

Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors

Abstract
1. Introduction
1.1. CAR T Cells
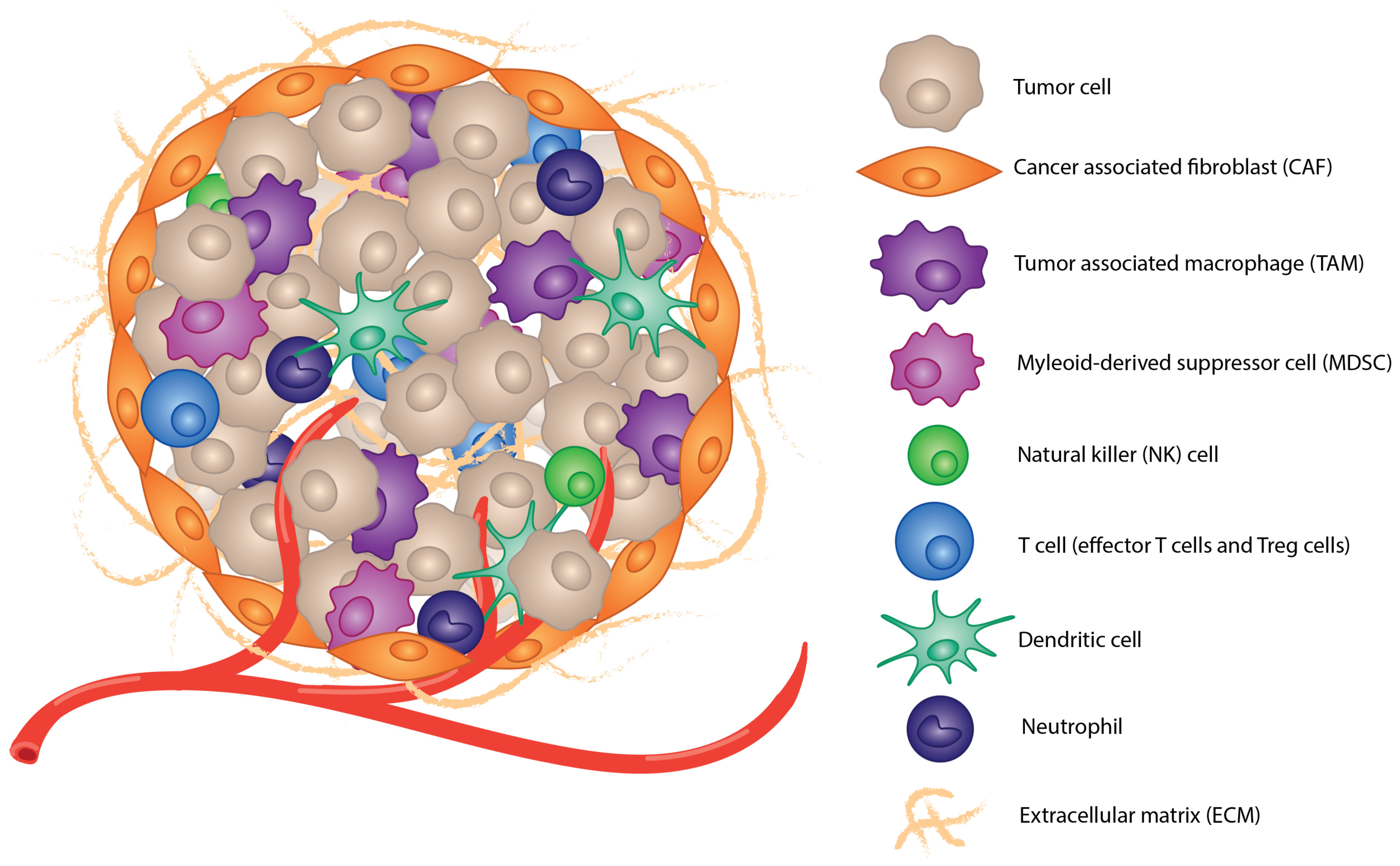
1.2. Tumor Microenvironment
2. Mechanisms for Immunosuppression
2.1. Immunosuppressive Cell Populations
2.2. Secreted Factors
2.3. Metabolic Influences on Immunosuppression
3. Effects of the Immunosuppressive TME on CAR T Cells
3.1. Homing and Solid Tumor Infiltration Difficulties
3.2. CAR T-Cell Exhaustion
3.3. Antigen Escape in CAR T-Cell Therapy
4. CAR T-Cell Advancements and Co-Treatments to Overcome Immunosuppression Roadblocks
4.1. Immune Checkpoint Inhibitors
4.2. CAR Variations
4.3. Oncolytic Viruses in Combination with CAR T Cells
4.4. Metabolic Modifications
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Guedan, S.; Calderon, H.; Posey, A.D.; Maus, M.V. Engineering and Design of Chimeric Antigen Receptors. Mol. Ther. Methods Clin. Dev. 2019, 12, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Clubb, J.D.; Chen, Y.Y. Engineering CAR-T Cells for Next-Generation Cancer Therapy. Cancer Cell 2020, 38, 473–488. [Google Scholar] [CrossRef]
- Graham, C.; Hewitson, R.; Pagliuca, A.; Benjamin, R. Cancer immunotherapy with CAR-T cells—Behold the future. Clin. Med. 2018, 18, 324–328. [Google Scholar] [CrossRef] [PubMed]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Haynie, C.; Johnson, A.; Weber, K.S. Alternative CAR Therapies: Recent Approaches in Engineering Chimeric Antigen Receptor Immune Cells to Combat Cancer. Biomedicines 2022, 10, 1493. [Google Scholar] [CrossRef] [PubMed]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Fujiwara, Y.; Kato, T.; Hasegawa, F.; Sunahara, M.; Tsurumaki, Y. The Past, Present, and Future of Clinically Applied Chimeric Antigen Receptor-T-Cell Therapy. Pharmaceuticals 2022, 15, 207. [Google Scholar] [CrossRef]
- Berdeja, J.G.; Madduri, D.; Usmani, S.Z.; Jakubowiak, A.; Agha, M.; Cohen, A.D.; Stewart, A.K.; Hari, P.; Htut, M.; Lesokhin, A.; et al. Ciltacabtagene autoleucel, a B-cell maturation antigen-directed chimeric antigen receptor T-cell therapy in patients with relapsed or refractory multiple myeloma (CARTITUDE-1): A phase 1b/2 open-label study. Lancet 2021, 398, 314–324. [Google Scholar] [CrossRef]
- Holstein, S.A.; Lunning, M.A. CAR T-Cell Therapy in Hematologic Malignancies: A Voyage in Progress. Clin. Pharmacol. Ther. 2020, 107, 112–122. [Google Scholar] [CrossRef]
- Lindo, L.; Wilkinson, L.H.; Hay, K.A. Befriending the Hostile Tumor Microenvironment in CAR T-Cell Therapy. Front. Immunol. 2020, 11, 618387. [Google Scholar] [CrossRef]
- Nallanthighal, S.; Heiserman, J.P.; Cheon, D.J. The Role of the Extracellular Matrix in Cancer Stemness. Front. Cell Dev. Biol. 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Paolillo, M.; Schinelli, S. Extracellular Matrix Alterations in Metastatic Processes. Int. J. Mol. Sci. 2019, 20, 4947. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Dai, Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017, 387, 61–68. [Google Scholar] [CrossRef]
- Kartikasari, A.E.R.; Huertas, C.S.; Mitchell, A.; Plebanski, M. Tumor-Induced Inflammatory Cytokines and the Emerging Diagnostic Devices for Cancer Detection and Prognosis. Front. Oncol. 2021, 11, 692142. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Zuazo-Gaztelu, I.; Casanovas, O. Unraveling the Role of Angiogenesis in Cancer Ecosystems. Front. Oncol. 2018, 8, 248. [Google Scholar] [CrossRef]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metastasis 2012, 29, 657–662. [Google Scholar] [CrossRef]
- Lei, X.; Lei, Y.; Li, J.-K.; Du, W.-X.; Li, R.-G.; Yang, J.; Li, J.; Li, F.; Tan, H.-B. Immune cells within the tumor microenvironment: Biological functions and roles in cancer immunotherapy. Cancer Lett. 2020, 470, 126–133. [Google Scholar] [CrossRef]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Ahrends, T.; Borst, J. The opposing roles of CD4+ T cells in anti-tumor immunity. Immunology 2018, 154, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Habif, G.; Crinier, A.; André, P.; Vivier, E.; Narni-Mancinelli, E. Targeting natural killer cells in solid tumors. Cell. Mol. Immunol. 2019, 16, 415–422. [Google Scholar] [CrossRef] [PubMed]
- Michielsen, A.J.; Hogan, A.E.; Marry, J.; Tosetto, M.; Cox, F.; Hyland, J.M.; Sheahan, K.D.; O’Donoghue, D.P.; Mulcahy, H.E.; Ryan, E.J.; et al. Tumour tissue microenvironment can inhibit dendritic cell maturation in colorectal cancer. PLoS ONE 2011, 6, e27944. [Google Scholar] [CrossRef] [PubMed]
- Aras, S.; Zaidi, M.R. TAMeless traitors: Macrophages in cancer progression and metastasis. Br. J Cancer 2017, 117, 1583–1591. [Google Scholar] [CrossRef] [PubMed]
- Coffelt, S.B.; Wellenstein, M.D.; de Visser, K.E. Neutrophils in cancer: Neutral no more. Nat. Rev. Cancer 2016, 16, 431–446. [Google Scholar] [CrossRef]
- Croci, D.O.; Zacarías Fluck, M.F.; Rico, M.J.; Matar, P.; Rabinovich, G.A.; Scharovsky, O.G. Dynamic cross-talk between tumor and immune cells in orchestrating the immunosuppressive network at the tumor microenvironment. Cancer Immunol. Immunother. 2007, 56, 1687–1700. [Google Scholar] [CrossRef] [PubMed]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J. Leukoc. Biol 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- DeNardo, D.G.; Ruffell, B. Macrophages as regulators of tumour immunity and immunotherapy. Nat. Rev. Immunol. 2019, 19, 369–382. [Google Scholar] [CrossRef]
- Huang, X.; Li, Y.; Fu, M.; Xin, H.B. Polarizing Macrophages In Vitro. Methods Mol. Biol. 2018, 1784, 119–126. [Google Scholar] [CrossRef]
- Lewis, C.; Murdoch, C. Macrophage responses to hypoxia: Implications for tumor progression and anti-cancer therapies. Am. J. Pathol. 2005, 167, 627–635. [Google Scholar] [CrossRef]
- Bingle, L.; Lewis, C.E.; Corke, K.P.; Reed, M.W.; Brown, N.J. Macrophages promote angiogenesis in human breast tumour spheroids in vivo. Br. J. Cancer 2006, 94, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Nishie, A.; Ono, M.; Shono, T.; Fukushi, J.; Otsubo, M.; Onoue, H.; Ito, Y.; Inamura, T.; Ikezaki, K.; Fukui, M.; et al. Macrophage infiltration and heme oxygenase-1 expression correlate with angiogenesis in human gliomas. Clin. Cancer Res. 1999, 5, 1107–1113. [Google Scholar] [PubMed]
- Hotchkiss, K.A.; Ashton, A.W.; Klein, R.S.; Lenzi, M.L.; Zhu, G.H.; Schwartz, E.L. Mechanisms by which tumor cells and monocytes expressing the angiogenic factor thymidine phosphorylase mediate human endothelial cell migration. Cancer Res. 2003, 63, 527–533. [Google Scholar]
- Dvorak, H.F. Tumors: Wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar]
- Qian, B.Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Law, A.M.K.; Valdes-Mora, F.; Gallego-Ortega, D. Myeloid-Derived Suppressor Cells as a Therapeutic Target for Cancer. Cells 2020, 9, 561. [Google Scholar] [CrossRef]
- Rodriguez-Garcia, A.; Palazon, A.; Noguera-Ortega, E.; Powell, D.J.; Guedan, S. CAR-T Cells Hit the Tumor Microenvironment: Strategies to Overcome Tumor Escape. Front. Immunol. 2020, 11, 1109. [Google Scholar] [CrossRef]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef]
- Ostrand-Rosenberg, S. Myeloid-derived suppressor cells: More mechanisms for inhibiting antitumor immunity. Cancer Immunol. Immunother. 2010, 59, 1593–1600. [Google Scholar] [CrossRef]
- Enblad, G.; Karlsson, H.; Gammelgård, G.; Wenthe, J.; Lövgren, T.; Amini, R.M.; Wikstrom, K.I.; Essand, M.; Savoldo, B.; Hallböök, H.; et al. A Phase I/IIa Trial Using CD19-Targeted Third-Generation CAR T Cells for Lymphoma and Leukemia. Clin. Cancer Res. 2018, 24, 6185–6194. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Kim, J.M.; Rasmussen, J.P.; Rudensky, A.Y. Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol. 2007, 8, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Samstein, R.M.; Arvey, A.; Josefowicz, S.Z.; Peng, X.; Reynolds, A.; Sandstrom, R.; Neph, S.; Sabo, P.; Kim, J.M.; Liao, W.; et al. Foxp3 exploits a pre-existent enhancer landscape for regulatory T cell lineage specification. Cell 2012, 151, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Liyanage, U.K.; Moore, T.T.; Joo, H.G.; Tanaka, Y.; Herrmann, V.; Doherty, G.; Drebin, J.A.; Strasberg, S.M.; Eberlein, T.J.; Goedegebuure, P.S.; et al. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J. Immunol. 2002, 169, 2756–2761. [Google Scholar] [CrossRef] [PubMed]
- Wolf, D.; Wolf, A.M.; Rumpold, H.; Fiegl, H.; Zeimet, A.G.; Muller-Holzner, E.; Deibl, M.; Gastl, G.; Gunsilius, E.; Marth, C. The expression of the regulatory T cell-specific forkhead box transcription factor FoxP3 is associated with poor prognosis in ovarian cancer. Clin. Cancer Res. 2005, 11, 8326–8331. [Google Scholar] [CrossRef]
- Wolf, D.; Sopper, S.; Pircher, A.; Gastl, G.; Wolf, A.M. Treg(s) in Cancer: Friends or Foe? J. Cell. Physiol. 2015, 230, 2598–2605. [Google Scholar] [CrossRef]
- Facciabene, A.; Motz, G.T.; Coukos, G. T-regulatory cells: Key players in tumor immune escape and angiogenesis. Cancer Res. 2012, 72, 2162–2171. [Google Scholar] [CrossRef]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef]
- Sahai, E.; Astsaturov, I.; Cukierman, E.; DeNardo, D.G.; Egeblad, M.; Evans, R.M.; Fearon, D.; Greten, F.R.; Hingorani, S.R.; Hunter, T.; et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat. Rev. Cancer 2020, 20, 174–186. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. A peek into cancer-associated fibroblasts: Origins, functions and translational impact. Dis. Model. Mech. 2018, 11, dmm029447. [Google Scholar] [CrossRef]
- Cannone, S.; Greco, M.R.; Carvalho, T.M.A.; Guizouarn, H.; Soriani, O.; Di Molfetta, D.; Tomasini, R.; Zeeberg, K.; Reshkin, S.J.; Cardone, R.A. Cancer Associated Fibroblast (CAF) Regulation of PDAC Parenchymal (CPC) and CSC Phenotypes Is Modulated by ECM Composition. Cancers 2022, 14, 3737. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, L.; Jungwirth, U.; Avgustinova, A.; Iravani, M.; Mills, A.; Haider, S.; Harper, J.; Isacke, C.M. Cancer-Associated Fibroblasts Suppress CD8+ T-cell Infiltration and Confer Resistance to Immune-Checkpoint Blockade. Cancer Res. 2022, 82, 2904–2917. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, R.; Cai, T.; Chen, Z.; Lan, M.; Zou, T.; Wang, B.; Wang, Q.; Zhao, Y.; Cai, Y. Effects of immune cells and cytokines on inflammation and immunosuppression in the tumor microenvironment. Int. Immunopharmacol. 2020, 88, 106939. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, K.; Karin, M. NF-κB, inflammation, immunity and cancer: Coming of age. Nat. Rev. Immunol. 2018, 18, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Kelly-Spratt, K.S.; Pitteri, S.J.; Gurley, K.E.; Liggitt, D.; Chin, A.; Kennedy, J.; Wong, C.-H.; Zhang, Q.; Buson, T.B.; Wang, H.; et al. Plasma proteome profiles associated with inflammation, angiogenesis, and cancer. PLoS ONE 2011, 6, e19721. [Google Scholar] [CrossRef]
- Kim, R.; Emi, M.; Tanabe, K. Cancer cell immune escape and tumor progression by exploitation of anti-inflammatory and pro-inflammatory responses. Cancer Biol. Ther. 2005, 4, 924–933. [Google Scholar] [CrossRef]
- Dennis, K.L.; Blatner, N.R.; Gounari, F.; Khazaie, K. Current status of interleukin-10 and regulatory T-cells in cancer. Curr. Opin. Oncol. 2013, 25, 637–645. [Google Scholar] [CrossRef]
- Syed, V. TGF-β Signaling in Cancer. J. Cell. Biochem. 2016, 117, 1279–1287. [Google Scholar] [CrossRef]
- Barrow, A.D.; Edeling, M.A.; Trifonov, V.; Luo, J.; Goyal, P.; Bohl, B.; Bando, J.K.; Kim, A.H.; Walker, J.; Andahazy, M.; et al. Natural Killer Cells Control Tumor Growth by Sensing a Growth Factor. Cell 2018, 172, 534–548.e519. [Google Scholar] [CrossRef]
- Pavlova, N.N.; Thompson, C.B. The Emerging Hallmarks of Cancer Metabolism. Cell Metab. 2016, 23, 27–47. [Google Scholar] [CrossRef] [PubMed]
- Pavlova, N.N.; Zhu, J.; Thompson, C.B. The hallmarks of cancer metabolism: Still emerging. Cell Metab. 2022, 34, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Otto, W. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Shi, R.; Tang, Y.Q.; Miao, H. Metabolism in tumor microenvironment: Implications for cancer immunotherapy. MedComm 2020, 1, 47–68. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 749. [Google Scholar] [CrossRef]
- Hayes, C.; Donohoe, C.L.; Davern, M.; Donlon, N.E. The oncogenic and clinical implications of lactate induced immunosuppression in the tumour microenvironment. Cancer Lett. 2021, 500, 75–86. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Wang, Z.; Quinn, W.J.; Kopinski, P.K.; Wang, L.; et al. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293.e1287. [Google Scholar] [CrossRef]
- Elia, I.; Haigis, M.C. Metabolites and the tumour microenvironment: From cellular mechanisms to systemic metabolism. Nat. Metab. 2021, 3, 21–32. [Google Scholar] [CrossRef]
- Jing, X.; Yang, F.; Shao, C.; Wei, K.; Xie, M.; Shen, H.; Shu, Y. Role of hypoxia in cancer therapy by regulating the tumor microenvironment. Mol. Cancer 2019, 18, 157. [Google Scholar] [CrossRef] [PubMed]
- Hatfield, S.M.; Kjaergaard, J.; Lukashev, D.; Schreiber, T.H.; Belikoff, B.; Abbott, R.; Sethumadhavan, S.; Philbrook, P.; Ko, K.; Cannici, R.; et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci. Transl. Med. 2015, 7, 277ra230. [Google Scholar] [CrossRef] [PubMed]
- Parodi, M.; Raggi, F.; Cangelosi, D.; Manzini, C.; Balsamo, M.; Blengio, F.; Eva, A.; Varesio, L.; Pietra, G.; Moretta, L.; et al. Hypoxia Modifies the Transcriptome of Human NK Cells, Modulates Their Immunoregulatory Profile, and Influences NK Cell Subset Migration. Front. Immunol. 2018, 9, 2358. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Elly, C.; Park, Y.; Liu, Y.-C. E3 Ubiquitin Ligase VHL Regulates Hypoxia-Inducible Factor-1α to Maintain Regulatory T Cell Stability and Suppressive Capacity. Immunity 2015, 42, 1062–1074. [Google Scholar] [CrossRef]
- Corbet, C.; Feron, O. Tumour acidosis: From the passenger to the driver’s seat. Nat. Rev. Cancer 2017, 17, 577–593. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Cyrus, N.; Brokowski, C.E.; Eisenbarth, S.C.; Phillips, G.M.; et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef]
- Safarzadeh Kozani, P.; Rahbarizadeh, F. Addressing the obstacles of CAR T cell migration in solid tumors: Wishing a heavy traffic. Crit. Rev. Biotechnol. 2022, 42, 1079–1098. [Google Scholar] [CrossRef]
- Skovgard, M.S.; Hocine, H.R.; Saini, J.K.; Moroz, M.; Bellis, R.Y.; Banerjee, S.; Morello, A.; Ponomarev, V.; Villena-Vargas, J.; Adusumilli, P.S. Imaging CAR T-cell kinetics in solid tumors: Translational implications. Mol. Ther. Oncolytics 2021, 22, 355–367. [Google Scholar] [CrossRef]
- Bernhard, H.; Neudorfer, J.; Gebhard, K.; Conrad, H.; Hermann, C.; Nährig, J.; Fend, F.; Weber, W.; Busch, D.H.; Peschel, C. Adoptive transfer of autologous, HER2-specific, cytotoxic T lymphocytes for the treatment of HER2-overexpressing breast cancer. Cancer Immunol. Immunother. 2008, 57, 271–280. [Google Scholar] [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Salmon, H.; Franciszkiewicz, K.; Damotte, D.; Dieu-Nosjean, M.C.; Validire, P.; Trautmann, A.; Mami-Chouaib, F.; Donnadieu, E. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J. Clin. Investig. 2012, 122, 899–910. [Google Scholar] [CrossRef]
- Piali, L.; Fichtel, A.; Terpe, H.J.; Imhof, B.A.; Gisler, R.H. Endothelial vascular cell adhesion molecule 1 expression is suppressed by melanoma and carcinoma. J. Exp. Med. 1995, 181, 811–816. [Google Scholar] [CrossRef]
- Griffioen, A.W.; Damen, C.A.; Blijham, G.H.; Groenewegen, G. Tumor angiogenesis is accompanied by a decreased inflammatory response of tumor-associated endothelium. Blood 1996, 88, 667–673. [Google Scholar] [CrossRef]
- Klein, D. The Tumor Vascular Endothelium as Decision Maker in Cancer Therapy. Front. Oncol. 2018, 8, 367. [Google Scholar] [CrossRef]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H.; He, Y.Z.; Lin, X.Y.; Ren, F.X.; Zhu, H.B.; Cheng, Y.; Nan, Z.; Liu, Z.B.; Yu, J.Y.; Guo, X.J. Regional Injection of CAR-T Cells for the Treatment of Refractory and Recurrent Diffuse Large B Cell Lymphoma: A Case Report. Front. Cell Dev. Biol. 2020, 8, 333. [Google Scholar] [CrossRef] [PubMed]
- Grosskopf, A.K.; Labanieh, L.; Klysz, D.D.; Roth, G.A.; Xu, P.; Adebowale, O.; Gale, E.C.; Jons, C.K.; Klich, J.H.; Yan, J.; et al. Delivery of CAR-T cells in a transient injectable stimulatory hydrogel niche improves treatment of solid tumors. Sci. Adv. 2022, 8, eabn8264. [Google Scholar] [CrossRef]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef]
- DePeaux, K.; Delgoffe, G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021, 21, 785–797. [Google Scholar] [CrossRef] [PubMed]
- Scharping, N.E.; Rivadeneira, D.B.; Menk, A.V.; Vignali, P.D.A.; Ford, B.R.; Rittenhouse, N.L.; Peralta, R.; Wang, Y.; DePeaux, K.; Poholek, A.C.; et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat. Immunol. 2021, 22, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-R.; Imrichova, H.; Wang, H.; Chao, T.; Xiao, Z.; Gao, M.; Rincon-Restrepo, M.; Franco, F.; Genolet, R.; Cheng, W.-C.; et al. Disturbed mitochondrial dynamics in CD8. Nat. Immunol. 2020, 21, 1540–1551. [Google Scholar] [CrossRef]
- Vardhana, S.A.; Hwee, M.A.; Berisa, M.; Wells, D.K.; Yost, K.E.; King, B.; Smith, M.; Herrera, P.S.; Chang, H.Y.; Satpathy, A.T.; et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020, 21, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Patsoukis, N.; Bardhan, K.; Chatterjee, P.; Sari, D.; Liu, B.; Bell, L.N.; Karoly, E.D.; Freeman, G.J.; Petkova, V.; Seth, P.; et al. PD-1 alters T-cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat. Commun. 2015, 6, 6692. [Google Scholar] [CrossRef]
- Ogando, J.; Sáez, M.E.; Santos, J.; Nuevo-Tapioles, C.; Gut, M.; Esteve-Codina, A.; Heath, S.; González-Pérez, A.; Cuezva, J.M.; Lacalle, R.A.; et al. PD-1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8. J. Immunother. Cancer 2019, 7, 151. [Google Scholar] [CrossRef]
- Tkachev, V.; Goodell, S.; Opipari, A.W.; Hao, L.Y.; Franchi, L.; Glick, G.D.; Ferrara, J.L.; Byersdorfer, C.A. Programmed death-1 controls T cell survival by regulating oxidative metabolism. J. Immunol. 2015, 194, 5789–5800. [Google Scholar] [CrossRef]
- Riley, J.L. PD-1 signaling in primary T cells. Immunol. Rev. 2009, 229, 114–125. [Google Scholar] [CrossRef]
- Chan, J.D.; Lai, J.; Slaney, C.Y.; Kallies, A.; Beavis, P.A.; Darcy, P.K. Cellular networks controlling T cell persistence in adoptive cell therapy. Nat. Rev. Immunol. 2021, 21, 769–784. [Google Scholar] [CrossRef]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef]
- Majzner, R.G.; Mackall, C.L. Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov. 2018, 8, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, J.; Ruella, M.; Houot, R. Born to survive: How cancer cells resist CAR T cell therapy. J. Hematol. Oncol. 2021, 14, 199. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Monti, S.; Rodig, S.J.; Juszczynski, P.; Currie, T.; O’Donnell, E.; Chapuy, B.; Takeyama, K.; Neuberg, D.; Golub, T.R.; et al. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood 2010, 116, 3268–3277. [Google Scholar] [CrossRef] [PubMed]
- Feucht, J.; Kayser, S.; Gorodezki, D.; Hamieh, M.; Döring, M.; Blaeschke, F.; Schlegel, P.; Bösmüller, H.; Quintanilla-Fend, L.; Ebinger, M.; et al. T-cell responses against CD19+ pediatric acute lymphoblastic leukemia mediated by bispecific T-cell engager (BiTE) are regulated contrarily by PD-L1 and CD80/CD86 on leukemic blasts. Oncotarget 2016, 7, 76902–76919. [Google Scholar] [CrossRef] [PubMed]
- Park, S.H.; You, E.; Park, C.J.; Cho, Y.-U.; Jang, S.; Im, H.-J.; Seo, J.-J.; Park, H.-S.; Lee, J.-H. Increased expression of immune checkpoint programmed cell death protein-1 (PD-1) on T cell subsets of bone marrow aspirates in patients with B-Lymphoblastic leukemia, especially in relapse and at diagnosis. Cytom. B Clin. Cytom. 2020, 98, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef]
- Jacobson, C.A.; Chavez, J.C.; Sehgal, A.R.; William, B.M.; Munoz, J.; Salles, G.; Munshi, P.N.; Casulo, C.; Maloney, D.G.; de Vos, S.; et al. Axicabtagene ciloleucel in relapsed or refractory indolent non-Hodgkin lymphoma (ZUMA-5): A single-arm, multicentre, phase 2 trial. Lancet Oncol. 2022, 23, 91–103. [Google Scholar] [CrossRef]
- Rafiq, S.; Yeku, O.O.; Jackson, H.J.; Purdon, T.J.; van Leeuwen, D.G.; Drakes, D.J.; Song, M.; Miele, M.M.; Li, Z.; Wang, P.; et al. Targeted delivery of a PD-1-blocking scFv by CAR-T cells enhances anti-tumor efficacy in vivo. Nat. Biotechnol. 2018, 36, 847–856. [Google Scholar] [CrossRef]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-mediated PD-1 disruption enhances anti-tumor efficacy of human chimeric antigen receptor T cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef]
- Stadtmauer, E.A.; Fraietta, J.A.; Davis, M.M.; Cohen, A.D.; Weber, K.L.; Lancaster, E.; Mangan, P.A.; Kulikovskaya, I.; Gupta, M.; Chen, F.; et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020, 367, eaba7365. [Google Scholar] [CrossRef]
- Ren, J.; Liu, X.; Fang, C.; Jiang, S.; June, C.H.; Zhao, Y. Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 2017, 23, 2255–2266. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Lei, W.; Zhang, C.; Yang, C.; Wei, J.; Guo, Q.; Guo, X.; Chen, Z.; Lu, Y.; Young, K.H.; et al. CD19-specific CAR T Cells that Express a PD-1/CD28 Chimeric Switch-Receptor are Effective in Patients with PD-L1-positive B-Cell Lymphoma. Clin. Cancer Res. 2021, 27, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Abbott, R.C.; Hughes-Parry, H.E.; Jenkins, M.R. To go or not to go? Biological logic gating engineered T cells. J. Immunother. Cancer 2022, 10, e004185. [Google Scholar] [CrossRef] [PubMed]
- Miao, L.; Zhang, J.; Huang, B.; Zhang, Z.; Wang, S.; Tang, F.; Teng, M.; Li, Y. Special Chimeric Antigen Receptor (CAR) Modifications of T Cells: A Review. Front. Oncol. 2022, 12, 832765. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Wang, Y.; Wei, J.; Han, W. Multi-antigen-targeted chimeric antigen receptor T cells for cancer therapy. J. Hematol. Oncol. 2019, 12, 128. [Google Scholar] [CrossRef]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Yeku, O.O.; Brentjens, R.J. Armored CAR T-cells: Utilizing cytokines and pro-inflammatory ligands to enhance CAR T-cell anti-tumour efficacy. Biochem. Soc. Trans. 2016, 44, 412–418. [Google Scholar] [CrossRef]
- Yeku, O.O.; Purdon, T.J.; Koneru, M.; Spriggs, D.; Brentjens, R.J. Armored CAR T cells enhance antitumor efficacy and overcome the tumor microenvironment. Sci. Rep. 2017, 7, 10541. [Google Scholar] [CrossRef]
- Hawkins, E.R.; D’Souza, R.R.; Klampatsa, A. Armored CAR T-Cells: The Next Chapter in T-Cell Cancer Immunotherapy. Biologics 2021, 15, 95–105. [Google Scholar] [CrossRef]
- Wang, S.; Yang, Y.; Ma, P.; Zha, Y.; Zhang, J.; Lei, A.; Li, N. CAR-macrophage: An extensive immune enhancer to fight cancer. EBioMedicine 2022, 76, 103873. [Google Scholar] [CrossRef]
- Morrissey, M.A.; Williamson, A.P.; Steinbach, A.M.; Roberts, E.W.; Kern, N.; Headley, M.B.; Vale, R.D. Chimeric antigen receptors that trigger phagocytosis. Elife 2018, 7, e36688. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Vadgama, J.V.; Wang, P. CCL2/CCR2 signaling in cancer pathogenesis. Cell Commun. Signal. 2020, 18, 82. [Google Scholar] [CrossRef] [PubMed]
- Klichinsky, M.; Ruella, M.; Shestova, O.; Lu, X.M.; Best, A.; Zeeman, M.; Schmierer, M.; Gabrusiewicz, K.; Anderson, N.R.; Petty, N.E.; et al. Human chimeric antigen receptor macrophages for cancer immunotherapy. Nat. Biotechnol. 2020, 38, 947–953. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L.; Lorenzini, M.H.; Chen, X.; Tran, U.; Bangayan, N.J.; Chen, Y.Y. Rewiring T-cell responses to soluble factors with chimeric antigen receptors. Nat. Chem. Biol. 2018, 14, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Chang, Z.L.; Hou, A.J.; Chen, Y.Y. Engineering primary T cells with chimeric antigen receptors for rewired responses to soluble ligands. Nat. Protoc. 2020, 15, 1507–1524. [Google Scholar] [CrossRef]
- Kasahara, Y.; Shin, C.; Kubo, N.; Mihara, K.; Iwabuchi, H.; Takachi, T.; Imamura, M.; Saitoh, A.; Imai, C. Development and characterisation of NKp44-based chimeric antigen receptors that confer T cells with NK cell-like specificity. Clin. Transl. Immunol. 2020, 9, e1147. [Google Scholar] [CrossRef]
- Parodi, M.; Favoreel, H.; Candiano, G.; Gaggero, S.; Sivori, S.; Mingari, M.C.; Moretta, L.; Vitale, M.; Cantoni, C. NKp44-NKp44 Ligand Interactions in the Regulation of Natural Killer Cells and Other Innate Lymphoid Cells in Humans. Front. Immunol. 2019, 10, 719. [Google Scholar] [CrossRef]
- Murayama, Y.; Kasahara, Y.; Kubo, N.; Shin, C.; Imamura, M.; Oike, N.; Ariizumi, T.; Saitoh, A.; Baba, M.; Miyazaki, T.; et al. NKp44-based chimeric antigen receptor effectively redirects primary T cells against synovial sarcoma. Transl. Oncol. 2022, 25, 101521. [Google Scholar] [CrossRef]
- Mardi, A.; Shirokova, A.V.; Mohammed, R.N.; Keshavarz, A.; Zekiy, A.O.; Thangavelu, L.; Mohamad, T.A.M.; Marofi, F.; Shomali, N.; Zamani, A.; et al. Biological causes of immunogenic cancer cell death (ICD) and anti-tumor therapy; Combination of Oncolytic virus-based immunotherapy and CAR T-cell therapy for ICD induction. Cancer Cell Int. 2022, 22, 168. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kohlhapp, F.J.; Zloza, A. Oncolytic viruses: A new class of immunotherapy drugs. Nat. Rev. Drug Discov. 2016, 15, 660. [Google Scholar] [CrossRef]
- Galluzzi, L.; Buqué, A.; Kepp, O.; Zitvogel, L.; Kroemer, G. Immunogenic cell death in cancer and infectious disease. Nat. Rev. Immunol. 2017, 17, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Harrington, K.; Freeman, D.J.; Kelly, B.; Harper, J.; Soria, J.C. Optimizing oncolytic virotherapy in cancer treatment. Nat. Rev. Drug Discov. 2019, 18, 689–706. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Zhao, Y.; Hu, X.; Jiao, J.; Wang, W.; Yao, H. Advances in the clinical development of oncolytic viruses. Am. J. Transl. Res. 2022, 14, 4192–4206. [Google Scholar] [PubMed]
- Garmaroudi, G.A.; Karimi, F.; Naeini, L.G.; Kokabian, P.; Givtaj, N. Therapeutic Efficacy of Oncolytic Viruses in Fighting Cancer: Recent Advances and Perspective. Oxidative Med. Cell. Longev. 2022, 2022, 3142306. [Google Scholar] [CrossRef] [PubMed]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients with Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef]
- Senzer, N.N.; Kaufman, H.L.; Amatruda, T.; Nemunaitis, M.; Reid, T.; Daniels, G.; Gonzalez, R.; Glaspy, J.; Whitman, E.; Harrington, K.; et al. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J. Clin. Oncol. 2009, 27, 5763–5771. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Bines, S.D. OPTIM trial: A Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010, 6, 941–949. [Google Scholar] [CrossRef]
- Kaufman, H.L.; Kim, D.W.; DeRaffele, G.; Mitcham, J.; Coffin, R.S.; Kim-Schulze, S. Local and distant immunity induced by intralesional vaccination with an oncolytic herpes virus encoding GM-CSF in patients with stage IIIc and IV melanoma. Ann. Surg. Oncol. 2010, 17, 718–730. [Google Scholar] [CrossRef]
- Wang, G.; Zhang, Z.; Zhong, K.; Wang, Z.; Yang, N.; Tang, X.; Li, H.; Lu, Q.; Wu, Z.; Yuan, B.; et al. CXCL11-armed oncolytic adenoviruses enhance CAR-T cells therapeutic efficacy and reprogram tumor microenvironment in glioblastoma. Mol. Ther. 2022, 31, 1. [Google Scholar] [CrossRef]
- Tang, N.; Cheng, C.; Zhang, X.; Qiao, M.; Li, N.; Mu, W.; Wei, X.F.; Han, W.; Wang, H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight 2020, 5, e133977. [Google Scholar] [CrossRef] [PubMed]
- Cattley, R.T.; Lee, M.; Boggess, W.C.; Hawse, W.F. Transforming growth factor β (TGF-β) receptor signaling regulates kinase networks and phosphatidylinositol metabolism during T-cell activation. J. Biol. Chem. 2020, 295, 8236–8251. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.; Ji, T.; Zhang, H.; Dong, W.; Chen, X.; Xu, P.; Chen, D.; Liang, X.; Yin, X.; Liu, Y.; et al. A Pck1-directed glycogen metabolic program regulates formation and maintenance of memory CD8. Nat. Cell Biol. 2018, 20, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Doedens, A.L.; Phan, A.T.; Stradner, M.H.; Fujimoto, J.K.; Nguyen, J.V.; Yang, E.; Johnson, R.S.; Goldrath, A.W. Hypoxia-inducible factors enhance the effector responses of CD8+ T cells to persistent antigen. Nat. Immunol. 2013, 14, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Jin, X.; Sun, R.; Xiong, X.; Wang, J.; Xie, D.; Zhao, M. Optimization of metabolism to improve efficacy during CAR-T cell manufacturing. J. Transl. Med. 2021, 19, 499. [Google Scholar] [CrossRef]
- Rad SM, A.H.; Halpin, J.C.; Mollaei, M.; Smith Bell, S.W.J.; Hirankarn, N.; McLellan, A.D. Metabolic and Mitochondrial Functioning in Chimeric Antigen Receptor (CAR)-T Cells. Cancers 2021, 13, 1229. [Google Scholar] [CrossRef]
- Nabe, S.; Yamada, T.; Suzuki, J.; Toriyama, K.; Yasuoka, T.; Kuwahara, M.; Shiraishi, A.; Takenaka, K.; Yasukawa, M.; Yamashita, M. Reinforce the antitumor activity of CD8. Cancer Sci. 2018, 109, 3737–3750. [Google Scholar] [CrossRef]
- Amini, A.; Veraitch, F. Glucose deprivation enriches for central memory T cells during chimeric antigen receptor-T cell expansion. Cytotherapy 2019, 21, S30–S31. [Google Scholar] [CrossRef]
- Saravia, J.; Raynor, J.L.; Chapman, N.M.; Lim, S.A.; Chi, H. Signaling networks in immunometabolism. Cell Res. 2020, 30, 328–342. [Google Scholar] [CrossRef]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 26. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; O’Hear, C.E.; Alli, R.; Basham, J.H.; Abdelsamed, H.A.; Palmer, L.E.; Jones, L.L.; Youngblood, B.; Geiger, T.L. PI3K orchestration of the in vivo persistence of chimeric antigen receptor-modified T cells. Leukemia 2018, 32, 1157–1167. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cytokine/Chemokine | Secreted by | Function |
|---|---|---|
| IL-1β | MDSCs TAMs | Mediates the inflammatory response through inflammasomes Induces cancer cell proliferation |
| IL-2 | T cells NK cells | Promotes T-cell growth Influences Treg differentiation Promotes production of TNF-α and INF-γ |
| IL-4 | CD4+ T cells B cells | Influences M2 macrophage polarization Creates a positive feedback loop in CD4+ T cells to produce more IL-4 |
| IL-6 | TAMs MDSCs | Promotes inflammation Can lead to accelerated tumor growth by promoting self-renewal in tumor cells |
| IL-10 | TAMs MDSCs Treg cells CD4+ T cells B cells | Influences M2 macrophage polarization Suppresses dendritic cells Negatively regulates the inflammatory response Stimulates T reg cells |
| IL-12 | TAMs Dendritic cells | Promotes differentiation of T cells Enhances production of INF-γ Influences M1 macrophage polarization |
| IL-13 | CD4+ T cells | Influences M2 macrophage polarization |
| TNF-α | TAMs T cells NK cells | Influences signaling pathways to promote apoptosis or necrosis Promotes cancer cell proliferation |
| TGF-β | TAMs MDSCs Treg cells CAFs | Dual role in tumor suppression and promotion Inhibits CD8+ T-cell cytotoxic function Reduces MHC presentation in tumor cells Mediates tumor migration and immune cell infiltration |
| IFN-γ | T cells B cells NK cells Dendritic cells | Dual role in tumor suppression and promotion Activates macrophages Promotes apoptosis Inhibits angiogenesis in the TME |
| Immunosuppressive TME Element | Effect on CAR T Cells | Methods of Overcoming the Limitation |
|---|---|---|
| Insufficient vasculature and “shell” surrounding tumors | CAR T-cell homing and tumor infiltration failure | Injection of CAR T cells into tumors, rather than intravenous delivery CAR macrophages |
| Immunosuppressive cytokine profiles | CAR T-cell exhaustion and anergy | Cytokine secreting CAR T cells and soluble ligand binding T cells |
| Tumor expression of PDL-1 | CAR T-cell exhaustion and death | Checkpoint inhibitor co-treatment and CAR T cells modified to block or alter PD-1/PDL-1 response |
| High levels of tumor antigen | CAR T-cell exhaustion and anergy | Use of less differentiated T cells in CAR T cells production |
| Mutation to alter surface expression of neoantigens | Antigen escape | Bispecific and altered binding domain CAR T cells |
| Hostile metabolic environment | CAR T-cell exhaustion and decreased cytotoxicity | Genetic modification to CAR T-cell’s metabolic processes Expansion phase media modifications (nutrient-limited media, inclusion of metabolic inhibitors) |
| High levels of immunosuppressive cell populations (Tregs, MDSCs, TAMs) | CAR T-cell anergy | Oncolytic virus co-treatment in conjunction with CAR T cells CAR macrophages |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cheever, A.; Townsend, M.; O’Neill, K. Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells 2022, 11, 3626. https://doi.org/10.3390/cells11223626
Cheever A, Townsend M, O’Neill K. Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells. 2022; 11(22):3626. https://doi.org/10.3390/cells11223626
Chicago/Turabian StyleCheever, Abigail, Michelle Townsend, and Kim O’Neill. 2022. "Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors" Cells 11, no. 22: 3626. https://doi.org/10.3390/cells11223626
APA StyleCheever, A., Townsend, M., & O’Neill, K. (2022). Tumor Microenvironment Immunosuppression: A Roadblock to CAR T-Cell Advancement in Solid Tumors. Cells, 11(22), 3626. https://doi.org/10.3390/cells11223626