



Pharmacological Characterization of Purified Full-Length Dopamine Transporter from Drosophila melanogaster


Abstract
1. Introduction
2. Materials and Methods
2.1. Constructs
2.2. Expression and Purification of dDAT
2.3. Radioligand Binding to Purified dDAT
2.4. Expression of Transporters in COS-7 Cells
2.5. Competition Binding in COS-7 Cells
2.6. Statistical Analyses
3. Results
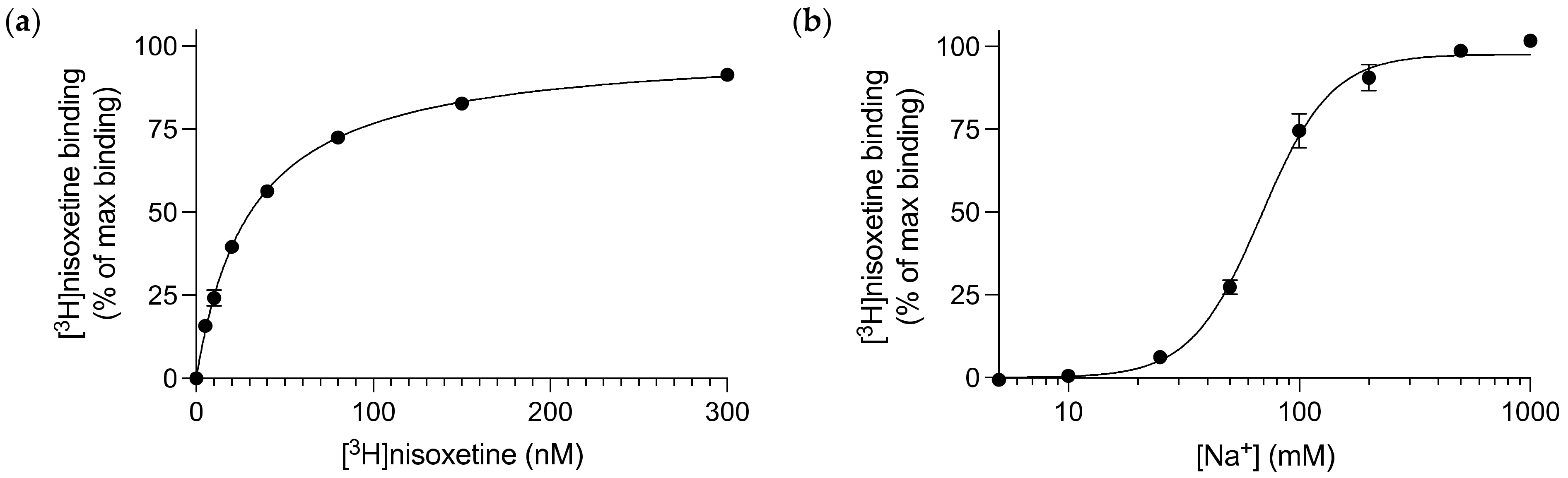
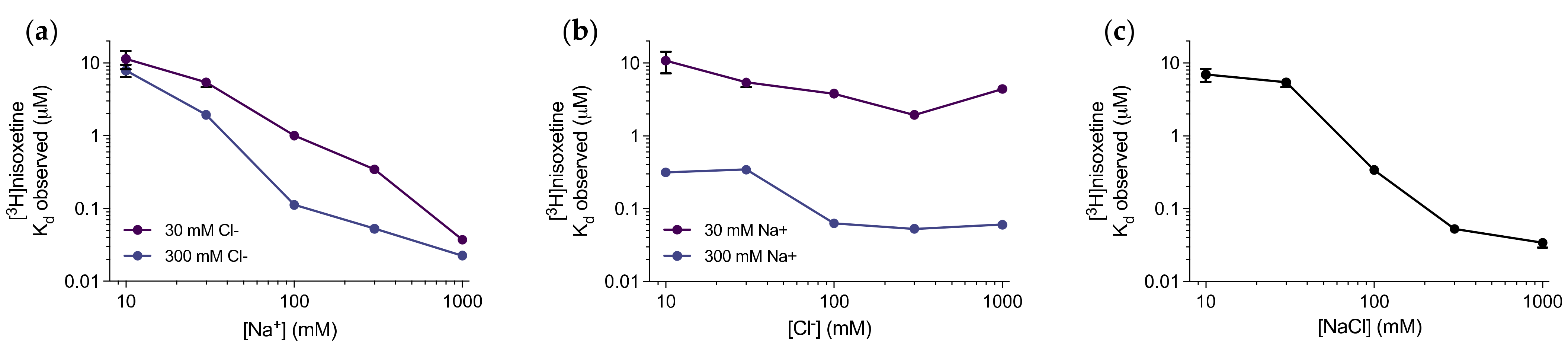
3.1. Nisoxetine Binds to Purified dDAT in a Na+-dependent Manner
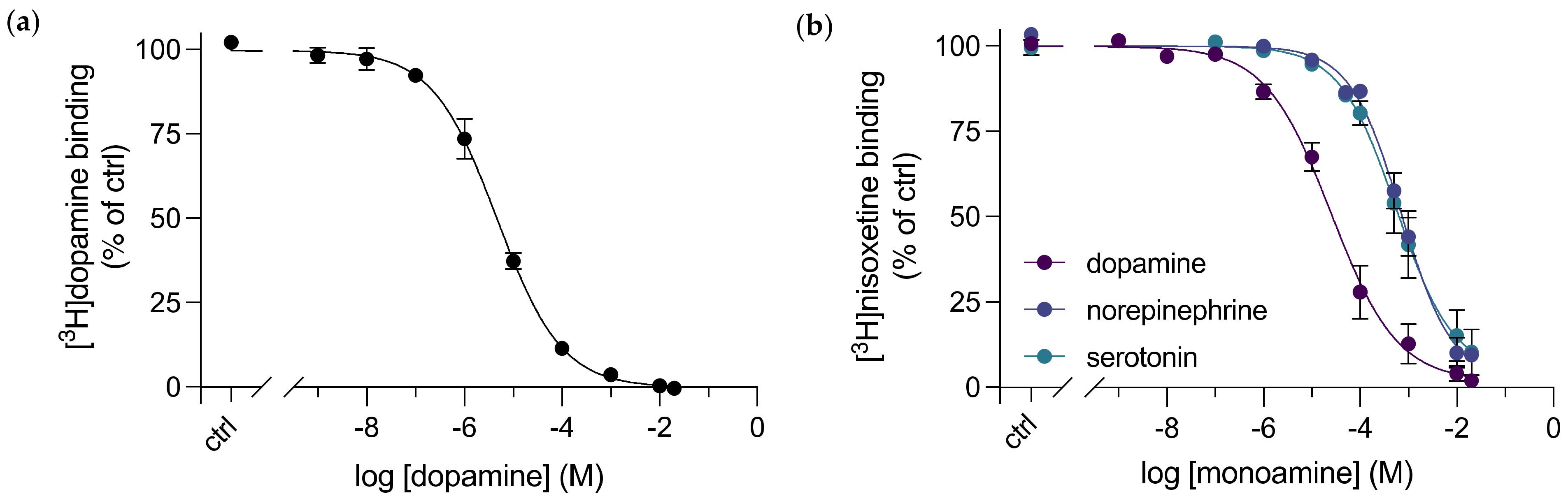
3.2. A Direct Binding Affinity for Dopamine to dDAT was Determined
3.3. Purified dDAT Displays Substrate Selectivity for Dopamine over Other Monoamine Neurotransmitters
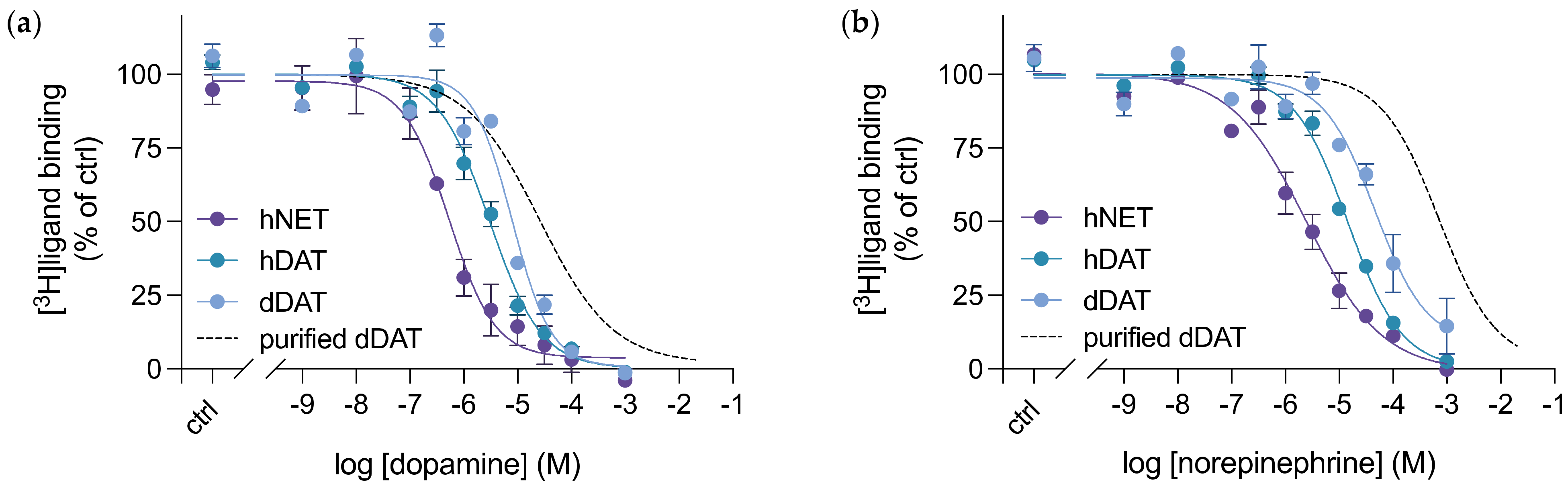
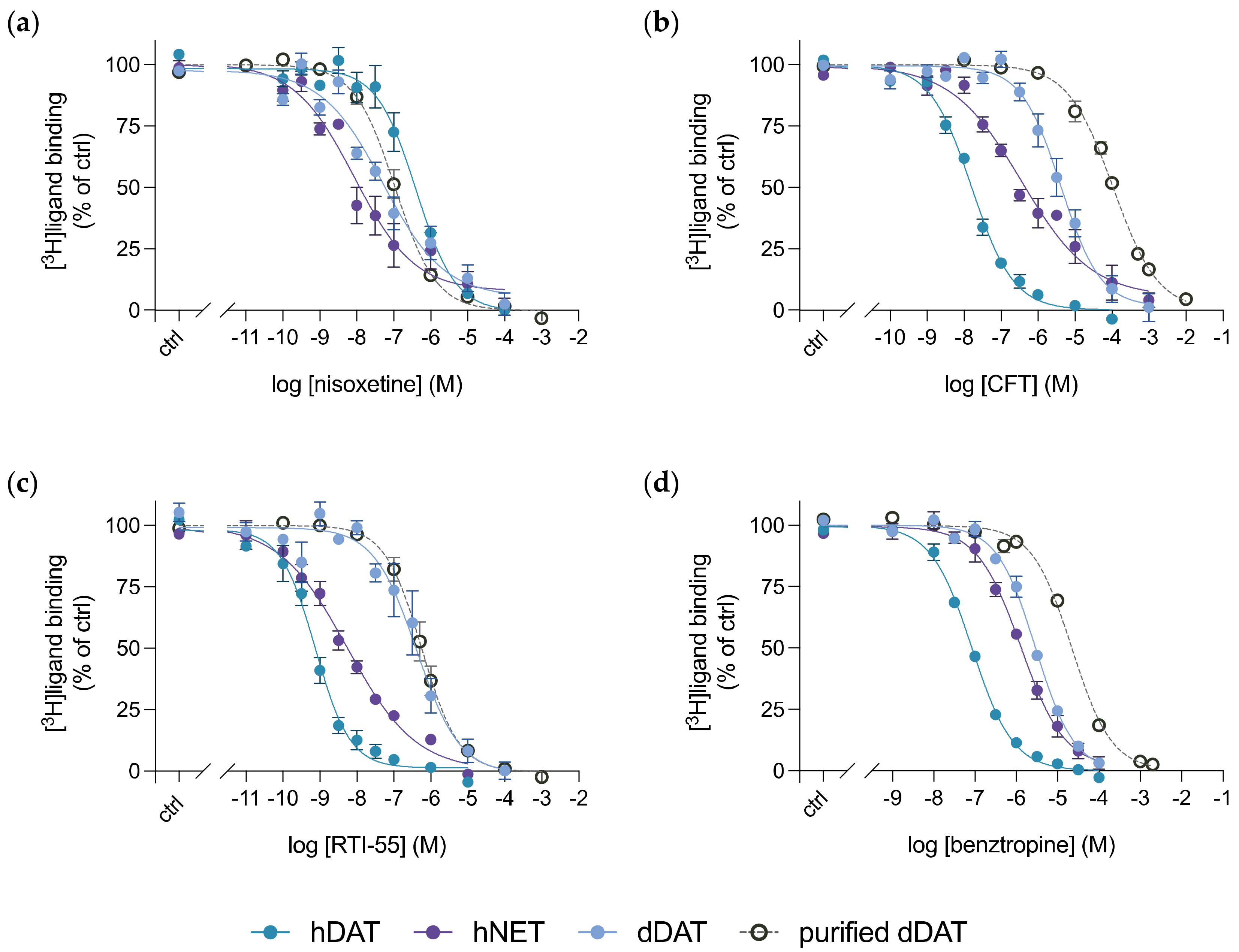
3.4. Purified dDAT Harbours an Inhibitory Profile Similar to hNET
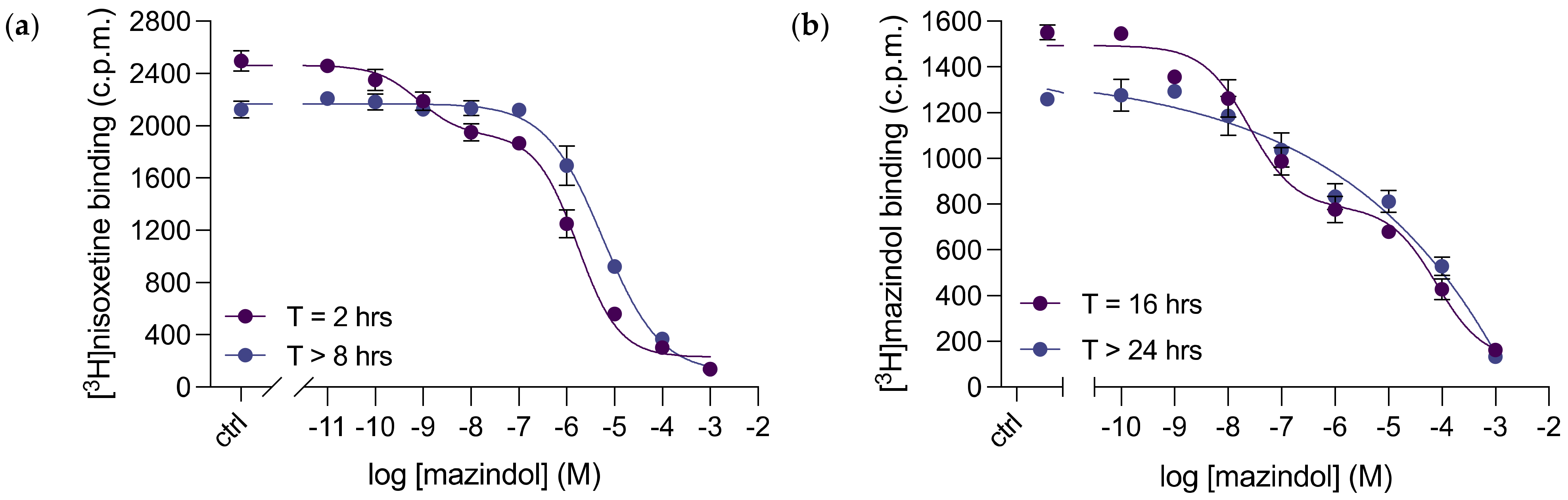
3.5. Mazindol Displays Time-Dependent, Biphasic Binding Kinetics to dDAT
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chinta, S.J.; Andersen, J.K. Dopaminergic Neurons. Int. J. Biochem. Cell Biol. 2005, 37, 942–946. [Google Scholar] [CrossRef] [PubMed]
- Gether, U.; Andersen, P.H.; Larsson, O.M.; Schousboe, A. Neurotransmitter Transporters: Molecular Function of Important Drug Targets. Trends Pharmacol. Sci. 2006, 27, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, A.S.; Andersen, J.; Jorgensen, T.N.; Sorensen, L.; Eriksen, J.; Loland, C.J.; Stromgaard, K.; Gether, U. SLC6 Neurotransmitter Transporters: Structure, Function, and Regulation. Pharmacol. Rev. 2011, 63, 585–640. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.H.; Reith, M.E.A.; Quick, M.W. Synaptic Uptake and beyond: The Sodium- and Chloride-Dependent Neurotransmitter Transporter Family SLC6. Pflugers Arch. Eur. J. Physiol. 2004, 447, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Sitte, H.H.; Freissmuth, M. Amphetamines, New Psychoactive Drugs and the Monoamine Transporter Cycle. Trends Pharmacol. Sci. 2015, 36, 41–50. [Google Scholar] [CrossRef]
- Beuming, T.; Kniazeff, J.; Bergmann, M.L.; Shi, L.; Gracia, L.; Raniszewska, K.; Newman, A.H.; Javitch, J.A.; Weinstein, H.; Gether, U.; et al. The Binding Sites for Cocaine and Dopamine in the Dopamine Transporter Overlap. Nat. Neurosci. 2008, 11, 780–789. [Google Scholar] [CrossRef]
- Zou, M.-F.; Cao, J.; Abramyan, A.M.; Kopajtic, T.; Zanettini, C.; Guthrie, D.A.; Rais, R.; Slusher, B.S.; Shi, L.; Loland, C.J.; et al. Structure–Activity Relationship Studies on a Series of 3α-[Bis(4-Fluorophenyl)Methoxy]Tropanes and 3α-[Bis(4-Fluorophenyl)Methylamino]Tropanes As Novel Atypical Dopamine Transporter (DAT) Inhibitors for the Treatment of Cocaine Use Disorders. J. Med. Chem. 2017, 60, 10172–10187. [Google Scholar] [CrossRef]
- Giros, B.; Jaber, M.; Jones, S.R.; Wightman, R.M.; Caron, M.G. Hyperlocomotion and Indifference to Cocaine and Amphetamine in Mice Lacking the Dopamine Transporter. Nature 1996, 379, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Tilley, M.R.; Wei, H.; Zhou, F.; Zhou, F.M.; Ching, S.; Quan, N.; Stephens, R.L.; Hill, E.R.; Nottoli, T.; et al. Abolished Cocaine Reward in Mice with a Cocaine-Insensitive Dopamine Transporter. Proc. Natl. Acad. Sci. USA 2006, 103, 9333–9338. [Google Scholar] [CrossRef]
- Saier, M.H. TCDB: The Transporter Classification Database for Membrane Transport Protein Analyses and Information. Nucleic Acids Res. 2006, 34, D181–D186. [Google Scholar] [CrossRef]
- Masson, J.; Sagné, C.; Hamon, M.; El Mestikawy, S. Neurotransmitter Transporters in the Central Nervous System. Pharmacol. Rev. 1999, 51, 439–464. [Google Scholar] [PubMed]
- Yamashita, A.; Singh, S.K.; Kawate, T.; Jin, Y.; Gouaux, E. Crystal Structure of a Bacterial Homologue of Na+/Cl--Dependent Neurotransmitter Transporters. Nature 2005, 437, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Gouaux, E. Illumination of Serotonin Transporter Mechanism and Role of the Allosteric Site. Sci. Adv. 2021, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.H.; Penmatsa, A.; Gouaux, E. Neurotransmitter and Psychostimulant Recognition by the Dopamine Transporter. Nature 2015, 521, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Pidathala, S.; Mallela, A.K.; Joseph, D.; Penmatsa, A. Structural Basis of Norepinephrine Recognition and Transport Inhibition in Neurotransmitter Transporters. Nat. Commun. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Ortore, G.; Orlandini, E.; Betti, L.; Giannaccini, G.; Mazzoni, M.R.; Camodeca, C.; Nencetti, S. Focus on Human Monoamine Transporter Selectivity. New Human DAT and NET Models, Experimental Validation, and SERT Affinity Exploration. ACS Chem. Neurosci. 2020, 11, 3214–3232. [Google Scholar] [CrossRef]
- Coleman, J.A.; Gouaux, E. Structural Basis for Recognition of Diverse Antidepressants by the Human Serotonin Transporter. Nat. Struct. Mol. Biol. 2018, 25, 170–175. [Google Scholar] [CrossRef]
- Bisgaard, H.; Larsen, M.A.B.; Mazier, S.; Beuming, T.; Newman, A.H.; Weinstein, H.; Shi, L.; Loland, C.J.; Gether, U. The Binding Sites for Benztropines and Dopamine in the Dopamine Transporter Overlap. Neuropharmacology 2011, 60, 182–190. [Google Scholar] [CrossRef]
- Jardetzky, O. Simple Allosteric Model for Membrane Pumps. Nature 1966, 211, 969–970. [Google Scholar] [CrossRef]
- Drew, D.; Boudker, O. Shared Molecular Mechanisms of Membrane Transporters. Annu. Rev. Biochem. 2016, 85, 543–572. [Google Scholar] [CrossRef]
- Navratna, V.; Tosh, D.K.; Jacobson, K.A.; Gouaux, E. Thermostabilization and Purification of the Human Dopamine Transporter (HDAT) in an Inhibitor and Allosteric Ligand Bound Conformation. PLoS ONE 2018, 13, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.G.; Malle, M.G.; Nielsen, A.K.; Bohr, S.S.-R.; Pugh, C.F.; Nielsen, J.C.; Poulsen, I.H.; Rand, K.D.; Hatzakis, N.S.; Loland, C.J. The Dopamine Transporter Antiports Potassium to Increase the Uptake of Dopamine. Nat. Commun. 2022, 13, 2446. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, A.K.; Möller, I.R.; Wang, Y.; Rasmussen, S.G.F.; Lindorff-Larsen, K.; Rand, K.D.; Loland, C.J. Substrate-Induced Conformational Dynamics of the Dopamine Transporter. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-Ray Structure of Dopamine Transporter Elucidates Antidepressant Mechanism. Nature 2013, 503, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Pörzgen, P.; Park, S.K.; Hirsh, J.; Sonders, M.S.; Amara, S.G. The Antidepressant-Sensitive Dopamine Transporter in Drosophila Melanogaster: A Primordial Carrier for Catecholamines. Mol. Pharmacol. 2001, 59, 83–95. [Google Scholar] [CrossRef]
- Penmatsa, A.; Wang, K.H.; Gouaux, E. X-Ray Structures of Drosophila Dopamine Transporter in Complex with Nisoxetine and Reboxetine. Nat. Struct. Mol. Biol. 2015, 22, 506–508. [Google Scholar] [CrossRef] [PubMed]
- Goehring, A.; Lee, C.-H.; Wang, K.H.; Michel, J.C.; Claxton, D.P.; Baconguis, I.; Althoff, T.; Fischer, S.; Garcia, K.C.; Gouaux, E. Screening and Large-Scale Expression of Membrane Proteins in Mammalian Cells for Structural Studies. Nat. Protoc. 2014, 9, 2574–2585. [Google Scholar] [CrossRef] [PubMed]
- Quick, M.; Javitch, J.A. Monitoring the Function of Membrane Transport Proteins in Detergent-Solubilized Form. Proc. Natl. Acad. Sci. USA 2007, 104, 3603–3608. [Google Scholar] [CrossRef]
- Pedersen, A.V.; Andreassen, T.F.; Loland, C.J. A Conserved Salt Bridge between Transmembrane Segments 1 and 10 Constitutes an Extracellular Gate in the Dopamine Transporter. J. Biol. Chem. 2014, 289, 35003–35014. [Google Scholar] [CrossRef]
- Velez-Pardo, C.; Jimenez del Rio, M.; Ebinger, G.; Vauquelin, G. Manganese and Copper Promote the Binding of Dopamine to “Serotonin Binding Proteins” in Bovine Frontal Cortex. Neurochem. Int. 1995, 26, 615–622. [Google Scholar] [CrossRef]
- Rehmani, N.; Zafar, A.; Arif, H.; Hadi, S.M.; Wani, A.A. Copper-Mediated DNA Damage by the Neurotransmitter Dopamine and L-DOPA: A pro-Oxidant Mechanism. Toxicol Vitr. 2017, 40, 336–346. [Google Scholar] [CrossRef]
- Tatsumi, M.; Groshan, K.; Blakely, R.D.; Richelson, E. Pharmacological Profile of Antidepressants and Related Compounds at Human Monoamine Transporters. Eur. J. Pharmacol. 1997, 340, 249–258. [Google Scholar] [CrossRef] [PubMed]
- Agoston, G.E.; Wu, J.H.; Izenwasser, S.; George, C.; Katz, J.; Kline, R.H.; Newman, A.H. Novel N-Substituted 3α-[Bis(4’-Fluorophenyl)Methoxy]Tropane Analogues: Selective Ligands for the Dopamine Transporter. J. Med. Chem. 1997, 40, 4329–4339. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Slack, R.D.; Bakare, O.M.; Burzynski, C.; Rais, R.; Slusher, B.S.; Kopajtic, T.; Bonifazi, A.; Ellenberger, M.P.; Yano, H.; et al. Novel and High Affinity 2-[(Diphenylmethyl)Sulfinyl]Acetamide (Modafinil) Analogues as Atypical Dopamine Transporter Inhibitors. J. Med. Chem. 2016, 59, 10676–10691. [Google Scholar] [CrossRef]
- Owens, M.J.; Knight, D.L.; Nemeroff, C.B. Second-Generation SSRIs: Human Monoamine Transporter Binding Profile of Escitalopram and R-Fluoxetine. Biol. Psychiatry 2001, 50, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Loland, C.J.; Grånäs, C.; Javitch, J.A.; Gether, U. Identification of Intracellular Residues in the Dopamine Transporter Critical for Regulation of Transporter Conformation and Cocaine Binding. J. Biol. Chem. 2004, 279, 3228–3238. [Google Scholar] [CrossRef]
- Loland, C.J.; Mereu, M.; Okunola, O.M.; Cao, J.; Prisinzano, T.E.; Mazier, S.; Kopajtic, T.; Shi, L.; Katz, J.L.; Tanda, G.; et al. R-Modafinil (Armodafinil): A Unique Dopamine Uptake Inhibitor and Potential Medication for Psychostimulant Abuse. Biol. Psychiatry 2012, 72, 405–413. [Google Scholar] [CrossRef]
- Schmitt, K.C.; Rothman, R.B.; Reith, M.E.A. Nonclassical Pharmacology of the Dopamine Transporter: Atypical Inhibitors, Allosteric Modulators, and Partial Substrates. J. Pharmacol. Exp. Ther. 2013, 346, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Douglas, J.; Munro, J. Drug Treatment and Obesity. Pharmacol. Ther. 1982, 18, 351–373. [Google Scholar] [CrossRef]
- Arnulf, I.; Leu-Semenescu, S.; Dodet, P. Precision Medicine for Idiopathic Hypersomnia. Sleep Med. Clin. 2019, 14, 333–350. [Google Scholar] [CrossRef]
- Jacobs, M.T.; Zhang, Y.W.; Campbell, S.D.; Rudnick, G. Ibogaine, a Noncompetitive Inhibitor of Serotonin Transport, Acts by Stabilizing the Cytoplasm-Facing State of the Transporter. J. Biol. Chem. 2007, 282, 29441–29447. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.A.; Yang, D.; Zhao, Z.; Wen, P.C.; Yoshioka, C.; Tajkhorshid, E.; Gouaux, E. Serotonin Transporter–Ibogaine Complexes Illuminate Mechanisms of Inhibition and Transport. Nature 2019, 569, 141–145. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | dDAT Ki (nM) | dDAT-COS Ki (nM) | dDAT-MDCK IC50 (nM) | DAT Ki (nM) | NET Ki (nM) |
|---|---|---|---|---|---|
| Nisoxetine | 31 [28; 34]* | 70 [42; 115] * | 5.6 ± 2.2 b | 223 [154; 323] | 8.8 [4.6; 15] * |
| Imipramine | 58 [51; 66] | 30 ± 10 b | 8500 ± 100 a | 37 ± 2 a | |
| Amitriptyline | 97 [80; 117] | 30 ± 1 b | 3250 ± 20 a | 35 ± 2 a | |
| RTI-55 | 130 [95; 178] | 299 [223; 400] | 66 ± 10 b | 0.5 [0.4; 0.63] | 4 [3.3; 4.9] |
| Desipramine | 141 [131; 151] | 18 ± 5 b | 3190 ± 40 a | 0.83 ± 0.05 a | |
| Nortriptyline | 292 [255; 335] | 1140 ± 30 a | 4.37 ± 0.07 a | ||
| Paroxetine | 322 [242; 428] | 22 ± 4 b | 490 ± 20 a | 40 ± 2 a | |
| Fluoxetine | 512 [438; 599] | 240 ± 60 b | 3600 ± 100 a | 240 ± 10 a | |
| Mazindol | 2800 [2200; 3700] | 4.4 ± 2.2 b | 8.1 ± 0.4 a | 0.45 ± 0.03 a | |
| JHW007 | 3100 [3000; 3300] | 24.6 ± 8 c | 1330 c | ||
| Benztropine | 5200 [4800; 5700] | 2800 [2600; 2900] | 56 [54; 59] | 989 [903; 1080] | |
| Bupropion | 7500 [7400; 7700] | 48000 ± 5000 b | 520 ± 20 a | 52000 ± 1000 a | |
| Cocaine | 20000 [18000; 23000] | 2660 ± 230 b | 220 ± 9 a | 1420 ± 50 a | |
| S-citalopram | 24000 [19000; 29000] | 8100 ± 2100 b | 27410 ± 3106 e | 7841 ± 998 e | |
| CFT | 25000 [23000; 27000] | 3900 [2800; 5400] | 11 [9; 12] * | 357 [251; 508] | |
| JJC8-088 | 33000 [30000; 35000] | 2.53 ± 0.25 d | 15000 ± 575 d | ||
| JJC8-091 | 199000 [188000; 211000] | 289 ± 43 d | |||
| Modafinil | 554000 [516000; 596000] | 2520 ± 204 d | > 100000 d | ||
| Dopamine | 6900 [4500; 10500] | 7700 [7200; 8300] | 2900 ± 500 b | 1900 [1600; 2200] | 433 [343; 546] |
| Methamphetamine | 13000 [10000; 16000] | 2800 ± 100 a | 660 ± 20 a | ||
| D-amphetamine | 18000 [16000; 21000] | 6600 ± 900 b | 2900 ± 200 a | 530 ± 40 a | |
| Serotonin | 173000 [97000; 311000] | 43000 ± 7000 b | > 100000 a | > 100000 a | |
| Norepinephrine | 184000 [141000; 239000] | 68000 [46000; 103000] | 49000 ± 9000 b | 9500 [8800; 10300] | 1800 [1200; 2600] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pugh, C.F.; DeVree, B.T.; Schmidt, S.G.; Loland, C.J. Pharmacological Characterization of Purified Full-Length Dopamine Transporter from Drosophila melanogaster. Cells 2022, 11, 3811. https://doi.org/10.3390/cells11233811
Pugh CF, DeVree BT, Schmidt SG, Loland CJ. Pharmacological Characterization of Purified Full-Length Dopamine Transporter from Drosophila melanogaster. Cells. 2022; 11(23):3811. https://doi.org/10.3390/cells11233811
Chicago/Turabian StylePugh, Ciara Frances, Brian Thomas DeVree, Solveig Gaarde Schmidt, and Claus Juul Loland. 2022. "Pharmacological Characterization of Purified Full-Length Dopamine Transporter from Drosophila melanogaster" Cells 11, no. 23: 3811. https://doi.org/10.3390/cells11233811
APA StylePugh, C. F., DeVree, B. T., Schmidt, S. G., & Loland, C. J. (2022). Pharmacological Characterization of Purified Full-Length Dopamine Transporter from Drosophila melanogaster. Cells, 11(23), 3811. https://doi.org/10.3390/cells11233811