.png)
Liver Injury and Cell Survival in Non-Alcoholic Steatohepatitis Regulated by Sex-Based Difference through B Cell Lymphoma 6

{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Mechanism of NAFLD and NASH
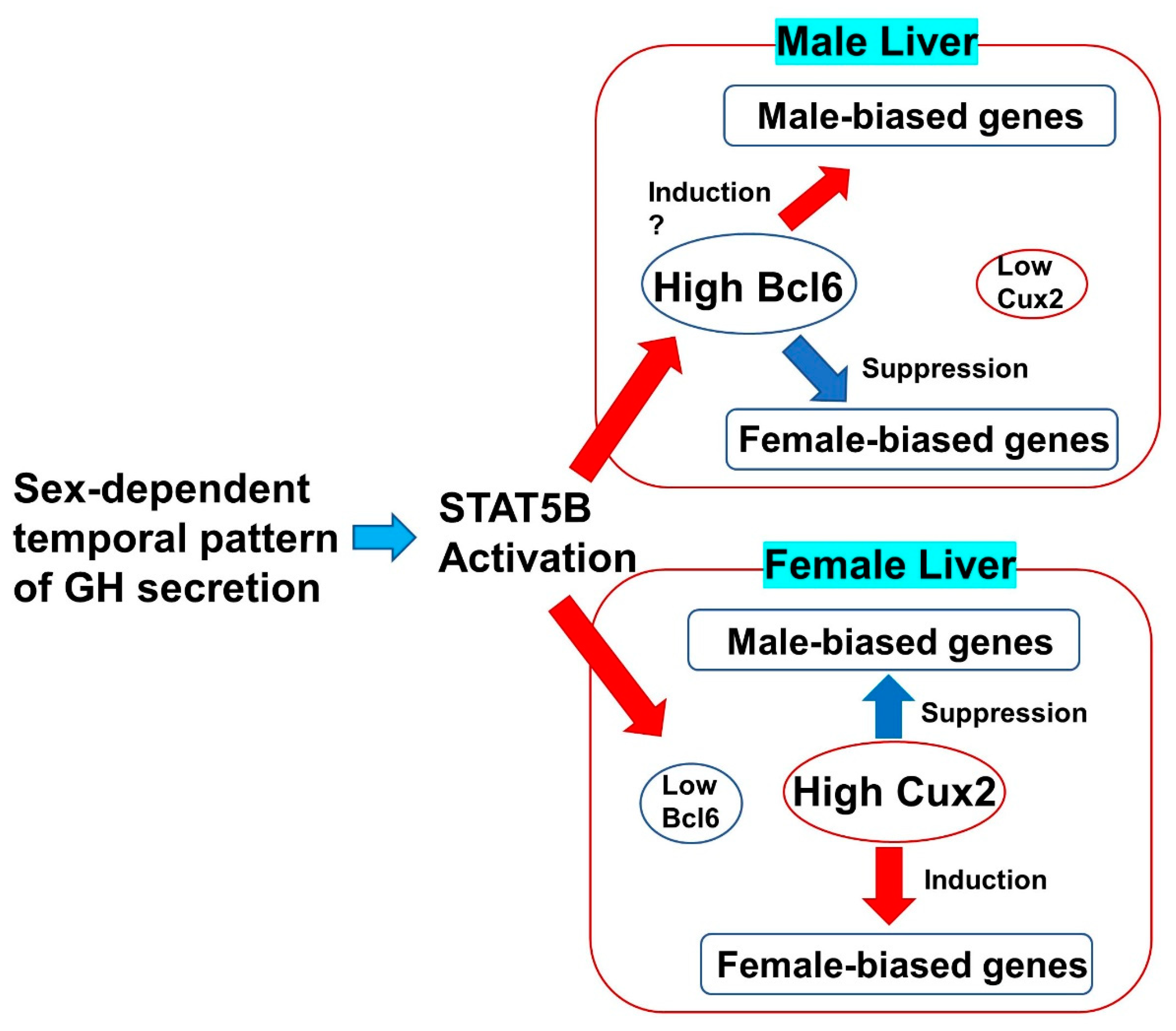
3. The Sex-Biased Liver Functions Regulated by B Cell Lymphoma 6
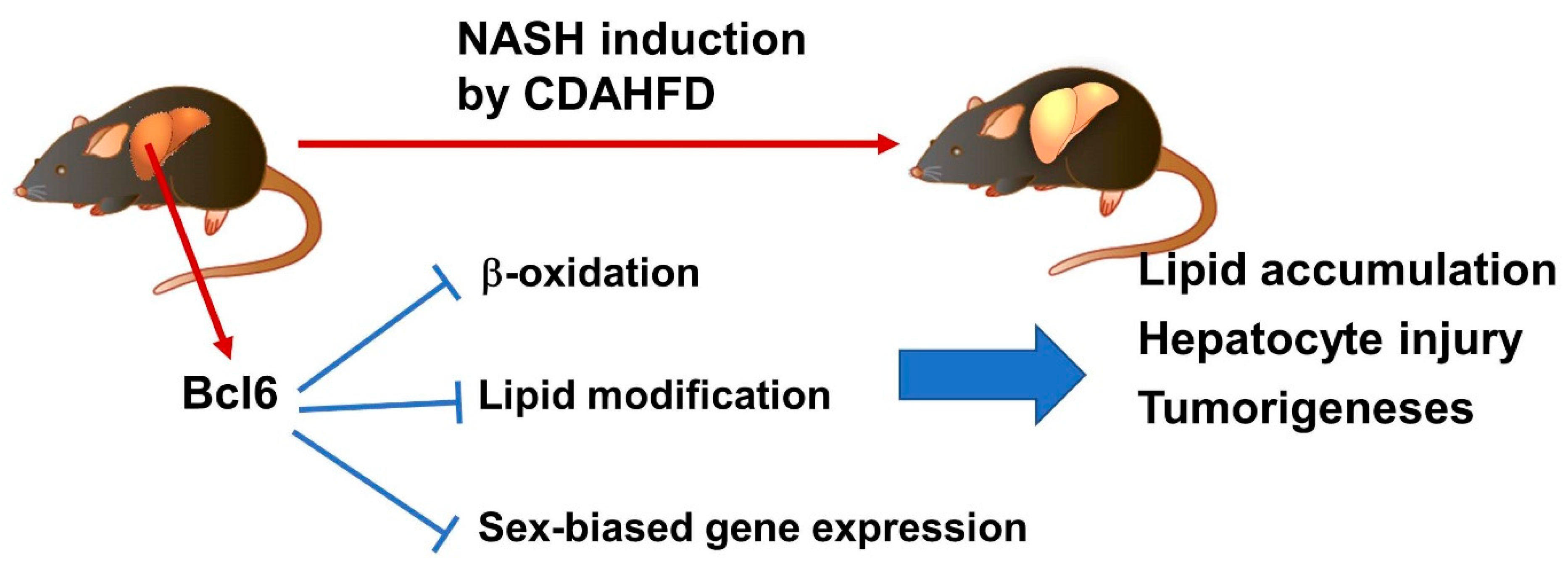
4. The Progression of NASH and Hepatocellular Carcinomas Is Regulated by Sex-Dependent Factors
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Michalopoulos, G.K.; Bhushan, B. Liver regeneration: Biological and pathological mechanisms and implications. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 40–55. [Google Scholar] [CrossRef]
- Leoni, S.; Casabianca, A.; Biagioni, B.; Serio, I. Viral hepatitis: Innovations and expectations. World J. Gastroenterol. 2022, 28, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Golbidi, S.; Mesdaghinia, A.; Laher, I. Exercise in the Metabolic Syndrome. Oxid. Med. Cell. Longev. 2012, 2012, 349710. [Google Scholar] [CrossRef]
- Keane, D.; Kelly, S.; Healy, N.; McArdle, M.; Holohan, K.; Roche, H. Diet and Metabolic Syndrome: An Overview. Curr. Vasc. Pharmacol. 2013, 11, 842–857. [Google Scholar] [CrossRef]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef] [PubMed]
- Rosso, N.; Chavez-Tapia, N.C.; Tiribelli, C.; Bellentani, S. Translational approaches: From fatty liver to non-alcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 9038–9049. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: The multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef] [PubMed]
- Taliento, A.E.; Dallio, M.; Federico, A.; Prati, D.; Valenti, L. Novel Insights into the Genetic Landscape of Nonalcoholic Fatty Liver Disease. Int. J. Environ. Res. Public Health 2019, 16, 2755. [Google Scholar] [CrossRef] [PubMed]
- Abdelmegeed, M.A.; Ha, S.-K.; Choi, Y.; Akbar, M.; Song, B.-J. Role of CYP2E1 in Mitochondrial Dysfunction and Hepatic Injury by Alcohol and Non-Alcoholic Substances. Curr. Mol. Pharmacol. 2017, 10, 207–225. [Google Scholar] [CrossRef]
- Masarone, M.; Rosato, V.; Dallio, M.; Gravina, A.G.; Aglitti, A.; Loguercio, C.; Federico, A.; Persico, M. Role of Oxidative Stress in Pathophysiology of Nonalcoholic Fatty Liver Disease. Oxid. Med. Cell. Longev. 2018, 2018, 9547613. [Google Scholar] [CrossRef]
- Kaufman, R.J. Orchestrating the unfolded protein response in health and disease. J. Clin. Investig. 2002, 110, 1389–1398. [Google Scholar] [CrossRef]
- Paganoni, R.; Lechel, A.; Spasic, M.V. Iron at the Interface of Hepatocellular Carcinoma. Int. J. Mol. Sci. 2021, 22, 4097. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An Iron-Dependent Form of Nonapoptotic Cell Death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Kok, C.Y.-Y.; Okochi, H.; Nakano, H.; et al. Hepatic ferroptosis plays an important role as the trigger for initiating inflammation in nonalcoholic steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Abu Shanab, A.; Scully, P.; Crosbie, O.; Buckley, M.; O’Mahony, L.; Shanahan, F.; Gazareen, S.; Murphy, E.; Quigley, E.M.M. Small Intestinal Bacterial Overgrowth in Nonalcoholic Steatohepatitis: Association with Toll-Like Receptor 4 Expression and Plasma Levels of Interleukin 8. Am. J. Dig. Dis. 2011, 56, 1524–1534. [Google Scholar] [CrossRef]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P.-Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- LaVine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of Vitamin E or Metformin for Treatment of Nonalcoholic Fatty Liver Disease in Children and Adolescents: The TONIC randomized controlled trial. JAMA J. Am. Med. Assoc. 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.N.; Wang, J.; Muralidharan, S.; Chalasani, S.; Fullenkamp, A.M.; Wilson, L.A.; Sanyal, A.J.; Kowdley, K.V.; Neuschwander-Tetri, B.A.; Brunt, E.M.; et al. Relationship between adipose tissue insulin resistance and liver histology in nonalcoholic steatohepatitis: A pioglitazone versus vitamin E versus placebo for the treatment of nondiabetic patients with nonalcoholic steatohepatitis trial follow-up study. Hepatology 2012, 56, 1311–1318. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Premkumar, M.; Kulkarni, A.V.; Kumar, P.; Reddy, D.N.; Rao, N.P. Drugs for Non-alcoholic Steatohepatitis (NASH): Quest for the Holy Grail. J. Clin. Transl. Hepatol. 2021, 9, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef] [PubMed]
- Puengel, T.; Lefere, S.; Hundertmark, J.; Kohlhepp, M.; Penners, C.; Van de Velde, F.; Lapauw, B.; Hoorens, A.; Devisscher, L.; Geerts, A.; et al. Combined Therapy with a CCR2/CCR5 Antagonist and FGF21 Analogue Synergizes in Ameliorating Steatohepatitis and Fibrosis. Int. J. Mol. Sci. 2022, 23, 6696. [Google Scholar] [CrossRef]
- Norstedt, G.; Palmiter, R. Secretory rhythm of growth hormone regulates sexual differentiation of mouse liver. Cell 1984, 36, 805–812. [Google Scholar] [CrossRef]
- Waxman, D.J.; O’Connor, C. Growth Hormone Regulation of Sex-Dependent Liver Gene Expression. Mol. Endocrinol. 2006, 20, 2613–2629. [Google Scholar] [CrossRef]
- Hunt, C.M.; Westerkam, W.R.; Stave, G.M. Effect of age and gender on the activity of human hepatic CYP3A. Biochem. Pharmacol. 1992, 44, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Kushlan, M.C.; Gollan, J.L.; Ma, W.L.; Ockner, R.K. Sex differences in hepatic uptake of long chain fatty acids in single-pass perfused rat liver. J. Lipid Res. 1981, 22, 431–436. [Google Scholar] [CrossRef]
- Khristi, V.; Ratri, A.; Ghosh, S.; Pathak, D.; Borosha, S.; Dai, E.; Roy, R.; Chakravarthi, V.P.; Wolfe, M.W.; Rumi, M.K. Disruption of ESR1 alters the expression of genes regulating hepatic lipid and carbohydrate metabolism in male rats. Mol. Cell. Endocrinol. 2019, 490, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, M.; Shimizu, I.; Shiba, M.; Ito, S. Suppressive effects of estradiol on dimethylnitrosamine-induced fibrosis of the liver in rats. Hepatology 1999, 29, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E.; Sakurai, T.; Kim, S.; Maeda, S.; Kim, K.; Elsharkawy, A.M.; Karin, M. Gender Disparity in Liver Cancer Due to Sex Differences in MyD88-Dependent IL-6 Production. Science 2007, 317, 121–124. [Google Scholar] [CrossRef] [PubMed]
- Dent, A.L.; Shaffer, A.L.; Yu, X.; Allman, D.; Staudt, L.M. Control of Inflammation, Cytokine Expression, and Germinal Center Formation by BCL-6. Science 1997, 276, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Fukuda, T.; Hatano, M.; Koseki, H.; Okabe, S.-I.; Ishibashi, K.; Kojima, S.; Arima, M.; Komuro, I.; Ishii, G.; et al. The role of Bcl6 in mature cardiac myocytes. Cardiovasc. Res. 1999, 42, 670–679. [Google Scholar] [CrossRef] [PubMed]
- LaPensee, C.R.; Lin, G.; Dent, A.L.; Schwartz, J. Deficiency of the Transcriptional Repressor B Cell Lymphoma 6 (Bcl6) Is Accompanied by Dysregulated Lipid Metabolism. PLoS ONE 2014, 9, e97090. [Google Scholar] [CrossRef]
- Udy, G.B.; Towers, R.P.; Snell, R.G.; Wilkins, R.J.; Park, S.-H.; Ram, P.A.; Waxman, D.J.; Davey, H.W. Requirement of STAT5b for sexual dimorphism of body growth rates and liver gene expression. Proc. Natl. Acad. Sci. USA 1997, 94, 7239–7244. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Laz, E.V.; Waxman, D.J. Dynamic, Sex-Differential STAT5 and BCL6 Binding to Sex-Biased, Growth Hormone-Regulated Genes in Adult Mouse Liver. Mol. Cell. Biol. 2012, 32, 880–896. [Google Scholar] [CrossRef]
- Kaji, T.; Ishige, A.; Hikida, M.; Taka, J.; Hijikata, A.; Kubo, M.; Nagashima, T.; Takahashi, Y.; Kurosaki, T.; Okada, M.; et al. Distinct cellular pathways select germline-encoded and somatically mutated antibodies into immunological memory. J. Exp. Med. 2012, 209, 2079–2097. [Google Scholar] [CrossRef]
- Postic, C.; Magnuson, M.A. DNA excision in liver by an albumin-Cre transgene occurs progressively with age. Genesis 2000, 26, 149–150. [Google Scholar] [CrossRef]
- Chikada, H.; Ida, K.; Ando, E.; Inagaki, Y.; Sakamoto, A.; Kamiya, A. Establishment and analysis of a mouse model that regulates sex-related differences in liver drug metabolism. Lab. Investig. 2018, 98, 1500–1511. [Google Scholar] [CrossRef] [PubMed]
- Sugathan, A.; Waxman, D.J. Genome-Wide Analysis of Chromatin States Reveals Distinct Mechanisms of Sex-Dependent Gene Regulation in Male and Female Mouse Liver. Mol. Cell. Biol. 2013, 33, 3594–3610. [Google Scholar] [CrossRef]
- Conforto, T.L.; Zhang, Y.; Sherman, J.; Waxman, D.J. Impact of CUX2 on the Female Mouse Liver Transcriptome: Activation of Female-Biased Genes and Repression of Male-Biased Genes. Mol. Cell. Biol. 2012, 32, 4611–4627. [Google Scholar] [CrossRef] [PubMed]
- Melia, T.; Hao, P.; Yilmaz, F.; Waxman, D.J. Hepatic Long Intergenic Noncoding RNAs: High Promoter Conservation and Dynamic, Sex-Dependent Transcriptional Regulation by Growth Hormone. Mol. Cell. Biol. 2016, 36, 50–69. [Google Scholar] [CrossRef]
- Chowen, J.A.; Frago, L.M.; Argente, J. The regulation of GH secretion by sex steroids. Eur. J. Endocrinol. 2004, 151 (Suppl. 3), U95–U100. [Google Scholar] [CrossRef]
- Wu, J.; Yao, X.-Y.; Shi, R.-X.; Liu, S.-F.; Wang, X.-Y. A potential link between polycystic ovary syndrome and non-alcoholic fatty liver disease: An update meta-analysis. Reprod. Health 2018, 15, 77. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Abudu, A.; Salinas, I.; Sinha, N.; Cline-Fedewa, H.; Yaw, A.M.; Qi, W.; Lydic, T.A.; Takahashi, D.L.; Hennebold, J.D.; et al. Androgen-mediated perturbation of the hepatic circadian system through epigenetic modulation promotes NAFLD in PCOS mice. Endocrinology 2022, 163, bqac127. [Google Scholar] [CrossRef]
- Bur, I.M.; Cohen-Solal, A.M.; Carmignac, D.; Abecassis, P.-Y.; Chauvet, N.; Martin, A.O.; van der Horst, G.T.; Robinson, I.C.; Maurel, P.; Mollard, P.; et al. The Circadian Clock Components CRY1 and CRY2 Are Necessary to Sustain Sex Dimorphism in Mouse Liver Metabolism. J. Biol. Chem. 2009, 284, 9066–9073. [Google Scholar] [CrossRef]
- Meda, C.; Barone, M.; Mitro, N.; Lolli, F.; Pedretti, S.; Caruso, D.; Maggi, A.; Della Torre, S. Hepatic ERα accounts for sex differences in the ability to cope with an excess of dietary lipids. Mol. Metab. 2020, 32, 97–108. [Google Scholar] [CrossRef]
- Zhuang, Q.K.-W.; Galvez, J.H.; Xiao, Q.; AlOgayil, N.; Hyacinthe, J.; Taketo, T.; Bourque, G.; Naumova, A.K. Sex Chromosomes and Sex Phenotype Contribute to Biased DNA Methylation in Mouse Liver. Cells 2020, 9, 1436. [Google Scholar] [CrossRef] [PubMed]
- AlOgayil, N.; Bauermeister, K.; Galvez, J.H.; Venkatesh, V.S.; Zhuang, Q.K.-W.; Chang, M.L.; Davey, R.A.; Zajac, J.D.; Ida, K.; Kamiya, A.; et al. Distinct roles of androgen receptor, estrogen receptor alpha, and BCL6 in the establishment of sex-biased DNA methylation in mouse liver. Sci. Rep. 2021, 11, 13766. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Falt, S.; Sandelin, A.; Gustafsson, J.-A.; Dahlman-Wright, K. Genome-Wide Identification of Estrogen Receptor α-Binding Sites in Mouse Liver. Mol. Endocrinol. 2008, 22, 10–22. [Google Scholar] [CrossRef]
- Villa, A.; Della Torre, S.; Stell, A.; Cook, J.; Brown, M.; Maggi, A. Tetradian oscillation of estrogen receptor α is necessary to prevent liver lipid deposition. Proc. Natl. Acad. Sci. USA 2012, 109, 11806–11811. [Google Scholar] [CrossRef]
- Hirano, K.-I.; Kuwasako, T.; Nakagawa-Toyama, Y.; Janabi, M.; Yamashita, S.; Matsuzawa, Y. Pathophysiology of human genetic CD36 deficiency. Trends Cardiovasc. Med. 2003, 13, 136–141. [Google Scholar] [CrossRef] [PubMed]
- Rojek, A.M.; Skowronski, M.T.; Füchtbauer, E.-M.; Füchtbauer, A.C.; Fenton, R.A.; Agre, P.; Frøkiær, J.; Nielsen, S. Defective glycerol metabolism in aquaporin 9 (AQP9) knockout mice. Proc. Natl. Acad. Sci. USA 2007, 104, 3609–3614. [Google Scholar] [CrossRef]
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.-J.; Kim, S.-W.; Kim, J.N.; Seo, Y.-S.; et al. Formyl peptide receptor 2 determines sex-specific differences in the progression of nonalcoholic fatty liver disease and steatohepatitis. Nat. Commun. 2022, 13, 578. [Google Scholar] [CrossRef]
- Zhang, J.; Powell, C.; Kay, M.; Sonkar, R.; Meruvu, S.; Choudhury, M. Effect of Chronic Western Diets on Non-Alcoholic Fatty Liver of Male Mice Modifying the PPAR-γ Pathway via miR-27b-5p Regulation. Int. J. Mol. Sci. 2021, 22, 1822. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.-H.; Choi, H.; Kim, H.-J.; Lee, M.-O. Chemotactic cytokines secreted from Kupffer cells contribute to the sex-dependent susceptibility to non-alcoholic fatty liver diseases in mice. Life Sci. 2022, 306, 120846. [Google Scholar] [CrossRef]
- Sommars, M.A.; Ramachandran, K.; Senagolage, M.D.; Futtner, C.R.; Germain, D.M.; Allred, A.L.; Omura, Y.; Bederman, I.R.; Barish, G.D. Dynamic repression by BCL6 controls the genome-wide liver response to fasting and steatosis. eLife 2019, 8, e43922. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Chikada, H.; Ida, K.; Nishikawa, Y.; Inagaki, Y.; Kamiya, A. Liver-specific knockout of B cell lymphoma 6 suppresses progression of non-alcoholic steatohepatitis in mice. Sci. Rep. 2020, 10, 9704. [Google Scholar] [CrossRef]
- Nikkanen, J.; Leong, Y.A.; Krause, W.C.; Dermadi, D.; Maschek, J.A.; Van Ry, T.; Cox, J.E.; Weiss, E.J.; Gokcumen, O.; Chawla, A.; et al. An evolutionary trade-off between host immunity and metabolism drives fatty liver in male mice. Science 2022, 378, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Li, H.; Lee, D.; Oliver, P.; Quarfordt, S.; Osada, J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem. 1994, 269, 23610–23616. [Google Scholar] [CrossRef] [PubMed]
- Salisbury, D.A.; Casero, D.; Zhang, Z.; Wang, D.; Kim, J.; Wu, X.; Vergnes, L.; Mirza, A.H.; Leon-Mimila, P.; Williams, K.J.; et al. Transcriptional regulation of N6-methyladenosine orchestrates sex-dimorphic metabolic traits. Nat. Metab. 2021, 3, 940–953. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kamiya, A.; Ida, K. Liver Injury and Cell Survival in Non-Alcoholic Steatohepatitis Regulated by Sex-Based Difference through B Cell Lymphoma 6. Cells 2022, 11, 3751. https://doi.org/10.3390/cells11233751
Kamiya A, Ida K. Liver Injury and Cell Survival in Non-Alcoholic Steatohepatitis Regulated by Sex-Based Difference through B Cell Lymphoma 6. Cells. 2022; 11(23):3751. https://doi.org/10.3390/cells11233751
Chicago/Turabian StyleKamiya, Akihide, and Kinuyo Ida. 2022. "Liver Injury and Cell Survival in Non-Alcoholic Steatohepatitis Regulated by Sex-Based Difference through B Cell Lymphoma 6" Cells 11, no. 23: 3751. https://doi.org/10.3390/cells11233751
APA StyleKamiya, A., & Ida, K. (2022). Liver Injury and Cell Survival in Non-Alcoholic Steatohepatitis Regulated by Sex-Based Difference through B Cell Lymphoma 6. Cells, 11(23), 3751. https://doi.org/10.3390/cells11233751