
Vagus Nerve Stimulation: A Personalized Therapeutic Approach for Crohn’s and Other Inflammatory Bowel Diseases

 ,
, 
 , ,
, ,  , ,
, ,  and
and 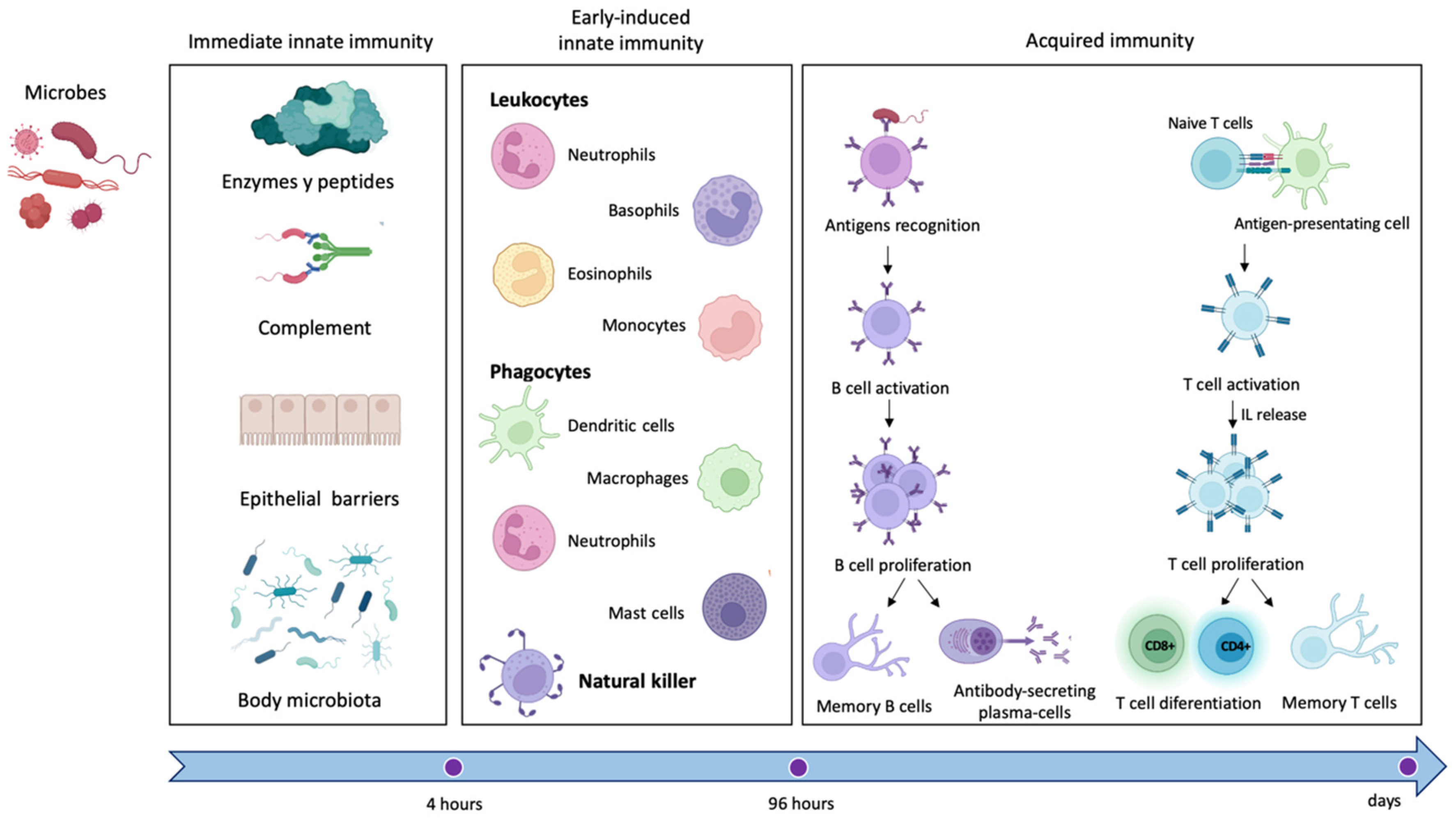{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Immune System
3. Interaction between the Immune System and the Enteric Nervous System
4. Gut Microbiota-Central Nervous System-Immune System Interaction
5. Efferent and Afferent Innervation of the Gut
5.1. Anatomo-Functional Organization of the Vagus Nerve
5.2. VN Innervation of Gastrointestinal Tract
5.3. Sympathetic Innervation
6. Immune-Mediated Process and Inflammation in Crohn’s Disease
6.1. Crohn’s Disease
6.2. The Innate Immune Response into Crohn’s Disease
- The intestinal epithelium normally blocks the paracellular passage of antigens present in the intestinal lumen through junctions between intestinal epithelial cells [5]. In Crohn’s disease, an abnormal increase in permeability allows the paracellular passage of antigens to the lamina propria, lying beneath the epithelium, in which resident macrophages and dendritic cells can identify antigens and present them to T cells, triggering an inflammatory response. In a high percentage of patients with Crohn’s disease, the increased permeability of the intestinal epithelium is produced by mutations in the NOD2 gene. Mutations in this gene allow antigens within the gastrointestinal tract to spread through intercellular spaces to the lamina propria and trigger inflammation [129].
- Paneth cells are highly specialized intestinal epithelial cells that excrete granules with antimicrobial peptides, such as α-defensins, and control the commensal microbiota [130]. Several studies have linked Crohn’s disease with dysfunctional Paneth cells, which generate dysmorphic and functionally altered granules, producing inflammation in the ileum [5,131]. Furthermore, the abnormal activity of Paneth cells can induce stress in their endoplasmic reticulum, which is correlated to autophagy and to unfolded protein response [1,5,132].
- In the absence of disease, ImS does not trigger immune responses to many antigens (tolerance). In Crohn’s disease, the immunological tolerance of dendritic cells and/or macrophages could be diminished, and once they recognize antigens, they polarize undifferentiated T lymphocytes (naive T lymphocytes) to differentiate into Th1 or Th17 by means of specific inflammatory mediators: IL-12 or IL-23, respectively [133]. This can increase the amount of other inflammatory mediators (IL-17) and/or induce the proliferation of differentiated immune cells such as Th17 lymphocytes [134]. IL-17, in turn, induces epithelial cells secretion of IL-8, a pro-inflammatory mediator that boosts the immune response by promoting the recruitment of neutrophils in the inflamed intestine, whereas the recruitment of neutrophils could be under the control of the neuro-immune response [135]. Additionally, there is evidence that Th17 cells are further amplified by IL-21, which controls IL-17 secretion by lymphocytes located in the lamina propria [134,136].
6.3. The Acquired Immune Response into Crohn’s Disease
7. Targeting Inflammation with Vagus Nerve Stimulation
- The CAP: STN, activated by general visceral efferent fibers of VN, project to the dorsal motor nucleus of the vagus (DMx), i.e., the parasympathetic nucleus of the VN, that synapses in celiac ganglia and sends sympathetic fibers to the spleen through the splenic nerve (Figure 7).Therefore, the spleen, as a key component of the cholinergic anti-inflammatory pathway, could provide a potential therapeutic target for immune-mediated diseases [158]. Splenic nerve endings in the spleen activate T lymphocytes expressing acetylcholine transferase (ChAT + T cells). When the traveling T cells identify macrophages, they secrete acetylcholine (ACh) that inhibits the release of macrophages inflammatory mediators [159] through (1) downregulation of nuclear translocation of NF-kB and (2) activating Janus kinase (JAK)/signal transducer and activator of transcription in macrophages and other cytokine-producing cells via nicotinic acetylcholine receptors (nAChR), which leads to suppression of proinflammatory cytokines [160,161,162].The importance of the spleen in the cholinergic anti-inflammatory pathway and in mediating the protective effect of efferent VNS was reported in an animal model in which the anti-inflammatory effect of VNS was abolished in animals lacking the α7 subunit of the nAChR (α7nAChR) due to splenectomy [159,163]. Moreover, an adoptive transfer of VNS-conditioned α7nAChR splenocytes conferred protection to recipient mice with ischemia-reperfusion injury (IRI) [161]. However, only recently the role of the spleen and α7nAChR has been proven for IBD [164,165]. In IBD, a decreased VN activity is observed, and it has been reported that central cholinergic activation in mice models of IBD might result in reduced mucosal inflammation. This is associated with decreased major histocompatibility complex II (MHC II) levels and pro-inflammatory cytokine secretion by splenic CD11c⁺ cells mediated by α7nAChR signaling. This anti-inflammatory cholinergic-mediated effect was abolished in mice with vagotomy, splenic neurectomy, or splenectomy [166]. The key role of the spleen has also become more intricate due to the discovery of the splenic glial cells, which may represent active partners mediating immune response [167]. Likely other glial cells, splenic glial cells are elongated cells of the white pulp expressing GFAP and S100β that ensheath and support both sympathetic and sympathetic fibers, guiding axons during outgrowth and response to injury and disease [168,169,170,171,172]. Moreover, through an extensive network, splenic glial cells make contact with lymphocytes, thus mediating the neuro–immune interaction. It is conceivable that adrenergic signaling of sympathetic fibers modulates immune reaction via splenic glial cells that express both adrenergic and purinergic receptors [173].
- The hypothalamic–pituitary–adrenal axis: the cholinergic anti-inflammatory pathway also involves a response of the hypothalamic–pituitary–adrenal axis (Figure 7). VN inputs to STN modulate the membrane potential of noradrenergic neurons within STN (group A2) that project to the parvicellular neurons of the paraventricular hypothalamic area (PVH) that, in turn, release a specific hormone, the corticotropin-stimulating factor (CRF) [174]. CRF binds to specific receptors expressed by cells of the pituitary gland, releasing adrenocorticotropin, a hormone that modulates the cells in the zona fasciculata in the adrenal glands that, in turn, release glucocorticoids, which play a strong anti-inflammatory role [153,155,156].
8. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, L.; Wang, F.-S.; Gershwin, M.E. Human autoimmune diseases: A comprehensive update. J. Intern. Med. 2015, 278, 369–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-Z. Inflammatory bowel disease: Pathogenesis. World J. Gastroenterol. 2014, 20, 91. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.J.; Hu, H. Enteric nervous system: Sensory transduction, neural circuits and gastrointestinal motility. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Niesler, B.; Kuerten, S.; Demir, I.E.; Schäfer, K.-H. Disorders of the enteric nervous system—A holistic view. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 393–410. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef]
- Bessac, A.; Cani, P.D.; Meunier, E.; Dietrich, G.; Knauf, C. Inflammation and Gut-Brain Axis during Type 2 Diabetes: Focus on the Crosstalk Between Intestinal Immune Cells and Enteric Nervous System. Front. Neurosci. 2018, 12, 725. [Google Scholar] [CrossRef]
- Pavlov, V.A.; Tracey, K.J. The vagus nerve and the inflammatory reflex--linking immunity and metabolism. Nat. Rev. Endocrinol. 2012, 8, 743–754. [Google Scholar] [CrossRef]
- Koopman, F.A.; Stoof, S.P.; Straub, R.H.; van Maanen, M.A.; Vervoordeldonk, M.J.; Tak, P.P. Restoring the Balance of the Autonomic Nervous System as an Innovative Approach to the Treatment of Rheumatoid Arthritis. Mol. Med. 2011, 17, 937–948. [Google Scholar] [CrossRef]
- Murray, K.; Reardon, C. The cholinergic anti-inflammatory pathway revisited. Neurogastroenterol. Motil. 2018, 30, e13288. [Google Scholar] [CrossRef] [PubMed]
- Korai, S.A.; Ranieri, F.; Di Lazzaro, V.; Papa, M.; Cirillo, G. Neurobiological After-Effects of Low Intensity Transcranial Electric Stimulation of the Human Nervous System: From Basic Mechanisms to Metaplasticity. Front. Neurol. 2021, 12, 587771. [Google Scholar] [CrossRef]
- Kaniusas, E.; Kampusch, S.; Tittgemeyer, M.; Panetsos, F.; Gines, R.F.; Papa, M.; Kiss, A.; Podesser, B.; Cassara, A.M.; Tanghe, E.; et al. Current Directions in the Auricular Vagus Nerve Stimulation I—A Physiological Perspective. Front. Neurosci. 2019, 13, 854. [Google Scholar] [CrossRef] [PubMed]
- Kaniusas, E.; Kampusch, S.; Tittgemeyer, M.; Panetsos, F.; Gines, R.F.; Papa, M.; Kiss, A.; Podesser, B.; Cassara, A.M.; Tanghe, E.; et al. Current Directions in the Auricular Vagus Nerve Stimulation II—An Engineering Perspective. Front. Neurosci. 2019, 13, 772. [Google Scholar] [CrossRef] [PubMed]
- Farmer, A.D.; Strzelczyk, A.; Finisguerra, A.; Gourine, A.V.; Gharabaghi, A.; Hasan, A.; Burger, A.M.; Jaramillo, A.M.; Mertens, A.; Majid, A.; et al. International Consensus Based Review and Recommendations for Minimum Reporting Standards in Research on Transcutaneous Vagus Nerve Stimulation (Version 2020). Front. Hum. Neurosci. 2021, 14, 568051. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.S.; Pavlov, V.A.; Tracey, K.J. Mechanisms and Therapeutic Relevance of Neuro-immune Communication. Immunity 2017, 46, 927–942. [Google Scholar] [CrossRef]
- Xiao, T.S. Innate immunity and inflammation. Cell. Mol. Immunol. 2017, 14, 1–3. [Google Scholar] [CrossRef]
- Chaplin, D.D. Overview of the immune response. J. Allergy Clin. Immunol. 2010, 125, S3–S23. [Google Scholar] [CrossRef]
- Sun, J.C.; Ugolini, S.; Vivier, E. Immunological memory within the innate immune system. EMBO J. 2014, 33, 1295–1303. [Google Scholar] [CrossRef]
- Iwasaki, A.; Medzhitov, R. Control of adaptive immunity by the innate immune system. Nat. Immunol. 2015, 16, 343–353. [Google Scholar] [CrossRef]
- Riera Romo, M.; Pérez-Martínez, D.; Castillo Ferrer, C. Innate immunity in vertebrates: An overview. Immunology 2016, 148, 125–139. [Google Scholar] [CrossRef]
- Beutler, B. Innate immunity: An overview. Mol. Immunol. 2004, 40, 845–859. [Google Scholar] [CrossRef] [PubMed]
- Flannigan, K.L.; Geem, D.; Harusato, A.; Denning, T.L. Intestinal Antigen-Presenting Cells: Key Regulators of Immune Homeostasis and Inflammation. Am. J. Pathol. 2015, 185, 1809–1819. [Google Scholar] [CrossRef] [PubMed]
- Bain, C.C.; Mowat, A.M. Macrophages in intestinal homeostasis and inflammation. Immunol. Rev. 2014, 260, 102–117. [Google Scholar] [CrossRef] [PubMed]
- Coombes, J.L.; Powrie, F. Dendritic cells in intestinal immune regulation. Nat. Rev. Immunol. 2008, 8, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria activate sensory neurons that modulate pain and inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef]
- Pongratz, G.; Straub, R.H. The sympathetic nervous response in inflammation. Arthritis Res. Ther. 2014, 16, 504. [Google Scholar] [CrossRef]
- Kaser, A.; Zeissig, S.; Blumberg, R.S. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010, 28, 573–621. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system and neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 286–294. [Google Scholar] [CrossRef]
- Grubišić, V.; Verkhratsky, A.; Zorec, R.; Parpura, V. Enteric glia regulate gut motility in health and disease. Brain Res. Bull. 2018, 136, 109–117. [Google Scholar] [CrossRef]
- Furness, J.B. The enteric nervous system: Normal functions and enteric neuropathies. Neurogastroenterol. Motil. 2008, 20, 32–38. [Google Scholar] [CrossRef]
- Ordovas-Montanes, J.; Rakoff-Nahoum, S.; Huang, S.; Riol-Blanco, L.; Barreiro, O.; von Andrian, U.H. The Regulation of Immunological Processes by Peripheral Neurons in Homeostasis and Disease. Trends Immunol. 2015, 36, 578–604. [Google Scholar] [CrossRef] [PubMed]
- Tracey, K.J. Reflex control of immunity. Nat. Rev. Immunol. 2009, 9, 418–428. [Google Scholar] [CrossRef] [PubMed]
- Veiga-Fernandes, H.; Pachnis, V. Neuroimmune regulation during intestinal development and homeostasis. Nat. Immunol. 2017, 18, 116–122. [Google Scholar] [CrossRef] [PubMed]
- Cabarrocas, J.; Savidge, T.C.; Liblau, R.S. Role of enteric glial cells in inflammatory bowel disease. Glia 2003, 41, 81–93. [Google Scholar] [CrossRef]
- Puzan, M.; Hosic, S.; Ghio, C.; Koppes, A. Enteric Nervous System Regulation of Intestinal Stem Cell Differentiation and Epithelial Monolayer Function. Sci. Rep. 2018, 8, 6313. [Google Scholar] [CrossRef]
- Margolis, K.G.; Gershon, M.D. Enteric Neuronal Regulation of Intestinal Inflammation. Trends Neurosci. 2016, 39, 614–624. [Google Scholar] [CrossRef]
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of gut microbiota in the development of low-grade inflammation and type 2 diabetes associated with obesity. Gut Microbes 2012, 3, 279–288. [Google Scholar] [CrossRef]
- Abot, A.; Cani, P.D.; Knauf, C. Impact of Intestinal Peptides on the Enteric Nervous System: Novel Approaches to Control Glucose Metabolism and Food Intake. Front. Endocrinol. 2018, 9, 328. [Google Scholar] [CrossRef] [PubMed]
- Buhner, S.; Schemann, M. Mast cell–nerve axis with a focus on the human gut. Biochim. Biophys. Acta-Mol. Basis Dis. 2012, 1822, 85–92. [Google Scholar] [CrossRef]
- Muller, P.A.; Koscsó, B.; Rajani, G.M.; Stevanovic, K.; Berres, M.-L.; Hashimoto, D.; Mortha, A.; Leboeuf, M.; Li, X.-M.; Mucida, D.; et al. Crosstalk between Muscularis Macrophages and Enteric Neurons Regulates Gastrointestinal Motility. Cell 2014, 158, 300–313. [Google Scholar] [CrossRef]
- Cailotto, C.; Gomez-Pinilla, P.J.; Costes, L.M.; van der Vliet, J.; Di Giovangiulio, M.; Némethova, A.; Matteoli, G.; Boeckxstaens, G.E. Neuro-Anatomical Evidence Indicating Indirect Modulation of Macrophages by Vagal Efferents in the Intestine but Not in the Spleen. PLoS ONE 2014, 9, e87785. [Google Scholar] [CrossRef]
- Xia, Y.; Hu, H.-Z.; Liu, S.; Ren, J.; Zafirov, D.H.; Wood, J.D. IL-1β and IL-6 excite neurons and suppress nicotinic and noradrenergic neurotransmission in guinea pig enteric nervous system. J. Clin. Investig. 1999, 103, 1309–1316. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekharan, B.; Jeppsson, S.; Pienkowski, S.; Belsham, D.D.; Sitaraman, S.V.; Merlin, D.; Kokkotou, E.; Nusrat, A.; Tansey, M.G.; Srinivasan, S. Tumor Necrosis Factor–Neuropeptide Y Cross Talk Regulates Inflammation, Epithelial Barrier Functions, and Colonic Motility. Inflamm. Bowel Dis. 2013, 19, 2535–2546. [Google Scholar] [CrossRef]
- Gougeon, P.-Y.; Lourenssen, S.; Han, T.Y.; Nair, D.G.; Ropeleski, M.J.; Blennerhassett, M.G. The Pro-Inflammatory Cytokines IL-1 and TNF Are Neurotrophic for Enteric Neurons. J. Neurosci. 2013, 33, 3339–3351. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Stevanovic, K.; Karamooz, N.; Li, Z.S.; Ahuja, A.; D’Autréaux, F.; Saurman, V.; Chalazonitis, A.; Gershon, M.D. Enteric Neuronal Density Contributes to the Severity of Intestinal Inflammation. Gastroenterology 2011, 141, 588–598.e2. [Google Scholar] [CrossRef] [PubMed]
- Nezami, B.G.; Srinivasan, S. Enteric Nervous System in the Small Intestine: Pathophysiology and Clinical Implications. Curr. Gastroenterol. Rep. 2010, 12, 358–365. [Google Scholar] [CrossRef]
- Rühl, A.; Franzke, S.; Collins, S.M.; Stremmel, W. Interleukin-6 expression and regulation in rat enteric glial cells. Am. J. Physiol. Liver Physiol. 2001, 280, G1163–G1171. [Google Scholar] [CrossRef]
- Ruhl, A. Glial cells in the gut. Neurogastroenterol. Motil. 2005, 17, 777–790. [Google Scholar] [CrossRef]
- Gulbransen, B.D.; Sharkey, K.A. Novel functional roles for enteric glia in the gastrointestinal tract. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 625–632. [Google Scholar] [CrossRef]
- Kabouridis, P.S.; Lasrado, R.; McCallum, S.; Chng, S.H.; Snippert, H.J.; Clevers, H.; Pettersson, S.; Pachnis, V. Microbiota Controls the Homeostasis of Glial Cells in the Gut Lamina Propria. Neuron 2015, 85, 289–295. [Google Scholar] [CrossRef]
- Savidge, T.C.; Newman, P.; Pothoulakis, C.; Ruhl, A.; Neunlist, M.; Bourreille, A.; Hurst, R.; Sofroniew, M.V. Enteric Glia Regulate Intestinal Barrier Function and Inflammation Via Release of S-Nitrosoglutathione. Gastroenterology 2007, 132, 1344–1358. [Google Scholar] [CrossRef] [PubMed]
- Ruhl, A.; Nasser, Y.; Sharkey, K.A. Enteric glia. Neurogastroenterol. Motil. 2004, 16, 44–49. [Google Scholar] [CrossRef]
- Neunlist, M.; Van Landeghem, L.; Mahé, M.M.; Derkinderen, P.; des Varannes, S.B.; Rolli-Derkinderen, M. The digestive neuronal–glial–epithelial unit: A new actor in gut health and disease. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 90–100. [Google Scholar] [CrossRef]
- Seguella, L.; Gulbransen, B.D. Enteric glial biology, intercellular signalling and roles in gastrointestinal disease. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 571–587. [Google Scholar] [CrossRef] [PubMed]
- Walsh, K.T.; Zemper, A.E. The Enteric Nervous System for Epithelial Researchers: Basic Anatomy, Techniques, and Interactions with the Epithelium. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Marietta, E.; Horwath, I.; Taneja, V. Microbiome, Immunomodulation, and the Neuronal System. Neurotherapeutics 2018, 15, 23–30. [Google Scholar] [CrossRef]
- Rutsch, A.; Kantsjö, J.B.; Ronchi, F. The Gut-Brain Axis: How Microbiota and Host Inflammasome Influence Brain Physiology and Pathology. Front. Immunol. 2020, 11, 604179. [Google Scholar] [CrossRef]
- Nishida, A.; Inoue, R.; Inatomi, O.; Bamba, S.; Naito, Y.; Andoh, A. Gut microbiota in the pathogenesis of inflammatory bowel disease. Clin. J. Gastroenterol. 2018, 11, 1–10. [Google Scholar] [CrossRef]
- Kaniusas, E.; Szeles, J.C.; Kampusch, S.; Alfageme-Lopez, N.; Yucuma-Conde, D.; Li, X.; Mayol, J.; Neumayer, C.; Papa, M.; Panetsos, F. Non-invasive Auricular Vagus Nerve Stimulation as a Potential Treatment for Covid19-Originated Acute Respiratory Distress Syndrome. Front. Physiol. 2020, 11, 890. [Google Scholar] [CrossRef]
- Grigg, J.B.; Sonnenberg, G.F. Host-Microbiota Interactions Shape Local and Systemic Inflammatory Diseases. J. Immunol. 2017, 198, 564–571. [Google Scholar] [CrossRef]
- Asadi, A.; Shadab Mehr, N.; Mohamadi, M.H.; Shokri, F.; Heidary, M.; Sadeghifard, N.; Khoshnood, S. Obesity and gut–microbiota–brain axis: A narrative review. J. Clin. Lab. Anal. 2022, 36, e24420. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; la Serre, C.; Lartigue, G. Gut microbiota sPARk vagus nerve excitation. J. Physiol. 2020, 598, 2043–2044. [Google Scholar] [CrossRef] [PubMed]
- Perez-Burgos, A.; Wang, B.; Mao, Y.-K.; Mistry, B.; Neufeld, K.-A.M.; Bienenstock, J.; Kunze, W. Psychoactive bacteria Lactobacillus rhamnosus (JB-1) elicits rapid frequency facilitation in vagal afferents. Am. J. Physiol. Liver Physiol. 2013, 304, G211–G220. [Google Scholar] [CrossRef]
- Buckley, M.M.; O’Malley, D. Development of an ex Vivo Method for Multi-unit Recording of Microbiota-Colonic-Neural Signaling in Real Time. Front. Neurosci. 2018, 12, 112. [Google Scholar] [CrossRef]
- Pradhananga, S.; Tashtush, A.A.; Allen-Vercoe, E.; Petrof, E.O.; Lomax, A.E. Protease-dependent excitation of nodose ganglion neurons by commensal gut bacteria. J. Physiol. 2020, 598, 2137–2151. [Google Scholar] [CrossRef]
- Ask, T.F.; Lugo, R.G.; Sütterlin, S. The Neuro-Immuno-Senescence Integrative Model (NISIM) on the Negative Association between Parasympathetic Activity and Cellular Senescence. Front. Neurosci. 2018, 12, 726. [Google Scholar] [CrossRef]
- Thayer, J.F.; Lane, R.D. A model of neurovisceral integration in emotion regulation and dysregulation. J. Affect. Disord. 2000, 61, 201–216. [Google Scholar] [CrossRef]
- Thayer, J.F.; Åhs, F.; Fredrikson, M.; Sollers, J.J.; Wager, T.D. A meta-analysis of heart rate variability and neuroimaging studies: Implications for heart rate variability as a marker of stress and health. Neurosci. Biobehav. Rev. 2012, 36, 747–756. [Google Scholar] [CrossRef]
- Woody, A.; Figueroa, W.S.; Benencia, F.; Zoccola, P.M. Stress-Induced Parasympathetic Control and Its Association with Inflammatory Reactivity. Psychosom. Med. 2017, 79, 306–310. [Google Scholar] [CrossRef]
- Sloan, R.P.; McCreath, H.; Tracey, K.J.; Sidney, S.; Liu, K.; Seeman, T. RR Interval Variability Is Inversely Related to Inflammatory Markers: The CARDIA Study. Mol. Med. 2007, 13, 178–184. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, H.; Suzuki, S.; Kawasaki, N.; Nakano, H.; Okazaki, T.; Chino, A.; Doi, T.; Saiki, I. Tumor Necrosis Factor-α-induced IKK Phosphorylation of NF-κB p65 on Serine 536 Is Mediated through the TRAF2, TRAF5, and TAK1 Signaling Pathway. J. Biol. Chem. 2003, 278, 36916–36923. [Google Scholar] [CrossRef] [PubMed]
- Feng, W.; Liu, H.; Luo, T.; Liu, D.; Du, J.; Sun, J.; Wang, W.; Han, X.; Yang, K.; Guo, J.; et al. Combination of IL-6 and sIL-6R differentially regulate varying levels of RANKL-induced osteoclastogenesis through NF-κB, ERK and JNK signaling pathways. Sci. Rep. 2017, 7, 41411. [Google Scholar] [CrossRef] [PubMed]
- Behrens, M.M.; Ali, S.S.; Dugan, L.L. Interleukin-6 Mediates the Increase in NADPH-Oxidase in the Ketamine Model of Schizophrenia. J. Neurosci. 2008, 28, 13957–13966. [Google Scholar] [CrossRef]
- Schulze-Osthoff, K.; Bakker, A.C.; Vanhaesebroeck, B.; Beyaert, R.; Jacob, W.A.; Fiers, W. Cytotoxic activity of tumor necrosis factor is mediated by early damage of mitochondrial functions. Evidence for the involvement of mitochondrial radical generation. J. Biol. Chem. 1992, 267, 5317–5323. [Google Scholar] [CrossRef]
- Zalli, A.; Carvalho, L.A.; Lin, J.; Hamer, M.; Erusalimsky, J.D.; Blackburn, E.H.; Steptoe, A. Shorter telomeres with high telomerase activity are associated with raised allostatic load and impoverished psychosocial resources. Proc. Natl. Acad. Sci. USA 2014, 111, 4519–4524. [Google Scholar] [CrossRef]
- Streltsova, L.I.; Tkacheva, O.N.; Plokhova, E.V.; Akasheva, D.U.; Strajesko, I.D.; Dudinskaya, E.N.; Boytsov, S.A. Age-related changes in heart rate variability and their relation with leucocyte telomere length. Cardiovasc. Ther. Prev. 2017, 16, 54–60. [Google Scholar] [CrossRef]
- Campisi, J. Cancer and ageing: Rival demons? Nat. Rev. Cancer 2003, 3, 339–349. [Google Scholar] [CrossRef]
- Campisi, J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol. 2001, 11, S27–S31. [Google Scholar] [CrossRef]
- Hatoum, O.A.; Binion, D.G.; Otterson, M.F.; Gutterman, D.D. Acquired microvascular dysfunction in inflammatory bowel disease: Loss of nitric oxide-mediated vasodilation. Gastroenterology 2003, 125, 58–69. [Google Scholar] [CrossRef]
- Wang, H.; Foong, J.P.P.; Harris, N.L.; Bornstein, J.C. Enteric neuroimmune interactions coordinate intestinal responses in health and disease. Mucosal Immunol. 2022, 15, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Neuhuber, W.L.; Berthoud, H.R. Functional anatomy of the vagus system—Emphasis on the somato-visceral interface. Auton. Neurosci. Basic Clin. 2021, 236, 102887. [Google Scholar] [CrossRef] [PubMed]
- Tekdemir, I.; Aslan, A.; Elhan, A. A clinico-anatomic study of the auricular branch of the vagus nerve and Arnold’s ear-cough reflex. Surg. Radiol. Anat. 1998, 20, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Kiyokawa, J.; Yamaguchi, K.; Okada, R.; Maehara, T.; Akita, K. Origin, course and distribution of the nerves to the posterosuperior wall of the external acoustic meatus. Anat. Sci. Int. 2014, 89, 238–245. [Google Scholar] [CrossRef]
- Peuker, E.T.; Filler, T.J. The nerve supply of the human auricle. Clin. Anat. 2002, 15, 35–37. [Google Scholar] [CrossRef]
- Ellrich, J. Transcutaneous Vagus Nerve Stimulation. Eur. Neurol. Rev. 2011, 6, 254. [Google Scholar] [CrossRef]
- Cheyuo, C.; Jacob, A.; Wu, R.; Zhou, M.; Coppa, G.F.; Wang, P. The Parasympathetic Nervous System in the Quest for Stroke Therapeutics. J. Cereb. Blood Flow Metab. 2011, 31, 1187–1195. [Google Scholar] [CrossRef]
- He, W.; Wang, X.; Shi, H.; Shang, H.; Li, L.; Jing, X.; Zhu, B. Auricular Acupuncture and Vagal Regulation. Evid.-Based Complement. Altern. Med. 2012, 2012, 786839. [Google Scholar] [CrossRef]
- Janis, J.E.; Hatef, D.A.; Ducic, I.; Ahmad, J.; Wong, C.; Hoxworth, R.E.; Osborn, T. Anatomy of the Auriculotemporal Nerve: Variations in Its Relationship to the Superficial Temporal Artery and Implications for the Treatment of Migraine Headaches. Plast. Reconstr. Surg. 2010, 125, 1422–1428. [Google Scholar] [CrossRef]
- Kemp, W.J.; Tubbs, R.S.; Cohen-Gadol, A.A. The Innervation of the Cranial Dura Mater: Neurosurgical Case Correlates and a Review of the Literature. World Neurosurg. 2012, 78, 505–510. [Google Scholar] [CrossRef]
- Butt, M.F.; Albusoda, A.; Farmer, A.D.; Aziz, Q. The anatomical basis for transcutaneous auricular vagus nerve stimulation. J. Anat. 2020, 236, 588–611. [Google Scholar] [CrossRef] [PubMed]
- Cutsforth-Gregory, J.K.; Benarroch, E.E. Nucleus of the solitary tract, medullary reflexes, and clinical implications. Neurology 2017, 88, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Câmara, R.; Griessenauer, C.J. Chapter 27—Anatomy of the Vagus Nerve. In Nerves and Nerve Injuries; Tubbs, R.S., Rizk, E., Shoja, M.M., Loukas, M., Barbaro, N., Spinner, R., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 385–397. ISBN 978-0-12-410390-0. [Google Scholar]
- Liddle, R.A. Neuropods. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 739–747. [Google Scholar] [CrossRef] [PubMed]
- Bohórquez, D.V.; Shahid, R.A.; Erdmann, A.; Kreger, A.M.; Wang, Y.; Calakos, N.; Wang, F.; Liddle, R.A. Neuroepithelial circuit formed by innervation of sensory enteroendocrine cells. J. Clin. Investig. 2015, 125, 782–786. [Google Scholar] [CrossRef] [PubMed]
- Zanos, T.P.; Silverman, H.A.; Levy, T.; Tsaava, T.; Battinelli, E.; Lorraine, P.W.; Ashe, J.M.; Chavan, S.S.; Tracey, K.J.; Bouton, C.E. Identification of cytokine-specific sensory neural signals by decoding murine vagus nerve activity. Proc. Natl. Acad. Sci. USA 2018, 115, E4843–E4852. [Google Scholar] [CrossRef]
- Powley, T.L. Brain-gut communication: Vagovagal reflexes interconnect the two “brains”. Am. J. Physiol. Liver Physiol. 2021, 321, G576–G587. [Google Scholar] [CrossRef]
- Breit, S.; Kupferberg, A.; Rogler, G.; Hasler, G. Vagus Nerve as Modulator of the Brain–Gut Axis in Psychiatric and Inflammatory Disorders. Front. Psychiatry 2018, 9, 44. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The gut-brain axis: Interactions between enteric microbiota, central and enteric nervous systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar]
- Lomax, A.E.; O’Reilly, M.; Neshat, S.; Vanner, S.J. Sympathetic vasoconstrictor regulation of mouse colonic submucosal arterioles is altered in experimental colitis. J. Physiol. 2007, 583, 719–730. [Google Scholar] [CrossRef]
- Vanner, S.; Surprenant, A. Neural reflexes controlling intestinal microcirculation. Am. J. Physiol. Liver Physiol. 1996, 271, G223–G230. [Google Scholar] [CrossRef]
- Reid, I.A. Interactions between ANG II, sympathetic nervous system, and baroreceptor reflexes in regulation of blood pressure. Am. J. Physiol. Metab. 1992, 262, E763–E778. [Google Scholar] [CrossRef]
- Bruno, R.M.; Ghiadoni, L.; Seravalle, G.; Dell’Oro, R.; Taddei, S.; Grassi, G. Sympathetic regulation of vascular function in health and disease. Front. Physiol. 2012, 3, 284. [Google Scholar] [CrossRef]
- Grassi, G. Renin–angiotensin–sympathetic crosstalks in hypertension: Reappraising the relevance of peripheral interactions. J. Hypertens. 2001, 19, 1713–1716. [Google Scholar] [CrossRef] [PubMed]
- Phillips, R.J.; Powley, T.L. Innervation of the gastrointestinal tract: Patterns of aging. Auton. Neurosci. Basic Clin. 2007. [CrossRef] [PubMed]
- Rosas-Ballina, M.; Ochani, M.; Parrish, W.R.; Ochani, K.; Harris, Y.T.; Huston, J.M.; Chavan, S.; Tracey, K.J. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc. Natl. Acad. Sci. USA 2008, 105, 11008–11013. [Google Scholar] [CrossRef] [PubMed]
- Willemze, R.A.; Welting, O.; van Hamersveld, H.P.; Meijer, S.L.; Folgering, J.H.A.; Darwinkel, H.; Witherington, J.; Sridhar, A.; Vervoordeldonk, M.J.; Seppen, J.; et al. Neuronal control of experimental colitis occurs via sympathetic intestinal innervation. Neurogastroenterol. Motil. 2018, 30, e13163. [Google Scholar] [CrossRef]
- Nijhuis, L.E.; Olivier, B.J.; Dhawan, S.; Hilbers, F.W.; Boon, L.; Wolkers, M.C.; Samsom, J.N.; de Jonge, W.J. Adrenergic β2 Receptor Activation Stimulates Anti-Inflammatory Properties of Dendritic Cells In Vitro. PLoS ONE 2014, 9, e85086. [Google Scholar] [CrossRef]
- Duan, H.; Cai, X.; Luan, Y.; Yang, S.; Yang, J.; Dong, H.; Zeng, H.; Shao, L. Regulation of the Autonomic Nervous System on Intestine. Front. Physiol. 2021, 12, 700129. [Google Scholar] [CrossRef]
- Willemze, R.A.; Welting, O.; van Hamersveld, P.; Verseijden, C.; Nijhuis, L.E.; Hilbers, F.W.; Meijer, S.L.; Heesters, B.A.; Folgering, J.H.A.; Darwinkel, H.; et al. Loss of intestinal sympathetic innervation elicits an innate immune driven colitis. Mol. Med. 2019, 25, 1. [Google Scholar] [CrossRef]
- Theochari, N.A.; Stefanopoulos, A.; Mylonas, K.S.; Economopoulos, K.P. Antibiotics exposure and risk of inflammatory bowel disease: A systematic review. Scand. J. Gastroenterol. 2018, 53, 1–7. [Google Scholar] [CrossRef]
- Vojdani, A. Antibodies as Predictors of Complex Autoimmune Diseases and Cancer. Int. J. Immunopathol. Pharmacol. 2008, 21, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Kikut, J.; Konecka, N.; Ziętek, M.; Kulpa, D.; Szczuko, M. Diet supporting therapy for inflammatory bowel diseases. Eur. J. Nutr. 2021, 60, 2275–2291. [Google Scholar] [CrossRef]
- Cai, Z.; Wang, S.; Li, J. Treatment of Inflammatory Bowel Disease: A Comprehensive Review. Front. Med. 2021, 8, 2681. [Google Scholar] [CrossRef] [PubMed]
- Ban, Q.-Y.; Liu, M.; Ding, N.; Chen, Y.; Lin, Q.; Zha, J.-M.; He, W.-Q. Nutraceuticals for the Treatment of IBD: Current Progress and Future Directions. Front. Nutr. 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Cardona, F.; Andrés-Lacueva, C.; Tulipani, S.; Tinahones, F.J.; Queipo-Ortuño, M.I. Benefits of polyphenols on gut microbiota and implications in human health. J. Nutr. Biochem. 2013, 24, 1415–1422. [Google Scholar] [CrossRef] [PubMed]
- Dueñas, M.; Cueva, C.; Muñoz-González, I.; Jiménez-Girón, A.; Sánchez-Patán, F.; Santos-Buelga, C.; Moreno-Arribas, M.V.; Bartolomé, B. Studies on modulation of gut microbiota by wine polyphenols: From isolated cultures to omic approaches. Antioxidants 2015, 4, 1–21. [Google Scholar] [CrossRef]
- Cueva, C.; Gil-Sánchez, I.; Ayuda-Durán, B.; González-Manzano, S.; González-Paramás, A.M.; Santos-Buelga, C.; Bartolomé, B.; Victoria Moreno-Arribas, M. An integrated view of the effects of wine polyphenols and their relevant metabolites on gut and host health. Molecules 2017, 22, 99. [Google Scholar] [CrossRef]
- Selma, M.V.; Espín, J.C.; Tomás-Barberán, F.A. Interaction between phenolics and gut microbiota: Role in human health. J. Agric. Food Chem. 2009, 57, 6485–6501. [Google Scholar] [CrossRef]
- Tzounis, X.; Rodriguez-Mateos, A.; Vulevic, J.; Gibson, G.R.; Kwik-Uribe, C.; Spencer, J.P.E. Prebiotic evaluation of cocoa-derived flavanols in healthy humans by using a randomized, controlled, double-blind, crossover intervention study. Am. J. Clin. Nutr. 2011, 93, 62–72. [Google Scholar] [CrossRef]
- Van Duynhoven, J.; Vaughan, E.E.; Van Dorsten, F.; Gomez-Roldan, V.; De Vos, R.; Vervoort, J.; Van Der Hooft, J.J.J.; Roger, L.; Draijer, R.; Jacobs, D.M. Interactions of black tea polyphenols with human gut microbiota: Implications for gut and cardiovascular health1-4. Am. J. Clin. Nutr. 2013, 98, 1631S–1641S. [Google Scholar] [CrossRef]
- Santos-Buelga, C.; González-Paramás, A.M.; Oludemi, T.; Ayuda-Durán, B.; González-Manzano, S. Plant phenolics as functional food ingredients. Adv. Food Nutr. Res. 2019, 90, 183–257. [Google Scholar] [PubMed]
- Kaulmann, A.; Bohn, T. Bioactivity of Polyphenols: Preventive and Adjuvant Strategies toward Reducing Inflammatory Bowel Diseases—Promises, Perspectives, and Pitfalls. Oxid. Med. Cell. Longev. 2016, 2016, 9346470. [Google Scholar] [CrossRef] [PubMed]
- Santos-Buelga, C.; Feliciano, A.S.; McPhee, D.J. Flavonoids: From structure to health issues. Molecules 2017, 22, 477. [Google Scholar] [CrossRef] [PubMed]
- Manach, C.; Williamson, G.; Morand, C.; Scalbert, A.; Rémésy, C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am. J. Clin. Nutr. 2005, 81, 230S–242S. [Google Scholar] [CrossRef] [PubMed]
- Westfall, S.; Pasinetti, G.M. The Gut Microbiota Links Dietary Polyphenols with Management of Psychiatric Mood Disorders. Front. Neurosci. 2019, 13, 1196. [Google Scholar] [CrossRef]
- Khan, N.; Adhami, V.M.; Mukhtar, H. Apoptosis by dietary agents for prevention and treatment of prostate cancer. Endocr. Relat. Cancer 2010, 17, R39–R52. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Y.; Li, J.; Fu, C.; Zhang, X. The Neuroprotective Effect of Tea Polyphenols on the Regulation of Intestinal Flora. Molecules 2021, 26, 3692. [Google Scholar] [CrossRef]
- Di Sabatino, A.; Rovedatti, L.; Vidali, F.; MacDonald, T.T.; Corazza, G.R. Recent advances in understanding Crohn’s disease. Intern. Emerg. Med. 2013, 8, 101–113. [Google Scholar] [CrossRef]
- Adolph, T.E.; Tomczak, M.F.; Niederreiter, L.; Ko, H.-J.; Böck, J.; Martinez-Naves, E.; Glickman, J.N.; Tschurtschenthaler, M.; Hartwig, J.; Hosomi, S.; et al. Paneth cells as a site of origin for intestinal inflammation. Nature 2013, 503, 272–276. [Google Scholar] [CrossRef]
- Salzman, N.H.; Hung, K.; Haribhai, D.; Chu, H.; Karlsson-Sjöberg, J.; Amir, E.; Teggatz, P.; Barman, M.; Hayward, M.; Eastwood, D.; et al. Enteric defensins are essential regulators of intestinal microbial ecology. Nat. Immunol. 2010, 11, 76–82. [Google Scholar] [CrossRef]
- Kaser, A.; Blumberg, R.S. Endoplasmic reticulum stress and intestinal inflammation. Mucosal Immunol. 2010, 3, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Strober, W.; Fuss, I.J. Proinflammatory Cytokines in the Pathogenesis of Inflammatory Bowel Diseases. Gastroenterology 2011, 140, 1756–1767.e1. [Google Scholar] [CrossRef]
- Abraham, C.; Cho, J. Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 2009, 15, 1090–1100. [Google Scholar] [CrossRef] [PubMed]
- Ge, Y.; Huang, M.; Yao, Y. Biology of Interleukin-17 and Its Pathophysiological Significance in Sepsis. Front. Immunol. 2020, 11, 1558. [Google Scholar] [CrossRef] [PubMed]
- Rescigno, M.; Di Sabatino, A. Dendritic cells in intestinal homeostasis and disease. J. Clin. Investig. 2009, 119, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Weaver, C.T.; Hatton, R.D. Interplay between the TH17 and TReg cell lineages: A (co-) evolutionary perspective. Nat. Rev. Immunol. 2009, 9, 883–889. [Google Scholar] [CrossRef]
- Imam, T.; Park, S.; Kaplan, M.H.; Olson, M.R. Effector T Helper Cell Subsets in Inflammatory Bowel Diseases. Front. Immunol. 2018, 9, 1212. [Google Scholar] [CrossRef]
- Fiocchi, C. Susceptibility Genes and Overall Pathogenesis of Inflammatory Bowel Disease: Where Do We Stand? Dig. Dis. 2009, 27, 226–235. [Google Scholar] [CrossRef]
- Li, N.; Shi, R.-H. Updated review on immune factors in pathogenesis of Crohn’s disease. World J. Gastroenterol. 2018, 24, 15–22. [Google Scholar] [CrossRef]
- Gonzalez-Correa, C.A.; Mulett-Vásquez, E.; Miranda, D.A.; Gonzalez-Correa, C.H.; Gómez-Buitrago, P.A. The colon revisited or the key to wellness, health and disease. Med. Hypotheses 2017, 108, 133–143. [Google Scholar] [CrossRef]
- Chow, A.K.; Gulbransen, B.D. Potential roles of enteric glia in bridging neuroimmune communication in the gut. Am. J. Physiol. Liver Physiol. 2017, 312, G145–G152. [Google Scholar] [CrossRef] [PubMed]
- Jauregui-Amezaga, A.; Somers, M.; De Schepper, H.; Macken, E. Next generation of biologics for the treatment of Crohn’s disease: An evidence-based review on ustekinumab. Clin. Exp. Gastroenterol. 2017, 10, 293–301. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, T.T. Inside the microbial and immune labyrinth: Totally gutted. Nat. Med. 2010, 16, 1194–1195. [Google Scholar] [CrossRef]
- Ha, F.; Khalil, H. Crohn’s disease: A clinical update. Therap. Adv. Gastroenterol. 2015, 8, 352–359. [Google Scholar] [CrossRef]
- Bonaz, B.; Sinniger, V.; Pellissier, S. Vagus nerve stimulation: A new promising therapeutic tool in inflammatory bowel disease. J. Intern. Med. 2017, 282, 46–63. [Google Scholar] [CrossRef]
- Bonaz, B. Is-there a place for vagus nerve stimulation in inflammatory bowel diseases? Bioelectron. Med. 2018, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Silberstein, S.D.; Mechtler, L.L.; Kudrow, D.B.; Calhoun, A.H.; McClure, C.; Saper, J.R.; Liebler, E.J.; Rubenstein Engel, E.; Tepper, S.J. Non–Invasive Vagus Nerve Stimulation for the ACute Treatment of Cluster Headache: Findings From the Randomized, Double-Blind, Sham-Controlled ACT1 Study. Headache 2016, 56, 1317–1332. [Google Scholar] [CrossRef] [PubMed]
- Howland, R.H. Vagus Nerve Stimulation. Curr. Behav. Neurosci. Rep. 2014, 1, 64–73. [Google Scholar] [CrossRef]
- Berthoud, H.R.; Neuhuber, W.L. Vagal mechanisms as neuromodulatory targets for the treatment of metabolic disease. Ann. N. Y. Acad. Sci. 2019, 1454, 42–55. [Google Scholar] [CrossRef]
- Dietrich, S.; Smith, J.; Scherzinger, C.; Hofmann-Preiß, K.; Freitag, T.; Eisenkolb, A.; Ringler, R. A novel transcutaneous vagus nerve stimulation leads to brainstem and cerebral activations measured by functional MRI/Funktionelle Magnetresonanztomographie zeigt Aktivierungen des Hirnstamms und weiterer zerebraler Strukturen unter transkutaner Vagusnervstimulation. Biomed. Tech. Eng. 2008, 53, 104–111. [Google Scholar] [CrossRef]
- Kraus, T.; Hösl, K.; Kiess, O.; Schanze, A.; Kornhuber, J.; Forster, C. BOLD fMRI deactivation of limbic and temporal brain structures and mood enhancing effect by transcutaneous vagus nerve stimulation. J. Neural Transm. 2007, 114, 1485–1493. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.; Picq, C.; Sinniger, V.; Mayol, J.F.; Clarençon, D. Vagus nerve stimulation: From epilepsy to the cholinergic anti-inflammatory pathway. Neurogastroenterol. Motil. 2013, 25, 208–221. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Wang, H.; Czura, C.J.; Friedman, S.G.; Tracey, K.J. The Cholinergic Anti-inflammatory Pathway: A Missing Link in Neuroimmunomodulation. Mol. Med. 2003, 9, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Tsigos, C.; Chrousos, G.P. Hypothalamic–pituitary–adrenal axis, neuroendocrine factors and stress. J. Psychosom. Res. 2002, 53, 865–871. [Google Scholar] [CrossRef]
- Kelley, K.W.; McCusker, R.H. Getting nervous about immunity. Semin. Immunol. 2014, 26, 389–393. [Google Scholar] [CrossRef][Green Version]
- McKlveen, J.M.; Myers, B.; Herman, J.P. The Medial Prefrontal Cortex: Coordinator of Autonomic, Neuroendocrine and Behavioural Responses to Stress. J. Neuroendocrinol. 2015, 27, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Wang, T.; Liao, L.; Fan, X.; Chang, L.; Hashimoto, K. Brain-spleen axis in health and diseases: A review and future perspective. Brain Res. Bull. 2022, 182, 130–140. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor α7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Wang, D.-W.; Yin, Y.-M.; Yao, Y.-M. Vagal Modulation of the Inflammatory Response in Sepsis. Int. Rev. Immunol. 2016, 35, 415–433. [Google Scholar] [CrossRef]
- Inoue, T.; Abe, C.; Sung, S.J.; Moscalu, S.; Jankowski, J.; Huang, L.; Ye, H.; Rosin, D.L.; Guyenet, P.G.; Okusa, M.D. Vagus nerve stimulation mediates protection from kidney ischemia-reperfusion injury through α7nAChR+ splenocytes. J. Clin. Investig. 2016, 126, 1939–1952. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Tracey, K.J. Cholinergic control of inflammation. J. Intern. Med. 2009, 265, 663–679. [Google Scholar] [CrossRef] [PubMed]
- de Jonge, W.J.; Ulloa, L. The alpha7 nicotinic acetylcholine receptor as a pharmacological target for inflammation. Br. J. Pharmacol. 2007, 151, 915–929. [Google Scholar] [CrossRef] [PubMed]
- Seyedabadi, M.; Rahimian, R.; Ghia, J.-E. The role of alpha7 nicotinic acetylcholine receptors in inflammatory bowel disease: Involvement of different cellular pathways. Expert Opin. Ther. Targets 2018, 22, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Hoover, D.B. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol. Ther. 2017, 179, 1–16. [Google Scholar] [CrossRef]
- Ji, H.; Rabbi, M.F.; Labis, B.; Pavlov, V.A.; Tracey, K.J.; Ghia, J.E. Central cholinergic activation of a vagus nerve-to-spleen circuit alleviates experimental colitis. Mucosal Immunol. 2014, 7, 335–347. [Google Scholar] [CrossRef]
- Lucas, T.A.; Zhu, L.; Buckwalter, M.S. Spleen glia are a transcriptionally unique glial subtype interposed between immune cells and sympathetic axons. Glia 2021, 69, 1799–1815. [Google Scholar] [CrossRef]
- Colangelo, A.M.; Cirillo, G.; Lavitrano, M.L.; Alberghina, L.; Papa, M. Targeting reactive astrogliosis by novel biotechnological strategies. Biotechnol. Adv. 2012, 30, 261–271. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; Bianco, M.R.; Cavaliere, C.; Zaccaro, L.; Sarmientos, P.; Alberghina, L.; Papa, M. BB14, a Nerve Growth Factor (NGF)-like peptide shown to be effective in reducing reactive astrogliosis and restoring synaptic homeostasis in a rat model of peripheral nerve injury. Biotechnol. Adv. 2012, 30, 223–232. [Google Scholar] [CrossRef]
- Papa, M.; De Luca, C.; Petta, F.; Alberghina, L.; Cirillo, G. Astrocyte-neuron interplay in maladaptive plasticity. Neurosci. Biobehav. Rev. 2014, 42, 35–54. [Google Scholar] [CrossRef]
- Cirillo, G.; Colangelo, A.M.; De Luca, C.; Savarese, L.; Barillari, M.R.; Alberghina, L.; Papa, M. Modulation of Matrix Metalloproteinases Activity in the Ventral Horn of the Spinal Cord Re-stores Neuroglial Synaptic Homeostasis and Neurotrophic Support following Peripheral Nerve Injury. PLoS ONE 2016, 11, e0152750. [Google Scholar] [CrossRef]
- Virtuoso, A.; Colangelo, A.M.; Korai, S.A.; Izzo, S.; Todisco, A.; Giovannoni, R.; Lavitrano, M.; Papa, M.; Cirillo, G. Inhibition of plasminogen/plasmin system retrieves endogenous nerve growth factor and adaptive spinal synaptic plasticity following peripheral nerve injury. Neurochem. Int. 2021, 148, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Lecca, D.; Ceruti, S.; Fumagalli, M.; Abbracchio, M.P. Purinergic trophic signalling in glial cells: Functional effects and modulation of cell proliferation, differentiation, and death. Purinergic Signal. 2012, 8, 539–557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Miller, W.L. The Hypothalamic-Pituitary-Adrenal Axis: A Brief History. Horm. Res. Paediatr. 2018, 89, 212–223. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kong, X.; Xiu, H.; Dou, Y.; Wu, Z.; Sun, P. Combination of curcumin and vagus nerve stimulation attenuates cerebral ischemia/reperfusion injury-induced behavioral deficits. Biomed. Pharmacother. 2018, 103, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef]
- Kapadia, R.; Yi, J.-H.; Vemuganti, R. Mechanisms of anti-inflammatory and neuroprotective actions of PPAR-gamma agonists. Front. Biosci. 2008, 13, 1813–1826. [Google Scholar] [CrossRef]
- Martin, H.L.; Mounsey, R.B.; Mustafa, S.; Sathe, K.; Teismann, P. Pharmacological manipulation of peroxisome proliferator-activated receptor γ (PPARγ) reveals a role for anti-oxidant protection in a model of Parkinson’s disease. Exp. Neurol. 2012, 235, 528–538. [Google Scholar] [CrossRef]
- Zhou, H.; Liang, H.; Li, Z.-F.; Xiang, H.; Liu, W.; Li, J.-G. Vagus Nerve Stimulation Attenuates Intestinal Epithelial Tight Junctions Disruption in Endotoxemic Mice Through α7 Nicotinic Acetylcholine Receptors. Shock 2013, 40, 144–151. [Google Scholar] [CrossRef] [PubMed]
- Kager, H.; Likar, R.; Jabarzadeh, H.; Sittl, R.; Breschan, C.; Szeles, J. Electrical punctual stimulation (P-STIM) with ear acupuncture following tonsillectomy, a randomised, controlled pilot study. Acute Pain 2009, 11, 101–106. [Google Scholar] [CrossRef]
- De Ferrari, G.M.; Schwartz, P.J. Vagus nerve stimulation: From pre-clinical to clinical application: Challenges and future directions. Heart Fail. Rev. 2011, 16, 195–203. [Google Scholar] [CrossRef]
- Adewole, D.O.; Serruya, M.D.; Harris, J.P.; Burrell, J.C.; Petrov, D.; Chen, H.I.; Wolf, J.A.; Cullen, D.K. The Evolution of Neuroprosthetic Interfaces. Crit. Rev. Biomed. Eng. 2016, 44, 123–152. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.B.; Mignardot, J.-B.; Le Goff-Mignardot, C.G.; Demesmaeker, R.; Komi, S.; Capogrosso, M.; Rowald, A.; Seáñez, I.; Caban, M.; Pirondini, E.; et al. Targeted neurotechnology restores walking in humans with spinal cord injury. Nature 2018, 563, 65–71. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cirillo, G.; Negrete-Diaz, F.; Yucuma, D.; Virtuoso, A.; Korai, S.A.; De Luca, C.; Kaniusas, E.; Papa, M.; Panetsos, F. Vagus Nerve Stimulation: A Personalized Therapeutic Approach for Crohn’s and Other Inflammatory Bowel Diseases. Cells 2022, 11, 4103. https://doi.org/10.3390/cells11244103
Cirillo G, Negrete-Diaz F, Yucuma D, Virtuoso A, Korai SA, De Luca C, Kaniusas E, Papa M, Panetsos F. Vagus Nerve Stimulation: A Personalized Therapeutic Approach for Crohn’s and Other Inflammatory Bowel Diseases. Cells. 2022; 11(24):4103. https://doi.org/10.3390/cells11244103
Chicago/Turabian StyleCirillo, Giovanni, Flor Negrete-Diaz, Daniela Yucuma, Assunta Virtuoso, Sohaib Ali Korai, Ciro De Luca, Eugenijus Kaniusas, Michele Papa, and Fivos Panetsos. 2022. "Vagus Nerve Stimulation: A Personalized Therapeutic Approach for Crohn’s and Other Inflammatory Bowel Diseases" Cells 11, no. 24: 4103. https://doi.org/10.3390/cells11244103
APA StyleCirillo, G., Negrete-Diaz, F., Yucuma, D., Virtuoso, A., Korai, S. A., De Luca, C., Kaniusas, E., Papa, M., & Panetsos, F. (2022). Vagus Nerve Stimulation: A Personalized Therapeutic Approach for Crohn’s and Other Inflammatory Bowel Diseases. Cells, 11(24), 4103. https://doi.org/10.3390/cells11244103