; Belge, H.; Bindels, R.J.M.; Devuyst, O. Learning physiology from inherited kidney disorders. Physiol. Rev. 2019, 99, 1575–1653. [Google Scholar] [CrossRef] [PubMed]
- Festa, B.P.; Berquez, M.; Nieri, D.; Luciani, A. Endolysosomal Disorders Affecting the Proximal Tubule of the Kidney: New Mechanistic Insights and Therapeutics. Rev. Physiol. Biochem. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Luciani, A.; Sirac, C.; Terryn, S.; Javaugue, V.; Prange, J.A.; Bender, S.; Bonaud, A.; Cogné, M.; Aucouturier, P.; Ronco, P.; et al. Impaired lysosomal function underlies monoclonal light chain–associated renal Fanconi syndrome. J. Am. Soc. Nephrol. 2016, 27, 2049–2061. [Google Scholar] [CrossRef]
- Gu, X.; Yang, H.; Sheng, X.; Ko, Y.A.; Qiu, C.; Park, J.; Huang, S.; Kember, R.; Judy, R.L.; Park, J.; et al. Kidney disease genetic risk variants alter lysosomal beta-mannosidase (MANBA) expression and disease severity. Sci. Transl. Med. 2021, 13, eaaz1458. [Google Scholar] [CrossRef] [PubMed]
- Pattaro, C.; Teumer, A.; Gorski, M.; Chu, A.Y.; Li, M.; Mijatovic, V.; Garnaas, M.; Tin, A.; Sorice, R.; Li, Y.; et al. Genetic associations at 53 loci highlight cell types and biological pathways relevant for kidney function. Nat. Commun. 2016, 7, 10023. [Google Scholar] [CrossRef] [PubMed]
- Wuttke, M.; Li, Y.; Li, M.; Sieber, K.B.; Feitosa, M.F.; Gorski, M.; Tin, A.; Wang, L.; Chu, A.Y.; Hoppmann, A.; et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 2019, 51, 957–972. [Google Scholar] [CrossRef] [Green Version]
- Christensen, E.I.; Birn, H. Megalin and cubilin: Multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 256–266. [Google Scholar] [CrossRef]
- Eshbach, M.L.; Weisz, O.A. Receptor-Mediated Endocytosis in the Proximal Tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: From experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, S.K.; Keyel, P.A.; Hawryluk, M.J.; Agostinelli, N.R.; Watkins, S.C.; Traub, L.M. Disabled-2 exhibits the properties of a cargo-selective endocytic clathrin adaptor. EMBO J. 2002, 21, 4915–4926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gekle, M. Renal tubule albumin transport. Annu. Rev. Physiol. 2005, 67, 573–594. [Google Scholar] [CrossRef] [PubMed]
- Kaksonen, M.; Roux, A. Mechanisms of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol. 2018, 19, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Faundez, V.; Hartzell, H.C. Intracellular chloride channels: Determinants of function in the endosomal pathway. Sci. STKE 2004, 2004, re8. [Google Scholar] [CrossRef] [Green Version]
- Abbas, Y.M.; Wu, D.; Bueler, S.A.; Robinson, C.V.; Rubinstein, J.L. Structure of V-ATPase from the mammalian brain. Science 2020, 367, 1240–1246. [Google Scholar] [CrossRef]
- Devuyst, O.; Luciani, A. Chloride transporters and receptor-mediated endocytosis in the renal proximal tubule. J. Physiol. 2015, 593, 4151–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Ren, D. Lysosomal physiology. Annu. Rev. Physiol. 2015, 77, 57–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurtado-Lorenzo, A.; Skinner, M.; El Annan, J.; Futai, M.; Sun-Wada, G.H.; Bourgoin, S.; Casanova, J.; Wildeman, A.; Bechoua, S.; Ausiello, D.A.; et al. V-ATPase interacts with ARNO and Arf6 in early endosomes and regulates the protein degradative pathway. Nat. Cell Biol. 2006, 8, 124–126. [Google Scholar] [CrossRef]
- Cullen, P.J.; Steinberg, F. To degrade or not to degrade: Mechanisms and significance of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2018, 19, 679–696. [Google Scholar] [CrossRef]
- Pohl, C.; Dikic, I. Cellular quality control by the ubiquitin-proteasome system and autophagy. Science 2019, 366, 818–822. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Mizushima, N. Autophagy and human diseases. Cell Res. 2014, 24, 69–79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, C.; Livingston, M.J.; Liu, Z.; Dong, Z. Autophagy in kidney homeostasis and disease. Nat. Rev. Nephrol. 2020, 16, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Petroni, G.; Amaravadi, R.K.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cadwell, K.; Cecconi, F.; Choi, A.M.K.; et al. Autophagy in major human diseases. EMBO J. 2021, 40, e108863. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef] [PubMed]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi1, L.F.; Bottino1, A.M.S.; Dueholm1, R.; Niehaus1, A.; Overwijn1, D.; Priso Fils1, A.C.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef] [PubMed]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Cho, K.F.; Rappold, R.; Thrun, A.; Tofaute, M.; Kim, D.J.; Moldavski, O.; Hurley, J.H.; Zoncu, R. A nutrient-induced affinity switch controls mTORC1 activation by its Rag GTPase–Ragulator lysosomal scaffold. Nat. Cell Biol. 2018, 20, 1052–1063. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Jiang, X.; Li, B.; Yang, H.J.; Miller, M.; Yang, A.; Dhar, A.; Pavletich, N.P. Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 2017, 552, 368–373. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Settembre, C.; Di Malta, C.; Polito, V.A.; Arencibia, M.G.; Vetrini, F.; Erdin, S.; Erdin, S.U.; Huynh, T.; Medina, D.; Colella, P.; et al. TFEB links autophagy to lysosomal biogenesis. Science 2011, 332, 1429–1433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, L.; McPhee, C.K.; Zheng, L.; Mardones, G.A.; Rong, Y.; Peng, J.; Mi, N.; Zhao, Y.; Liu, Z.; Wan, F.; et al. Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature 2010, 465, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J.; et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.C.; Ysselstein, D.; Krainc, D. Mitochondria-lysosome contacts regulate mitochondrial fission via RAB7 GTP hydrolysis. Nature 2018, 554, 382–386. [Google Scholar] [CrossRef]
- Grahammer, F.; Ramakrishnan, S.K.; Rinschen, M.M.; Larionov, A.A.; Syed, M.; Khatib, H.; Roerden, M.; Sass, J.O.; Helmstaedter, M.; Osenberg, D.; et al. MTOR regulates endocytosis and nutrient transport in proximal tubular cells. J. Am. Soc. Nephrol. 2017, 28, 230–241. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Mizushima, N.; Levine, B. Autophagy in Human Diseases. N. Engl. J. Med. 2020, 383, 1564–1576. [Google Scholar] [CrossRef] [PubMed]
- Sica, V.; Galluzzi, L.; Bravo-San Pedro, J.M.; Izzo, V.; Maiuri, M.C.; Kroemer, G. Organelle-Specific Initiation of Autophagy. Mol. Cell 2015, 59, 522–539. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Jung, C.H.; Seo, M.; Otto, N.M.; Grunwald, D.; Kim, K.H.; Moriarity, B.; Kim, Y.M.; Starker, C.; Nho, R.S.; et al. The ULK1 complex mediates MTORC1 signaling to the autophagy initiation machinery via binding and phosphorylating ATG14. Autophagy 2016, 12, 547–564. [Google Scholar] [CrossRef]
- Park, J.M.; Seo, M.; Jung, C.H.; Grunwald, D.; Stone, M.; Otto, N.M.; Toso, E.; Ahn, Y.; Kyba, M.; Griffin, T.J.; et al. ULK1 phosphorylates Ser30 of BECN1 in association with ATG14 to stimulate autophagy induction. Autophagy 2018, 14, 584–597. [Google Scholar] [CrossRef] [Green Version]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 153, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Wang, Q.J.; Li, X.; Yan, Y.; Backer, J.M.; Chait, B.T.; Heintz, N.; Yue, Z. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nat. Cell Biol. 2009, 11, 468–476. [Google Scholar] [CrossRef]
- Walczak, M.; Martens, S. Dissecting the role of the Atg12-Atg5-Atg16 complex during autophagosome formation. Autophagy 2013, 9, 424–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havasi, A.; Dong, Z. Autophagy and Tubular Cell Death in the Kidney. Semin. Nephrol. 2016, 36, 174–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamoto, T.; Takabatake, Y.; Kimura, T.; Takahashi, A.; Namba, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; Kitamura, H.; et al. Time-dependent dysregulation of autophagy: Implications in aging and mitochondrial homeostasis in the kidney proximal tubule. Autophagy 2016, 12, 801–813. [Google Scholar] [CrossRef] [Green Version]
- Dikic, I.; Elazar, Z. Mechanism and medical implications of mammalian autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 349–364. [Google Scholar] [CrossRef]
- Kaushik, S.; Cuervo, A.M. The coming of age of chaperone-mediated autophagy. Nat. Rev. Mol. Cell Biol. 2018, 19, 365–381. [Google Scholar] [CrossRef]
- Mijaljica, D.; Prescott, M.; Devenish, R.J. Microautophagy in mammalian cells: Revisiting a 40-year-old conundrum. Autophagy 2011, 7, 673–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lawrence, R.E.; Zoncu, R. The lysosome as a cellular centre for signalling, metabolism and quality control. Nat. Cell Biol. 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Eckardt, K.U.; Coresh, J.; Devuyst, O.; Johnson, R.J.; Köttgen, A.; Levey, A.S.; Levin, A. Evolving importance of kidney disease: From subspecialty to global health burden. Lancet 2013, 382, 158–169. [Google Scholar] [CrossRef] [Green Version]
- Platt, F.M. Emptying the stores: Lysosomal diseases and therapeutic strategies. Nat. Rev. Drug Discov. 2018, 17, 133–150. [Google Scholar] [CrossRef]
- Town, M.; Jean, G.; Cherqui, S.; Attard, M.; Forestier, L.; Whitmore, S.A.; Gallen, D.F.; Gribouval, O.; Broyer, M.; Bates, G.P.; et al. A novel gene encoding an integral membrane protein is mutated in nephropathic cystinosis. Nat. Genet. 1998, 18, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Cherqui, S.; Courtoy, P.J. The renal Fanconi syndrome in cystinosis: Pathogenic insights and therapeutic perspectives. Nat. Rev. Nephrol. 2017, 13, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Gahl, W.A.; Thoene, J.G.; Schneider, J.A. Cystinosis. N. Engl. J. Med. 2002, 347, 111–121. [Google Scholar] [CrossRef]
- Nesterova, G.; Gahl, W. Nephropathic cystinosis: Late complications of a multisystemic disease. Pediatr. Nephrol. 2008, 23, 863–878. [Google Scholar] [CrossRef] [PubMed]
- Trauner, D.A.; Williams, J.; Ballantyne, A.O.; Spilkin, A.M.; Crowhurst, J.; Hesselink, J. Neurological impairment in nephropathic cystinosis: Motor coordination deficits. Pediatr. Nephrol. 2010, 25, 2061–2066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viltz, L.; Trauner, D.A. Effect of age at treatment on cognitive performance in patients with cystinosis. J. Pediatr. 2013, 163, 489–492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Festa, B.P.; Chen, Z.; Berquez, M.; Debaix, H.; Tokonami, N.; Prange, J.A.; van de Hoek, G.; Alessio, C.; Raimondi, A.; Nevo, N.; et al. Impaired autophagy bridges lysosomal storage disease and epithelial dysfunction in the kidney. Nat. Commun. 2018, 9, 161. [Google Scholar] [CrossRef] [PubMed]
- Brodin-Sartorius, A.; Tête, M.J.; Niaudet, P.; Antignac, C.; Guest, G.; Ottolenghi, C.; Charbit, M.; Moyse, D.; Legendre, C.; Lesavre, P.; et al. Cysteamine therapy delays the progression of nephropathic cystinosis in late adolescents and adults. Kidney Int. 2012, 81, 179–189. [Google Scholar] [CrossRef] [Green Version]
- Jamalpoor, A.; Othman, A.; Levtchenko, E.N.; Masereeuw, R.; Janssen, M.J. Molecular Mechanisms and Treatment Options of Nephropathic Cystinosis. Trends Mol. Med. 2021, 27, 673–686. [Google Scholar] [CrossRef] [PubMed]
- Raggi, C.; Luciani, A.; Nevo, N.; Antignac, C.; Terryn, S.; Devuyst, O. Dedifferentiation and aberrations of the endolysosomal compartment characterize the early stage of nephropathic cystinosis. Hum. Mol. Genet. 2014, 23, 2266–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaide Chevronnay, H.P.; Janssens, V.; Van Der Smissen, P.; N’Kuli, F.; Nevo, N.; Guiot, Y.; Levtchenko, E.; Marbaix, E.; Pierreux, C.E.; Cherqui, S.; et al. Time course of pathogenic and adaptation mechanisms in cystinotic mouse kidneys. J. Am. Soc. Nephrol. 2014, 25, 1256–1269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galluzzi, L.; Baehrecke, E.H.; Ballabio, A.; Boya, P.; Bravo-San Pedro, J.M.; Cecconi, F.; Choi, A.M.; Chu, C.T.; Codogno, P.; Colombo, M.I.; et al. Molecular definitions of autophagy and related processes. EMBO J. 2017, 36, 1811–1836. [Google Scholar] [CrossRef]
- Fougeray, S.; Pallet, N. Mechanisms and biological functions of autophagy in diseased and ageing kidneys. Nat. Rev. Nephrol. 2015, 11, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Berquez, M.; Krohn, P.; Luciani, A.; Devuyst, O. Receptor-mediated endocytosis and differentiation in proximal tubule cell systems. J. Am. Soc. Nephrol. 2021, 32, 1265–1267. [Google Scholar] [CrossRef] [PubMed]
- Luciani, A.; Festa, B.P.; Chen, Z.; Devuyst, O. Defective autophagy degradation and abnormal tight junction-associated signaling drive epithelial dysfunction in cystinosis. Autophagy 2018, 14, 1157–1159. [Google Scholar] [CrossRef]
- Raben, N.; Hill, V.; Shea, L.; Takikita, S.; Baum, R.; Mizushima, N.; Ralston, E.; Plotz, P. Suppression of autophagy in skeletal muscle uncovers the accumulation of ubiquitinated proteins and their potential role in muscle damage in Pompe disease. Hum. Mol. Genet. 2008, 17, 3897–3908. [Google Scholar] [CrossRef] [Green Version]
- Raben, N.; Schreiner, C.; Baum, R.; Takikita, S.; Xu, S.; Xie, T.; Myerowitz, R.; Komatsu, M.; Van Der Meulen, J.H.; Nagaraju, K.; et al. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—Murine Pompe disease. Autophagy 2010, 6, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, Y.; Guhde, G.; Suter, A.; Eskelinen, E.L.; Hartmann, D.; Lüllmann-Rauch, R.; Janssen, P.M.L.; Blanz, J.; Von Figura, K.; Saftig, P. Accumulation of autophagic vacuoles and cardiomyopathy LAMP-2-deficient mice. Nature 2000, 406, 902–906. [Google Scholar] [CrossRef]
- Settembre, C.; Fraldi, A.; Jahreiss, L.; Spampanato, C.; Venturi, C.; Medina, D.; de Pablo, R.; Tacchetti, C.; Rubinsztein, D.C.; Ballabio, A. A block of autophagy in lysosomal storage disorders. Hum. Mol. Genet. 2008, 17, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Fraldi, A.; Annunziata, F.; Lombardi, A.; Kaiser, H.J.; Medina, D.L.; Spampanato, C.; Fedele, A.O.; Polishchuk, R.; Sorrentino, N.C.; Simons, K.; et al. Lysosomal fusion and SNARE function are impaired by cholesterol accumulation in lysosomal storage disorders. EMBO J. 2010, 29, 3607–3620. [Google Scholar] [CrossRef] [Green Version]
- Tessitore, A.; Pirozzi, M.; Auricchio, A. Abnormal autophagy, ubiquitination, inflammation and apoptosis are dependent upon lysosomal storage and are useful biomarkers of mucopolysaccharidosis VI. Pathogenetics 2009, 2, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, D.C.; Milenkovic, L.; Beier, S.M.; Manuel, H.; Buchanan, J.A.; Scott, M.P. Cell-autonomous death of cerebellar purkinje neurons with autophagy in niemann-pick type C disease. PLoS Genet. 2005, 1, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, C.D.; Elrick, M.J.; Lieberman, A.P. Tau deletion exacerbates the phenotype of Niemann-Pick type C mice and implicates autophagy in pathogenesis. Hum. Mol. Genet. 2009, 18, 956–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, Y.; Liou, B.; Ran, H.; Skelton, M.R.; Williams, M.T.; Vorhees, C.V.; Kitatani, K.; Hannun, Y.A.; Witte, D.P.; Xu, Y.H.; et al. Neuronopathic Gaucher disease in the mouse: Viable combined selective saposin C deficiency and mutant glucocerebrosidase (V394L) mice with glucosylsphingosine and glucosylceramide accumulation and progressive neurological deficits. Hum. Mol. Genet. 2010, 19, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chévrier, M.; Brakch, N.; Lesueur, C.; Genty, D.; Ramdani, Y.; Moll, S.; Djavaheri-Mergny, M.; Brasse-Lagnel, C.; Laquerrière, A.; Barbey, F.; et al. Autophagosome maturation is impaired in Fabry disease. Autophagy 2010, 6, 589–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vergarajauregui, S.; Connelly, P.S.; Daniels, M.P.; Puertollano, R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet. 2008, 17, 2723–2737. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, B.; Mesires, N.T.; Kennedy, J.C.; Curcio-Morelli, C.; Laplante, J.M.; Dice, J.F.; Slaugenhaupt, S.A. Chaperone-mediated autophagy is defective in mucolipidosis type IV. J. Cell. Physiol. 2009, 219, 344–353. [Google Scholar] [CrossRef]
- Curcio-Morelli, C.; Charles, F.A.; Micsenyi, M.C.; Cao, Y.; Venugopal, B.; Browning, M.F.; Dobrenis, K.; Cotman, S.L.; Walkley, S.U.; Slaugenhaupt, S.A. Macroautophagy is defective in mucolipin-1-deficient mouse neurons. Neurobiol. Dis. 2010, 40, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Kinghorn, K.J.; Grönke, S.; Castillo-Quan, J.I.; Woodling, N.S.; Li, L.; Sirka, E.; Gegg, M.; Mills, K.; Hardy, J.; Bjedov, I.; et al. A Drosophila model of neuronopathic gaucher disease demonstrates lysosomal-autophagic defects and altered mTOR signalling and is functionally rescued by rapamycin. J. Neurosci. 2016, 36, 11654–11670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, R.A.; Voit, A.; Srikanth, M.P.; Thayer, J.A.; Kingsbury, T.J.; Jacobson, M.A.; Lipinski, M.M.; Feldman, R.A.; Awad, O. MTOR hyperactivity mediates lysosomal dysfunction in Gaucher’s disease iPSC-neuronal cells. DMM Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukuda, T.; Ahearn, M.; Roberts, A.; Mattaliano, R.J.; Zaal, K.; Ralston, E.; Plotz, P.H.; Raben, N. Autophagy and Mistargeting of Therapeutic Enzyme in Skeletal Muscle in Pompe Disease. Mol. Ther. 2006, 14, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Nascimbeni, A.C.; Fanin, M.; Masiero, E.; Angelini, C.; Sandri, M. Impaired autophagy contributes to muscle atrophy in glycogen storage disease type II patients. Autophagy 2012, 8, 1697–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chi, C.; Leonard, A.; Knight, W.E.; Beussman, K.M.; Zhao, Y.; Cao, Y.; Londono, P.; Aune, E.; Trembley, M.A.; Small, E.M.; et al. LAMP-2B regulates human cardiomyocyte function by mediating autophagosome–lysosome fusion. Proc. Natl. Acad. Sci. USA 2019, 116, 556–565. [Google Scholar] [CrossRef] [Green Version]
- Davis, O.B.; Shin, H.R.; Lim, C.Y.; Wu, E.Y.; Kukurugya, M.; Maher, C.F.; Perera, R.M.; Ordonez, M.P.; Zoncu, R. NPC1-mTORC1 Signaling Couples Cholesterol Sensing to Organelle Homeostasis and Is a Targetable Pathway in Niemann-Pick Type C. Dev. Cell 2021, 56, 260–276. [Google Scholar] [CrossRef]
- Ivanova, E.A.; De Leo, M.G.; Van Den Heuvel, L.; Pastore, A.; Dijkman, H.; De Matteis, M.A.; Levtchenko, E.N. Endo-Lysosomal Dysfunction in Human Proximal Tubular Epithelial Cells Deficient for Lysosomal Cystine Transporter Cystinosin. PLoS ONE 2015, 10, e0120998. [Google Scholar] [CrossRef] [Green Version]
- Rega, L.R.; Polishchuk, E.; Montefusco, S.; Napolitano, G.; Tozzi, G.; Zhang, J.; Bellomo, F.; Taranta, A.; Pastore, A.; Polishchuk, R.; et al. Activation of the transcription factor EB rescues lysosomal abnormalities in cystinotic kidney cells. Kidney Int. 2016, 89, 862–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Napolitano, G.; Johnson, J.L.; He, J.; Rocca, C.J.; Monfregola, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. Impairment of chaperone-mediated autophagy leads to selective lysosomal degradation defects in the lysosomal storage disease cystinosis. EMBO Mol. Med. 2015, 7, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Massey, A.C.; Kaushik, S.; Sovak, G.; Kiffin, R.; Cuervo, A.M. Consequences of the selective blockage of chaperone-mediated autophagy. Proc. Natl. Acad. Sci. USA 2006, 103, 5805–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Massey, A.C.; Mizushima, N.; Cuervo, A.M. Constitutive activation of chaperone-mediated autophagy in cells with impaired macroautophagy. Mol. Biol. Cell 2008, 19, 2179–2192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Johnson, J.L.; He, J.; Napolitano, G.; Ramadass, M.; Rocca, C.; Kiosses, W.B.; Bucci, C.; Xin, Q.; Gavathiotis, E.; et al. Cystinosin, the small GTPase Rab11, and the Rab7 effector RILP regulate intracellular trafficking of the chaperone-mediated autophagy receptor LAMP2A. J. Biol. Chem. 2017, 292, 10328–10346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rahman, F.; Johnson, J.L.; Zhang, J.; He, J.; Pestonjamasp, K.; Cherqui, S.; Catz, S.D. DYNC1LI2 regulates localization of the chaperone-mediated autophagy receptor LAMP2A and improves cellular homeostasis in cystinosis. Autophagy 2021, 1–19. [Google Scholar] [CrossRef]
- Sansanwal, P.; Sarwal, M.M. Abnormal mitochondrial autophagy in nephropathic cystinosis. Autophagy 2010, 6, 971–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sansanwal, P.; Sarwal, M.M. P62/SQSTM1 prominently accumulates in renal proximal tubules in nephropathic cystinosis. Pediatr. Nephrol. 2012, 27, 2137–2144. [Google Scholar] [CrossRef] [PubMed]
- De Leo, E.; Elmonem, M.A.; Berlingerio, S.P.; Berquez, M.; Festa, B.P.; Raso, R.; Bellomo, F.; Starborg, T.; Janssen, M.J.; Abbaszadeh, Z.; et al. Cell-based phenotypic drug screening identifies luteolin as candidate therapeutic for nephropathic cystinosis. J. Am. Soc. Nephrol. 2020, 31, 1522–1537. [Google Scholar] [CrossRef]
- Zhang, J.; He, J.; Johnson, J.L.; Rahman, F.; Gavathiotis, E.; Cuervo, A.M.; Catz, S.D. Chaperone-mediated autophagy upregulation rescues megalin expression and localization in cystinotic proximal tubule cells. Front. Endocrinol. 2019, 10, 21. [Google Scholar] [CrossRef]
- De Leo, M.G.; Staiano, L.; Vicinanza, M.; Luciani, A.; Carissimo, A.; Mutarelli, M.; Di Campli, A.; Polishchuk, E.; Di Tullio, G.; Morra, V.; et al. Autophagosome-lysosome fusion triggers a lysosomal response mediated by TLR9 and controlled by OCRL. Nat. Cell Biol. 2016, 18, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Festa, B.P.; Berquez, M.; Gassama, A.; Amrein, I.; Ismail, H.M.; Samardzija, M.; Staiano, L.; Luciani, A.; Grimm, C.; Nussbaum, R.L.; et al. OCRL deficiency impairs endolysosomal function in a humanized mouse model for Lowe syndrome and Dent disease. Hum. Mol. Genet. 2019, 28, 1931–1946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lima, W.R.; Parreira, K.S.; Devuyst, O.; Caplanusi, A.; N’Kuli, F.; Marien, B.; Van Der Smissen, P.; Alves, P.M.S.; Verroust, P.; Christensen, E.I.; et al. ZONAB promotes proliferation and represses differentiation of proximal tubule epithelial cells. J. Am. Soc. Nephrol. 2010, 21, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Beaudry, S.; Negoro, H.; Boucher, I.; Tran, M.; Kong, T.; Denker, B.M. H 2O 2 activates G protein, α 12 to disrupt the junctional complex and enhance ischemia reperfusion injury. Proc. Natl. Acad. Sci. USA 2012, 109, 6680–6685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeźégou, A.; Llinares, E.; Anne, C.; Kieffer-Jaquinod, S.; O’Regan, S.; Aupetit, J.; Chabli, A.; Sagné, C.; Debacker, C.; Chadefaux-Vekemans, B.; et al. Heptahelical protein PQLC2 is a lysosomal cationic amino acid exporter underlying the action of cysteamine in cystinosis therapy. Proc. Natl. Acad. Sci. USA 2012, 109, E3434–E3443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anguiano, J.; Garner, T.P.; Mahalingam, M.; Das, B.C.; Gavathiotis, E.; Cuervo, A.M. Chemical modulation of chaperone-mediated autophagy by retinoic acid derivatives. Nat. Chem. Biol. 2013, 9, 374–382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNeill, A.; Magalhaes, J.; Shen, C.; Chau, K.Y.; Hughes, D.; Mehta, A.; Foltynie, T.; Cooper, J.M.; Abramov, A.Y.; Gegg, M.; et al. Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 2014, 137, 1481–1495. [Google Scholar] [CrossRef] [Green Version]
- Johnson, J.L.; Napolitano, G.; Monfregola, J.; Rocca, C.J.; Cherqui, S.; Catz, S.D. Upregulation of the Rab27a-Dependent Trafficking and Secretory Mechanisms Improves Lysosomal Transport, Alleviates Endoplasmic Reticulum Stress, and Reduces Lysosome Overload in Cystinosis. Mol. Cell. Biol. 2013, 33, 2950–2962. [Google Scholar] [CrossRef] [Green Version]
- Hollywood, J.A.; Przepiorski, A.; D’Souza, R.F.; Sreebhavan, S.; Wolvetang, E.J.; Harrison, P.T.; Davidson, A.J.; Holm, T.M. Use of human induced pluripotent stem cells and kidney organoids to develop a cysteamine/mtor inhibition combination therapy for cystinosis. J. Am. Soc. Nephrol. 2020, 31, 962–982. [Google Scholar] [CrossRef]
- Andrzejewska, Z.; Nevo, N.; Thomas, L.; Chhuon, C.; Bailleux, A.; Chauvet, V.; Courtoy, P.J.; Chol, M.; Chiara Guerrera, I.; Antignac, C. Cystinosin is a component of the vacuolar H+-ATPase-ragulator-rag complex controlling mammalian target of rapamycin complex 1 signaling. J. Am. Soc. Nephrol. 2016, 27, 1678–1688. [Google Scholar] [CrossRef] [Green Version]
- Bellomo, F.; De Leo, E.; Taranta, A.; Giaquinto, L.; Di Giovamberardino, G.; Montefusco, S.; Rega, L.R.; Pastore, A.; Medina, D.L.; Di Bernardo, D.; et al. Drug repurposing in rare diseases: An integrative study of drug screening and transcriptomic analysis in nephropathic cystinosis. Int. J. Mol. Sci. 2021, 22, 12829. [Google Scholar] [CrossRef]
- Bellomo, F.; Medina, D.L.; De Leo, E.; Panarella, A.; Emma, F. High-content drug screening for rare diseases. J. Inherit. Metab. Dis. 2017, 40, 601–607. [Google Scholar] [CrossRef]
- Galarreta, C.I.; Forbes, M.S.; Thornhill, B.A.; Antignac, C.; Gubler, M.C.; Nevo, N.; Murphy, M.P.; Chevalier, R.L. The swan-neck lesion: Proximal tubular adaptation to oxidative stress in nephropathic cystinosis. Am. J. Physiol. Ren. Physiol. 2015, 308, F1155–F1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonam, S.R.; Wang, F.; Muller, S. Lysosomes as a therapeutic target. Nat. Rev. Drug Discov. 2019, 18, 923–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nielsen, R.; Courtoy, P.J.; Jacobsen, C.; Dom, G.; Lima, W.R.; Jadot, M.; Willnow, T.E.; Devuyst, O.; Christensen, E.I. Endocytosis provides a major alternative pathway for lysosomal biogenesis in kidney proximal tubular cells. Proc. Natl. Acad. Sci. USA 2007, 104, 5407–5412. [Google Scholar] [CrossRef] [Green Version]
- Mindell, J.A. Lysosomal acidification mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [Green Version]
- Arunachalam, B.; Phan, U.T.; Geuze, H.J.; Cresswell, P. Enzymatic reduction of disulfide bonds in lysosomes: Characterization of a gamma-interferon-inducible lysosomal thiol reductase (GILT). Proc. Natl. Acad. Sci. USA 2000, 97, 745–750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).